

Abstract
Background
Computational network biology is an emerging interdisciplinary research area. Among many other network approaches, probabilistic graphical models provide a comprehensive probabilistic characterization of interaction patterns between molecules and the associated uncertainties.
Results
In this article, we first review graphical models, including directed, undirected, and reciprocal graphs (RG), with an emphasis on the RG models that are curiously under-utilized in biostatistics and bioinformatics literature. RG’s strictly contain chain graphs as a special case and are suitable to model reciprocal causality such as feedback mechanism in molecular networks. We then extend the RG approach to modeling molecular networks by integrating DNA-, RNA- and protein-level data. We apply the extended RG method to The Cancer Genome Atlas multi-platform ovarian cancer data and reveal several interesting findings.
Conclusions
This study aims to review the basics of different probabilistic graphical models as well as recent development in RG approaches for network modeling. The extension presented in this paper provides a principled and efficient way of integrating DNA copy number, DNA methylation, mRNA gene expression and protein expression.
Keywords: Directed graph, Undirected graph, Chain graph, Reciprocal graph, Causality
Background
This article starts with a comprehensive review of graphical models and recent development in constructing biological networks using reciprocal graphs (RG’s). In order to integrate multi-omics data including proteomics, we extend the work in [1] utilizing fundamental biological knowledge and the factorization of the joint distribution.
Computational network biology (CNB) is an emerging research field that encompasses theory and applications of network models to systematically study different molecules (DNA, RNA, proteins, metabolites and small molecules) and their complex interactions in living cells. CNB provides new insights in diverse research areas including system biology, molecular biology, genetics, pharmacology and precision medicine. The development of high-throughput profiling technologies allows for the interrogation of the status of many molecules in a cell at the same time, which makes it possible to jointly model these cell components with network modeling approaches. CNB is an interdisciplinary research area and has received contributions from researchers with various background. In this paper, we focus on network modeling approaches from a statistical perspective, namely, probabilistic graphical models (PGM) which allow a complete probabilistic description of interaction patterns between molecules and the associated uncertainties when estimating such interactions from noisy genomic and/or proteomic data.
PGM are probabilistic models for multivariate random variables whose conditional independence (also known as Markov property) structure is characterized by an underlying graph. PGM provides a concise, complete and explicit representation of joint distribution and allows for convenient Gibbs factorization of the density (if it exists) and hence local computations. The Markov property can be directly read off the graph through the notion of graph separation. PGM is closely related to causal inference, path analysis and expert system and finds a wide range of applications in political sciences, economics, genetics, biology, physics and psychology.
The most commonly used graphs are undirected graphs (UG) and directed acyclic graphs (DAG). UG represents symmetric interactions between variables whereas DAG allows for asymmetric ones that can be potentially interpreted as cause-effect relationships. Both UG and DAG are a special case of a more general graphical model known as chain graphs (CG) [2, 3] which allow for both symmetric and asymmetric conditional independence relationships. Despite of the flexibility of CG, the directed relationships are still constrained to be (block-) recursive, that is, reciprocal causality is prohibited. However, cyclic causal relationship are fundamental and ubiquitous in science, including, for example, the law of supply and demand in economics or feedback mechanism in gene regulatory networks. Spirtes [4] and Koster [5], in their respective seminal work, independently proposed coherent non-recursive graphical models including directed cycles of which the joint distribution was previously thought ill-defined ([6], p. 72). Spirtes [4] developed directed cyclic graphical (DCG) models which include DAG’s as a special case, and [5] proposed an even more general class of graphical models, reciprocal graphical (RG) models, which include CG (hence UG and DAG) and DCG as special cases. The relationships between the aforementioned graphs are depicted as a Venn’s diagram in Fig. 1.
Fig. 1.
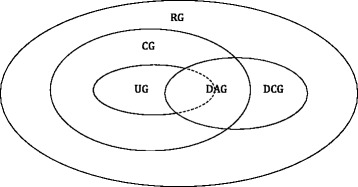
Venn’s diagram of different graphs. UG: undirected graph. DAG: directed acyclic graph. DCG: directed cyclic graph. CG: chain graph. RG: reciprocal graph
In this paper, we focus on RG’s due to their statistical generality, the capability of modeling feedback loops and their still underappreciated popularity in the biostatistics and bioinformatics literature. The rest of this paper is structured as follows. We first review the basics of UG, DAG and RG in this section. In the next section, we provide a comprehensive review of novel Bayesian approaches in modeling molecular networks using RG. Then, we extend the RG approach in [1] to modeling The Cancer Genome Atlas (TCGA) ovarian cancer data that are measured on three different levels: DNA, RNA and proteins. The extension is non-trivial: we factorize an RG into two separate RG’s (one for DNA and RNA and the other for RNA and protein) and make coherent joint inference by exploiting two known biochemical processes: DNA transcribes to RNA and RNA in turn translates to protein, the latter also allowing us to estimate the direction of the relationship between RNA and protein which informs the directionality of the regulatory relationships between proteins. To the best of our knowledge, we are among the first to reconstruct multi-omics functional networks that consider mass spectrometry proteomics data generated by the CPTAC.
Overview of directed, undirected and reciprocal graphical models. Graphical models are a class of statistical models for a set of random variables Z=(Z1,…,Zp)T that provide a graphic representation of conditional independence relationships among the variables. Using graphical models allow practitioners to simplify inferences, obtain parsimonious solutions via variable selection, enable local computations, and define a framework for causal inference.
In a graphical model, variables are represented by a set of nodes and their associated interactions are represented by edges. A missing edge usually represents the conditional independence between the corresponding pair of random variables conditional on a certain set of other variables. The conditioning set depends on the chosen type of graph. Here, we provide a minimal set of terminologies required to describe the Markov properties in later sections.
A graph is defined by a set of nodes V={1,…,p} and a set of directed and undirected edges E=Ed∪Eu. For a pair of nodes i,j∈V, we denote an undirected edge by i−j or {i,j}∈Eu and a directed edge by i→j or (i,j)∈Ed. We write i↦j if there exists a path from i to j. A path is an ordered sequence (i0,…,iK) of distinct nodes except possibly i0=iK such that {ik−1,ik}∈E or (ik−1,ik)∈E for k=1,…,K. A cycle is then a path with i0=iK. A path component is a set of nodes that are all connected by an undirected path. The boundary of a node i is bd(i)={j∣{j,i}∈E or (j,i)∈E} and the boundary of a subset A⊆V is . A node i is said to be a parent of node j, if (i,j)∈E. The set of parents of j is denoted by pa(j). The ancestors of node i are an(i)={i}∪{j|j↦i}. The descendants of node i, denoted by de(i), are the nodes i such that there is a path from i to j. The non-descendants of i are nd(i)=V∖(de(i)∪{i}). For A⊆V, the induced subgraph by A is where EA=E∩(A×A). A subset A⊆V is anterior if bd(A)=∅ and the minimal anterior set of A is denoted by an(A). Note that we use an() to denote both ancestors and minimal anterior set; it should not cause any confusion because in fact an({i})=an(i). Notice that (minimal) anterior set [5] is equivalent to (minimal) ancestral set in [7]. A graph is complete if all nodes are joined by an edge. A complete subgraph that is maximal with respect to the subset relation is called clique. A useful procedure in defining Markov properties of graphs with directed edges is moralization. The moralization is performed in two steps. Make the boundary of each path component complete by adding undirected edges and then replace all directed edges by undirected ones. We call the resulting undirected graph moral graph, denoted by .
We say that graph is a undirected graph (UG) if E=Eu contains only undirected edges. And graph is said to be a directed acyclic graph (DAG) if E=Ed contains only directed edges and no cycles. A reciprocal graph (RG) is a graph that can have both directed and undirected edges and cycles, with the only restriction being no directed edges between nodes in the same path component [5]. In the following, we provide a concise overview of each type of graph. More comprehensive and technical details of the contents can be found in [7] and [6].
Undirected graphical models
Let P denote the joint distribution of Z=ZV=(Z1,…,Zp)T. We say P has a Gibbs factorization (or simply factorization) with respect to a UG if its density f(Z) can be written as
| 1 |
where is the collection of cliques of and ψC is an arbitrary nonnegative function on the domain of ZC for .
Not surprisingly, the factorization property of P is closely related to its Markov properties; in fact, the former implies the latter. The Markov property of a UG relies on a notion of graph separation. For three disjoint subsets A,B,C∈V of nodes, A is said to be separated from B by C if every path between A and B includes a node in C. The global Markov property of a UG is given in the following definition.
Definition 1
(Global Markov property) Given a UG , the joint distribution P of ZV is global Markov with respect to if for any three disjoint subsets A, B and C of V, ZA is independent of ZB given ZC, written as ZA ⊥ ⊥ZB∣ZC whenever C separates A and B.
The global Markov property always implies the local Markov property.
Definition 2
(Local Markov property) Given a UG , the joint distribution P of ZV is local Markov with respect to if for any node i∈V, Zi ⊥ ⊥ZV∖{bd(i),i}∣Zbd(i).
The local Markov property in turn always implies the pairwise Markov property.
Definition 3
(Pairwise Markov property) Given a UG , the joint distribution P of ZV is pairwise Markov with respect to if for any pair of nodes i,j∈V, Zi ⊥ ⊥Zj∣ZV∖{i,j}.
In summary, factorization in (1) ⇒ global Markov property ⇒ local Markov property ⇒ pairwise Markov property. A natural question to ask is: is the relation also true backwards? The answer is yes when P has a positive and continuous density, which is summarized in the following theorem, often referred to as Hammersley-Clifford theorem [8].
Theorem 1
(Hammersley-Clifford) Suppose a probability distribution P has a positive and continuous density f. Then P satisfies the pairwise Markov property with respect to a UG if and only if f factorizes according to .
When P=N(0,Λ−1) is multivariate Gaussian with precision matrix Λ, undirected graphical models are defined by zero constraints on Λ. There is a one-to-one correspondence between the graph structure and the zero patterns in Λ. Specifically, the precision matrix Λ is constrained to the cone of positive definite matrices with off-diagonal entry Λij=0 if and only if there is a missing edge in between nodes i and j. Therefore, in the Gaussian case, learning the graph structure is equivalent to recovering the zero patterns in Λ. Interested readers are referred to recent Bayesian approaches [9–11] and non-Bayesian approaches [12, 13].
Directed graphical models
We say the distribution P factorizes with respect to a DAG if its density f(Z) can be written as
| 2 |
Similar to UG’s, the Markov property of a DAG can be also defined by the concept of graph separation. However, unlike UG’s, the separation is more complicated in DAG’s. There are two different but equivalent approaches in describing separation in a DAG. Pearl [14] introduced the notion of d-separation characterized by several conditions and then defined the global Markov property of a DAG in the same way as that of an UG except that separation is replaced by d-separation. Another approach [3] is based on the moralization and graph separation in UG’s. We elaborate more on the latter because it will be used again in defining Markov properties of RG’s. Similar to the previous section, we introduce global, local and pairwise Markov properties of DAG and then state the relationships between them. Recall that denotes the moralized graph.
Definition 4
(Directed global Markov property) Given a DAG , the joint distribution P of ZV is global Markov with respect to if for any three disjoint subsets A, B and C of V, ZA is independent of ZB given ZC, written as ZA ⊥ ⊥ZB∣ZC whenever C separates A and B in .
Definition 5
(Directed local Markov property) Given a DAG , the joint distribution P of ZV is local Markov with respect to if for any node i∈V, Zi ⊥ ⊥Znd(i)∣Zpa(i).
Definition 6
(Directed pairwise Markov property) Given a DAG , the joint distribution P of ZV is pairwise Markov with respect to if for any pair of nodes i,j∈V, Zi ⊥ ⊥Zj∣Znd(i)∖{j}.
Similar to UG’s, it is easy to show that factorization in (2) ⇒ directed global Markov property ⇒ directed local Markov property ⇒ directed pairwise Markov property. In fact, directed global and local Markov properties are equivalent [15]. Moreover, a theorem similar to Hammersley-Clifford’s theorem holds for DAG’s as well.
Theorem 2
([7]) Suppose a probability distribution P has a continuous density f. Then P satisfies the local Markov property with respect to a DAG if and only if f factorizes according to . Furthermore, if the density f is positive, local and pairwise Markov properties are equivalent.
In a Gaussian DAG, each conditional distribution in (2) is a linear model
where N(·∣μ,σ2) is a Gaussian density with mean μ and variance σ2. Essentially, a DAG is decomposed as a system of recursive and independent linear regressions for which the graph structure inference can be performed using standard model selection technique.
However, there is an important caveat in using DAG’s for statistical inference. That is the the notion of Markov equivalence – different DAG’s may induce the same set of conditional independence relationships. For example, the graphs i←j→k and i←j←k induce the same conditional independence relationship, i ⊥ ⊥k∣j. Both are equivalent to the UG i−j−k. In fact, any perfect DAG (i.e. a DAG without configuration i→j←k, that is, all parents are married) is Markov equivalent to a decomposable UG (i.e. UG in which all cycles of length four or more have a chord, which is an edge that is not part of the cycle but connects two nodes of the cycle). Conversely, given an undirected decomposable graph, there exists a Markov-equivalent perfect DAG; see [7] or [16]. One approach to avoid repeatedly analyzing Markov equivalent DAG’s is adopting a prior ordering of the nodes. The ordering may be obtained from subjective prior knowledge such as a reference network [17] or more objectively from an ordering learning algorithm such as order-MCMC [18]. For other recent DAG approaches, see [19–21] or [22].
Reciprocal graphical models, simultaneous equation models and path diagrams
We introduce the global Markov property for RG’s in the following definition and omit the local and pairwise Markov properties since they are not as practically useful as in UG and DAG.
Definition 7
(Reciprocal global Markov property) Given a RG , the joint distribution P of ZV is global Markov with respect to if for any three disjoint subsets A, B and C of V, ZA is independent of ZB given ZC, written as ZA ⊥ ⊥ZB∣ZC whenever C separates A and B in .
Similarly to UG and DAG, there is also a Hammersley-Clifford-type theorem for RG’s ([5], Theorem 3.4).
However, while it is a beautiful theoretical result on the equivalence between the global Markov property and the Gibbs factorization in RG, the latter is difficult to work with in practice due to its complex form. Fortunately, in the Gaussian case, there is an easy connection between RG and simultaneous equation models (SEMs). To describe this connection, let Z=(Y,X) be divided into two sets of variables Y and X and consider an SEM,
| 3 |
where we assume the following five conditions:
(Y,X)∼N(0,Σ);
E ⊥ ⊥X;
I−A is invertible;
Cov(E) is diagonal;
Ψ=Cov(X) is block diagonal with full diagonal blocks.
To link an SEM to an RG, we draw a path diagram of SEM by the following rules:
-
(i)
define nodes V={1,…,p,p+1,…,p+q} which represent (Y,X)=(Y1,…,Yp,X1,…,Xq);
-
(ii)
draw directed edges Ed={(j,i)∣aij≠0 or bi,j−p≠0}; and
-
(iii)
draw undirected edges Eu{{i,j}∣Ψi−p,j−p≠0}.
In words, (i) we introduce a node for each variable in (Y,X) with nodes j=1,…,p corresponding to Yj and p+j corresponding to Xj for j=1,…,q; (ii) nodes i=1,…,p (i.e. Yi nodes) become targets of directed edges from node j if the corresponding aij≠0 or bi,j−p≠0; (iii) we introduce undirected edges between Xi and Xj (i.e. nodes i+p and j+p) if Ψi,j≠0. Figure 2a shows an example of an RG with p=2 and q=3,
| 4 |
Fig. 2.
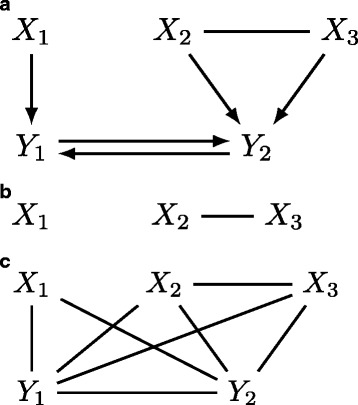
Illustration. a Path diagram for SEM with configuration in (4). b Moral subgraph induced by an(X1,X2,X3)={X1,X2,X3}. c Moral graph
with ∗ indicating non-zero elements. With conditions C1-C5, the Markov properties of an SEM can be read off its path diagram.
Theorem 3
([5]) Let Y=AY+BX+E be an SEM satisfying conditions C1-C5. The joint distribution P of (Y,X) is global Markov with respect to the path diagram .
For example, the SEM with configuration in (4) implies X1 ⊥ ⊥X2,X3 which is evident by looking at the moral subgraph in Fig. 2b. Clearly, the empty set ∅ separates X1 and X2,X3. We can also find X1 ⊥ ⊥X2,X3∣Y1,Y2 by moralizing the entire path diagram (Fig. 2c) since set {Y1,Y2} separates X1 and X2,X3 in . However, we remark that the class of SEMs satisfying conditions C1-C5 is a subclass of RG. For instance, i→j−k←l is a CG (hence an RG) but does not correspond to an SEM. Similarly to DAG’s, DCG’s (or more generally RG’s) can also be divided into Markov equivalence classes; see [23–28] for characterization and learning of Markov equivalence classes of DCG’s.
Methods
In this section, we will review recent RG approaches in modeling molecular networks including [29, 30] and [1].
Modeling signaling pathways through latent variables
Telesca et al. [29] considered gene expressions yij for p genes measured over n samples, i=1,…,n and j=1,…,p. They follow [31] and introduce trinary latent variables eij∈{−1,0,1} defining three possible categories of each expression yij,
| 5 |
The eij can be interpreted as an underlying biologic signal of under-, over- or normal expression. The motivation of the proposed inference is that only eij should be modeled without over-interpreting the additional noise in yij as biologically meaningful. Given eij, a Gaussian-uniform mixture model is assumed for gene expression yij,
| 6 |
where and is the normalized relative abundance, corrected for a sample-specific effect αi and a gene-specific effect μj. Conditional conjugate priors are assigned to : . Note that αi and μj are not identifiable because αi+μj=(αi−c)+(μj+c) for any constant c. If desired, the identifiability problem can be resolved by setting . For posterior simulation, the constraint can be implemented by setting αi←αi−c and μj←μj+c with at each iteration.
The dependence of gene expressions is modeled through the prior model for the latent variables eij. In [29], they further introduced another set of latent Gaussian variables zij as probit scores for each trinary eij,
The continuous latent probit scores are then modeled by an SEM
with and for covariates xi. Conjugate normal priors are assumed for regression coefficients βj and ajk. By (3) this defines an RGM. Let A be the p×p matrix of which the diagonal elements are zeros and the off-diagonal (j,k) entries are ajk. Then the joint distribution of Zi=(zi1,…,zip)T is given by
where mi=(mi1,…,mip)T and S=diag(s1,…,sp). The structure of the path diagram for this SEM is determined by the zero patterns in A, i.e. ajk=0 if and only if there is a missing edge from k to j. They define the prior over graph to be a subgraph of some fixed graph . The idea is to specify as some known maximum pathway. That is, where ψ∼Beta(aψ,bψ). Restricting to subgraphs of very effectively incorporates the prior information that might be available about an established biological pathway, using, for example, a public database such as KEGG. This restriction to subgraphs of is important to mitigate nonidentifiability due to Markov equivalence between distinct graphs. Moreover, it also greatly reduces the graph search space, which allows for efficient posterior simulation through trans-dimensional Markov chain Monte Carlo (MCMC). Further discussion and detailed treatment of this topic can be found in [29].
The network structure in [29] is built on the latent Gaussian variables which are linked to the observed gene expressions through another layer of latent trinary variables. Therefore it defines an indirect dependence structure on the actual gene expression. [30] extend this work to allow for a more direct characterization of the dependence structure of the observed protein expressions. Particularly, they proposed a fully general dependence prior between latent binary variables and developed a parsimonious framework for Bayesian model determination that allows for local computation.
Telesca et al. [30] start with the same sampling model as in (5) and (6), but then deviate substantially from the earlier model by reducing the trinary variable eij to a binary indicator variable zij=2I(eij=1)−1. That is,
The motivation of this reduction is the simplicity of the prior model on the resulting binary vector Zi=(zij). In fact, it is possible [8] to describe all possible probability models p(Zi) that comply with a given conditional independence structure. See below. Also, investigators usually focus on protein activations in RPPA studies. The joint distribution of Zi=(zi1,…,zip)T conditioned on the graph is by an Ising model,
The model is completed with normal priors for αj and βjk. In essence, the approach first maps an RG to its moral graph and then defines the joint probability model for Zi based on . The prior model for the graph is again hinged upon a known reference network . However, it is not constrained to be a subgraph of the reference network as before. Instead, they assume with ψ∼Beta(aψ,bψ) where d(·,·) is a discrepancy measure. They choose with δ>1 which is a weighted sum of edges dropped from and added to the reference graph . With δ>1, the discrepancy measure allows for parsimonious inference by imposing a heavier penalty for adding edges than removing edges. A distinctive feature of this construction is that the prior decreases to zero exponentially fast as discrepancy measure increases, which allows the posterior simulation to be concentrated around the reference network. See [30] for more modeling and inference details of this topic.
Integrative network analysis from multi-platform genomic data
Different types of molecules in a living cell do not act in isolation; in fact, DNA, RNA and proteins work closely together to carry out various functions for each cell. Studies that consider only one specific type of molecules remain unnecessarily restricted. In [1] they integrate DNA and RNA molecules for a more robust and biologically interpretable estimate of a gene network. In particular, they consider p gene expressions Y=(Y1,…,Yp)T, together with corresponding copy number and methylation, collectively denoted by X=(X1,…,X2p)T. They then exploit the central dogma of molecular biology that gene expression is produced by transcription from segments of DNA on which the copy number and methylation are measured, but the reverse processes are rare and biologically uninterpretable. The importance of this restriction is that in a network of Z=(Y,X) some edges have fixed pre-determined directions corresponding this observation. This in turn allows us to report meaningful inference on the directions of the remaining edges. Similar idea but in a different context has been proposed by [32–34]. With this assumption, they then model the regulatory relationships between copy number, methylation and gene expression with the following SEM
| 7 |
where with zeros on the diagonal, and E=(ε1,…,εp)T∼Np(0,Σ). The SEM in (7) immediately prohibits gene expression from regulating copy number or methylation since there is no X on the right-hand side of the equation. It also explicitly allows for feedback loops between the genes which are quite common motifs in molecular networks and have key functional roles in many cellular processes such as regulating gene expressions and acting as bistable switches [35]. Such feature is missing in DAG’s, UG’s and CG’s.
Next they assign thresholded priors [36] for A and B,
for i=1,…,p, j≠i and k=2i−1,2i. The threshold parameter ti controls the minimum effect sizes of aij and bik and is assigned a uniform hyperprior ti∼Uniform(0,t0). Normal priors are given to and , and and conjugate hyperpriors and . The thresholded priors enjoy nice theoretical properties as they are closely related to spike-and-slab prior and non-local prior as shown in [36–38]. We refer readers to [1] for a more comprehensive description of the work.
Reverse engineering gene networks with heterogeneous samples
Genomic data are often heterogeneous in the sense that the samples can be naturally divided into observed or hidden groups. For example, in a pan-cancer study, patients are normally grouped by the known cancer types whereas when studying a specific cancer, patients can be split into homogeneous latent subtypes. The inferential goals of these two cases are typically quite different. In the case with observed groups, much of the statistical work has been devoted to building models that allow information to be shared across groups. Such approaches improve the statistical power in graph estimation especially for groups with very limited samples by borrowing strength from other groups. For hidden groups, the underlying task becomes a clustering problem which aims to partition subjects into homogeneous clusters. In this section, we introduce two hierarchical RG models that are suitable for heterogeneous datasets. Interested readers can find more details in [38].
Suppose there are C known groups in the data. Let {si=c} be the group indicator for patient i in group c∈{1,…,C}. Denote Yc={Yi; si=c} and Xc={Xi; si=c} the set of observations for patients in group c. Then [38] entertain a group-specific SEM
where Ac = (ac,ij),Bc = (bc,ij)and Ec = (εc,1,…,εc,p)T ∼ Np(0,Σc) with and . To link the graphs across groups, they impose multivariate normal priors on the edge strength
The covariance matrix connects edge strength and graph structure across groups. When is close to ±1, and tend to have similar absolute values, which gives rise to a high probability that the edge i←j is included or excluded in groups c and c′ simultaneously since the threshold ti is shared across groups. Therefore, graphs from group c and c′ are more likely to share common edges. On the other hand, when is close to 0, the association between groups c and c′ is negligible. They do not fix Ω. Instead, they use an inverse-Wishart prior, Ω∼IW(ν,Ψ) and let the data dictate how strong the associations should be.
The Bayesian hierarchical model above allows for easy extension to the case where the groups are unknown. Ni et al. [38] simply augment the model with a Dirichlet-multinomial (DM) allocation model on the group indicator c|π,C∼Multinomial(1,π1,…,πC) and π|C∼Dir(η,…,η). The number of clusters C is usually unknown; they assume a geometric prior for C∼Geo(ρ), which eliminates the need to fix C a priori. They set the covariance matrix Ω to be diagonal since the goal of clustering is to split samples into groups with disparate networks rather than encourage similarity across the clusters. Alternatively, one can also set Ω to be a Stieltjes matrix (i.e. a positive definite matrix with nonpositive off-diagonal entries) to induce repulsive graphs.
Results
In this section, we generalize the RG approach of [1] by incorporating protein expressions in modeling the molecular network. Proteins provide important and orthogonal information in reverse engineering the network because they represent the downstream cumulative effect of changes that happen at the DNA and RNA levels and are more directly related to the phenotypical changes in cancer cells. Clinical utilization of genomic data alone show limited benefits, which is partly due to the poor concordance between gene and protein expressions [39, 40]. Many factors such as complex interactions between different types of molecules are responsible for the discrepancy between gene and protein abundance. The goal of this analysis is to explicitly model such complex interactions. We choose RG because it allows for efficient computation and great interpretability and is suitable to model reciprocal causality such as feedback mechanism.
We use the same notations Y=(Y1,…,Yp)T and X=(X1,…,X2p)T to denote gene expressions and the matched copy number and methylation. Importantly, additionally, for each gene Yj, there is also the associated protein expression, denoted by Zj for j=1,…,p. We extend the SEM in (7) to
| 8 |
where and have zeros on the diagonal, , and independent errors EY∼Np(0,ΣY) and EZ∼Np(0,ΣZ) with diagonal covariance matrices ΣY=diag(σ1,…,σp) and ΣZ=diag(λ1,…,λp). In essence, model (8) defines the following joint probability model for (Y,Z) given X,
| 9 |
The network structure is embedded in the parameters A,B,C and D. In this analysis, we restrict our attention to cis-regulatory relationships between DNAs and RNAs by constraining bij to 0 for j≠2i−1 or 2i for i=1,…,p. That is, copy number and methylation of gene i can only affect gene expression i. The interpretation of model (9) is given as follows: bi,2i−1≠0 if and only if copy number i is associated with its gene expression; bi,2i≠0 if and only if methylation i is associated with its gene expression; aij≠0 if and only if gene j regulates gene i; cij≠0 if and only if protein j regulates protein j; and dij≠0 if and only if gene j regulates protein i. We use the thresholded prior for A,B,C and D. For error variances, we assume inverse-gamma priors, σj,λj∼IG(a,b) for j=1,…,p which imply an inverse gamma conditional posterior distribution which in turn allows the use of a Gibbs sampling transition probability by sampling from the respective inverse gamma. The complete MCMC algorithm is provided in Algorithm 1.

Using TCGA-Assembler ([41], version 2), we acquired, processed and combined TCGA ovarian cancer data, including copy number, methylation and gene expression from the Genomic Data Commons (GDC) and mass spectrometry proteomic samples generated by the Clinical Proteomic Tumor Analysis Consortium (CPTAC). The resulting dataset contains 104 samples. In this case study, we focus on p=10 genes that are core members of the PI3K pathway [42], as shown in Fig. 3a, by playing a critical role in cell cycle progression, survival, motility, angiogenesis and immune surveillance [43]. It is one of the most frequently altered pathways in ovarian cancer [44, 45]. For each gene, we have matched copy number, methylation, gene expression and protein expression (i.e. 40 nodes in total). We report inference based on 100,000 MCMC samples (thinned out to every 10th iteration) after discarding the first 50,000 iterations as burn-in.
Fig. 3.
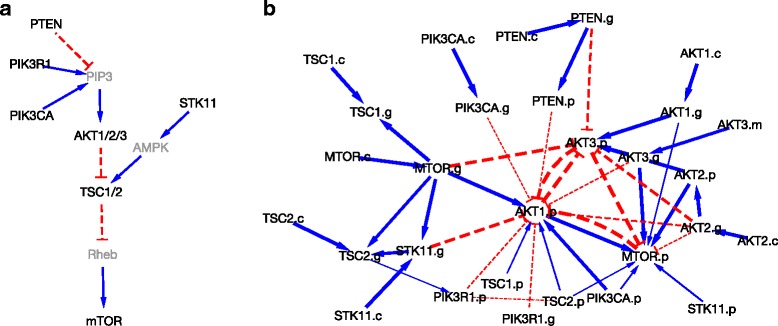
Genomic networks. a Reference network of core members of PI3K pathway. Molecules that are not used in our analysis are in gray. The blue solid lines with arrow heads are activations; red dashed lines with horizontal bars are inactivations. b Recovered network. The suffixes represent: c=copy number, m=methylation, g=gene and p=protein. Edge width is proportional to the posterior probability of inclusion. Disconnected molecules are not shown
The recovered network is shown in Fig. 3b where the blue solid lines represent activation whereas the red dashed lines are inactivations. The edge width is proportional to the posterior probabilities of inclusion p(aij≠0|X,Y,Z), p(bij≠0|X,Y,Z), p(cij≠0|X,Y,Z) and p(dij≠0|X,Y,Z), which can be approximated by MCMC sample averages, for example, where the superscript (l) denotes the lth MCMC sample and L is the total number of samples. The suffixes represent four different type of molecules: c=copy number, m=methylation, g=gene and p=protein. For clarity we do not show disconnected molecules. The degree of each node in Fig. 3b is reported in Table 1. The top three highly connected nodes are the protein AKT1 which is involved in 15 regulatory activities, the protein mTOR with 10 activities and the protein AKT3 with 9 activities. AKT1 and AKT3 belong to the same AKT family which is known to have shown strong oncogenic function and is a key mediator of PI3K pathway function [46]. AKT isoforms are often phosphorylated in ovarian cancers and may play a role in mediating the progression of late-stage serous ovarian carcinomas [47]. mTOR plays a critical role in regulating cell growth and proliferation by integrating signals including growth factors, nutrients, energy and stress [48, 49]. We also capture the well-known positive regulatory relationship between AKT1 and its downstream target mTOR [45, 49]. Other findings that are consistent with the biological literature include the inhibitory relationship between proteins PTEN and AKT1 and stimulatory relationship between proteins PIK3CA and AKT1 [50, 51]. Interestingly, the recovered network includes a negative feedback from proteins of mTOR to AKT1. Recent studies [45, 52–54] confirmed that such feedback mechanism can be either positive or negative depending on many factors including cell types and conditions.
Table 1.
| Molecule | Degree | Molecule | Degree |
|---|---|---|---|
| AKT1.p | 15 | PTEN.p | 2 |
| mTOR.p | 10 | PIK3CA.p | 2 |
| AKT3.p | 9 | AKT1.c | 1 |
| mTOR.g | 6 | AKT2.c | 1 |
| AKT2.g | 5 | AKT3.m | 1 |
| AKT3.g | 4 | mTOR.c | 1 |
| STK11.g | 4 | PIK3CA.c | 1 |
| TSC2.g | 4 | PTEN.c | 1 |
| AKT1.g | 3 | STK11.c | 1 |
| PTEN.g | 3 | TSC1.c | 1 |
| AKT2.p | 3 | TSC2.c | 1 |
| PIK3R1.p | 3 | PIK3R1.g | 1 |
| TSC2.p | 3 | STK11.p | 1 |
| PIK3CA.g | 2 | TSC1.p | 1 |
| TSC1.g | 2 |
Inference includes some surprising findings. For example, PIK3R1 is expected to activate rather than inactivate AKT1. Although the unexpected result may simply be a false positive, it deserves further experimental validation.
Discussion and conclusions
We have reviewed the basics of UG’s, DAG’s and RG’s and discussed recent RG-based approaches in modeling molecular networks. The extension of the approach in [1] allows to integrate multi-platform data including copy number, methylation, gene expression and protein expression. Our approach can be further be generalized to incorporating other data types such as single nucleotide polymorphisms (SNPs), mutation status and microRNAs. SEM-based RG approaches, however, cannot be directly applied to SNPs and mutation status because they have discrete support and the condition C1 requires the data to be multivariate normal. This limitation can be addressed by imputing latent normal variables. Although we do not find serious violation of normality assumption in our data, if desired, one could adopt the approach of [29] and introduce additional layers of hidden variables when the data are seemingly non-normal.
Another important assumption of the discussed approaches in this paper is homogeneity across samples. It is an unrealistic assumption in many applications, especially in oncology where the consensus is that tumors are extremely heterogeneous. Characterizing tumor heterogeneity can fundamentally improve our understanding of the cancer biology and practically allow us to divide heterogeneous patient population into homogeneous subpopulations so that refined personalized treatments can be developed targeting specific subgroups of cancer patients. Several approaches have been proposed for modeling networks with heterogeneous samples [38, 55–57].
Acknowledgements
The authors appreciate the instructive suggestions from the Editor and anonymous reviewers.
Funding
This work and the publication costs of this article were supported by NIH/NCI grant RO1 CA 132897.
Availability of data and materials
The TCGA ovarian cancer dataset is publicly available in Genomic Data Commons (https://gdc.cancer.gov/).
About this supplement
This article has been published as part of BMC Bioinformatics Volume 19 Supplement 3, 2018: Selected original research articles from the Fourth International Workshop on Computational Network Biology: Modeling, Analysis, and Control (CNB-MAC 2017): bioinformatics. The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-19-supplement-3.
Authors’ contributions
YN, PM and YJ conceived the idea of this article. YN carried out the literature reviews, extended the model and drafted the manuscript. PM and YJ made critical revisions. LW and YJ provided the data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yang Ni, Email: yangni87@gmail.com.
Peter Müller, Email: pmueller@math.utexas.edu.
Lin Wei, Email: kylin9wei@gmail.com.
Yuan Ji, Email: koaeraser@gmail.com.
References
- 1.Ni Y, Ji Y, Müller P. Reciprocal Graphical Models for Integrative Gene Regulatory Network Analysis. Bayesian Anal. 2017.
- 2.Lauritzen SL, Wermuth N. Graphical models for associations between variables, some of which are qualitative and some quantitative. Ann Stat. 1989;17(1):31–57. doi: 10.1214/aos/1176347003. [DOI] [Google Scholar]
- 3.Frydenberg M. The chain graph markov property. Scand J Stat. 1990;17(4):333–53. [Google Scholar]
- 4.Spirtes P. Proceedings of the Eleventh Conference on Uncertainty in Artificial Intelligence. San Francisco: Morgan Kaufmann Publishers Inc.; 1995. Directed Cyclic Graphical Representations of Feedback Models. [Google Scholar]
- 5.Koster JT. Markov properties of nonrecursive causal models. Ann Stat. 1996;24(5):2148–77. doi: 10.1214/aos/1069362315. [DOI] [Google Scholar]
- 6.Whittaker J. Graphical models in applied multivariate statistics. New York: Wiley Publishing; 2009. [Google Scholar]
- 7.Lauritzen SL. Graphical models. Oxford: Clarendon Press; 1996. [Google Scholar]
- 8.Besag J. Spatial interaction and the statistical analysis of lattice systems. J R Stat Soc Series B. 1974;36(2):192–236. [Google Scholar]
- 9.Dobra A, Lenkoski A, Rodriguez A. Bayesian inference for general gaussian graphical models with application to multivariate lattice data. J Am Stat Assoc. 2011;106(496):1418–33. doi: 10.1198/jasa.2011.tm10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green PJ, Thomas A. Sampling decomposable graphs using a markov chain on junction trees. Biometrika. 2013;100(1):91. doi: 10.1093/biomet/ass052. [DOI] [Google Scholar]
- 11.Wang H, Li SZ, et al. Efficient gaussian graphical model determination under g-wishart prior distributions. Electron J Stat. 2012;6:168–98. doi: 10.1214/12-EJS669. [DOI] [Google Scholar]
- 12.Meinshausen N, Bühlmann P. High-dimensional graphs and variable selection with the lasso. Ann Stat. 2006;34(3):1436–62. doi: 10.1214/009053606000000281. [DOI] [Google Scholar]
- 13.Friedman J, Hastie T, Tibshirani R. Sparse inverse covariance estimation with the graphical lasso. Biostatistics. 2008;9(3):432–41. doi: 10.1093/biostatistics/kxm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearl J. Probabilistic inference in intelligent systems. San Mateo: Morgan Kaufmann; 1988.
- 15.Lauritzen SL, Dawid AP, Larsen BN, Leimer HG. Independence properties of directed markov fields. Networks. 1990;20(5):491–505. doi: 10.1002/net.3230200503. [DOI] [Google Scholar]
- 16.Ni Y, Stingo FC, Baladandayuthapani V. Sparse Multi-Dimensional Graphical Models: A Unified Bayesian Framework. J Am Stat Assoc. 2017;112(518):779–93. doi: 10.1080/01621459.2016.1167694. [DOI] [Google Scholar]
- 17.Ni Y, Stingo FC, Baladandayuthapani V. Bayesian nonlinear model selection for gene regulatory networks. Biometrics. 2015;71(3):585–95. doi: 10.1111/biom.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedman N, Koller D. Being bayesian about network structure. a bayesian approach to structure discovery in bayesian networks. Mach Learn. 2003;50(1-2):95–125. doi: 10.1023/A:1020249912095. [DOI] [Google Scholar]
- 19.Shojaie A, Michailidis G. Penalized likelihood methods for estimation of sparse high-dimensional directed acyclic graphs. Biometrika. 2010;97:519–38. doi: 10.1093/biomet/asq038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stingo FC, Chen YA, Vannucci M, Barrier M, Mirkes PE. A bayesian graphical modeling approach to microrna regulatory network inference. Ann Appl Stat. 2010;4(4):2024. doi: 10.1214/10-AOAS360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altomare D, Consonni G, La Rocca L. Objective bayesian search of gaussian directed acyclic graphical models for ordered variables with non-local priors. Biometrics. 2013;69(2):478–87. doi: 10.1111/biom.12018. [DOI] [PubMed] [Google Scholar]
- 22.Yajima M, Telesca D, Ji Y, Müller P. Detecting differential patterns of interaction in molecular pathways. Biostatistics. 2015;16(2):240–51. doi: 10.1093/biostatistics/kxu054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richardson T. Proceedings of the Twelfth International Conference on Uncertainty in Artificial Intelligence. San Francisco: Morgan Kaufmann Publishers Inc.; 1996. A Discovery Algorithm for Directed Cyclic Graphs. [Google Scholar]
- 24.Richardson T. Proceedings of the Twelfth International Conference on Uncertainty in Artificial Intelligence. San Francisco: Morgan Kaufmann Publishers Inc.; 1996. A Polynomial-time Algorithm for Deciding Markov Equivalence of Directed Cyclic Graphical Models. [Google Scholar]
- 25.Richardson T. A characterization of markov equivalence for directed cyclic graphs. Int J Approx Reason. 1997;17(2-3):107–62. doi: 10.1016/S0888-613X(97)00020-0. [DOI] [Google Scholar]
- 26.Mooij JM, Janzing D, Heskes T, Schölkopf B. Proceedings of the 24th International Conference on Neural Information Processing Systems. USA: Curran Associates Inc.; 2011. On Causal Discovery with Cyclic Additive Noise Models. [Google Scholar]
- 27.Mooij JM, Heskes T. Proceedings of the Twenty-Ninth Conference on Uncertainty in Artificial Intelligence. Arlington: AUAI Press; 2013. Cyclic Causal Discovery from Continuous Equilibrium Data. [Google Scholar]
- 28.Lacerda G, Spirtes P, Ramsey J, Hoyer P. Proceedings of the Twenty-Fourth Conference Annual Conference on Uncertainty in Artificial Intelligence (UAI-08) Corvallis: AUAI Press; 2008. Discovering Cyclic Causal Models by Independent Components Analysis. [Google Scholar]
- 29.Telesca D, Müller P, Parmigiani G, Freedman RS. Ann Appl Stat. 2012;6(2):542–60. doi: 10.1214/11-AOAS525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Telesca D, Müller P, Kornblau SM, Suchard MA, Ji Y. Modeling protein expression and protein signaling pathways. J Am Stat Assoc. 2012;107(500):1372–84. doi: 10.1080/01621459.2012.706121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parmigiani G, Garrett ES, Anbazhagan R, Gabrielson E. A statistical framework for expression-based molecular classification in cancer. J R Stat Soc Series B. 2002;64(4):717–36. doi: 10.1111/1467-9868.00358. [DOI] [Google Scholar]
- 32.Cai X, Bazerque JA, Giannakis GB. Inference of gene regulatory networks with sparse structural equation models exploiting genetic perturbations. PLoS Comput Biol. 2013;9(5):1003068. doi: 10.1371/journal.pcbi.1003068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni Y, Stingo FC, Baladandayuthapani V. Integrative bayesian network analysis of genomic data. Cancer Inf. 2014;13(Suppl 2):39. doi: 10.4137/CIN.S13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L, Kim S. Learning gene networks under snp perturbations using eqtl datasets. PLoS Comput Biol. 2014;10(2):1003420. doi: 10.1371/journal.pcbi.1003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin SY, Rath O, Zebisch A, Choo SM, Kolch W, Cho KH. Functional roles of multiple feedback loops in extracellular signal-regulated kinase and wnt signaling pathways that regulate epithelial-mesenchymal transition. Cancer Res. 2010;70(17):6715–24. doi: 10.1158/0008-5472.CAN-10-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ni Y, Stingo F, Baladandayuthapani V. Bayesian Graphical Regression. J Am Stat Assoc. 2018. [DOI] [PMC free article] [PubMed]
- 37.Rossell D, Telesca D. Nonlocal Priors for High-Dimensional Estimation. J Am Stat Assoc. 2017;112(517):254–65. doi: 10.1080/01621459.2015.1130634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ni Y, Müller P, Zhu Y, Ji Y. Heterogeneous reciprocal graphical models. Biometrics. 2018. [DOI] [PubMed]
- 39.Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mrna abundance in yeast. Mol Cellular Biol. 1999;19(3):1720–30. doi: 10.1128/MCB.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akbani R, Ng PKS, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang J-Y, Yoshihara K, Li J, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun. 2014;5:3887. doi: 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu Y, Qiu P, Ji Y. Tcga-assembler: open-source software for retrieving and processing tcga data. Nat Methods. 2014;11(6):599–600. doi: 10.1038/nmeth.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–57. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 43.Liliac L, Amalinei C, Balan R, Grigoras A, Caruntu ID. Ovarian cancer: insights into genetics and pathogeny. Histol Histopathol. 2012;27(6):707–19. doi: 10.14670/HH-27.707. [DOI] [PubMed] [Google Scholar]
- 44.Kotsopoulos IC, Papanikolaou A, Lambropoulos AF, Papazisis KT, Tsolakidis D, Touplikioti P, Tarlatzis BC. Serous ovarian cancer signaling pathways. Int J Gynecol Cancer. 2014;24(3):410–7. doi: 10.1097/IGC.0000000000000079. [DOI] [PubMed] [Google Scholar]
- 45.Cheaib B, Auguste A, Leary A. The pi3k/akt/mtor pathway in ovarian cancer: therapeutic opportunities and challenges. Chin J Cancer. 2015;34(1):4. doi: 10.5732/cjc.014.10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bast RC, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9(6):415–28. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanrahan AJ, Schultz N, Westfal ML, Sakr RA, Giri DD, Scarperi S, Janikariman M, Olvera N, Stevens EV, She QB. Genomic complexity and akt dependence in serous ovarian cancer. Cancer Discov. 2012;2(1):56–67. doi: 10.1158/2159-8290.CD-11-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wullschleger S, Loewith R, Hall MN. Tor signaling in growth and metabolism. Cell. 2006;124(3):471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 49.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vara JÁF, Casado E, de Castro j, Cejas P, Belda-Iniesta C, González-Barón M. Pi3k/akt signalling pathway and cancer. Cancer Treat Rev. 2004;30(2):193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 51.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the pten tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 52.Hay N. The akt-mtor tango and its relevance to cancer. Cancer Cell. 2005;8(3):179–83. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 53.Dormond O, Madsen JC, Briscoe DM. The effects of mtor-akt interactions on anti-apoptotic signaling in vascular endothelial cells. J Biol Chem. 2007;282(32):23679–86. doi: 10.1074/jbc.M700563200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mead H, Zeremski M, Guba M. mTOR Signaling in Angiogenesis. In: Polunovsky VA, Houghton PJ, editors. mTOR Pathway and mTOR Inhibitors in Cancer Therapy. Totowa: Humana Press; 2010. [Google Scholar]
- 55.Ickstadt K, Bornkamp B, Grzegorczyk M, Wieczorek J, Sheriff MR, Grecco HE, Zamir E. Nonparametric bayesian networks. Bayesian Stat 9. 2011;9:283. [Google Scholar]
- 56.Rodriguez A, Lenkoski A, Dobra A. Sparse covariance estimation in heterogeneous samples. Electron J Stat. 2011;5:981. doi: 10.1214/11-EJS634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mukherjee C, Rodriguez A. Gpu-powered shotgun stochastic search for dirichlet process mixtures of gaussian graphical models. J Comput Graph Stat. 2016;25(3):762–88. doi: 10.1080/10618600.2015.1037883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The TCGA ovarian cancer dataset is publicly available in Genomic Data Commons (https://gdc.cancer.gov/).