

Abstract
Background
A single base pair mutation in the genome can result in many congenital disorders in humans. The recent gene editing approach using CRISPR/Cas9 has rapidly become a powerful tool to replicate or repair such mutations in the genome. These approaches rely on cleaving DNA, while presenting unexpected risks.
Results
In this study, we demonstrate a modified CRISPR/Cas9 system fused to Cytosine DeAminase (Cas9-DA), which induces a single nucleotide conversion in the genome. Cas9-DA was introduced into sea urchin eggs with sgRNAs targeted for SpAlx1, SpDsh, or SpPks, each of which is critical for skeletogenesis, embryonic axis formation, or pigment formation, respectively. We found that both Cas9 and Cas9-DA edit the genome, and cause predicted phenotypic changes at a similar efficiency. Cas9, however, resulted in significant deletions in the genome centered on the gRNA target sequence, whereas Cas9-DA resulted in single or double nucleotide editing of C to T conversions within the gRNA target sequence.
Conclusion
These results suggest that the Cas9-DA approach may be useful for manipulating gene activity with decreased risks of genomic aberrations.
Keywords: CRISPR, Cas9, deaminase, genome editing, sea urchin
Introduction
Several congenital diseases in humans are caused by single base-pair mutations that can compromise fetal development, decrease live birth rates, reduce life span, and/or decrease quality of life. For example, a single point mutation in the β-globin chain of hemoglobin results in the debilitating disease of sickle cell anemia. Sickle cell anemia is an autosomal recessive disorder that occurs in 1 of every 365 African-Americans, and exists as one of the most frequent blood disorders in the United States (Vidaud et al., 1990). Other known point mutations that result in genetic diseases include a single mutation in the ABC1 gene for coronary heart disease, in the cardiac transcription factor CSX/NKX2.5, which leads to defects in heart development, and in the CFTR gene for Cystic Fibrosis (Berolini et al., 2001; Ikeda et al., 2002; Riordan et al., 1989). While these single nucleotide changes in the genome alter only one amino acid in the protein chain, the results are life-threatening. With increased technology and the growth of personalized treatments, it is imperative to explore ways to obviate the causes of such genetic diseases with minimal risk.
Genetic approaches using homologous recombination and mutational screens have been mainstream to delineate which genes are involved disease-related events. Such approaches have the potential to also correct genomic mutations. Recent advances in genome-editing mechanisms offer approaches that increase genetic access to more organisms to study basic and clinically relevant processes. With its associated Cas9 nuclease, clustered regularly interspaced short palindromic repeats (CRISPR) is one of many innovations that allows one to edit specific sequences of a genome. CRISPR is a native nucleic-acid-based adaptive immune response found in a wide range of bacteria and archaea (Jinek et al., 2012). The CRISPR system functions to catalog genomic information from invading bacteriophages, and to utilize this DNA library in subsequent generations to protect the cell from repeated invasion with specific DNA targeting with guide RNA (sgRNA) and DNA cleavage with Cas9 (Carte et al., 2008; Deltcheva et al., 2011; Gesner et al., 2011; Haurwitz et al., 2010; Sashital et al., 2011; Wang et al., 2011; Barrangou et al., 2007).
The Cas9 endonuclease possesses a recognition domain that associates with the sgRNAs to form a CRISPR-associated heteroduplex that can target and cleave nucleic acids through base-pair interactions between the sgRNA sequence and the complementary sequence of the intruder (Wiedenheft et al., 2012; Garneau et al., 2010; Makarova et al., 2011). Protospacer adjacent motif (PAM) sequences are located immediately downstream of target DNA sequences, and constrain the site of endonuclease cleavage. For example, Cas9 of Streptococcus pyogenes recognizes a PAM sequence corresponding to 5′-NGG-3′ (Mojica et al., 2009; Hirano et al., 2016). After the binding of Cas9, both strands of DNA are cleaved within the complementary region by the two-nickase sites of the Cas9 endonuclease. CRISPR technology has been used to modify the genome and gene expressions of living organisms by pairing Cas9 with specially designed sgRNA, which targets and cleaves intact genomes of organisms at specific and predefined sites.
The double stranded breaks in DNA caused by Cas9 are repaired by non-homologous end joining (NHEJ), where nucleotides are inserted or deleted in an error-prone process (Fig. 1A, left). The resulting mutations compromise gene integrity, and either knocks down or knocks out gene functionality. The NHEJ mediated repair approach functions well in certain cell types and organisms. However, it comes with variability in repair, and can cause changes in cellular physiology in response to the activation of the dsDNA repair mechanisms. On the other hand, inactivation of these two Cas9 endonuclease domains (RuvC and HNH) results in protein targeting, but not cleavage of DNA (Jinek et al., 2012; Femandes et al., 2016). The nickase-inactivated Cas9 can then be fused to a variety of other enzymes, such as transcription effectors and epigenetic modifiers, to carry out site-specific modifications without altering the genome (Fernandes et al., 2016). In this study, we employed a strategy to edit the genome and gene expression independent of DNA cleaving.
Figure 1. Cas9-Deaminase (DA)-mediated genome editing.
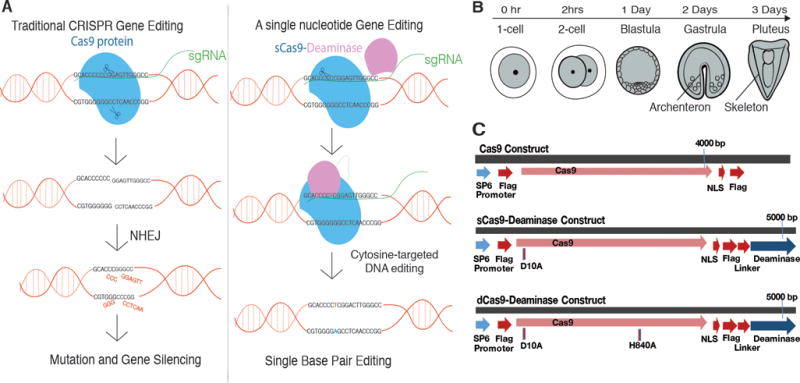
A, A Model for Cas9 and Cas9-DA-mediated gene editing. CRISPR/Cas9 alters the genome through Non Homologous End Joining (NHEJ), whereas CRISPR/Cas9-DA is designed to edit specific C to T conversions on one DNA strand through deamination. The complementary G will be then converted to an A (labeled in blue) during replication or through the internal mis-matching repair mechanism in the subsequent cell cycle. Not all changes may be retained due to this repair. B, A schematic depicting normal sea urchin development. The embryo hatches at Day1, gastrulates, and starts forming skeletons at Day2 and reaches the larval stage with extended skeletons at Day3. C, Cas9 (top), sCas9-DA (middle), and dCas9-DA (bottom) construct designs. sCas9-DA has a mutation (D10A) in one of the nickase sites, whereas dCas9-DA has double mutations (D10A and H840A) to inactivate both nickase sites. Each construct possesses a SP6 promoter for IVT. Flag, Flag-tag; NLS, Nuclear Localization Signal.
DNA/RNA deaminases (DAs), such as the APOBEC/AID and ADAR families of enzymes, are candidates for an engineered genome-editing tool that do not cut double-stranded DNA. Instead, these DAs modify the base of a nucleotide by deamination, and modify the DNA or RNA sequence without cleaving the polymer. APOBEC/AID enzymes natively protect against viral agents, in addition to contributing to protein diversity in other immune responses and mitochondrial function through mRNA editing (Seplyarskiy et al., 2016). Several deaminases that catalyze single nucleotide editing have been identified and two are the best understood. One is cytosine deaminase, an APOBEC/AID enzyme that catalyzes C to U, then allows replication as a T in DNA (Smith et al., 2012; Harris et al., 2002; Harris et al., 2003; Chen et al., 2008; Petersen Mahrt et al., 2002). The other is adenosine deaminase such as ADAR that catalyzes A to I on RNA, and then read as a G during translation and post-transcriptional events (Wang et al., 1998). In this study, we utilized PmCDA1, a cytosine deaminase identified in Petromyzon marinus (sea lamprey) that has previously been reported to have a high mutagenic effect in yeast (Lada et al., 2011). PmCDA1 is a DNA/RNA editing AID/APOBEC deaminase, and is involved in the diversification of genes for immunoglobulin analogs in sea lampreys (Kato et al., 2012). AID catalyzes cytosine deamination and results in a thymidine (through a uracil intermediate), causing a C to T conversion, or a G to A conversion in the complementary strand in the genome (Smith et al., 2012; Harris et al., 2002; Harris et al., 2003; Chen et al., 2008; Petersen Mahrt et al., 2002).
By fusing PmCDA1 to an inactivated Cas9 (to prevent nickase activity), one can exploit the targeting power of Cas9-sgRNA to promote deaminase activity without cleaving DNA (Fig. 1A, right). Recent studies in yeast demonstrated that fusing PmCDA1 to a Cas9 with inactivated nickase activity caused mutations to occur dominantly within a range of three to five bases surrounding the 18 nucleotides upstream of the PAM sequence on the strand of DNA non-complementary to the gRNA (Komor et al., 2016; Nishida et al., 2016). However, its activity in an embryo and its potential application to manipulate animal development has not been tested. Here, we use Strongylocentrotus purpuratus, more commonly known as the purple sea urchin, as a model system to test the effectiveness of Cas9-deaminase (Cas9-DA) technology in embryonic development. Several features of the sea urchin embryo make this animal an ideal in vivo system to test the quality and efficiency of new technologies. A single female produces millions of eggs that can be easily microinjected, development is highly synchronized, reproducible, and rapid (millions of embryos become gastrulae at the same time within two days and pluteus larvae within three days post fertilization (dpf); see also Fig. 1B); and each cell in development can be observed under the microscope due to its optical transparency. Sea urchin development is also well studied with detailed genomic and transcriptomic information (see e.g. echinobase.org).
Results and Discussion
CRISPR/Cas9 genome editing reduced skeleton formation: Targeting SpAlx1
To test CRISPR/Cas9 efficiency in Strongylocentrotus purpuratus, we first targeted the endogenous gene locus of SpAlx1. SpAlx1 is a transcription factor essential for skeletogenesis during embryonic/larval development (Sharma and Ettensohn, 2010). Biomineralization for skeletogenesis starts when embryos reach the gastrula stage at 2 dpf. Therefore, successful CRISPR genome editing is expected to compromise biomineralization (skeletal granule formation) and/or extension of skeletal rods in embryos/larvae after that developmental stage. Larvae resulting from eggs injected with Cas9 mRNA and Alx1 sgRNAs were observed at 3 dpf under the microscope and scored for larvae with skeletal granule formation and rod extension.
At 3 dpf, approximately 98% of larvae in the control group (Fig. 2A & C, Cas9-only) developed extended skeletal rods, while the experimental group did so significantly less, at 81.5% (Cas9+Alx1 sgRNA in Fig. 2A & C; see also Fig. 2A′ for the representative images of each phenotype). Only 1% of controls demonstrated a complete lack of skeletal formation, while approximately 10% of the experimental group showed this phenotype. In these larvae, a significant difference was found in developmental progress between the control and sample groups (Cas9 only and Cas9+Alx1 sgRNA in Fig. 2B; see also Fig. 2B′ for the representative phenotypes), each of these cohorts showed an overall delay in development when compared to the Cas9-DA treatment (described in the next section). Although these larvae were cultured for up to 4 dpf, skeleton formation never fully recovered in embryos/larvae injected with Cas9 and Alx1 sgRNAs, which suggest a specific inhibition in skeletogenesis, but not a general delay in development (data not shown). Since genome editing occurs at random time points after the introduction of CRISPR to fertilized eggs and through random deletions and NHEJ, it is reasonable to see a variety of embryos/larvae with different levels of compromised skeleton formation. This is particularly true for the skeletogenic cell lineage, which are cells that fuse to form a syncytium, enabling the diffusion of a variety of factors important for skeletogenesis between cells. We would anticipate self-rescue in cases where not all skeletal cells in the embryo/larva are edited. Although complete inhibition of skeleton formation only occurred 10% of the time, these results suggest that the CRIPSR/Cas9 system appears to interfere with Alx1 gene activity in this organism (Nakayama et al., 2013; Bassett et al., 2013; Lin and Su, 2016; Oulhen and Wessel., 2016; Sugi, 2016). The CRISPR/Cas9 approach has been reported to cause mild cytotoxicity, especially when overdosed (Nakayama et al., 2013). This cytotoxicity was demonstrated in our study when we administered a higher dose (a stock concentration of 500ng/μl of Cas9 mRNA and 600ng/μl total of sgRNAs) resulting in delays in development, or some potentially non-specific cytotoxicity.
Figure 2.
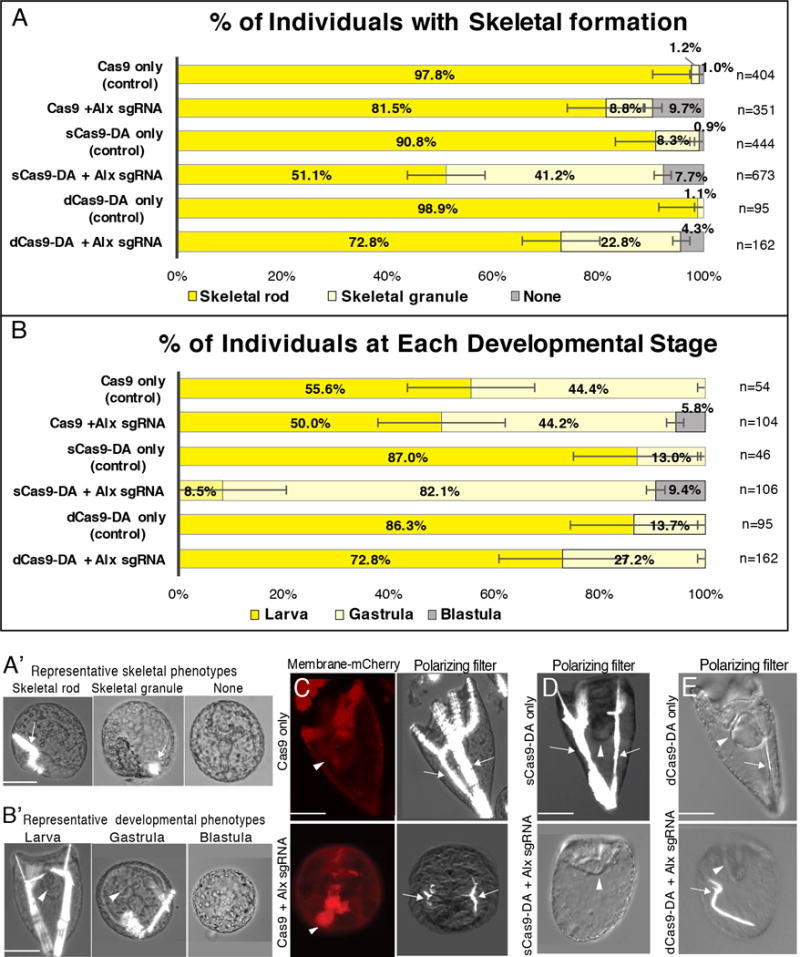
CRISPR/Cas9/Deaminase targeting of Alx1 evaluated at 3 dpf. A–B′, Proportion of injected individuals with skeleton formation (A and A′) and with each developmental stage (B and B′) at 3 dpf. n= total number of embryos/larvae observed. A′ and B′, Representative images of Cas9+Alx1 sgRNAs introduced embryos that show each phenotype used for scoring in A and B. A′, Embryos/larvae were scored in A for formation of skeletal rods, skeletal granule, or no-skeleton. B′, Embryos/larvae were scored in B for extended skeletal rod formation and elongated body shape as “larva”, for archenteron appearance with no extended skeletal rod as “gastrula”, and for blastocoel presence with no archenteron formation as “blastula”. Inhibition of full skeleton formation typically correlated with halted development and embryos were scored as gastrula. C-E, 3 dpf embryos/larvae injected with Cas9 (C), sCas9-DA (D) or dCas9-DA (E) with Alx1 sgRNAs. Membrane mCherry mRNA (red) was used as an injection marker. Brightfield was used with a polarizing-filter to identify skeleton formation (arrows). Arrowheads indicate archenteron. Cas9/sCas9/dCas9-only embryos/larvae were injected without sgRNAs as controls. Scale bars = 50um.
CRISPR/Cas9-DA genome editing reduced skeleton formation during development: Targeting SpAlx1
Using the same injection conditions and the sgRNAs tested above, we next determined if the CRISPR/Cas9-Deaminase (DA) system is effective in the sea urchin embryo. Zygotes injected with sCas9-DA (containing a single mutated nickase site) developed to larvae at 3 dpf, and resulted in 51% of the experimental group forming extended skeletal rods compared to 91% of the control group (sCas9-only and sCas9+Alx1 sgRNA in Fig. 2). While these sCas9-DA groups appeared more normal in general development and archenteron formation compared to the Cas9 groups (Fig. 2C and D), 87% of the “sCas9-DA only” group reached the larval stage, whereas only 56% of the “Cas9-only” group reached the same stage (Fig. 2B), even when the same amount of Cas9/Cas9-DA was injected. It should be noted that complete skeletal rod formation is essential to form “ arms” and thus reach the larval stage. Therefore, only 8.5% of the individuals injected with sCas9-DA+Alx1sgRNA were counted as larvae due to a significant lack of extended skeletons in the population (see also Fig. 2A′ and B′ for the representative phenotype for each group). However, it is interesting to note that these larvae were as healthy as their corresponding sibling controls: these individuals showed fully differentiated tripartite guts and elongated bodies despite the lack of complete skeletons (compare “sCas9-DA only” and “sCas9-DA+Alx1sgRNA” in Fig. 2D). We conclude from these results that sCas9-DA is similarly effective to the CRISPR/Cas9 system, but the sCas9-DA yields healthier development within the multiple batches of larvae tested. This may be a result of spurious CRISPR/Cas9 activity upon Cas9 and sgRNA overdosing in the genome which does not occur when a nickase site of Cas9 has been inactivated.
Non-cleaving Cas9, lacking both functional nickase sites (double, dCas9-DA), was also tested to investigate the editing capacities of the attached DA independent of Cas9’s DNA cleaving activities. At 3 dpf, approximately 99% of the control group (dCas9-DA in Fig. 2A and E) demonstrated extended skeletal rod formation, while similar phenotypes were present in only 73% of the experimental group (dCas9-DA+Alx1 sgRNA in Fig. 2A and E). All control larvae had skeletons, while 4% of the experimental group showed a complete lack of skeleton formation. Notably, general development, such as archenteron formation, was optimal in all of the control groups without sgRNAs (Cas9/sCas9-DA/dCas9-DA only) as well as in the dCas9-DA + Alx1 sgRNA group, compared to the Cas9/sCas9-DA+Alx1 sgRNA groups (Fig. 2B). These results suggest that CRISPR/dCas9-DA edited the gene in a targeted manner within the embryo, and general developmental health also improved. Thus, the nickase activities of Cas9 may cause some cytotoxicity when applied with sgRNAs in these embryos/larvae with this relatively higher dose.
CRISPR/Cas9 and CRISPR/Cas9-DA reduced archenteron formation: Targeting genomic DNA- Dishevelled (SpDsh)
Dishevelled (Dsh) is a critical player in the Wnt pathway in diverse organisms. In the sea urchin, SpDsh is essential for the establishment of embryonic polarity and endoderm formation during embryogenesis (Byrum et al., 2009). Thus, successful CRISPR-mediated gene inactivation is expected to compromise gastrulation and archenteron formation, which starts from 2 dpf. Dsh gene inactivation is also expected to indirectly inhibit skeleton formation through a general compromise in development. As for the conventional CRISPR/Cas9-mediated gene inactivation, the experimental group (Cas9+Dsh sgRNA in Fig. 3A–C) showed reduced skeleton and archenteron extension compared to the control group (Cas9-only in Fig. 3A–C) at 3 dpf. For the sCas9-DA groups, the phenotype was mild especially in skeleton formation, although archenteron formation was reduced (Fig. 3A, B, D) (see also Fig. 2A′ and B′ for representative phenotypes used for scoring). Overall, these results show similar efficiency between CRISPR/Cas9 and CRISPR/Cas9-DA but with significant differences between target gene sequences.
Figure 3.
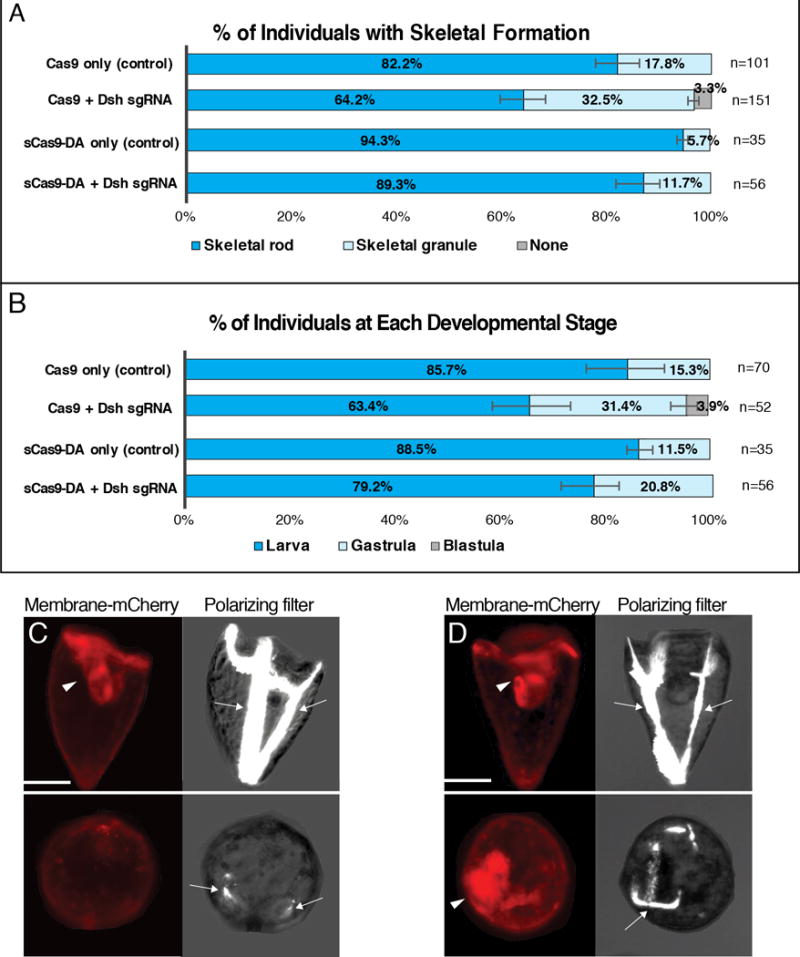
CRISPR/Cas9/Deaminase targeting Dsh evaluated at 3 dpf. A & B, Proportion of injected individuals with skeleton formation (A) and with each developmental stage (B) at 3 dpf. n= total number of embryos/larvae observed. Inhibition of gastrulation (archenteron formation) results in delay of overall development. C & D, 3 dpf embryos/larvae injected at fertilization with Cas9 (C), or sCas9-DA (D) with Dsh sgRNAs. Membrane mCherry mRNA (red) was used as an injection marker. Brightfield was used with a polarizing-filter to identify skeleton formation (arrows). Arrowheads indicate archenteron. Cas9/sCas9-only embryos/larvae were injected without sgRNAs as controls. Scale bars = 50um.
CRISPR/Cas9-DA edits genomic DNA in a targeted manner
To test if CRISPR/Cas9 and/or CRISPR/Cas9-DA has indeed edited the targeted site in the genome, we sequenced the targeted genomic site after phenotypic analysis of the larvae. We collected larvae targeted for the Alx1 and Dsh genes, subjected them to genome DNA extraction, followed by PCR to amplify and sequence the genomic region of the gRNA target site and its flanking sequence. Since dCas9-DA embryos/larvae showed little phenotypic variation, we randomly selected individuals for each sample group. The gRNA1 of both Alx1 and Dsh were designed within a short exon sequence (~150bp) and we found it technically difficult to amplify this gRNA targeted site and its flanking sequence using primers designed in the introns, perhaps because of the high polymorphism frequency of this animal (echinobase.org). We therefore focused only on the gRNA2 target sequence of each gene locus for PCR amplification and sequencing. Further, despite introduction of Cas9 or s/dCas9-DA mRNA at the one-cell stage, the timing of editing may vary, and not all cells in the larva may be equally edited at the same time. This results in mutational variations amongst cells, even within a single larva.
All control samples for CRISPR/Cas9 and CRISPR/Cas9-DA showed no significant mutations within the gRNA target sequence, as well as its immediate ~30bp flanking sequence (Fig. 4A and B). Of the larvae targeted for Alx1 with the standard CRISPR/Cas9 and sgRNA system, 34% of clones showed a large deletion (>100b; Table S2), 41% of them showed smaller deletion (<100bp) around the gRNA target region, for a total of 74% of the samples containing deleted sequence. Only 17% of the samples showed no mutation (Fig. 4A). For larvae injected with sCas9-DA+Alx sgRNAs, a short deletion around the gRNA sequence was unexpectedly found quite frequently (31%), as well as the C to T conversion at 8bp upstream of the PAM sequence, which induced a stop codon (a black squared region in Fig. 4A). This may explain the specific phenotype with otherwise healthy development. Another C to T conversion site was found at 8bp (11%) upstream of the PAM sequence, (Fig. 4A), indicating specific editing at a predictable site. It should be noted that this C to T conversion did not induce a stop codon. In the dCas9-DA+Alx1 sgRNA group, 31% of the sample showed C to T conversion at 8bp and/or 20bp upstream of the PAM sequence, which is consistent with the results of the sCas9-DA group. Since 23% of the samples exhibited double mutations at 8bp and 20bp upstream of the PAM sequence, dCas9-DA may have edited these sites at the same time. No deletion of the DNA fragment was found except for one sample (this deletion also induced a stop codon at 70bp downstream of the PAM sequence), and 46% of the samples showed no editing in the targeted sequence. The overall editing efficiency by dCas9-DA was thus less efficient in this case compared to that by Cas9, and which is consistent with the mild phenotype of the dCas9-DA group. Overall, these results suggest that while DNA nicking by sCas9 is not essential for deaminase to function in editing, it may still increase DA’s accessibility to the DNA. This may, however, result in a small deletion in some cases (e.g. potentially dependent on the chromatin structure of the target gene locus) such as in the embryonic cell.
Figure 4.

Sequencing results of Alx1-gRNA2 (A) and Dsh-gRNA2 (B) target and flanking sequences (~200bp) were amplified by PCR from the extracted genome of each single embryo/larva, and the PCR products were subcloned and subjected to individual sequencing. ~80bp of each PCR product is shown in this figure. The original gRNA target sequence is indicated in red, the PAM target within the genome in green, mutated nucleotides in blue, and deleted sequences with a dashed line. Blue boxes indicate representative target cytosine mutated by Cas9-DA. Yellow boxes indicate cytosine that could induce a stop codon when converted to thymidine, while no mutations were found in those regions. # of samples (%) indicates number and proportion of each mutational pattern. * The deletion and C-to-T conversion formed a stop codon (TAA) at the squared region. ** The deletion formed a stop codon (TAA) at 70bp downstream from the PAM sequence.
Larvae targeted for Dsh by CRISPR/Cas9 also showed various deletions centered on the gRNA site in ~40% of the larvae (Fig. 4B) in spite of minimum effects on the embryo phenotype. This result suggests that either the Dsh maternal protein or another compensatory mechanism in the embryo might have masked the effect of Dsh gene inactivation. The more dramatic phenotype exhibited when using morpholino anti-sense oligonucleotide knockdown likely results from blocking ongoing translation of maternal mRNAs, whereas the CRISPR/Cas9 targeting would only interfere with the construction of new transcripts. Larvae that were injected with sCas9-DA and Dsh gRNAs also revealed C to T conversions at the DNA strand 7 and 20 nucleotides upstream of PAM sequence in 36% of the samples. It should be noted that these mutations did not induce a stop codon despite altering the amino acid from proline to leucine. It is not apparent how these amino acid changes may impact the function of the Dsh gene product. Additionally, 45% of this sample group showed no mutation, although T to G conversions at 8 nucleotides upstream of PAM sequence were found in the genome. This may have been mediated indirectly through its adjacent C’s conversion to T. C to T conversion outside of the gRNA-targeted sequence was rarely observed, suggesting CRISPR/Cas9-DA edits single nucleotides in a predictable site.
Testing the efficiency and consensus of CRISPR/Cas9-DA editing
Based on the above results, CRISPR/Cas9-DA appears to edit the genome in a targeted manner. However, we realize predicting a promising single gRNA site for Cas9-DA to cause a stop codon or changed protein function is challenging at this point. We therefore introduced 16 sgRNAs targeting a single (3rd) exon of PKS (SPU_ 002895) to test the extent of Cas9-DA’s mutational capabilities with near saturation screening (Fig. 5A). With this approach, we also aimed to test differences in mutational efficiency amongst different gRNA sites. The PKS gene was chosen for this test because the gene possesses a large exon, enabling high density gRNA target construction. It was also recently shown to cause albinism upon mutation (gRNA#6 and #10 in Fig. 5A) but does not affect development, providing an easy visual phenotype for scoring (Oulhen and Wessel, 2016).
Figure 5.
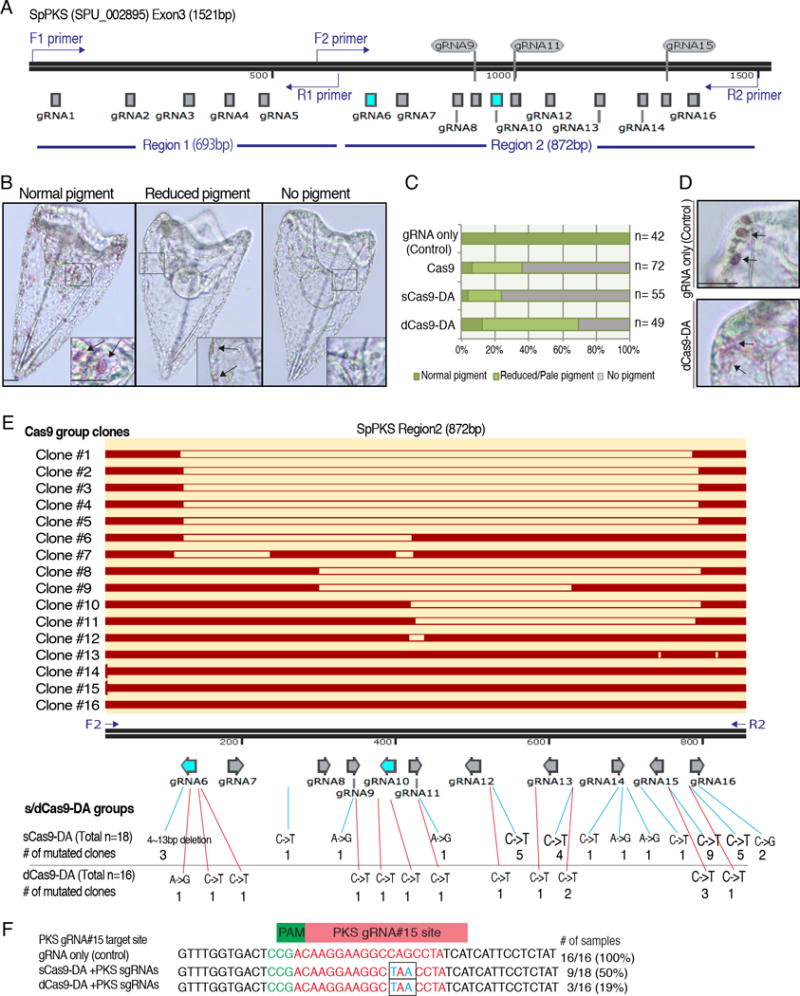
sgRNA efficiency and consensus test targeting the polyketide synthase (PKS) gene. A, A schematic diagram of the PKS 3rd exon showing the 16 gRNA target sites. gRNAs #6 and #10 were previously used (highlighted in blue), and shown to cause an albino phenotype when introduced with Cas9 mRNA (Oulhen and Wessel, 2016). Primers (indicated in blue) for the genomic PCR were designed at the end of 5′ and 3′ ends, respectively. B–D, Three resulting phenotypic categories of the PKS mutation. (B) Captured images of 3 dpf larvae injected with Cas9-DA mRNA and 16 PKS sgRNAs. Larvae with multiple pigmented cells (arrows) were counted as “Normal pigment”, followed by “Reduced pigment”, and “No pigment” groups as shown in graph C. The insert at the bottom corner of each image is a magnified view of each squared region. (C) Graph showing average group allocations (normal pigment, reduced pigment, no pigment) between injection groups. The results are the average of two independent experiments. n= total number observed. (D) A magnified view of the arms of pluteus larvae. Pigment cells in the dCas9-DA group were often pale in color compared the control (arrows). E, Sequencing results of the PKS 3rd exon. Since the primer for the Region1 was designed within a close proximity to the gRNA#1 target site, only the Region 2 was analyzed for sequencing to avoid any confusion. Original Region2 sequence is indicated with the black bar; each gRNA site is indicated with arrows directing toward each PAM sequence; matched/unedited sequence of each clone from the Cas9 group is shown in red bar. Blank spaces of each clone indicate a deletion in the sequence. The diagram was constructed by SnapGene software. Mutations found in the sCas9-DA and dCas9-DA groups are summarized underneath each gRNA site. For details regarding the mutations, please refer to Tables. S5–S7. F, Sequencing results of the gRNA#15 target site of PKS exon3 sequence. gRNA#15 target site is shown in red, PAM in green, mutations in blue. The C->T and G-A mutations (black squares) formed a stop codon in each mutated clone. # of samples (%) indicates number and proportion of mutation patterns. Due to the prevalence of polymorphisms in these wild-type animals, it should be noted that the original PKS (SPU#002895) sequence was modified based on the sequencing result of the control (gRNA only) group.
The 16 sgRNAs mixture (a total of 300ng/μl as a final injection mixture; approximately 19ng/μl each of sgRNA) was co-injected with Cas9, sCas9-DA, dCas9-DA, or no Cas9 mRNA (control) into fertilized eggs. The resultant larvae were then scored at 3 dpf to observe pigment formation (Fig. 5B, C). As reported in Oulhen and Wessel (2016), over 90% of the larvae introduced with Cas9 mRNA and the sgRNAs showed marked reduction in pigment formation. Remarkably, the sCas9-DA group showed an even higher reduction at over 95%. On the other hand, in the dCas9-DA group, approximately 70% of the larvae showed pigment formation. The color of pigment in these larvae often appeared pale compared to the control group (Fig. 5D), implying a possible partial inhibition of PKS gene function by DA as might be expected for nucleotides’ change in the gene. We also noted that with this lower concentration of gRNAs (approximately, 19ng/μl each of sgRNA) compared to the condition used above for Alx1 and Dsh experiments (Figs. 2–4; 300ng/μl each of gRNAs was used), fewer larvae showed developmental defects in all groups, suggesting that titrating the minimum amount of sgRNA is critical, especially to minimize cytotoxicity as well as precise Cas9 activity.
To analyze mutational efficiency, the genomic DNA of 8 individual larvae from each sample group was extracted, and the PKS exon3 region was amplified by PCR. Due to the long exon sequence (approximately, 1.5kbp in total), the first half (Region 1) and the latter half (Region 2) of the Eexon 3 was individually amplified (Fig. 5A). The resultant PCR product was subcloned and sequenced individually. In all groups, region 1, which contained gRNA#1~5, showed no significant mutations (n= 16 clones for each sample group). The gRNA#1 site was designed only within 25bp downstream of the forward PCR primer (Fig. 5A, F1 primer), which may have prevented amplification of the deleted/edited genomic sequence. We therefore decided to focus only on the region 2 for to avoid any possible confusion for the further analysis.
Region 2, which contained gRNA#6~16, showed several patterns of deletions in the Cas9 group (Fig. 5E; Table S5): 8 (50%) out of 16 clones showed the DNA double-strand cut at the gRNA#16 site, 7 (44%) at the gRNA#6 site, 4 (25%) at the gRNA#11 site, 2 at the gRNA#8 site, 1 at the gRNA#10 site, and 1 at the gRNA#13 site. Most clones showed two or more independent mutations (e.g. cut both at the gRNA#16 and #6 sites). Although it is hard to test for efficiency of the gRNA sites between gRNAs #6 and #16 as the entire Region 2 was often cut out, we conclude that gRNAs #6 and #16 sites were favored by Cas9/Cas9-DA. In the sCas9-DA group that showed a strong albino phenotype, a small deletion (4~13bp) was found at the gRNA#6 site in 3 (17%) out of 18 clones, each of which caused a frame shift and stop codons in the downstream sequence.
The remaining clones in the sCas9-DA group showed no deletion. However, single nucleotide mutations were found in most clones (83%) of the sCas9-DA group, often at multiple sites and at different gRNA sites for each clone. Slightly less than half of the clones (7/16 clones; 44%) exhibited mutations in the dCas9-DA group (Fig. 5E; Table S6), reflecting the modest phenotype in this group. In both groups, the most frequent mutation was found at the gRNA#15 site (50% for sCas9-DA and 19% for dCas9-DA groups; Fig. 5F). A combination of the C->T mutation at 12bp and G->A mutation (read as C->T mutation at the complementary strand) at 14bp upstream from the PAM sequence appeared to cause a stop codon in those clones. On the other hand, no significant mutation was found in any gRNA target site in the control group (gRNA only; Table S7).
Through these results, we conclude that small deletions in the sCas9-DA group, as well as an induced stop codon at the gRNA#15 site in the both s/dCas9-DA groups may have led to an albino phenotype. While other nucleotide mutations also caused amino acid alterations, those alterations did not result in a stop codon. Since it is unclear at present what amino acids of PKS are critical for its enzymatic function, it is difficult to be more specific about what mutations were essential for the albino phenotype.
CRISPR/Cas9-DA technology as a promising method to manipulate a genome
In this study, CRISPR/Cas9 and CRISPR/sCas9-DA demonstrated similar genome editing efficiencies for endogenous targets at around 50% ~90% with fewer side effects in the CRISPR/sCas9-DA groups, even with high dosage (e.g. Injection stock of 500ng/μl Cas9/Cas9-DA mRNA and total 600ng/μl sgRNAs used in Figs. 2–4 experiments). Sequencing results suggest that conventional CRISPR/Cas9 targeting to Alx1 induced a large deletion, whereas CRISPR/sCas9-DA induced C to T conversion with comparatively smaller deletions. These results suggest that strong nuclease activities may be present in the embryonic cells of sea urchins, which may have caused higher cell toxicity in the CRISPR/Cas9 groups under this experimental condition. Interestingly, several mutations and insertions of random nucleotides were also noted around Cas9-mediated DNA digestion sites (Fig 4A, Cas9+Alx1 sgRNA). Although we currently do not know the direct cause or mechanism of this event, similar phenomena of extra digestion and filling-in were also observed when DNA digestion was induced by the Minos transposon in the sea urchin embryo (Sasakura et al., 2010). It is thus intriguing to speculate that the sea urchin embryo could show strong nuclease and filling-in activities to repair the genome, perhaps, when they are over-accessed by the DNA-digestion enzymes such as Cas9 or transposon insertion. Further, even a single nickase activity of Cas9 (sCas9) in some cases led to a deletion in the genome when combined with DA. Although the detailed mechanism requires future testing, we speculate that DA’s binding to the nicked DNA strand likely opened the double strands, and induced endogenous nuclease activities, resulting in a small deletion of the target DNA. To avoid these unexpected deletions, we also tested a lower amount of sgRNAs for the PKS gene (300ng/μl in total of 16 gRNAs), in which we saw lower frequency of deletion in the sCas9-DA group, or no deletion in the dCas9-DA group (Fig. 5). These results suggest that careful titration of sgRNAs is key to avoid cytotoxicity, and for precise genome editing.
Sequencing results also revealed that the CRISPR/Cas9-DA system appears to favorably edit positions between 7~20 bases upstream of the PAM sequence to the gRNA. Other nucleotide changes were found less frequently. Although we did not sequence entire genomes in this study, we found no evidence of consistent mutations (other than potential polymorphisms) outside of the gRNA target sequence overall, indicating the controlled nature of DA’s editing capacity. However, multiple point mutations from a single clone of Dsh and PKS PCR products were found in this study (Figs. 4B and 5F), suggesting that DA may have functioned on multiple sites of the gRNA site. This may have occurred because a relatively long linker sequence (279 bp) was used in this study, which likely enabled flexibility in the DA to reach nearby DNA sequences. However, this does not answer why other cytosines within the gRNA target sequence were not edited in this study, which also differs from results obtained in yeast (Nishida et al., 2016; Komor et al., 2016).
Other questions to be explored include identifying the three-dimensional orientation of CRISPR/Cas9-DA activity. We initially only observed mutations upstream of the PAM sequence when a single sgRNA site was targeted for each exon. However, this did not appear to be the case when 16 gRNA sites were targeted simultaneously within a single PKS exon (Fig. 5, Table S6). This may be caused by the simultaneous binding of multiple CRISPR/Cas9-DAs within a single exon, which might have altered the conformation of the PKS exon3 genomic locus. Further detailed analysis with titrated amounts of gRNA targeting, along with utilizing Cas9-DA with variable lengths in the linker domain, may provide the opportunity to engineer a system that will allow for increasingly refined gene editing.
At this stage, the CRISPR/Cas9-DA system requires further refinement for more general use in animal development. However, this system is a straightforward approach compared to methods that require the cutting and pasting of the genome, and indeed shows marked reduction in cytotoxicity, even in applications of higher doses of Cas9-DA and sgRNAs. Through further studies and refinement of methodologies, this proposed technology holds the potential to be advantageous in numerous applications, including human gene therapies that prefer minimum modifications to the genome.
Experimental Procedures
Animals
Strongylocentrotus purpuratus (sea urchins) were obtained by Pat Leahy, Kerchoff Marine Laboratories, California Institute of Technology or by Josh Ross, South Coast Bio-Marine LLC. Long Beach, California, USA. Eggs were collected in seawater (SW) and sperm was collected dry by injections of 0.5 M KCl to induce spawning. Eggs and embryos/larvae were cultured in SW at 16 °C. For microinjections, fertilization was performed in the presence of 1mM 3-aminotriazol (3AT; Sigma, St. Louis, MO, USA) to inhibit crosslinking of the fertilization envelopes.
Preparation of Cas9 and Cas9-Deaminase Constructs
The Cas9 construct was obtained from Addgene (plasmid #51307), which has been previously reported to function in Xenopus tropicalis (Nakayama et al., 2013). This Cas9 construct showed increased translation efficiency, as well as nuclear localization in our system compared to other Cas9 constructs optimized for yeast or mammalian cells (data not shown). This construct was used for basic CRISPR approaches, as well as for CRISPR-deaminase experiments. Petromyzon marinus cytosine deaminase 1 (PmCDA; GenBank accession #A5H718) was obtained as a courtesy of Drs. Keiji Nishida and Akihiko Kondo at Kobe University (Nishida et al., 2016). To create the Cas9-deaminase (DA) constructs, DA and its flanking SH3 linker sequences were fused to Cas9 at the C-terminus (Fig. 1C) (Panjarian et al., 2013) by the In-Fusion HD Cloning kit with primers summarized in Table S1 (#639648, Clontech, USA).
Two different Cas9-DA constructs were prepared (Fig. 1C). The first construct was mutated at one of two nickase sites (D10A) to construct sCas9-DA, which prevents double stranded breaks and thereby limits NHEJ activity and causes a nick, allowing the loosening of the DNA strand to increase deaminase’s accessibility to the target site. Another construct, dCas9-DA, was mutated at both nickase sites to completely inactivate Cas9’s cleaving activities (H840A). To construct these plasmids, two separate rounds of mutagenesis were performed with specifically designed primers (Table S1) using the Agilent online tool: http://www.genomics.agilent.com/primerDesignProgram.jsp. Mutagenesis was performed using QuikChange II Site-Directed Mutagenesis Kit (cat. #200523-5) by following the manufacturer’s protocol.
For in vitro transcription (IVT), Cas9 was linearized with XhoI (Cas9) or NotI (sCas9, dCas9-DA) and transcribed using the mMESSAGE mMACHINE® SP6 Transcription Kit (Thermo Fisher; catalog #AM1340) by following manufacturer’s protocol, with incubation for 5 hours at 37°C, followed by DNaseI treatment for 15 minutes at 37°C. RNA was then purified using the RNeasy Micro Kit from Qiagen (Cat. #74004) following the manufacturer’s protocol.
sgRNA designing and synthesis
The following genes were targeted: SpAlx1 (SPU#025302), which is essential for skeletogenesis (Sharma and Ettensohn, 2010) and SpDsh (SPU#025646), which is critical for endomesodermal specification and gastrulation (Byrum et al., 2009). Two individual sgRNAs were designed per target gene. Due technical difficulties to find promising targets both work for Cas9 and Cas9-DA, Alx1-gRNA#1 and -gRNA#2 were designed to target 500–519bp and 1128–1147bp of SpAlx1 ORF (Table S3) respectively, and Dsh-gRNA#1 and -gRNA#2 were designed to target 173–192bp and 1299–1318bp of SpDsh ORF (Table S4) respectively. For the gRNA saturation assay targeting PKS (SPU#002895), 16 gRNA target sites were designed within the single 3rd exon (Fig. 5A) using CRISPRscan software (crisprscan.org). For each gRNA design, twenty base-pair gRNA targets were designed with enriched guanine residues, as well as cytosine that may be converted to thymine by deaminase and induce stop codon(s). Additionally, these sequences were flanked at the 3′ end by a PAM sequence (NGG) where the Cas9 binds. Each forward primer of length 40–70 base pairs (Table S1) included the T7 promoter and the designated gRNA sequence. Each forward primer and partner reverse primer for all sgRNA templates were used to amplify 119 base pairs of sgRNA templates using pUC57 vector as a template (Addgene # 4509) (Table S1). Each sgRNA template was directly amplified by PCR from the pUC57 vector using the forward primer that contained the T7 promoter sequence and each gRNA target sequence. The reverse primer contains the gRNA scaffold sequence. sgRNA templates for in vitro transcription (IVT) were run on a 1.5% gel and purified to yield linear DNA fragments around 120 base pairs, which were transcribed using the MEGAscript T7 Transcription Kit from Ambion (Cat No. AM1334) by following the manufacturer’s protocol. The resulting sgRNAs were purified using the Qiagen MiRNeasy kit (Cat No./ID 217004). The same sgRNAs were used both for conventional CRISPR and CRISPR-DA analyses.
Microinjection
To check the quality of mRNA and sgRNAs prior to each microinjection, 1 μl each of the final injection mix containing Cas9/sCas9-DA/dCas9-DA mRNA, sgRNAs, membrane-mCherry-mRNA were run on a RNase-free 2% agarose gel, alongside with the stock mRNA/sgRNAs control treatments. Only injection mixes that produced specific bands of mRNAs/sgRNAs at the expected sizes were used for injection. Upon fertilization in 1 mM 3-aminotriazol (3AT) SW, zygotes were injected with approximately 8~12 pL of the injection mixture, containing a final concentration of 500 ng/μl of Cas9, sCas9-DA or dCas9-DA mRNAs, 300ng/μl each of sgRNA1 and sgRNA2, and 500ng/μl of membrane-mCherry as an injection marker. For PKS gene targeting, 300ng/μl of 16 sgRNA mixture was used instead. Injected embryos were incubated at 16°C until they were analyzed at 3 dpf at the pluteus larval stage. Negative controls consisted of the same conditions, but without sgRNAs or Cas9/sCas9-DA/dCas9-DA mRNAs. To minimize possible technical variations of microinjections as well as batch differences, all sample groups were injected and analyzed at the same time for each cycle of experiments.
Phenotype observation, DNA extraction, and Sequencing
Injected embryos/larvae were observed for developmental phenotypes and scored manually under the microscope at 3 dpf during the time point in which each of the targeted genes is expected to have a critical function. The embryos/larvae were imaged by wide-field microscopy (Zeiss Axioplan). Scoring results were averaged from at least three independent experiments.
To extract genomic DNA from an individual embryo/larva, each embryo/larva expressing membrane-mCherry fluorescence for injection was treated with a final concentration of 2mg/mL proteinase K (ThermoFisher Cat. No. EO0491) in TE at 50 °C for 3 hours, followed by heat inactivation at 94 °C for 10 minutes. Each gRNA-targeted genomic region and its flanking sequence (approximately ~200bp in total) was amplified by PCR followed with high fidelity platinum Taq polymerase (Promega, USA) using primers summarized in Table S1. The amplified PCR products were then subcloned into the pGEM-T vector (Promega) by following the manufacturer’s protocol, and sequenced to detect in/del mutations. 8 individual embryo/larva of each sample group were randomly selected from each experimental cycle (24 genomic DNA samples as a total of 3 injection cycles unless individually indicated) for genomic extraction. Among those genomic DNA samples, 3~5 samples was subjected to PCR and subcloning in to the pGEM-T vector, and 1~3 clones per embryonic/larval genome sample were subjected to sequencing. To compare results quantitatively among the sample groups, injection, phenotype scoring and genomic extraction were always performed at once for the same target groups (e.g. Alx1, Dsh, PKS targeting groups).
Supplementary Material
Highlights.
Cas9-Deaminase modifies single nucleotides by conversion without dsDNA cleavage.
Cas9-Deaminase most effectively converts cytosine to thymine between 7–20 bases upstream of the PAM sequences of the targeted sgRNA site.
CRISPR/Cas9-Deaminase edits the genome at a similar rate to standard CRISPR/Cas9 systems.
Gene modification by Cas9-Deaminase was demonstrated by targeting Alx1, Dishevelled, and polyketide synthetase (Pks).
Acknowledgments
We thank Dr. Keiji Nishida at Kobe University for sharing a Deaminase construct, Yiannis Savvi and Robert Reenan at Brown University for many helpful discussions, as well as members of PRIMO at Brown University for generous support.
Funding: This work was supported by an American Heart Association Scientist Development Grant to MY (14SDG18350021), and by NIH (2R01HD028152; R21HD075561) grants to GMW.
Footnotes
Author contributions: S.S. and N.S were responsible for concepts, experimental design and implementation, data analysis, manuscript preparation; G.M.W. was responsible for conceptualization, experimental design, manuscript editing. M.Y. was responsible for conceptualization, experimental design and implementation, data analysis, manuscript construction and editing.
References
- Bassett AR, Tibbit C, Ponting CP, Liu JL. Highly Efficient Targeted Mutagenesis of Drosophila with the CRISPR/Cas9 System. Cell Reports. 2013;4:220–228. doi: 10.1016/j.celrep.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Bertolini S, Pisciotta L, Seri M, Cusano R, Cantafora A, Calabresi L, Franceschini G, Ravazzolo R, Calandra S. A point mutation in ABC1 gene in a patient with severe premature coronary heart disease and mild clinical phenotype of Tangier disease. Atherosclerosis. 2001;154:599–605. doi: 10.1016/s0021-9150(00)00587-6. [DOI] [PubMed] [Google Scholar]
- Byrum CA, Xu R, Bince JM, McClay DR, Wikramanayake AH. Blocking Dishevelled signaling in the noncanonical Wnt pathway in sea urchins disrupts endoderm formation and spiculogenesis, but not secondary mesoderm formation. Dev Dyn. 2009;238:1649–1665. doi: 10.1002/dvdy.21978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carte J, Wang R, Li H, Terns RM, Terns MP. Cas6isanendoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–3496. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier E, Doudna JA. Biotechnology: Rewriting a genome. Nature. 2013;495:50–51. doi: 10.1038/495050a. [DOI] [PubMed] [Google Scholar]
- Chen KM, Harjes E, Gross PJ, Fahmy A, Lu Y, Shindo K, Harris RS, Matsuo H. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature. 2008;452(7183):116–119. doi: 10.1038/nature06638. [DOI] [PubMed] [Google Scholar]
- Clancy S. Genetic mutation. Nature Education. 2008;1:1. [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes H, Pastor M, Bochtler M. Type II and type V CRISPR effector nucleases from a structural biologist’s perspective. Postepy Biochem. 2016;62:315–326. [PubMed] [Google Scholar]
- Garneau JE, Dupuis MÈ, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- Gesner EM, Schellenberg MJ, Garside EL, George MM, MacMillan AM. Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nature Struct Mol Biol. 2011;18:688–692. doi: 10.1038/nsmb.2042. [DOI] [PubMed] [Google Scholar]
- Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell. 2002;10(5):1247–1253. doi: 10.1016/s1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S, Nishimasu H, Ishitani R, Nureki O. Structural Basis for the Altered PAM Specificities of Engineered CRISPR-Cas9. Molecular Cell. 2016;61:886–894. doi: 10.1016/j.molcel.2016.02.018. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Hiroi Y, Hosoda T, Utsunomiya T, Matsuo S, Ito T, Inoue J, Sumiyoshi T, Takano H, Nagai R, Komuro I. Novel point mutation in the cardiac transcription factor CSX/NKX2.5 associated with congenital heart disease. Circ J. 2002;66:561–563. doi: 10.1253/circj.66.561. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato L, Stanlie A, Begum NA, Kobayashi M, Aida M, Honjo T. An Evolutionary View of the Mechanism for Immune and Genome Diversity. J Immunol. 2012;188:3559–3566. doi: 10.4049/jimmunol.1102397. [DOI] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNAcleavage. Nature. 2016;533:420–4. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Su YH. Genome editing in sea urchin embryos by using a CRISPR/Cas9 system. Dev Biol. 2016;409(2):420–428. doi: 10.1016/j.ydbio.2015.11.018. [DOI] [PubMed] [Google Scholar]
- Lada AG, Krick CF, Kozmin SG, Mayorov VI, Karpova TS, Rogozin IB, Pavlov YI. Mutator effects and mutation signatures of editing deaminases produced in bacteria and yeast. Biochemistry Moscow. 2011;76:131–146. doi: 10.1134/s0006297911010135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, D́ıez-Villaseñor C, Garćıa-Martínez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Fish MB, Fisher M, Oomen-Hajagos J, Thomsen GH, Grainger RM. Simple and efficient CRISPR/Cas9-mediated targeted mutagenesis in Xenopus tropicalis. Genesis. 2013;51:835–843. doi: 10.1002/dvg.22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, Mochizuki M, Miyabe A, Araki M, Hara KY, Shimatani Z, Kondo A. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353:6305. doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- Oulhen N, Wessel GM. Albinism as a visual, in vivo guide for CRISPR/Cas9 functionality in the sea urchin embryo. Mol Reprod Dev. 2016;83(12):1046–1047. doi: 10.1002/mrd.22757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Sasakura Y, Yaguchi J, Yaguchi S, Yajima M. Excision and Transposition Activity of Tc1/mariner Superfamily Transposons in Sea Urchin Embryos. Zool Sci. 2010;27:256–262. doi: 10.2108/zsj.27.256. [DOI] [PubMed] [Google Scholar]
- Sashital DG, Jinek M, Doudna JA. An RNA induced conformational change required for CRISPR RNA cleavage by the endonuclease Cse3. Nature Struct Mol Biol. 2011;18:680–687. doi: 10.1038/nsmb.2043. [DOI] [PubMed] [Google Scholar]
- Sugi T. Genome Editing in C. elegans and Other Nematode Species. Int J Mol Sci. 2016;17(3):295. doi: 10.3390/ijms17030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjarian S, Iacob RE, Chen S, Wales TE, Engen JR, Smithgall TE. Enhanced SH3/Linker Interaction Overcomes Abl Kinase Activation by Gatekeeper and Myristic Acid Binding Pocket Mutations and Increases Sensitivity to Small Molecule Inhibitors. J Biol Chem. 2013;288:6116–6129. doi: 10.1074/jbc.M112.431312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418(6893):99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016;26:174–182. doi: 10.1101/gr.197046.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma T, Ettensohn CA. Activation of the skeletogenic gene regulatory network in the early sea urchin embryo. Development. 2010;137:1149–1157. doi: 10.1242/dev.048652. [DOI] [PubMed] [Google Scholar]
- Smith HC, Bennett RP, Kizilyer A, McDougall WM, Prohaska KM. Functions and regulation of the APOBEC family of proteins. Semin Cell Dev Biol. 2012;23:258–268. doi: 10.1016/j.semcdb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, Hatanaka F, Yamamoto M, Araoka T, Li Z, Kurita M, Hishida T, Li M, Aizawa E, Guo S, Chen S, Goebl A, Soligalla RD, Qu J, Jiang T, Fu X, Jafari M, Esteban CR, Berggren WT, Lajara J, Nuñez-Delicado E, Guillen P, Campistol JM, Matsuzaki F, Liu GH, Magistretti P, Zhang K, Callaway EM, Zhang K, Belmonte JC. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540(7631):144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Preamplume G, Terns MP, Terns RM, Li H. Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure. 2011;19:257–264. doi: 10.1016/j.str.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Quiocho FA. Complexes of adenosine deaminase with two potent inhibitors: X-ray structures in four independent molecules at pH of maximum activity. Biochemistry. 1998;37:8314–8324. doi: 10.1021/bi980324o. [DOI] [PubMed] [Google Scholar]
- Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.