

Abstract
Voltage-gated channels are normally opened by depolarization and closed by repolarization of the membrane. Despite sharing significant sequence homology with voltage-gated K+ channels, the gating of hyperpolarization-activated, cyclic-nucleotide-gated (HCN) pacemaker channels has the opposite dependence on membrane potential: hyperpolarization opens, whereas depolarization closes, these channels. The mechanism and structural basis of the process that couples voltage sensor movement to HCN channel opening and closing is not understood. On the basis of our previous studies of a mutant HERG (human ether-a-go-go-related gene) channel, we hypothesized that the intracellular linker that connects the fourth and fifth transmembrane domains (S4–S5 linker) of HCN channels might be important for channel gating. Here, we used alanine-scanning mutagenesis of the HCN2 S4–S5 linker to identify three residues, E324, Y331, and R339, that when mutated disrupted normal channel closing. Mutation of a basic residue in the S4 domain (R318Q) prevented channel opening, presumably by disrupting S4 movement. However, channels with R318Q and Y331S mutations were constitutively open, suggesting that these channels can open without a functioning S4 domain. We conclude that the S4–S5 linker mediates coupling between voltage sensing and HCN channel activation. Our findings also suggest that opening of HCN and related channels corresponds to activation of a gate located near the inner pore, rather than recovery of channels from a C-type inactivated state.
Keywords: ion channels, mutagenesis, Ih, Xenopus
The slow depolarization of specialized pacemaker cells is mediated in part by a hyperpolarization-activated, monovalent cation-selective inward current called If in the heart (1, 2) and Ih in neurons (3). The molecular basis of the pacemaker current was discovered in 1998. Four different genes were cloned that encode mammalian pacemaker channels (4–6), now most commonly referred to as hyperpolarization-activated, cyclic-nucleotide-gated (HCN) channels (7). These currents contribute to changes in heart rate in response to neural input, generate oscillatory activity in neural networks, and contribute to the facilitation of repetitive neural activity (8).
The overall structure of HCN channels is very similar to that of voltage-gated K+ (Kv) channels (see Fig. 1A), yet HCN channels open in response to membrane hyperpolarization rather than depolarization, resulting in strong inward rectification of the current–voltage relationship. Two competing models have been proposed to account for this unique gating behavior based on studies of SPIH (6), KAT1 (9, 10), and a mutant Shaker K+ channel (11). One model proposes that at normal cellular resting membrane potentials, HCN channels are in an inactivated state and that membrane hyperpolarization allows channels to recover from inactivation and enter an open state (6, 11), causing inward rectification in a manner similar to that observed for human ether-a-go-go-related gene (HERG) K+ channels (12, 13). A second model proposes that the gating of HCN channels is the inverse of Kv channel gating. In other words, channels are normally in a closed state and hyperpolarization results in channel activation.
Figure 1.

Alanine-scanning mutagenesis of the S4–S5 linker of ntHCN2 channel. (A) Schematic of a single HCN2 subunit showing location and amino acid sequence of the S4–S5 linker. (B) Representative whole-cell recordings of WT, F327A, and W323A ntHCN2 channel currents elicited with 3-s pulses, applied from a holding potential of −30 mV in 10-mV increments to potentials ranging from −140 to −30 mV. The tail currents were measured at −130 mV. Arrow indicates zero current level. (C) Voltage dependence of ntHCN2 channel activation. Currents recorded at −130 mV immediately after each test pulse were normalized to the largest current, then plotted as a function of test potential. The resulting data were fitted to a Boltzmann function to obtain the V1/2 and slope factor (k) for the relationship (see Table 1 for values). (D) Bar graph of min-Po for all S4–S5 linker Ala mutant channels. The number of oocytes is indicated above each bar.
Channels with gating characteristics similar to HCN have been cloned from sea urchin sperm (SPIH) (6) and plants (KAT1) (14). Several studies of KAT1 have provided indirect evidence that favors the second model for hyperpolarization-dependent opening of these channels. KAT1 appears to undergo inactivation at very negative membrane potentials (15). Moreover, gating of KAT1 is not dependent on factors that normally alter C-type inactivation in Kv channels (9), and activation appears to be coupled to movement of S4 upon membrane hyperpolarization (10). Thus, whereas most indirect evidence suggests that KAT1 channels activate at negative membrane potentials, the mechanism of gating of other hyperpolarization-activated cation channels such as SPIH (6, †) and HCN remains controversial and its structural basis is unknown.
Extensive structure–function studies have established that the positively charged S4 domain is the major voltage sensor of voltage-gated channels (16, 17). From site-directed mutagenesis studies, it is clear that the S4 domain of HCN channels has a similar role (18, 19). Although outward rotation or translocation of S4 clearly precedes opening of Shaker K+ channels (20, 21), a recent study of KAT1‡ suggests that channel opening is associated with inward movement of a voltage-sensing domain. The structure that links movement of S4 to opening of the activation gate is unknown and has been the subject of much speculation. On the basis of its location at the intracellular end of the S4 domain, the S4–S5 linker is a potential candidate for the structural link between the voltage sensor and the activation gate in Kv and HCN channels. Mutations in the S4–S5 linker of Shaker (22), Kv2 and Kv3 (23), and HERG channels (24) alter the voltage dependence and kinetics of activation gating.
A clue to a potential mechanism for the “inverted” gating scheme of HCN channels was revealed in our previous study of mutant HERG K+ channels (24). Mutation of basic residues in the S4–S5 linker of HERG greatly accelerated the rate of channel deactivation, and, depending upon the specific mutation, either increased or decreased the rate of activation. In addition, one mutation (D540K) disrupted channel closure and caused the channel to open slowly in response to hyperpolarization, similar to activation of HCN channel current. Hyperpolarization-dependent gating of D540K HERG channels was steeply voltage-dependent and saturated at −160 mV (24), suggesting that inward movement of the S4 domain mediated the hyperpolarization-induced channel opening. We hypothesized that, similar to D540K HERG, the S4–S5 linker might be a crucial link in the hyperpolarization-dependent gating pathway of HCN channels. We used an alanine-scanning mutagenesis approach to identify residues in the S4–S5 linker of HCN2 (Fig. 1A) that when mutated, interfered with normal channel closure. Some S4–S5 linker mutations nearly abolished voltage-dependent gating as though the link between voltage sensing and channel activation was severed. We conclude that the S4–S5 linker physically couples the movement of the S4 domain to channel opening and closing, and that hyperpolarization-dependent opening of HCN channels corresponds to opening of an activation gate and not to recovery from an inactivated state.
Materials and Methods
HCN2 channel cDNA was cloned from Marathon-Ready (CLONTECH) mouse brain cDNA into the pSP64T oocyte expression vector (19). To facilitate subcloning, the very G+C-rich N-terminal portion of the HCN2 channel (encoding amino acids 2–130) was deleted. The wild-type N-terminal-truncated channel is referred to as WT ntHCN2. The biophysical properties of ntHCN2 channels were similar to full-length HCN2 channels, except that the voltage dependence of activation was shifted by −12 mV (19). All mutations were introduced into ntHCN2 cDNA as described previously (25). Restriction mapping and DNA sequencing of the PCR-amplified segment were used to confirm the presence of the desired mutation and the lack of extra mutations. Complementary RNA (cRNA) for injection into oocytes was prepared with SP6 Capscribe (Roche) after linearization with EcoRI. RNA concentration was quantified by UV spectroscopy and gel electrophoresis.
Stage IV and V Xenopus laevis oocytes were isolated and injected (26, 27) with 30 ng of cRNA encoding WT or mutant ntHCN2 channels. The oocytes were cultured in Barth's solution supplemented with 50 μg/ml gentamycin and 1 mM pyruvate at 18°C for 1–3 days before use in voltage-clamp experiments.
For voltage-clamp experiments, oocytes were bathed in a modified ND96 solution containing (in mM): 96 NaCl, 4 KCl, 1 MgC12, 1 CaC12, 5 Hepes; pH 7.6. Currents were recorded at room temperature (23–25°C) by using standard two-microelectrode voltage-clamp techniques (28). The voltage dependence of ntHCN2 channel activation was determined from a holding potential of −30 or 0 mV. Instantaneous tail currents measured at −110 or −130 mV were corrected for leak by subtracting the average value of leak (less than −150 nA at −130 mV) recorded from a matched set of uninjected oocytes for each experiment. Normalized tail current amplitude (It) was plotted vs. test potential and fitted with a Boltzmann function,
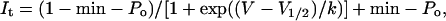 |
to obtain the voltage required for half-maximal activation (V1/2) and the slope factor (k), a measure of the voltage dependence of channel gating. This equation was also used to obtain the minimum open probability (min-Po, defined as the minimum value of relative tail current). Data are expressed as mean ± SEM (n = number of oocytes).
Results
Alanine-Scanning Mutagenesis of the S4–S5 Linker.
HCN channel function was assayed by recording properties of current induced by 3-s hyperpolarizing pulses from a holding potential of −30 mV, a potential where WT ntHCN2 channels were nearly completely closed. Representative currents for an oocyte expressing WT ntHCN2 channels are shown in Fig. 1B. WT ntHCN2 channels were activated, after a short delay, by hyperpolarization to potentials more negative than −60 mV. The activation of WT ntHCN2 channels after the delay phase was monoexponential, having a time constant of 342 ± 24 ms (n = 18) at −120 mV. Current reached a steady-state level in less than 1 s at a test potential of −140 mV. The voltage dependence of channel activation was assessed by plotting the instantaneous current measured at −130 mV immediately after each test pulse. The half-point (V1/2) and slope factor for this relationship were, respectively, −84 ± 0.2 mV and 7.7 ± 0.1 mV (n = 8; Fig. 1C, filled squares). Note that the minimum normalized tail current (min-Po) was greater than zero, indicating that a small proportion of HCN2 channels remained open at these potentials. For WT ntHCN2 channels, min-Po was 0.08 ± 0.002 (n = 8).
The amino acid sequences of HCN channels are most similar to the eag/erg family of K+ channels. On the basis of our observation that HERG channels containing a single mutation of the S4–S5 linker (D540K) disrupt the closed state and allow reopening in response to hyperpolarization (24), we hypothesized that mutations in the S4–S5 linker of HCN2 channels might also alter channel gating. To test this hypothesis, we individually mutated to Ala 17 of the 19 residues (Q322–I340) located in the ntHCN2 S4–S5 linker. The two Ala residues (A334, A336) located in this region were not mutated. In most cases, mutation of a single S4–S5 linker amino acid to Ala affected the voltage dependence or kinetics of ntHCN2 current activation (e.g., F327A, W323A, Fig. 1 B and C). The effect of individual mutations on the voltage dependence of activation and the time constant for current activation at −120 mV is summarized in Table 1. Mutation of three different residues, E324, Y331, and R339, to Ala caused an increase in min-Po (Fig. 1D). The min-Po was 0.31 ± 0.003 for E324A (n = 11), 0.58 ± 0.005 for Y331A (n = 9), and 0.34 ± 0.003 for R339A (n = 11) ntHCN2 channels. The effects of point mutations in these three residues were studied in greater detail.
Table 1.
Voltage dependence and kinetics of current activation for HCN2 channels containing Ala point mutations in the S4–S5 linker
| Channel | V1/2, mV | k, mV | n | τact, ms | n |
|---|---|---|---|---|---|
| ntHCN2 | −84.2 ± 0.2 | 7.7 ± 0.1 | 8 | 342 ± 24 | 18 |
| Q322A | −81.8 ± 0.7 | 8.3 ± 0.1 | 9 | 489 ± 27 | 9 |
| W323A | −68.1 ± 1.2 | 6.4 ± 0.3 | 8 | 180 ± 19 | 8 |
| E324A | −55.0 ± 0.9 | 8.4 ± 0.4 | 10 | 172 ± 7 | 8 |
| E325A | −82.2 ± 1.0 | 7.7 ± 0.3 | 8 | 311 ± 19 | 8 |
| I326A | −80.8 ± 0.5 | 8.5 ± 0.3 | 10 | 365 ± 14 | 8 |
| F327A | −98.8 ± 0.8 | 8.2 ± 0.3 | 7 | 721 ± 54 | 7 |
| H328A | −109 ± 0.3 | 7.1 ± 0.7 | 5 | 826 ± 80 | 5 |
| M329A | −80.1 ± 1.2 | 8.4 ± 0.5 | 4 | 185 ± 16 | 6 |
| T330A | −83.7 ± 0.2 | 7.5 ± 0.2 | 10 | 313 ± 13 | 10 |
| Y331A | −62.9 ± 1.2 | 13.4 ± 1.0 | 9 | 203 ± 10 | 6 |
| D332A | −72.3 ± 0.6 | 7.0 ± 0.2 | 12 | 160 ± 5 | 8 |
| L333A | −73.8 ± 0.9 | 7.1 ± 0.8 | 8 | 223 ± 9 | 9 |
| S335A | −81.6 ± 0.4 | 7.5 ± 0.3 | 5 | 275 ± 78 | 6 |
| V337A | −90.0 ± 0.8 | 8.0 ± 0.4 | 4 | 454 ± 11 | 6 |
| M338A | −87.8 ± 1.1 | 8.9 ± 0.3 | 6 | 517 ± 22 | 8 |
| R339A | −72.5 ± 1.3 | 8.6 ± 0.4 | 11 | 248 ± 13 | 10 |
| I340A | −96.6 ± 1.1 | 7.3 ± 0.5 | 5 | 535 ± 16 | 11 |
V1/2, potential of half-maximal current activation; k, slope factor of the activation curve; τact, time constant for current activation at −120 mV; n, number of oocytes.
Aromatic Residue at Position 331 Is Required for Normal Channel Closure.
Y331A ntHCN2 channel currents were recorded at test voltages ranging from +50 mV to −120 mV from a holding potential of 0 mV. Tail currents were measured at −110 mV. Step changes in membrane potential elicited large instantaneous currents, and at potentials negative to about −40 mV, a further increase of time-dependent inward current (Fig. 2A). Unlike WT channels, Y331A ntHCN2 channels conducted significant outward currents at potentials positive to the reversal potential of −30 mV. The relatively large instantaneous component of current indicated that these channels failed to close completely even at potentials as positive as +50 mV. Additional mutations of Y331 were investigated to determine whether other amino acid substitutions also disrupted channel closure. The instantaneous currents conducted by Y331S, Y331D (Fig. 2A), and Y331K (not shown) ntHCN2 channels were even greater than observed for Y331A channels. The V1/2 for voltage-dependent activation of Y331A and Y331S channels was shifted to more positive potentials and the slope factor increased compared with WT ntHCN2 channels (Tables 1 and 2), but these changes could not account for the dramatic increase in min-Po (Fig. 2B). In contrast, Y331F channels opened and closed relatively normally (Fig. 2A), having a min-Po of 0.09 ± 0.001 (n = 7). Thus, Phe, but not Ser, could adequately replace Tyr at position 331. This finding indicates that an aromatic residue, and not a hydroxyl group, is important at position 331 for normal gating. In summary, an aromatic residue at position 331 in the S4–S5 linker of HCN2 channels was required for normal channel gating. On the basis of its position in the middle of the S4–S5 linker, it is unlikely that mutations of Y331 would directly affect the movement of the S4 domain. However, Y331 might be part of a structural linker that normally couples the movement of the S4 to opening of the activation gate.
Figure 2.

Mutation of Y331 disrupts ntHCN2 channel closure. (A) Representative current traces for Y331A, Y331S, Y331D, and Y331F ntHCN2 channels. Holding potential was 0 mV and test potentials were −120 to +40 mV for Y331A, Y331S, and Y331D channels. Holding potential was −30 mV and test potentials were −140 to −30 mV for Y331F channels. Arrow indicates zero current level. (B) Voltage dependence for activation of WT and Y331 mutant ntHCN2 channels. V1/2 and k for each relationship are shown in Table 2.
Table 2.
Voltage dependence of activation for Y331, R339, and E324 mutant ntHCN2 channels
| Mutant | V1/2, mV | k, mV | n |
|---|---|---|---|
| Y331S | −76.0 ± 1.0 | 10.5 ± 1.0 | 6 |
| Y331D | −80.7 ± 4.0 | 7.7 ± 4.1 | 22 |
| Y331K | −75.4 ± 1.3 | 7.7 ± 0.8 | 11 |
| Y331F | −99.8 ± 0.8 | 5.7 ± 0.1 | 7 |
| R339E | −56.6 ± 0.9 | 12.8 ± 0.8 | 12 |
| R339Q | −64.4 ± 1.1 | 12.8 ± 0.8 | 22 |
| R339C | −62.4 ± 1.5 | 12.4 ± 0.6 | 13 |
| R339D | −74.8 ± 0.5 | 7.5 ± 0.4 | 8 |
| E324Q | −75.4 ± 1.3 | 7.7 ± 0.4 | 6 |
| E324K | −78.7 ± 0.6 | 7.4 ± 0.2 | 13 |
Mutations of R339 and E324 Also Disrupt HCN2 Channel Closure.
The R339A mutation caused an increase in min-Po. If an electrostatic interaction between R339 and another residue near the gate of HCN2 were important for channel closure, then mutation of R339 to a nonbasic residue would also be predicted to disrupt channel closure. As expected, R339Q and R339C ntHCN2 channels could not close properly (Fig. 3A). We also expected that charge reversal of R339 would cause disruption of gating. While this was true for R339E, it was not the case for R339D (Fig. 3A). The min-Po for R339E was 0.65 ± 0.002 (n = 12). In contrast, the min-Po for R339D was 0.17 ± 0.004 (n = 8), only about twice as large as WT ntHCN2 channels. This was an unexpected result because Asp and Glu differ by only a single methylene group.
Figure 3.

Effects of R339 mutations on ntHCN2 channel gating. (A) Representative current traces for R339A, R339Q, R339C, R339E, and R339D ntHCN2 channels. Holding potential was −30 mV and test potentials ranged from −140 to −30 mV for R339A and R339D. Holding potential was 0 mV and test potentials ranged from −120 to +40 mV for R339Q, R339C, and R339E ntHCN2 channels. Arrow indicates zero current level. (B) Voltage dependence for activation of WT and R339 mutant ntHCN2 channels. V1/2 and k for each relationship are shown in Table 2.
Because the min-Po of E324A ntHCN2 channels was increased and had a V1/2 for activation shifted by +30 mV, we explored the effects of other mutations of this residue (Fig. 4). The most conservative mutation, E324D, resulted in a nonfunctional channel as judged by lack of measurable currents. Replacement of Glu with Gln, a polar residue with a similar-sized side group, resulted in a channel with relatively normal gating (min-Po = 0.11 ± 0.002; V1/2 = −75 ± 1.3 mV; n = 6). Compared with E324A, a charge-reversing mutation (E324K) had a similar V1/2 of −79 ± 0.6 mV but a reduced min-Po of 0.25 ± 0.002 (n = 13). Compared with Y331 or R339, mutation of E324 had less effect on min-Po, indicating that E324 is less important than these other residues for normal gating of HCN2 channels.
Figure 4.

Effects of E324 mutations on ntHCN2 channel gating. (A) Representative current traces for E324A, E324Q, and E324K ntHCN2 channels. The holding potential was 0 mV and test potentials were from −140 to 0 mV for E324Q and −120 to 0 mV for E324A and E324K. Arrow indicates zero current level. (B) Voltage dependence for activation of WT and E324 mutant ntHCN2 channels. V1/2 and k for each relationship are shown in Table 2.
R318Q/Y331S ntHCN2 Channels Are Constitutively Open.
We hypothesized that elimination of the aromatic residue at position 331 in the S4–S5 linker disrupted channel closure by interfering with the normal coupling between voltage-dependent S4 movement and closure of the activation gate. The large instantaneous and small time-dependent component of current for Y331S, Y331A, Y331D, and Y331K ntHCN2 channels suggested that channels remained largely open at 0 mV. In a previous study, we found that channels having a mutation of a basic residue (R318Q) in the S4 domain of ntHCN2 failed to open in response to membrane depolarization, yet mutant protein was still expressed at the surface membrane about half as effectively as WT HCN2 protein (19). If Y331 mutations disrupt channel closure by interfering with the normal coupling between voltage-dependent S4 movement and closure of the activation gate, then we predicted that the second mutation Y331S could rescue the function of R318Q ntHCN2 channels. As we reported previously, R318 ntHCN2 channels do not express detectable currents (Fig. 5A). Addition of a second mutation (R318Q/Y331S) resulted in functional expression. However, unlike Y331S ntHCN2 channel current, steps in membrane potential elicited instantaneous currents without a time-independent component (Fig. 5B). Although R318Q/Y331S ntHCN2 channels appeared to be constitutively open, the I–V relationship still exhibited rectification (Fig. 5C). Rectification of whole-cell currents could result from either an intrinsic decrease in single channel conductance (29) or pore block by intracellular cations as described for Kir inward rectifier K+ channels (30, 31). These findings suggest that mutation of Y331 to a nonaromatic residue can lock most channels in an open state that circumvents the requirement for voltage-dependent movement of the S4 domain.
Figure 5.

R318Q/Y331S ntHCN2 channels are constitutively open. (A) R318Q ntHCN2 channel currents were undetectable. (B) R318Q/Y331S ntHCN2 channel currents activate instantaneously and have no time-dependent component, unlike Y331S channel currents (Fig. 2A). Currents were elicited from a holding potential of −30 mV with 1-s pulses to potentials of −140 to +20 mV, applied in 10-mV increments. (C) Current–voltage relationships for R318Q and R318Q/Y331S HCN2 channel currents.
Discussion
We found that mutation of several specific residues located in the S4–S5 linker of ntHCN2 channels disrupted channel closure. At least three mechanisms could explain mutation-induced disruption of channel closure. First, the voltage sensor could be trapped in a position that favors the open state. Second, mutation of residues that form the activation gate could trap or stabilize the channel in the open state (32, §), despite normal voltage-dependent translocation of the voltage sensor. Third, the mechanism that couples voltage sensing to channel opening could be disrupted in a manner that favors the open state. We found that mutation of only three residues (E324, Y331, R339) of the S4–S5 linker affected min-Po. On the basis of the location of these residues, it is unlikely that their mutation would have any direct affect on movement of the S4 domain, or configuration of the activation gate, a structure believed to be formed by the S6 domains (33, 34). Mutation of specific residues in the S4–S5 linker apparently bias channels toward an open state even in the absence of voltage sensor movement. Thus, we favor the third mechanism, implying that the S4–S5 linker constitutes a link between the voltage sensor movement and the opening and closing of the activation gate as previously proposed for Shaker channels (34, 35).
Most mutations of R339 disrupted channel closure and increased min-Po, including replacement with a small hydrophobic residue (Ala), a polar residue (Gln), a cysteine, or an acidic residue (Glu). However, replacement with the other basic amino acid (Asp) only slightly increased min-Po. Clearly, R339 is a crucial residue, but simple features such as the charge or bulk of its side chain cannot account for its role in gating. Mutation of Y331 to nonaromatic residues (Ala, Ser, Lys, or Asp) disrupted channel closure. Only replacement of Tyr with another aromatic residue, Phe, resulted in channels with normal gating behavior. In addition, mutation of single amino acids near this Tyr residue had no significant affect on gating (e.g., T330A, D332A). The requirement for an aromatic residue at position 331 for normal channel function suggests the possibility that Y331 may interact by π-orbital stacking with another aromatic residue, perhaps one located within the activation gate.
If the S4–S5 linker constitutes the link between voltage sensor movement and HCN2 channel opening and closing, then it should be possible to disconnect channel opening from voltage sensing. We tested this hypothesis by comparing the function of channels containing a single S4 domain mutation (R318Q) or a double mutation (R318Q/Y331S). In our previous study of the S4 domain of HCN2 channels (19), we found that the R318Q mutation abolished function, but did not prevent trafficking and insertion of the channels into the plasma membrane, suggesting that loss of function was caused by a failure of the S4 domain to move properly in response to a change in transmembrane potential. Introduction of a second mutation (Y331S) into R318Q HCN2 rescued function, but also changed the gating in an important way. R318Q/Y331S channels were always open, having no time-dependent component of current (i.e., min-Po = 1). By contrast, Y331S ntHCN2 channel current exhibited a time-dependent component of current, albeit reduced compared with wild-type current. The lack of time-dependent gating of R318Q/Y331S channels suggests a disconnection between the normal requirement for voltage sensing and channel activation; however, we cannot rule out the possibility that addition of the Y331S mutation to R318Q HCN2 locks the S4 domain in the activated position.
Taken together, our findings suggest that the S4–S5 linker couples S4 movement and channel opening. This model implies that hyperpolarization-dependent opening of HCN pacemaker channels is due to an activation process located near the inner pore region. A similar conclusion was reached for other hyperpolarization-activated cation channels. Block of HCN1 and SPIH channels by the drug ZD7288 also supports the idea that the activation gate for these channels is located near the intracellular side of the pore (36). The binding site for this drug is composed of residues on the S6 domain that line the inner pore. Analysis of a mutant channel revealed that access to this site and subsequent trapping inside the pore was modulated by activation, consistent with the idea that trapping occurred when the activation gate closed. Finally, modification by intracellular Cd2+ of Cys residues introduced near the intracellular end of the SPIH channel pore was also shown to be gated by activation.† These findings support the view that opening of HCN and related channels corresponds to activation of a gate located near the inner pore, rather than recovery of channels from a C-type inactivated state.
Our findings suggest a possible model for voltage-dependent gating of the HCN2 channel. At potentials near 0 mV, the S4–S5 linker stabilizes the activation gate in a closed conformation. Membrane hyperpolarization causes inward rotation or translocation of the S4 domain, twisting of the attached S4–S5 linker, and a destabilization of this interaction that in turn permits the S6 domains to twist apart and increase the aperture of the inner pore region (“opening” of the activation gate). Mutation of a critical residue in the S4–S5 linker (i.e., Y331, R339, or, to a lesser extent, E324) disrupts the stabilizing interaction between the two domains and energetically favors the open state of the channel.
Acknowledgments
We thank Peter Westinskow for technical assistance. This work was supported by National Institutes of Health Grant HL65299.
Abbreviations
- HCN
hyperpolarization-activated, cyclic-nucleotide-gated
- ntHCN2
N-terminal truncated HCN2 channel
- HERG
human ether-a-go-go-related gene
- Kv channel
voltage-gated potassium channel
- min-Po
minimum open probability
- WT
wild type
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Rothberg, B. S., Phale, P., Shin, K.-S. & Yellen, G. (2001) Biophys. J. 80, 16a (abstr.).
Latorre, R., Basso, C., Gonzalez, C., Alvarez, O. & Cosmelli, D. (2001) Biophys. J. 80, 436a (abstr.).
Hackos, D. H. & Swartz, K. J. (2000) Biophys. J. 78, 398A (abstr.).
References
- 1.DiFrancesco D, Noma A, Trautwein W. Pflügers Arch. 1979;381:271–279. doi: 10.1007/BF00583259. [DOI] [PubMed] [Google Scholar]
- 2.DiFrancesco D. Annu Rev Physiol. 1993;55:455–472. doi: 10.1146/annurev.ph.55.030193.002323. [DOI] [PubMed] [Google Scholar]
- 3.Pape H-C. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- 4.Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. Nature (London) 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- 5.Santoro B, Liu D T, Yao H, Bartsch D, Kandel E R, Siegelbaum S A, Tibbs G R. Cell. 1998;93:717–729. doi: 10.1016/s0092-8674(00)81434-8. [DOI] [PubMed] [Google Scholar]
- 6.Gauss R, Seifert R, Kaupp U B. Nature (London) 1998;393:583–591. doi: 10.1038/31248. [DOI] [PubMed] [Google Scholar]
- 7.Clapham D E. Neuron. 1998;21:5–7. doi: 10.1016/s0896-6273(00)80508-5. [DOI] [PubMed] [Google Scholar]
- 8.Santoro B, Tibbs G R. Ann NY Acad Sci. 1999;868:741–764. doi: 10.1111/j.1749-6632.1999.tb11353.x. [DOI] [PubMed] [Google Scholar]
- 9.Marten I, Hoshi T. Biophys J. 1998;74:2953–2962. doi: 10.1016/S0006-3495(98)78002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zei P C, Aldrich R W. J Gen Physiol. 1998;112:679–713. doi: 10.1085/jgp.112.6.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller A G, Aldrich R W. Neuron. 1996;16:853–858. doi: 10.1016/s0896-6273(00)80105-1. [DOI] [PubMed] [Google Scholar]
- 12.Sanguinetti M C, Jiang C, Curran M E, Keating M T. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 13.Trudeau M, Warmke J W, Ganetzky B, Robertson G A. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- 14.Anderson J A, Huprikar S S, Kochian L V, Lucas W J, Gaber R F. Proc Natl Acad Sci USA. 1992;89:3736–3740. doi: 10.1073/pnas.89.9.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moroni A, Gazzarrini S, Cerana R, Colombo R, Sutter J U, DiFrancesco D, Gradmann D, Thiel G. Biophys J. 2000;78:1862–1871. doi: 10.1016/S0006-3495(00)76735-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sigworth F J. Q Rev Biophys. 1993;27:1–40. doi: 10.1017/s0033583500002894. [DOI] [PubMed] [Google Scholar]
- 17.Bezanilla F. Physiol Rev. 2000;80:555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- 18.Vaca L, Stieber J, Zong X, Ludwig A, Hofmann F, Biel M. FEBS Lett. 2000;479:35–40. doi: 10.1016/s0014-5793(00)01837-8. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Mitcheson J S, Lin M, Sanguinetti M C. J Biol Chem. 2000;275:36465–36471. doi: 10.1074/jbc.M007034200. [DOI] [PubMed] [Google Scholar]
- 20.Cha A, Snyder G E, Selvin P R, Bezanilla F. Nature (London) 1999;402:809–813. doi: 10.1038/45552. [DOI] [PubMed] [Google Scholar]
- 21.Glauner K S, Mannuzza L M, Gandhi C S, Isacoff E Y. Nature (London) 1999;402:813–818. doi: 10.1038/45561. [DOI] [PubMed] [Google Scholar]
- 22.McCormack K, Tanouye M A, Iverson L E, Lin J W, Ramaswami M, McCormack T, Campanelli J T, Mathew M K, Rudy B. Proc Natl Acad Sci USA. 1991;88:2931–2935. doi: 10.1073/pnas.88.7.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shieh C-C, Klemic K G, Kirsch G E. J Gen Physiol. 1997;109:767–778. doi: 10.1085/jgp.109.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanguinetti M C, Xu Q P. J Physiol. 1999;514:667–675. doi: 10.1111/j.1469-7793.1999.667ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarkar G, Sommer S S. BioTechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- 26.Goldin A L. Methods Cell Biol. 1991;36:487–509. doi: 10.1016/s0091-679x(08)60293-9. [DOI] [PubMed] [Google Scholar]
- 27.Goldin A L, Sumikawa K. Methods Enzymol. 1992;207:279–296. doi: 10.1016/0076-6879(92)07018-j. [DOI] [PubMed] [Google Scholar]
- 28.Stuhmer W. Methods Enzymol. 1992;207:319–339. doi: 10.1016/0076-6879(92)07021-f. [DOI] [PubMed] [Google Scholar]
- 29.Zou A, Curran M E, Keating M T, Sanguinetti M C. Am J Physiol. 1997;272:H1309–H1314. doi: 10.1152/ajpheart.1997.272.3.H1309. [DOI] [PubMed] [Google Scholar]
- 30.Fakler B, Brandle U, Glowatzki E, Weidemann S, Zenner H-P, Ruppersberg J P. Cell. 1995;80:149–154. doi: 10.1016/0092-8674(95)90459-x. [DOI] [PubMed] [Google Scholar]
- 31.Lopatin A N, Makhina E N, Nichols C G. J Gen Physiol. 1995;106:923–955. doi: 10.1085/jgp.106.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis M W, Fleischhauer R, Dent J A, Joho R H, Avery L. Science. 1999;286:2501–2504. doi: 10.1126/science.286.5449.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doyle D A, Morais Cabral J, Pfuetzner R A, Kuo A, Gulbis J M, Cohen S L, Chait B T, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 34.Yellen G. Q Rev Biophys. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- 35.Yi B A, Jan L Y. Neuron. 2000;27:423–425. doi: 10.1016/s0896-6273(00)00052-0. [DOI] [PubMed] [Google Scholar]
- 36.Shin K S, Rothberg B S, Yellen G. J Gen Physiol. 2001;117:91–101. doi: 10.1085/jgp.117.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]