

Abstract
Quaternary ammonium analogues of atropine that are unable to cross the blood-brain barrier are used to alleviate peripheral muscarinic toxicity in animal models of epilepsy produced by systemic administration of pilocarpine or diisopropylfluorophosphate (DFP). Currently, methylatropine is the most popular and potent of these quaternary derivatives; however, it is expensive and produced in limited quantity. Here, we propose the use of ethylatropine bromide as an alternative to methylatropine. The synthesis of ethylatropine bromide is simple, inexpensive and has low environmental impact. We demonstrate the efficacy of ethylatropine bromide to antagonize the carbachol induced rise in intracellular calcium in a calcium mobilization assay, and its ability to prevent pilocarpine-induced total fluid secretions in mice without blocking pilocarpine-induced seizures. The ease of synthesis, cost effectiveness, and efficacy makes ethylatropine bromide a desirable alternative to methylatropine as a peripherally restricted acetylcholine receptor antagonist.
Keywords: Muscarinic receptor, nicotinic receptor, α7, acetylcholine, atropine, M1, pilocarpine, salivation, calcium, carbachol, ethylatropine, methylatropine
Graphical abstract
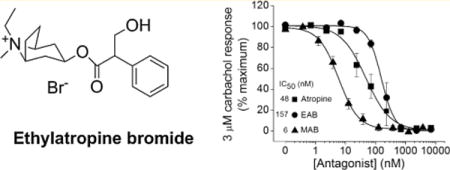
Atropine sulfate, listed by the World Health Organization (WHO) as an essential medication, is an antimuscarinic agent with a broad range of medical applications.1 For example, atropine is often used to elevate heart rate in vagal-induced bradycardia, to reduce production of saliva and bronchial fluids during anesthetic-induced surgery, and to relieve the peripheral and central effects of muscarinic receptor activation in patients suffering from organophosphorus (OP) agent poisoning.
Atropine sulfate is a competitive antagonist of the muscarinic acetylcholine receptors (mAChRs) that binds and inhibits all subtypes (M1, M2, M3, M4, and M5) with approximately equal affinity.2 Quaternary ammonium analogues of atropine (methyl, ethyl, propyl, and benzyl derivatives) were reported in 1956.3 Methylatropine has been the most widely used of these quaternary derivatives for animal studies in which the peripheral but not central muscarinic receptors must be blocked. Until recently, methylatropine nitrate and methylatropine bromide were produced in the United States. However, in 2005, the production of bromomethane, a key starting material in the synthesis of methylatropine bromide, was halted due to environmental concerns; thus, this analogue is no longer manufactured in the United States and is available in limited quantities via manufacturing in other countries. As a result, there has been an increase in demand for methylatropine nitrate and its cost has ballooned. Here we present an environmentally safe and inexpensive synthesis for ethylatropine bromide, and we report its potency and efficacy as a peripherally restricted antagonist of muscarinic receptors in vitro and in vivo.
RESULTS AND DISCUSSION
We synthesized ethylatropine bromide as shown in Figure 1. Atropine sulfate monohydrate was treated with 2N sodium hydroxide to produce neutral atropine, which was then treated with excess ethyl bromide in a sealed tube using acetone as a solvent at 100 °C for 6 h to furnish the final product in good yield. See Methods for characterization data and analysis of purity.
Figure 1.
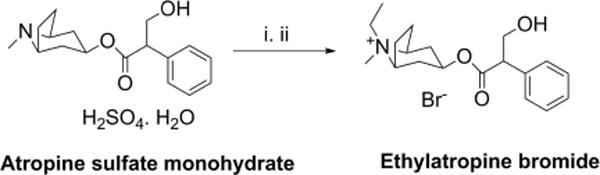
Synthesis of ethylatropine bromide. Reagents and conditions: (i) 2N NaOH and water, 97% recovery yield; (ii) ethyl bromide, acetone, 100 °C (sealed tube), 6 h, 70% yield.
Atropine is a potent muscarinic acetylcholine receptor antagonist with similar Ki values for receptors M1–M5.2 The specific functional role of muscarinic receptor subtypes in cholinergic parasympathetic responses in vivo have been investigated.4 Only mice lacking the M1 receptor were highly resistant to pilocarpine-induced seizures suggesting that the M1 muscarinic receptor mediates seizure induction in the pilocarpine model.4,5 The M1 receptor is coupled to the G-protein heterotrimeric subunit Gαq and agonist binding activates an intracellular second messenger cascade that involves activation of protein kinase C (PKC) and calcium mobilization in cultured cells.6–9 Knowing this we asked the following questions: Does agonist binding to the human M1 receptor lead to a detectable change in intracellular Ca2+ in our CHO-hM1 cells? If so, does ethylatropine bromide inhibit activation of the M1 receptor and how does its potency compare to atropine and methylatropine? Finally, does ethylatropine block peripheral muscarinic receptors in vivo? To address these questions, we initially measured intracellular Ca2+ in cultured CHO-hM1 cells using a fluorescent Ca2+ sensing dye (Fluo-4) and a FLEXStation II 96-well fluorometer. First, the features and limitations of the Ca2+ assay were determined. After baseline Ca2+ stabilization in the cells, bath application of HBSS (1X) had no significant effect on the calcium signal but ATP (100 μM) rapidly increased the fluorescent counts, which remained elevated for the remainder of the reading time (Figure 2A). Thapsigargin (a Ca2+-ATPase inhibitor) (1 μM) when applied in the same manner as ATP caused a smaller and slower increase in the fluorescent counts (Figure 2A). Although these two compounds both led to an increase in cytosolic Ca2+, their modes of action are quite different and suggest that changes in intracellular Ca2+ can be detected via different mechanisms in cultured CHO-hM1 cells. In a similar manner as ATP, bath application of carbachol (a nonsubtype-selective mAChR agonist) rapidly increased cytosolic Ca2+ (Figure 2B). The carbachol-induced rise in intracellular Ca2+ was concentration-dependent with a half maximally effective concentration (EC50) of 34 nM and could be long lasting (Figure 2B, C). Similarly, application of the pan mAChR agonist pilocarpine resulted in a concentration dependent increase in cytoplasmic calcium with an EC50 of 96 nM (Figure 2C).
Figure 2.
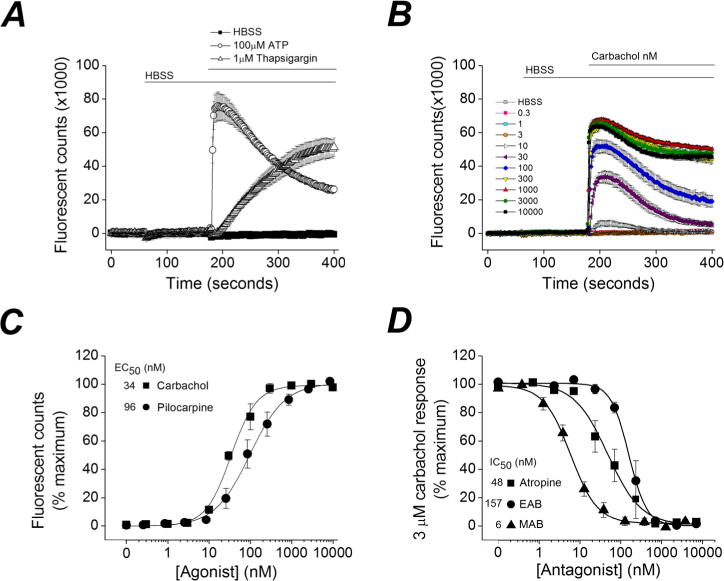
Ethylatropine bromide inhibits the human M1 receptor. (A) Ca2+-dependent fluorescent signal of CHO-hM1 cells loaded with Fluo-4 was monitored. With use of the integrated fluidics of the FLEXStation II, a bolus of HBSS was delivered at 60 s, resulting in a miniscule reduction in the fluorescent signal. Addition of ATP (100 μM) or thapsigargin (1 μM) at 180 s resulted in a long-lasting increase in [Ca2+]i. Data are the mean of eight replicates from a single plate. Bars indicate the duration of drug exposures. (B) A rapid increase in fluorescence was detected after application of the muscarinic receptor agonist carbachol at various concentrations. Data are the mean of eight replicates from a single plate for each concentration applied. (C) Concentration–response curves of the carbachol and pilocarpine-induced Ca2+ signal. The maximum Ca2+ signal was measured and the response was calculated for increasing concentrations of each agonist (carbachol, EC50 = 34 nM, n = 3 experiments in replicates of 8; pilocarpine, EC50 = 96 nM, n = 5 experiments in replicates of 8). (D) Carbachol induces a rapid rise in intracellular Ca2+ at 3 μM, which can be attenuated in a concentration-dependent manner by pretreatment with atropine (IC50 = 48 nM, n = 3 experiments in replicates of 4), methylatropine bromide (MAB) (IC50 = 6 nM, n = 7 experiments in replicates of 4) or ethylatropine bromide (EAB) (IC50 = 157 nM, n = 3 experiments in replicates of 4), but not by HBSS (not shown). In (C) and (D), data are mean ± SEM.
After establishing the agonist concentration response relationship, we then performed functional agonist–antagonist competition assays to determine whether ethylatropine bromide alters the activation of the M1 receptor by carbachol. The concentration of carbachol that resulted in the maximum response was 3 μM (Figure 2C), and thus, we used this concentration to investigate the antagonist efficacy of ethylatropine bromide. Activation of hM1 by carbachol in CHO cells was antagonized by atropine sulfate, ethylatropine bromide and methylatropine bromide with IC50s of 48 nM, 157 nM and 6 nM, respectively (Figure 2D). Taken together, these data indicate that ethylatropine bromide functionally inhibits the hM1 receptor in vitro.
Administration of subconvulsive doses of pilocarpine in mice can result in excessive salivation and diarrhea10–12 due to activation of the parasympathetic nervous system. Pilocarpine-induced secretions typically begin within minutes of administration and can be attenuated by systemic administration of methylatropine bromide,11 suggesting the secretions are at least partly mediated by peripheral muscarinic receptors. We determined whether ethylatropine bromide can attenuate pilocarpine-induced excessive salivation and diarrhea symptoms in mice. On the day of the experiment the mice were weighed, placed in individual containers, and allowed to freely explore. The mice exhibited normal exploratory behavior during the first 10 min in the containers and were deemed fit for the experiment. The mice were then administered saline or ethylatropine bromide at varying doses subcutaneously and were closely monitored. A subconvulsive dose of pilocarpine (50 mg/kg, i.p.) was administered 25 min later, resulting in excessive fluid secretion via salivation, urination, and diarrhea. Total fluid secretion (a combination of salivation, urination, and defecation) was measured for each mouse and expressed as a percentage of body weight. Mice injected with saline followed by pilocarpine displayed the highest total secretion (2.1 ± 0.2% of body weight, n = 4) over a 45 min time span. Mice administered ethylatropine bromide (1 mg/kg) dissolved in saline had almost no fluid secretions (0.4 ± 0.1%, n = 4; p < 0.01 compared with saline) during the same period. The effects of ethylatropine bromide were dose-dependent, with an approximate IC50 of 0.3 mg/kg (Figure 3A). None of the mice injected with 50 mg/kg pilocarpine experienced seizures. These data indicate that ethylatropine bromide is effective at attenuating pilocarpine-induced excessive secretions in mice.
Figure 3.
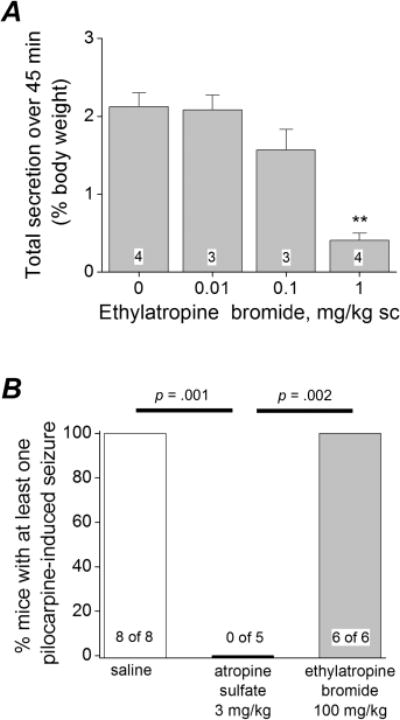
Ethylatropine bromide attenuates pilocarpine-induced secretions. (A) Total secretions (salivation, urination, and diarrhea) were measured and used to indicate in vivo efficacy of ethylatropine bromide. Mice were administered saline or ethylatropine bromide followed by pilocarpine 25 min later. Total secretion was measured 45 min after pilocarpine and calculated as a percent of the animal’s initial body weight. Data are mean ± SEM for each group. The number in the white box within the bar represents the total number of mice within each group. **p < 0.01, one-way ANOVA with Dunnett’s posthoc test compared to saline. (B) Mouse behavior was scored using a modified Racine scale and the number of mice with at least one pilocarpine-induced seizure was determined. Mice were administered saline, ethylatropine bromide, or atropine sulfate followed by pilocarpine (278 mg/kg) 25 min later. Fisher’s exact tests were performed comparing saline and ethylatropine bromide to atropine sulfate.
In mice, administration of a high dose of pilocarpine (>200 mg/kg) rapidly induces seizures and status epilepticus with clonic-tonic seizures.4,5 In order to determine whether ethylatropine bromide accesses the CNS, we investigated its effect on pilocarpine-induced seizures in mice. Six C57Bl/6 mice were administered ethylatropine bromide (100 mg/kg, s.c.) followed 25 min later by pilocarpine (278 mg/kg) to induce seizures. The motor behaviors of the mice were scored every 5 min. All six mice injected with ethylatropine bromide progressed to a score 3 (see scoring in Methods) within 30 min of pilocarpine administration and experienced at least one tonic seizure (Figure 3B). Similarly all eight saline injected mice experienced pilocarpine-induced tonic seizures. However, administration of atropine sulfate (3 mg/kg) 25 min prior to pilocarpine (278 mg/kg) blocked the development of seizures in every mouse (Figure 3B). These data suggest that atropine sulfate but not ethylatropine bromide can cross the blood-brain barrier and prevent seizure development.
The ability of ethylatropine bromide to inhibit nicotinic acetylcholine receptors was investigated in Xenopus oocytes. RNA encoding the human α7 nAChR was injected into oocytes and two-electrode voltage clamp was performed 2–5 days later. Large rapidly desensitizing inward currents were detected in oocytes upon activation with acetylcholine (1 mM) (Figure 4A). Bath application of high concentrations of ethylatropine bromide inhibited the acetylcholine induced inward current. The inhibition was partially reversed upon removal of ethyatropine bromide (EAB; Figure 4A) and was dose-dependent with 3 μM EAB producing a modest effect (1.2 ± 8% inhibition, n = 3). Higher doses resulted in a more pronounced inhibition (29 ± 3% for 30 μM, n = 5 and 46 ± 2% for 300 μM, n = 4) compared to oocytes not exposed to ethylatropine bromide (Figure 4B). Rundown of the nicotinic current accounted for the appearance of only a partial washout. The estimated IC50 of atropine sulfate to antagonize α7 acetylcholine (1 mM) induced currents was ∼1 μM,13 whereas the estimated IC50 for ethylatropine bromide to antagonize α7 acetylcholine (1 mM) induced currents is >300 μM (Figure 4). Taken together, these data suggest that ethylatropine bromide displays a much lower potency on nicotinic acetylcholine receptors compared to atropine sulfate.
Figure 4.
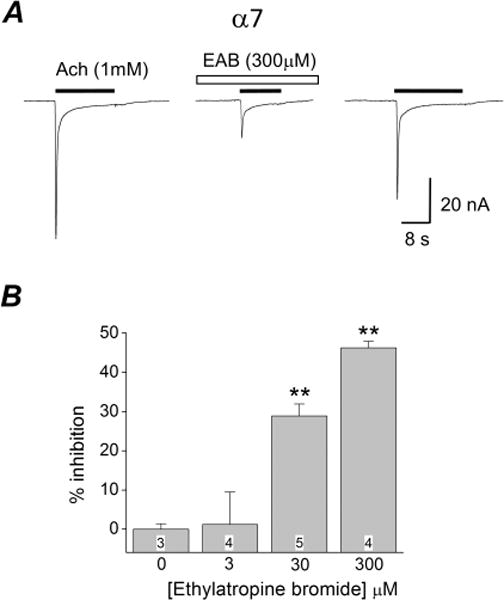
Inhibition of homomeric α7 nAChR by ethylatropine bromide. (A) Whole-cell currents were recorded from an oocyte 4 days after injection with cRNAs encoding α7. After application of EAB (300 μM), the peak current of the homomeric α7 receptor elicited by acetylcholine (1 mM) was reduced. The inhibition was reversed upon removal of EAB. (B) Varying the concentration of EAB revealed significant inhibition only at 30 μM and 300 μM, but not at 3 μM. **p < 0.01; one-way ANOVA with Dunnett’s posthoc tests. The number within the bar represents the total number of oocytes within each group. Data are mean ± SEM.
The anticholinergic drug atropine is considered an essential medication in hospitals as it is used to treat a vast array of clinical manifestations. Due to the high usefulness of atropine there is a constant demand for its production. The broad spectrum of action of atropine can be attributed to the ubiquitous expression of acetylcholine receptors throughout the body.14 More recently, there has been interest in the creation of quaternary derivatives of atropine that are unable to cross the blood-brain barrier and can be used to combat peripheral disorders associated with the regulation of mAChRs. One such quaternary derivative of atropine is methylatropine. Although methylatropine is clinically effective, there has been a dramatic reduction in its production partially caused by the cease in production of methylatropine bromide, which is synthesized using bromomethane gas as a precursor. The United States Environmental Protection Agency (EPA) considers bromomethane a priority air pollutant and highly regulates its production. Although methylatropine is commercially available, it is very expensive and can only be found in limited quantity. Here, we present the synthesis of ethylatropine bromide as an alternative to methylatropine; the synthesis of ethylatropine bromide is simple, environmentally safe and inexpensive. One key feature that makes the synthesis simple is the fact that the starting materials are readily available for commercial purchase, are inexpensive, and can be obtained in large quantities. Also, the synthesis steps are few and simple (Figure 1), resulting in a high yield of the product. None of the materials or byproducts of the synthesis of ethylatropine bromide is listed as a hazardous environmental pollutant on the Web site of the EPA.
Ethylatropine bromide was effective at antagonizing human M1 muscarinic receptors, and was approximately 3.3-fold less potent than atropine itself. By contrast ethylatropine was ∼300-fold less potent than atropine sulfate at α7 nicotinic receptors. Ethylatropine bromide did not prevent pilocarpine-induced seizures at a dose 300-fold higher than that needed to attenuate pilocarpine-induced secretions. Although the dose of pilocarpine to elicit central (seizure) effects was 5.6-fold higher than that used to elicit peripheral effects, these data support the conclusion that ethylatropine bromide is prevented from entering the brain. In the future, it will be important to investigate the action of ethylatropine bromide on other mAChRs and determine its pharmacokinetics and whether it is beneficial for the treatment of muscarinic receptor regulated clinical disorders.
CONCLUSION
Although ethylatropine bromide is not a novel compound, it last appeared in the literature in 196015 and our demonstration of its efficacy as a muscarinic receptor antagonist encourages its use to investigate peripheral muscarinic receptor physiology. The ease of synthesis, cost effectiveness, and efficacy of ethylatropine bromide makes it a viable alternative to methylatropine as a peripherally restricted muscarinic antagonist.
METHODS
Ethics Statement
All procedures concerning animals were approved by Emory University Institutional Animal Care and Use Committee and conformed to NIH guidelines. Every effort was made to minimize animal suffering.
Synthesis of Ethylatropine Bromide
Step 1
Atropine sulfate monohydrate (1 g) was dissolved in water (10 mL) and treated with 2 N NaOH (10 mL). The neutral compound was extracted with ethyl acetate, dried, and concentrated to provide 0.83 g of atropine (97% recovery), which was used for the next step.
Step 2
Ethyl bromide (5 mL, 67 mmol), atropine (0.83 g, 2.87 mmol), and acetone were combined in a sealed tube and heated for 6 h at 100 °C. The white precipitate formed was filtered and recrystallized with acetone, and finally vacuum-dried at 50 °C to furnish ethylatropine bromide (0.8 g, 70% yield).
Characterization Data
1H NMR (400 MHz, DMSO-d6). δ 7.24 (m, 5H), 5.06 (t, J = 4.8 Hz, 1H), 4.97 (t, J = 4.8 Hz, 1H), 3.93 (m, 1H), 3.81 (bs, 1H), 3.76 (m, 2H), 3.66 (m, 1H), 3.23 (m, 2H), 2.96 (s, 3H), 2.48 (m, 5H), 2.08 (m, 3H), 1.80 (m, 2H), 1.65 (m, 1H), 1.17 (m, 3H). LC-MS, 99% pure at λ 254 (only 0.0065% of atropine was present). MS (m/z) calculated for C19H28NO3: 318; found 318. Anal. Calcd for C19H28BrNO3: C, 57.29; H, 7.09; Br, 20.06; N, 3.52. Found. C, 56.74; H, 7.05; Br, 20.41; N, 3.40.
Cell Culture
Chinese hamster ovary cells stably expressing the human M1 mAChR (CHO-hM1) using a lentivirus as previously described6 were generously provided by Dr. Allan Levey (Emory University School of Medicine, Atlanta, GA). The CHO-hM1 cells were grown and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA), supplemented with 10% (v/v) FBS (Invitrogen), 100 units/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). The cultures were incubated at 37 °C in 95% air/5% CO2, and the culture medium was replaced every 3–4 days with fresh medium.
Measurement of [Ca2+]i in CHO-hM1 Cells Using a Ca2+-Sensitive Dye
CHO-hM1 cells were plated into 96-well plates (black with clear bottom) at a density of 40 000 cells/well (100 μL/well). After 4–5 days in culture, the cells grew to confluency and the level of intracellular calcium was assayed using the Fluo-4 no wash calcium assay kit according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA). In brief, the DMEM culture medium was removed and replaced with a calcium assay buffer (CAB) (100 μL/well) containing 1× HBSS (Invitrogen), 20 mM HEPES, 2.5 mM probenacid, and Fluo4-NW dye mix (pH 7.4, Invitrogen). The cells were then incubated for 45 min (37 °C, 5% CO2) for dye loading and then 15 min at 25 °C. The calcium fluorescence measurement was performed at 25 °C. For calcium fluorescence measurement of antagonist potency and generation of antagonist concentration–response curves HBSS (1×), atropine sulfate monohydrate, methylatropine bromide, or ethylatropine bromide was added (25 μL, 5× final concentration of the compound) after 60 s of recording with a FLEXStation II benchtop scanning fluorometer (Molecular Devices, Sunnyvale, CA). Then HBSS, ATP, thapsigargin, or the muscarinic receptor agonists (pilocarpine or carbachol) (25 μL, 6× final concentration) were added at 180 s. Fluorescence plate reading continued for a total of 400 s with excitation at 485 nm, emission at 538 nm, and a low-pass filter of 530 nm. All chemicals were added to the cell plate at a speed of 26 μL/s. The data were recorded using SoftMax Pro software (Molecular Devices).
Pilocarpine Secretion Assay
Adult male C57BL/6 mice (8–12 weeks old) purchased from Charles River Laboratories (Wilmington, MA) were housed in standard plastic cages (4–5 mice/cage) in a temperature controlled room (22 ± 2 °C) on a 12 h reverse light–dark cycle with food and water ad libitum. On the day of the experiment the mice were weighed and placed individually in plastic containers. Following a 10 min habituation period, the mice were injected with saline or ethylatropine bromide (0.01, 0.1, or 1 mg/kg in saline, s.c.). After 25 min, freshly prepared pilocarpine (50 mg/kg in saline, i.p.) was injected and the mice were placed in a clean labeled container. The mice were closely observed for 45 min. Each mouse received an injection volume based on weight (10 mL/kg).
Prior to the injection of pilocarpine, a square dry fiber pad the size of the container floor was weighed to the nearest 0.001 g. Within 10 s of the injection of pilocarpine, the mice were placed into the containers onto the fiber pads. After observing the behaviors induced by pilocarpine for 45 min the mice were removed from the containers and the fiber pads that contained saliva, urine and fecal boli were weighed by an observer blinded to the experimental treatment of the mice. The amount of total secretion was measured by the change in the weight of the fiber pad and calculated as a percent of the animal’s initial body weight. Each mouse was tested only once.
Pilocarpine-Induced Seizures
Adult male C57BL/6 mice (8–12 weeks old) obtained from Charles River Laboratories were injected with saline, ethylatropine bromide (100 mg/kg in saline, s.c.), or atropine sulfate (3 mg/kg in saline, s.c.). After 25 min, pilocarpine (278 mg/kg in saline, freshly prepared, i.p.) was injected to induce seizures. Each mouse received an injection volume based on weight (10 mL/kg). In mice, pilocarpine-induced seizures consist of distinct motor behaviors. Behavior was closely monitored and scored for 60 min. Seizures were classified as score 3 and 4 motor behaviors as outlined in Table 1.
Table 1.
| behavior score | observed motor behavior |
|---|---|
| 0 | normal behavior: walking, exploring, sniffing, grooming |
| 1 | freeze behavior: immobile, staring, curled-up posture |
| 2 | automatisms: repetitive blinking, chewing, head bobbing, vibrissae twitching, scratching, face washing |
| 3 | partial-body clonus, myoclonic jerks, shivering |
| 4 | whole-body clonus, “corkscrew” turning and flipping, loss of posture, rearing, falling |
| 5 | SE onset, nonintermittent seizure activity lasting at least 30 min |
| 6 | wild running, bouncing, tonic seizures |
| 7 | death |
Oocyte Injection
Human cDNA encoding the α7 nicotinic receptor was inserted into the pGEM-HE vector. cRNA was transcribed in vitro using a mMessage mMachine kit (Ambion, Waltham, MA) according to the manufacturer’s instructions and 15–30 ng of cRNA was injected into Xenopus laevis oocytes obtained from Ecocyte Bioscience (Austin, TX). Prior to electrophysiological recording, injected oocytes were maintained at 19 °C for 2–5 days in Barth’s solution containing 88 mM NaCl, 2.4 mM NaHCO3, 1 mM KCl, 0.33 mM Ca(NO3) 2, 0.91 mM CaCl2, 0.82 mM MgSO4, and 5 mM Tris/HCl, pH 7.4, and supplemented with gentamycin (100 mg/mL), penicillin (10 U/mL), and streptomycin (10 mg/mL).
Electrophysiology
Two-electrode voltage-clamp (TEVC) recordings were performed at room temperature (23–25 °C) from cells continually perfused (∼12 mL/min) in Barth’s solution (above). Recording electrodes were filled with 3 M KCl. α7 homomeric nAChRs were activated by bath application of acetylcholine (1 mM) as described previously.13 Application of agonist produced rapidly increasing and desensitizing currents in all oocytes. The oocytes were perfused with Barth’s solutions for a minimum of 3 min between agonist applications (minimum of three) for baseline stabilization and then perfused for 2 min with EAB. Multiple agonist applications were then carried out in the presence of EAB to measure the peak inhibition. Following inhibition by EAB the cells were bathed with Barth’s solution again for a minimum of 3 min prior to multiple agonist application for α7 recovery from the EAB effect. Currents were elicited from a holding potential of –80 mV. Current records were digitized at 1 kHz with a Digidata 1200 analogue to digital converter (Axon Instruments, Foster City, CA) under the control of pClamp 8 acquisition software (Molecular Devices, Sunnyvale, CA) and stored on a computer disk for later analysis. Results from oocytes in which the agonist-induced current was not stable under baseline conditions were discarded.
Chemicals and Drugs
Carbachol was purchased from Abcam (Cambridge, MA). Pilocarpine, methylatropine bromide, and ATP were purchased from Sigma-Aldrich (St. Louis, MO). Atropine sulfate monohydrate was purchased from Alfa Aesar (Ward Hill, MA). Thapsigargin was purchased from Tocris Bioscience (Minneapolis, MN).
Data Analysis
Data are presented as means ± standard error. All calcium responses (peak response) were normalized to that of the maximum response of the agonist used (carbachol or pilocarpine). Concentration–response curves were fitted using Origin 7.0 (Origin-Lab, Northampton, MA) to a four-parameter logistic equation to determine EC50 or IC50 values. Statistical analysis was performed with GraphPad Prism version 6 (GraphPad, San Diego, CA). One-way ANOVA (with Dunnett’s posthoc test) of selected means was performed as appropriate to examine differences of chemical effects. Fisher’s exact tests were performed to examine differences in the percent of mice with pilocarpine-induced seizures. The differences were considered to be statistically significant if p < 0.05. A Grubb’s test was performed in GraphPad to identify outliers; none were found.
Acknowledgments
The authors thank Dr. Allan Levey for generously sharing the CHO-hM1 cells, Dr. Stephen Traynelis for sharing the human α7 nicotinic acetylcholine receptor RNA, and Mr. Avery Hennigan for his helpful technical assistance.
Funding
This work was supported by NIH UO1 NS058158 and R01 NS097776 (RD).
ABBREVIATIONS
- AChR
acetylcholine receptor
- mAChR
muscarinic acetylcholine receptor
- nAChR
nicotinic acetylcholine receptor
- DMEM
Dulbecco’s modified Eagle’s medium
- EAB
ethylatropine bromide
- MAB
methylatropine bromide
- AS
atropine sulfate
- CHO
Chinese hamster ovary
- IP
intraperitoneal
- SC
subcutaneous
- Veh
vehicle
- mpk
mg/kg
- CCh
carbachol
- GPCR
G-protein coupled receptor
- CHO-hM1
Chinese hamster ovary cells overexpressing human M1 receptor
- HBSS
Hank’s balanced salt solution
- OP
organophosphorus
- EPA
environmental protection agency
- CNS
central nervous system
- TEVC
two-electrode voltage clamp
- PKC
protein kinase C
- Gαq
G-protein alpha
- IC50
half maximal inhibitory concentration
- EC50
half maximal effective concentration
Footnotes
ORCID
Asheebo Rojas: 0000-0003-0895-0839
Author Contributions
A.R., T.G., and R.D. designed and performed experiments, analyzed data, and wrote the paper. A.W. performed experiments and analyzed data.
Notes
The authors declare no competing financial interest.
References
- 1.Mehta DK, Ryan RSM, Hogerzeil HV, editors. WHO model formulary 2004. World health Organization; Geneva: 2004. [Google Scholar]
- 2.Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Southan C, Davies JA. The Concise Guide to PHARMACOLOGY 2015/16: G protein-coupled receptors. Br J Pharmacol. 2015;172:5744–5869. doi: 10.1111/bph.13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shea SM. Inhibition of gastric secretion in rats by some quaternary derivatives of atropine. Br J Pharmacol Chemother. 1956;11:171–174. doi: 10.1111/j.1476-5381.1956.tb01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bymaster FP, Carter PA, Yamada M, Gomeza J, Wess J, Hamilton SE, Nathanson NM, McKinzie DL, Felder CC. Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur J Neurosci. 2003;17:1403–1410. doi: 10.1046/j.1460-9568.2003.02588.x. [DOI] [PubMed] [Google Scholar]
- 5.Hamilton SE, Loose MD, Qi M, Levey AI, Hille B, McKnight GS, Idzerda RL, Nathanson NM. Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc Natl Acad Sci U S A. 1997;94:13311–13316. doi: 10.1073/pnas.94.24.13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis AA, Heilman CJ, Brady AE, Miller NR, Fuerstenau-Sharp M, Hanson BJ, Lindsley CW, Conn PJ, Lah JJ, Levey AI. Differential effects of allosteric M(1) muscarinic acetylcholine receptor agonists on receptor activation, arrestin 3 recruitment, and receptor downregulation. ACS Chem Neurosci. 2010;1:542–551. doi: 10.1021/cn100011e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haga T. Molecular properties of muscarinic acetylcholine receptors. Proc Jpn Acad, Ser B. 2013;89:226–256. doi: 10.2183/pjab.89.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura K, Hamada K, Terauchi A, Matsui M, Nakamura T, Okada T, Mikoshiba K. Distinct roles of M1 and M3 muscarinic acetylcholine receptors controlling oscillatory and non-oscillatory [Ca2+]i increase. Cell Calcium. 2013;54:111–119. doi: 10.1016/j.ceca.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Street M, Marsh SJ, Stabach PR, Morrow JS, Brown DA, Buckley NJ. Stimulation of Gαq-coupled M1 muscarinic receptor causes reversible spectrin redistribution mediated by PLC, PKC and ROCK. J Cell Sci. 2006;119:1528–1536. doi: 10.1242/jcs.02872. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Lu Y, Chen HZ. Differentiating effects of anisodamine on cognitive amelioration and peripheral muscarinic side effects induced by pilocarpine in mice. Neurosci Lett. 2003;344:173–176. doi: 10.1016/s0304-3940(03)00444-0. [DOI] [PubMed] [Google Scholar]
- 11.Takakura AC, Moreira TS, Laitano SC, De Luca LA, Renzi A, Menani JV. Central muscarinic receptors signal pilocarpine-induced salivation. J Dent Res. 2003;82:993–997. doi: 10.1177/154405910308201211. [DOI] [PubMed] [Google Scholar]
- 12.Renzi A, Colombari E, Mattos TR, Silveira JE, Saad WA, Camargo LA, de Luca LA, Derobio JG, Menani JV. Involvement of the central nervous system in the salivary secretion induced by pilocarpine in rats. J Dent Res. 1993;72:1481–1484. doi: 10.1177/00220345930720110401. [DOI] [PubMed] [Google Scholar]
- 13.Zwart R, Vijverberg HP. Potentiation and inhibition of neuronal nicotinic receptors by atropine: competitive and noncompetitive effects. Mol Pharmacol. 1997;52:886–895. doi: 10.1124/mol.52.5.886. [DOI] [PubMed] [Google Scholar]
- 14.Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol. 2008;154:1558–1571. doi: 10.1038/bjp.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soyka L, Gyermek L. Cholinergic blockade as related to chain length of monoquaternary derivatives of atropine and benzoyl-tropine. J Med Pharm Chem. 1960;2:361–373. doi: 10.1021/jm50011a002. [DOI] [PubMed] [Google Scholar]