

Abstract
Cofactor-mimetic aerobic oxidation has conceptually merged with catalysis of syngas reactions to form a wide range of Markovnikov-selective olefin radical hydrofunctionalizations. We cover the development of the field and review contributions to reaction invention, mechanism and application to complex molecule synthesis. We also provide a mechanistic framework for understanding this compendium of radical reactions.
Graphical Abstract
1. Introduction
1.1 Radicals from Alkenes: Relevance of TM HAT to Synthetic Chemistry
The occasion for this review is an invitation by Jack Norton and John Sowa to contribute an article to a thematic series on metal hydrides (MH). The canonical reactions of metal hydrides with unsaturated organic substrates – typical of the reactions taught in introductory organometallic courses1 – involve initial coordination of the Lewis acidic metal center to alkenes or alkynes.2,3 In contrast, Jack and many others have pioneered the understanding and application of alternative reaction pathways: the non-canonical radical reactions of metal hydrides, or hydrogen atom transfer (HAT) reactions.4
Many early explicit contributions to this area belong to the inorganic and bioinorganic literature (see Section 2). In retrospect, diverse examples of these reactions can be found more broadly distributed across fields, including synthetic organic chemistry. Slowly and circuitously, these MH HAT reactions have begun infiltrating the standard repertoire of synthetic transformations.5,6,7 From a synthetic organic chemistry perspective, their importance derives mainly from two attributes: unique chemoselectivity8 for known transformations and retrosynthetic possibility9 for new transforms.
Metal-hydride hydrogen atom transfer (MH HAT) reactions with alkenes exhibit Markovnikov selectivity, identical to the selectivity observed with Brønsted acid reactions (Figure 1). In the case of HAT, initial C-H bond formation at the less electronically stabilized position leads to carbon-centered radical generation instead of carbocation formation. The carbon-radical product of hydrogen atom transfer and the carbocation product of proton transfer can each be captured to generate, for example, hydration products. But alkene hydration is a 190-year old reaction,10,11 so why the interest in HAT? The answer is “chemoselectivity.”8 Carbocations, or carbenium ions, are high energy species that violate the octet rule. The lifetime of tert-butyl cation in water/TFE mixtures is close to that of a single bond vibration.12 Alkyl-substituted alkene protonation generates an unstabilized carbocation that has super-acidic C-H bonds, exceeding the acidity of fluorosulfonic acid and protonated benzene.13 Therefore alkene protonation in the midst of multiple functional groups can be disfavored versus other lower energy reactions, including the reverse reaction: alcohol ionization. In contrast, MH HAT can occur at ambient temperature or lower, at standard pressure, at low concentration of all reactants, and in the presence of functional groups that are reactive to acid, base, heat, reductant and/or oxidant. In other words, MH HAT conditions are mild, that ill-defined but intuitively meaningful word. The better word is chemoselective,8 meaning that MH HAT reagents select for reaction with an alkene when given the opportunity to react with other functional groups. So, even though both proton transfer and HAT can mediate the Markovnikov hydrofunctionalization of alkenes – hydration, hydroamination, hydroazidation, hydrocyanation, hydrohalogenation – the chemoselectivity of HAT commends it to the synthetic chemist.
Figure 1.

Proton transfer versus HAT Markovnikov hydrofunctionalization of alkenes.
Carbon-centered radicals can differ in relative rates of reactivity given the functional groups appended to them.14 Electron withdrawing groups (EWG) attached to a carbon radical render it electron-deficient, which decelerates its addition to electron-deficient alkenes but accelerates its reaction with electron-rich alkenes.15,16 The opposite is true of carbon-centered radicals with appended electron donating groups (EDG). These react faster with electron-deficient alkenes than with electron-rich alkenes.17 The consequence for HAT is that a polarity-reversal18 of reactivity can occur compared to Brønsted-acid alkene hydrofunctionalization. If a trisubstituted alkene reacts with a Brønsted-acid, the more-substituted carbon becomes a highly-electrophilic carbocation, which reacts with nucleophiles. If the same trisubstituted alkene reacts via HAT with a metal-hydride, the more-substituted carbon becomes an electron-rich tertiary radical, which can react with electrophiles or nucleophiles, and the efficiency depends on the relative rates of competitive reactions.
From the perspective of retrosynthetic analysis,9 complexity can be removed from a target (TGT) in new ways through predictable HAT radical transforms to access alkene intermediates (tgt). Because of the polarity-reversal available to HAT transforms, “dissonant”19 relationships that would be hard to establish between two functional groups can be severed directly, as in TGT of equation 1.20, 21, 22, 23 This dissection leads to a cyclohexene retron for a Diels-Alder transform. Because of the step-wise nature of HAT reactions, trans-selective hydrogenation is possible (eq. 2).24 Previously, the TGT of eq. 2 would not include an obvious partial retron for the Diels-Alder transform, whereas now the cyclohexene tgt and its diene-dienophile precursors are retrosynthetically accessible. Due to formal polarity-reversal across alkenes, now the more-substituted position on an alkene can be thought of as nucleophilic, leading by new disconnections to simple substrates (eq. 3). Furthermore, due to the non-necessity of carbon-metal bond formation in initial HAT and the early transition states of ensuing radical reactions, extremely hindered substrates and/or electrophiles can undergo productive reaction.25
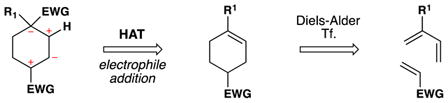 |
eq. 1 |
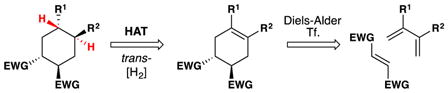 |
eq. 2 |
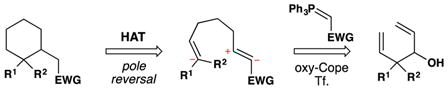 |
eq. 3 |
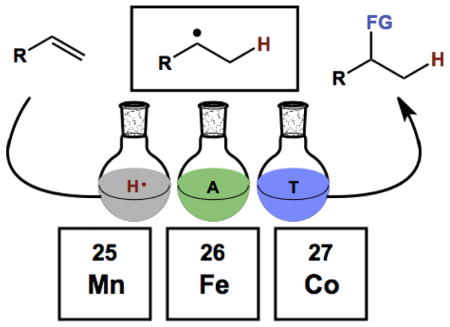
If the power of these HAT transforms to chemical synthesis is clear, it is only a recent acknowledgement. That is to say, characterization of reactions widely-used in synthetic organic chemistry as involving MH HAT is recent24,26,27,28 and supported by logical analogy to literature data rather than by direct characterization of reaction intermediates. In this review, we summarize reactions that appear to fit the HAT mechanistic paradigm and those that are widely accepted to. We also include many reactions for which little is known about the mechanism but their similarity in reagents merit inclusion. We trace the historical roots of the most commonly used reagents and catalysts. And we provide a mechanistic framework for understanding, predicting and applying this reactivity, in addition to the foregoing discussion on retrosynthetic analysis. This review focuses on the radical hydrofunctionalization of alkenes with putative first row transition metal hydrides (particularly those of manganese, iron and cobalt). Additionally, we focus on synthetic applications of these reagents and so restrict our discussion of physical organic chemistry studies to those that clarify the synthetic utility of these reactions.
It is just as important to catalog what will not be covered. If a reaction does not involve generation of a carbon-centered radical via reaction of a first row transition metal hydride (putative) with a carbon-carbon double bond, then this review does not discuss it. Thus, we do not discuss generation of carbon centered radicals from functionalities other than C=C bonds, such as epoxides, alcohols and carbonyls.29 We do not discuss hydrides of second and third row transition metals, nor of group 13 and 14 metalloid-hydrides (i.e. [Si]-H, [Ge]-H, [Sn]-H, [Pb]-H), hydrogen atom transfer from C-H bonds (e.g. 1,4-cyclohexadiene), thiol or selenium ene radical additions, Kharash reactions, dissolving metal reductions, and photochemical reactions. A vast body of radical polymerization literature is also excluded, but has been reviewed elsewhere.30
A number of excellent reviews which cover many aspects of the chemistry discussed herein have been published.5,29,31,32,33,34,35 The interested reader is directed to these for further reading.
1.2 Historical Context for the Modern Era
The mechanism of hydrogen atom transfer (HAT) from a transition metal hydride to an olefin was proposed by Jack Halpern of the University of Chicago in the mid-1970’s to explain the deuteration of anthracene using syngas and Co2(CO)8.36,37 The mechanistic relevance of these cobalt-mediated single electron reactions to bioinorganic chemistry, particularly that of vitamin B12, was clear from the outset.36 Separately during the 1970’s, the preparative value of open-shell metalloenzyme cofactors or their mimics began to be recognized. Early efforts sought to establish catalytic aerobic oxidation methods that did not proceed through high energy auto-oxidation pathways, i.e. hydrogen atom abstraction from an organic substrate (see Section 2.1 below). The simplest methods arose from the Mitsui Petrochemical Industries (MPI) who recruited Teruaki Mukaiyama in 1987 to head up its newly established Basic Research Facilities. 38 Also around this time, connection between Halpern’s HAT mechanisms and these oxidase cofactor mimetics was lost. Instead, papers describing these later aerobic reactions either proposed no specific mechanism or invoked more canonical hydrometallation pathways with occasional recognition of a radical intermediate. Mukaiyama’s work explicitly inspired Erick Carreira, who in the mid-2000’s further advanced this chemistry by the development of general C-N, C-C and C-X bond-forming reactions with his then graduate student, Jerome Waser, among others. Around the same time, the Boger lab developed a wide variety of hydrofunctionalization methods during the course of their synthetic studies on vindoline and vinblastine. Through an orthogonal path in the literature, Jack Norton began to study the HAT reactions of carbonyl metal hydrides and vitamin B12 mimics to electronically diverse alkenes, and established the first link between these metal complexes and the work of Mukaiyama by way of rate analyses conducted by Nojima. Finally, intimations by Boger that hydrogen atom radical addition, not hydrometallation, probably underlay his radical hydrofunctionalizations; and explicit connections by Herzon and Shenvi between Mukaiyama’s work and Halpern’s mechanistic paradigm, served to reunite these disparate areas.
The following discussion is organized by type of bond formation (C-O, C-N, C-C, etc.) and discusses the work of these aforementioned authors along with many others. We have sought to arrange the individual sub-sections in a loosely chronological manner to emphasize precedence, but recognize the inherent conflicts in this double-indexing.
2. C-O bonds
2.1 Hydration
The discovery of oxidase and oxygenase metalloenzymes that operate in the presence of air (O2) and a reductant, such as NAD(P)H or ascorbic acid, stimulated significant effort by synthetic chemists39,40,41 to investigate the aerobic oxidation of unsaturated carbon-carbon bonds by first-row transition metal complexes.42,43 These studies combined four components: a transition-metal, organic ligands, oxygen gas and a reductant, to effect aerobic oxygenation reactions. From this line of inquiry stems much of the synthetic work in radical functionalization of alkenes initiated by first-row transition metal hydrides.
The ability of first-row transition metals like FeII and CoII to promote high energy radical auto-oxidation chemistry was known even in the 19th century.44,45 While useful and important in environmental chemistry, these classic auto-oxidation reactions46 exhibit high reactivity but low selectivity and proceed through high energy oxygen-centered free radicals (such as hydroxyl, hydroperoxy and superoxide radicals). In the case of simple aliphatic hydrocarbons, single C-H abstraction products, oxidized polymers, cleavage products, and multiply-oxidized products can result from free radical chain processes.47 The low chemoselectivity of auto-oxidation in these early variants makes the radical-chain process of limited synthetic use. Yet metalloenzymes such as the heme-protein oxygenases exhibit exquisite control over the reactivity of their transition metal centers with O2.48 Indeed, hemocyanin and hemoglobin bind and transport molecular oxygen in vivo and can incorporate the oxygen atoms into organic substrates with high selectivity.49,50 These remarkable enzymes temper and direct the reactivity of their metal centers through ligand control.
Chemists took note of metalloenzymes and began modifying the cofactor ligands to temper the high reactivity of first row transition metal ions. Early work explored porphyrin, salen, oxime, and related ligand sets. These ligands were originally designed and synthesized to structurally model metalloprotein cofactors for exploration of their chemical reactivity (Figure 3).
Figure 3.

Co(dmg)2(pyr)Cl and Mn(TTP)Cl were structural models for cobalamin (Vitamin B12) and the heme cofactor of cytochrome P-450 respectively.
The hemeprotein cytochrome “P-450” was coined in 196451 subsequent to its initial discovery in 1958.52,53 In the particular case of the P-450 hemeprotein, the primary ligand of the FeII/III center is a porphyrin (Figure 3).48 The P-450 Fe-heme system operates with O2 as oxidant and NADH as reductant. In 1979, Tabushi and Koga reported a manganese(II/III) porphyrin system, which was able to oxidize cyclohexene with air in the presence of NaBH4, as a structural model for the P-450 Fe-heme system (Figure 4).52
Figure 4.
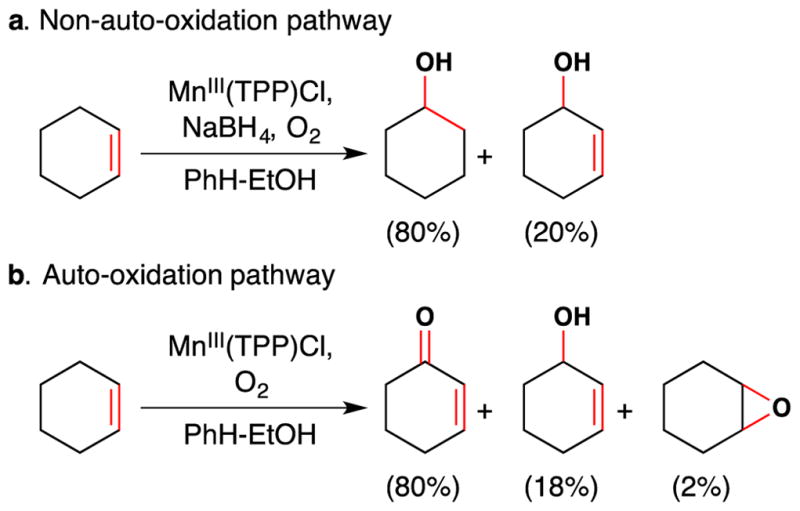
Unusual reactivity of Mn(TPP)Cl in the presence of NaBH4.
This artificial system exhibited remarkable behavior. It effected an oxidation with molecular oxygen but did not proceed through a typical auto-oxidation mechanism. Rather, the active reagent oxidized cyclohexene to give a product distribution distinct from what is observed when the auto-oxidation pathway is operative (Figure 4). Instead of reacting through allylic C-H abstraction, cyclohexene reacts directly through the olefin of cyclohexene itself. So cyclohexanol is produced as the major product in path a. This contrasts with path b, where the product distribution reflects an auto-oxidation mechanism. Additionally, autooxidation has a characteristic induction period and can be inhibited with radical inhibitors like BHT, but path a does not exhibit these characteristics.
Similar studies published in the late 1970’s showed that other first-row transition metal porphyrins could effect oxygen activation reactions with borohydride reductants and did not exhibit the characteristics of auto-oxidation.54,55,56 This work was not focused on unactivated olefin functionalization though.
Tabushi’s work with Mn(TPP)Cl52 was noticed by Perree-Fauvet and Gaudemer, who reported in 1981 the catalytic oxidation of cyclohexene and four other olefins (cyclooctene, styrene, 1-octene and 1-phenyl-prop-1-ene) to mixtures of the corresponding ketones and alcohols with a modification of Tabushi’s system [MnIII(TPP)Cl (5 mol%), NBu4+BH4− (0.4 equiv.), CH2Cl2 [0.1 M], air].57 Tabushi and Perre-Fauvet’s work mark early examples of unactivated olefin oxidation with air under first row transition metal catalysis that do not proceed via an auto-oxidation mechanistic pathway.
The 1980’s and 1990’s saw the publication of a number of related oxidation protocols with manganese and iron porphyrin complexes:
In 1988, Shimizu reported the oxidation of olefins with O2 and NaBH4, catalyzed by manganese meso-tetrakis(p-sulfonatophenyl)porphin in MeOH. Seven substrates are included in the study, including the “unactivated” alkenes – cyclopentene, cyclohexene and 1-methyl-cyclohexene – alongside variously substituted styrenes.58 Shimizu subsequently reported the oxidation of styrenes with chloro(meso-tetraphenylporphyrinato)-manganese(III), O2 and NaBH4.59 Sakurai and coworkers found a Mn(TPP)Cl/NaBH4/Avicel/O2 system to be an effective model for the cytochrome P-450 system60 (avicel is a microcrystalline cellulose powder). Takeuchi and Kano have studied the mechanism of these (porphinato)-manganese(III) oxidations of stryrenes.61 In brief, they found that this reaction proceeds via formation of a benzylic radical (that can be trapped with TEMPO), which reacts with O2 to give an alkyl peroxy radical. This nacent peroxy radical may be capped to form a MnIII porphinato peroxy-bridged alkyl species, which can give the corresponding alcohol or ketone. Reduction and homo-dimerization products were also observed, probably arising through benzylic radical dimerization and hydrogen atom abstraction, respectively. No evidence for formation of a MnIII alkyl species was found in this study, and benzylic radical formation occurs with reduction of MnIII to MnII. Although the authors do not propose HAT to the alkene from a MnIII hydride, both of these observations are consistent with a transition metal HAT mechanism.
In 1985, Hirobe and coworkers reported an Fe(TPP)Cl/O2/NaBH4/Me4NOH system as a model of cytochrome P-450 for alkene oxidations.62 Kano has studied iron(porphinato)-catalyzed oxygenations of styrenes63 and iron(porphinato) mediated reduction of alkenes and alkynes with NaBH4.64 Setsune examined the hydrometallation of alkynes with iron(II/III) porphyrins [FeIII(TPP)Cl and NaBH4].65,66 As they had done with the manganese(III) porphinato system,61 Takeuchi and coworkers explored the mechanism of these (porphyrinato)iron(III)-catalyzed styrene oxidations by O2 in the presence of BH4-.67 The product distribution of this reaction is similar to that observed with manganese (alcohol, ketone, dimerization, and reduction products) and is also thought to involve radical intermediates. Unlike the manganese case, however, the authors propose formation of an iron(III) alkyl intermediate, although no direct evidence of this species could be obtained. In their conclusion, the authors suggest that an iron(III) hydride may be a key species involved in formation of the assumed iron(III) alkyl intermediate, but do not further elaborate.
Another biomolecule that has served as an inspiration to synthetic chemists is coenzyme B12 (Figure 5), whose crystal structure was solved in 1965 through the prodigious skill of Nobel laureate, Dorothy Hodgkin.68 The red crystals contain a CoIII metal center in an octahedral ligand field consisting of a corrin ring with a small variety of axial ligands. Subsequent structural studies by Schrauzer and coworkers69,70,71,72 in the 1960’s revealed that the chemistry of this important cofactor could be modeled by CoIII cobaloximes, Co(dmg)2(pyr)Cl in particular, which bear two ‘dimethylglyoximato (dmg)’ ligands as mimics for the corrin ring (Figure 3).73
Figure 5.

Okamoto and Oka’s 1st generation hydration reaction with full substrate scope.
With an interest in directing the reactivity of this model cofactor Co(dmg)2(pyr)Cl towards selective alkene hydration with molecular oxygen, Okamoto and Oka reported in 1981 the first of several papers documenting the hydration of styrenes and other aryl-substituted olefins to the corresponding benzylic alcohols (Figure 5).74 Although these hydration reactions proceed in low yield, they are catalytic in Co(dmg)2(pyr)Cl. The authors suggest that this reaction is useful for producing Markovnikov hydration products under weakly basic conditions, in contrast to Brønsted acid mediated hydration. With respect to the mechanism, Okamoto et al. propose coordination of a CoI species to an alkene, followed by O2 insertion into a Co-C bond. They do not propose a pathway for hydrogen atom incorporation.
Okamoto and Oka continued to explore iron and cobalt complexes in order to improve this transformation and published a second generation reaction in 1984 (Figure 6).75 This improved reaction used the cobalt(II) porphyrin, Co(TPP), as catalyst, and Et4NBH4 as reductant. The catalyst hydrates aryl-conjugated alkenes in yields of 14–98% and the reactions are run for 2–10 days. In a screen of a variety of first row transition metal salts and complexes, the team noted that cobalt complexes bearing Schiff base and dimethylglyoximato (dmg) ligands also effected this transformation, as did FeII phthalocyanin (aka FePc). They noted that 1-hexene could be hydrated but only with poor regioselectivity (1-hexanol:2-hexanol = 45:55) with a TON = 32 in 12 hrs. Some of the substituted styrene substrates that were successfully hydrated under these conditions include p-chloro, o-methyl, p-methyl, p-methoxy, p-bromo, and m-nitrostyrene (Figure 6). The authors also observed dimerization at the benzylic position, olefin reduction, benzylic peroxygenation, and oxidation to the secondary ketone as reaction byproducts.
Figure 6.

Okamoto and Oka’s second generation styrene hydration reaction with full substrate scope.
In studies similar to Okamoto and Oka, Inoue and coworkers reported in 1985 the oxidation of styrene and styrene derivatives with CoII(TPP) and related complexes using NaBH4 as a reductant under an O2 atmosphere.76 Setsune and coworkers later studied the hydrometallation of alkenes and alkynes by the combination of cobalt(II) porphyrins, NaBH4, and oxidizing agents.77
As Okamoto and Oka had noted in their study,75 an iron phthalocyanin complex was also able to hydrate conjugated alkenes with molecular oxygen. In this vein, Kasuga and coworkers found that tetra-t-butylphthalocyanine complexes of manganese(III), iron(III) and cobalt(II) could all catalytically oxidize styrenes in the presence of O2 and NaBH4 (Figure 8).78 Notably, AlIII, NiII, CuII and ZnII phthalocyanin complexes gave no product. Styrene was converted into 1-phenylethanol in 88% yield by the MnIII complex with 100% conversion, in 63% yield by the FeIII complex with 84% conversion, and in 53% yield by the CoII complex with 100% conversion. 7% of ethylbenzene was also observed in the CoII case, and this yield increased to 90% under anaerobic conditions. The kinetic profiles of the manganese(III) and iron(III) complexes were similar, but the cobalt(II) complex displayed an induction period. TEMPO inhibited the CoII complex partially, while the MnIII and FeIII complexes were inhibited fully (see Section 7.6 for further discussion). Use of metal phthalocyanine complexes in catalysis has been reviewed previously.79
Figure 8.

CoII, FeIII and MnIII tetra-t-butylphthalocyanine. [The FeIII and MnIII complexes bear coordinated counterions, frequently halides].
During studies on the iron-mediated coupling of vindoline and catharanthine to form vinblastine in 2008,26,80 the the Boger group disclosed the use of stoichiometric iron(III) oxalate [Fe2ox3] with NaBH4 to effect the oxidation of an unactivated alkene in air and used it for the penultimate introduction of the vinblastine C-20′ alcohol. Subsequent to this work and while mapping out the scope of the reactions of the Fe2ox3/NaBH4 system, the Boger group also group significantly extended Kasuga’s78 work. FeII(phthalocyanin) was shown to catalytically oxidize unactivated alkenes with molecular oxygen (radical trap and metal reoxidant) in the presence of a variety of reductants, of which NaBH4 proved superior (Figure 9).81 Boger’s Fe2ox3/NaBH4 system has the advantage that the nascent radical could be quenched with a wide variety of radical traps to form a variety of carbon-heteroatom bonds [C, N, O (including TEMPO and O2), S, halogen] (see also Sections 3.3, 5.1). [The iron(III) center of iron(III) oxalate adopts an octahedral geometry but bears its oxalate (and water) ligands in a variety of coordination modes.]82 The group has applied this technology to the synthesis of vinblastine analogs (see Sections 2.5, 3.5, 5.4).
Figure 9.

Boger’s FePc-catalyzed and Fe2ox3-mediated aerobic hydration of alkenes.
Boger and coworkers note that their application of iron oxalate-mediated hydrofunctionalization towards vinblastine (2) derivatization26,80,81 (Figure 10, entries 1 and 2) was developed in part based on work conducted in the late 1980s and early ‘90s by Sakamoto and coworkers of Mitsui Petrochemical Incorporation. As exemplified by Figure 10, entry 3, the Mitsui chemists disclosed a method for the aerobic oxidation of anhydrovinblastine (1) to an epimeric mixture of vinblastine (2) and leurosidine (3), favoring the former.83 This disclosure examined a range of reaction conditions, varying solvent, iron source, coordinating ligands, hydride source and temperature. Among their findings was the discovery that yields of anhydrovinblastine (1) oxidation to vinblastine (2) with iron salts were improved by inclusion of buffered diacid additives, such as ammonium oxalate or malonate. This particular insight was disclosed in another patent from the same group,84 in which such a transformation is demonstrated on anhydrovinblastine generated in situ (Figure 10, entry 4), utilizing iron(III) salts for both oxidative coupling as well as aerobic redox-hydration. As an aside, it should be noted that discussion of anhydrovinblastine generation in situ (i.e. Figure 10, entries 2, 4 and 5) within this review are based upon the mechanistic insights made by Boger and coworkers rather than on any intimation to this effect within predecessor works. For these and other insights, the reader is directed to Section 7.6.
Figure 10.

Boger’s vinblastine redox hydration and representative examples from its predecessors the patent literature. aBoger’s conditions, ref 26. bMitsui chemists’ conditions, ref 83. cMitsui chemists’ conditions, ref 84. dAllelix, Inc. chemists’ conditions, ref 85. HPLC yields in parentheses.
The work of the Mitsui chemists, in turn, is preceded in the patent literature by a 1988 disclosure by Vukovic and Goodbody of Allelix, Inc. (Figure 10, entry 5),85 which dwells most heavily on maximizing yields of 1 or 2 during the coupling of their constitutive monomers, vindoline and catharanthine, respectively (Figure 11). The production of vinblastine under these familiar conditions suggest a HAT mechanism at play. Although we are hesitant to speculatively assign intent versus serendipity, the production of vinblastine in this particular patent appears to be consistent with the latter. This idea is further bolstered by an excerpt from Vukovic, Goodbody, Kutney and Misawa’s contemporaneous report86 detailing the same dimerization reaction to 1 in the chemical literature:
Figure 11.

Catharanthine and vindoline are the constitutive subunits of vinblastine.
“Whether this reaction is biomimetic is difficult to ascertain but … this fact is apparently substantiated by the discovery of trace amounts of what appears to be vinblastine amongst the incubation products. However, this evidence is very preliminary and requires further investigation.”
In 2010, Ishibashi and coworkers developed iron-catalyzed/mediated cyclizations of 1,6-dienes and enynes (Figure 12).87 By employing FePc as catalyst, with sodium borohydride as reductant and under an oxygen atmosphere, the authors found they could readily cyclize 1,6-dienes to form 5-membered rings via a 5-exo-dig radical cyclization, which was terminated by trapping with O2 to yield primary alcohols (Figure 12). Alternatively, stoichiometric FeCl3 or FeBr3 could be used to obtain halide-terminated compounds and permitted use of 1,6-enynes as substrates (see Section 5.2). The authors demonstrated the scope of their reaction with six substrates. All of the substrates contained either a heteroatom or a tetrasubstituted carbon atom within the formed pentacycles to accelerate cyclization and yielded mixtures of diastereomers in applicable cases. In the case of substrate 4, which contains both a terminal alkene and a 1,2-disubstituted alkene, initiation of the radical cyclization occurred from both alkenes, although the less hindered terminal alkene reacted more quickly. Consequently, a mixture of products was obtained (5 and 6).
Figure 12.

Ishibashi’s cyclization of 1,6-dienes with FePc as catalyst.
In 2014 Taniguchi and coworkers published a method for the direct synthesis of 1,4-diols from alkenes using FePc as a catalyst (Figure 13).88 Prior work had demonstrated that alcohols could be obtained from alkenes, presumably via formation of a metallo-peroxide such as 9 from alkene 7. Insightfully, the Taniguchi group considered that the intermediate peroxide 9 in this reaction could homolytically cleave at the O-O bond to generate a O-centered radical (10) and this in turn could undergo a [1,5]-hydrogen abstraction to generate a C-centered radical (11), which upon quenching with O2 and reduction of the nascent O-O bond would give a 1,4-diol (12). Eventually the team found that they could effect this 1,4-diol synthesis for a variety of alkenes in low to moderate yields. The use of Me2S in this reaction was found to improve the yield. The authors propose that Me2S serves as an electron-donating axial ligand on the Fe center, which has been shown89,90,91 to affect whether the [FeIII]O-O bond undergoes homolytic or heterolytic cleavage. The reaction was tolerant of 3-iodoanisole, 1-bromododecane, ethyl benzoate, and nitrobenzene as additives; and nitrile-, azide-, amide- and ester-containing substrates were successfully converted to the corresponding 1,4-diols. The authors propose intermediate formation of an FeIII alkyl species (8), but do not elaborate on the elementary steps involved in its formation except to say it is generated by reaction between a putative FeIII hydride and the olefin.
Figure 13.

Taniguchi’s method for 1,4-diol formation from alkenes. a. Selected substrates. b. Abridged mechanistic proposal.
Returning to work on cobalt complexes, Drago and his team reported in 1982 that a CoII Schiff base complex, CoSalMDPT [cobalt(II) bis(salicylidene-γ-iminopropyl)methylamine)], could promote the aerobic oxidation of linear unactivated olefins (1-hexene, 3-methyl-1-hexene, and 3-butenol-1-ol) and styrene to mixtures of the corresponding ketone and secondary alcohol products (Figure 14).92 Although this reaction may not see preparative use since ketone and alcohol products are formed in approximately equimolar amounts and in low yield (< 30%), the authors demonstrate with kinetic experiments that this reaction does not proceed through an auto-oxidation mechanism. Subsequent work from Drago and coworkers also explored the reactivity of these pentadentate Co(salen) complexes in the oxidation of phenols.93 In 1987, the team published an article detailing their mechanistic studies on this oxidation along with seven additional substrates (Figure 14).94
Figure 14.

Drago’s oxidation of linear alkenes with expanded scope.
In 1987, Teruaki Mukaiyama was asked by Shogo Takebayashi, president of Mitsui Petrochemical Industries, Ltd., to start a basic research laboratory at the newly built MPI research laboratories with the purpose of working exclusively on basic research topics.38 Consequently, Mukaiyama initiated a course of research involving the oxidation of olefins with first-row transition metals that could be performed under mild conditions. Aware of the aforementioned work involving metal complexes bearing porphyrin, salen and oxime ligands, Mukaiyama decided to explore instead the chemistry of β-diketonate ligands.38 As noted in his later account, β-diketonate ligands have the advantage of easily modifiable electronic and steric properties; they were already known to regulate the stereochemical and/or electrochemical characteristics of coordinated complexes; and with the exception of the most simple metal complexes, were unexplored in this aerobic oxidation chemistry.38 With respect to metals, cobalt was known to readily cycle between its +2 and +3 oxidation states and Co(acac)2 was known to absorb oxygen in the presence of pyridine,95,96 so the team at MPI began with exploration of Co(acac)2 reactivity with molecular oxygen and later expanded to other transition metals.38
Subsequently in 1989, Isayama and Mukaiyama of the Basic Research Laboratories for Organic Synthesis with the Mitsui Petrochemical Industries in Japan published their first results:97 an “oxidation-reduction hydration” reaction that used a CoII catalyst bearing acetylacetone (acac), with i-PrOH as both solvent and reductant (Figure 15). What set these reactions apart from prior contributions was effective use of α-olefins and other electron-neutral alkenes, generally high yields of a single product, and high chemoselectivity against the reactions of other functional groups.
Figure 15.

Mukaiyama’s first reduction-hydration reaction with full substrate scope.
However, this first generation variant of the “Mukaiyama hydration reaction” required 20 mol% of Co(acac)2 in an oxygen atmosphere (1 atm.) with heating at 75 C and resulted in a mixture of three different products: a ketone (A), an alcohol (B), and hydrogenated product (C). Unactivated (alkyl substituted) alkene substrates as well as aryl substituted alkenes (i.e. styrene) were competent substrates in this reaction. An α,β-unsaturated ester did not react, however. Only secondary alcohols such as isopropanol or cyclopentanol were competent as hydride sources; primary and tertiary alcohols did not provide the product alcohol. Moderate increases in yield were observed if 2,6-lutidine or 2-picoline were present (e.g. 60% vs 46% for 4-phenyl-1-butene). The proposed mechanism involves formation of the metallo-peroxide 13, which may undergo either O-O or C-O bond cleavage to give the observed products.
To improve the hydration reaction, the team made a variety of β-diketonate ligands with different electronic properties and the corresponding CoII complexes.98 With Co(tfa)2, a catalyst which bears the more electron deficient 1,1,1-trifluoroacetylacetonato (tfa) ligand (not trifluoroacetic acid), the authors were able to improve the yields of the desired Markovnikov alcohols to 70–90% (Figure 16). For example, the formation of decan-2-ol from 1-decene improved from 45% to 81% under these modified conditions. The remainder of the mass balance is attributed to ketone formation. Thus these conditions suppress formation of the hydrogenation product, and the highest yields (>90%) were observed for 2,2-disubstituted and tri-substituted alkenes for which Markovnikov hydration is the only product. CoIIsalen complexes were ineffective catalysts in these reactions.
Figure 16.

Mukaiyama’s second generation reaction with full substrate scope.
In a 1990 paper, Yamada and coworkers detailed further study of ligand and solvent effects involved in this hydration reaction and included a few more unactivated alkene substrates.99 They correlated the redox potentials (E1/2) of these complexes with their behavior in the hydration reaction and found that only complexes with redox potentials in the range of 0–0.5 V relative to a Ag0/I electrode in acetonitrile showed catalytic reactivity. The authors propose that complexes with redox potentials below 0 V are too electron rich and so are oxidized by molecular oxygen to the CoIII species after binding O2, while complexes with redox potentials above 0.5 V are too electron deficient to bind oxygen at all.38,99
In the course of further optimization studies, Mukaiyama’s team discovered that triethylsilane (Et3SiH) could replace i-PrOH as a reductant in their reduction-hydration reaction (Figure 17).100 Silanes were known to reduce organic compounds,101,102 so the team screened several silanes (Et3SiH, PhMe2SiH, Ph2SiH2, PhSiH3, i-Pr3SiH and (MeO)3SiH) with a variety of cobalt complexes to find that Et3SiH worked best with Co(tfa)2 (5 mol% catalyst loading). Although the yields of the desired alcohol generally drop by about 10% compared with use of i-PrOH (e.g. 73% vs. 81% for 1-decene oxidation), the desired alcohol was still obtained in good yield (53–87%) for the eight substrates examined.
Figure 17.

Use of Et3SiH in Mukaiyama’s reduction-hydration reaction.
Use of 1,2-dichloroethane (DCE) instead of n-PrOH as solvent in the above reaction (Figure 17) yielded instead the corresponding triethylsilyl peroxide in a useful and unprecedented reaction (see Section 2.2 for further details).103,104 The isolation of a peroxide supports Mukaiyama’s hypothesis that a metallo-peroxide (13 in Figure 15) is a common intermediate in this reaction.105
Shortly after, Isayama and Mukaiyama reported a significant advance towards the mild aerobic oxidation of olefins (Figure 18).105 They found by a screen of silanes and solvents that use of PhSiH3 with Co(acac)2 in polar aprotic solvents (DME, THF) under an O2 atmosphere permitted them to conduct the reduction-hydration reaction at ambient temperature in excellent yield (Figure 18). In addition to an improved yield, the catalyst loading was lowered from 20 mol% to 5 mol%. [Note that 5 mol% catalyst loading could already be obtained by use of Co(tfa)3 and Et3SiH]. These conditions furnished the desired alcohol (A in Figure 18) in 72–84% yield alongside the ketone B in 10–24% yield. In the case of 4-phenyl-1-butene, the hydroperoxide was also observed after 40 hours, but treatment with sodium thiosulfate during reaction work-up effected decomposition into the alcohol A (82%) and ketone B (10%). If diphenylsilane (Ph2SiH2) was used instead of phenylsilane, then the peroxysilane could be isolated directly (see Section 2.2 for further discussion of hydroperoxidation).
Figure 18.

Mildest conditions known for Mukaiyama’s reduction hydration reaction with full substrate scope.
In 1990, Mukaiyama published a method for the hydration of α,β-unsaturated esters to α-hydroxy esters (Figure 19).106 Instead of cobalt sources, the team found that use of MnII(dpm)2 with phenylsilane as reductant could effect this transformation in high yields. Alcohol regioselectivity appears to depend on intermediate radical stability, and so β-hydroxy products are obtained when the alkene is trisubstituted, especially if R1 or R2 are aryl groups (Figure 19). Generally, mixtures of α- and β-hydroxy ester products are obtained from β,β–disubstituted esters. This is probably due to similarities in the stabilization of an intermediate radical. With linear esters such as benzyl 2-butenoate (91%) however, the reaction gives solely α-hydroxy esters in good yield. The dipivaloyl methanato ligand (dpm) gave better yields on their test substrate than use of acetylacetonato ligand (91% vs. 62%). Use of the secondary alcohol i-PrOH gave better conversion than primary alcohols, while the reaction proceeded very slowly and in low yield in tertiary alcoholic and non-alcoholic solvents (for an explanation of this solvent requirement, see Sections 6.1, 7.5).
Figure 19.

Mukaiyama’s method for formation of α-hydroxy esters from α,β-unsaturated esters with full substrate scope. The reaction is assumed to proceed via radical intermediates.
Magnus and coworkers extended Mukaiyama’s procedure for α-hydroxylation of α,β-unsaturated esters to α,β-unsaturated ketones (Figure 20).107 Magnus’ conditions differ from Mukaiyama’s primarily in that Magnus employs Mn(dpm)3 instead of Mn(dpm)2 as the precatalyst. Magnus suggests that Mn(dpm)3 is also the active precatalyst in Mukaiyama’s method106 but that Mukaiyama had misassigned the oxidation state. Addition of triethyl phosphite facilitates reduction of intermediate peroxides. Magnus’ team found that a putative MnIII hydride, HMn(dpm)2, was capable of hydrating β-substituted linear and cyclic enones. In the absence of oxygen, β,β-disubstituted enones are not reduced; but when oxygen is introduced into the reaction, hydration occurs. Therefore, Magnus proposes an oxygen bound complex, HMnO2(dpm)2, as the active reducing agent, at least for cases involving β,β-disubstituted enones (see Section 7.7 for further discussion).
Figure 20.

Magnus’ modification of Mukaiyama’s method for formation of α-hydroxy ketones from α,β-unsaturated ketones.
In 2001, Magnus and coworkers reported a method for the direct conversion of αβ-unsaturated nitriles into cyanohydrins using Mn(dpm)3 as a catalyst with phenylsilane and molecular oxygen (Figure 21).108 The reaction yielded cyanohydrins (A) exclusively in cases such as 14 and 15, where the β position is monosubstituted with an alkyl group. In cases where the β position is substituted with an arene (16) or disubstituted (17), a mixture of regioisomeric alcohols results. The yields range from low to moderate.
Figure 21.

Magnus’ synthesis of α-cyanohydrins from α,β-unsaturated nitriles with selected substrate scope.
Yamada and coworkers reported in 2004 that they could stereoselectively prepare α-hydroxycarboxamides from αβ-unsaturated carboxamides using the 2,5-bis(2-naphthyl)-pyrrolidine chiral controller shown in Figure 22.109 The authors demonstrate the utility of their method on six substrates in which R1 and R2 are linear alkyl chains. The authors propose that an α-keto radical reacts with molecular oxygen in the enantiodetermining step.
Figure 22.

Yamada’s asymmetric α-hydroxylation.
Nam Ho Lee’s group of Cheju National university in Korea has reported several oxidation methods employing MnIII salen complexes as catalysts in the presence of molecular oxygen and sodium borohydride or phenylsilane which oxidize styrenyl alkenes to benzylic alcohols,110,111,112,113 and α,β-unsaturated esters to α-hydroxy esters.114 These papers were published from 1999 to 2006, as part of a program exploring the use of Schiff base-manganese complexes in organic synthesis. Unactivated alkenes could not be oxidized under these conditions.
Carreira’s method for hydrohydrazination and hydroazidation of olefins, published in 2006, may also be used to install alcohols (see Section 3.1–3 for further discussion).115
More recently, the Shenvi lab reported that Ph(i-PrO)SiH2 is generated and consumed rapidly under the Mukaiyama manganese-catalyzed hydration and improves the performance of a number of transition metal hydrogen atom transfer reactions (see Sections 6.1 and 7.6,8 for further discussion).116 In the hydration, 4-phenyl-1-butene was rapidly converted to 4-phenyl-2-butanol (Figure 23), whereas phenylsilane required a markedly longer reaction time. Significantly, use of Ph(i-PrO)SiH2 permitted the reaction to be run in THF, whereas previously only secondary alcoholic solvents could be used. Co(acac)2 behaved equally well with both PhSiH3 and Ph(i-PrO)SiH2.
Figure 23.

Manganese(III)-catalyzed Mukaiyama hydration with Ph(i-PrO)SiH2 in THF.
2.2 Hydroperoxidation
Whereas hydration of alkenes with molecular oxygen involves cleavage of the O-O bond of O2, hydroperoxidation preserves this bond. Hydroperoxide intermediates may be converted into alcohols and ketones, cyclized to form endoperoxides, or leveraged to functionalize adjacent C-H bonds.
In a seminal 1989 publication, Isayama and Mukaiyama reported that in the presence of catalytic amounts of CoII complexes, various alkenes could be converted to the corresponding Markovnikov triethylsilyl peroxides in high yields under remarkably mild conditions (Figure 24).103 This transformation was discovered upon conducting a solvent screen of the hydration reaction with triethylsilane as a reductant (see Figure 17, Section 2.1).100
Figure 24.

The Isayama/Mukaiyama hydroperoxidation with full substrate scope. Modp and dedp ligands are shown.
Isayama and Mukaiyama found that this reaction worked best on styrene with the Co(modp)2 or Co(dedp)2 as precatalysts (both > 90% yield) (Figure 24). Co(acac)2 and Co(tfa)2 could also effect hydrosilylperoxidation of 4-phenyl-1-butene, but in lower yields (30% and 73% yields respectively). In a subsequent paper,105 the team screened a variety of different silanes and, among others things, found that Co(acac)2 degrades the isolated silylperoxides under an argon atmosphere at room temperature. Other aprotic solvents such as benzene and ethyl acetate were generally compatible with this reaction but were lower yielding than 1,2-DCE. The substrate scope includes hydrosilylperoxidation of styrenes and alkyl substituted alkenes as shown in Figure 24. Silanes other than triethylsilane failed to give stable silylperoxides.
In 1990, Isayama reported his further study of this system and extension of the reaction to the α-triethylsilylperoxidation of α,β-unsaturated esters (Figure 25).104 Isayama had previously noticed an induction period and found that addition of catalytic tert-butyl hydroperoxide (TBHP) eliminated this induction period. Use of TBHP as an additive resulted in a significant reduction in reaction duration and an expansion in substrate scope to β-substituted, α,β-unsaturated esters. Additionally, Co(acac)2 could be used instead of the more exotic Co(modp)2 complex. For example, the silylperoxidation of styrene under the action of Co(acac)2 proceeded in 30% yield without TBHP present, but in a synthetically useful 85% yield with TBHP present. The product triethylsilylperoxy compounds (yields were not reported) could also be converted to the corresponding alcohol by treatment with acidic methanol and aqueous sodium thiosulfate.
Figure 25.

Isayama’s α-triethylsilylperoxidation of α,β-unsaturated esters.
O’Neill et al. reported in 2003 that Co(thd)2 [bis(2,2,6,6-tetramethyl-3,5-heptanedionato)cobalt(II), also known as Co(dpm)2] is a superior catalyst relative to Co(acac)2 for hydroperoxysilylation of unactivated alkenes and has applied their finding to the synthesis of spiro-1,2,4-trioxanes.117 These authors have shown that use of Co(dpm)2 instead of Co(acac)2 in the Mukaiyama/Isayama silylhydroperoxidation103,104 results in modest to significantly improved yields.
The Nojima group has studied the mechanism and selectivity of the Isayama/Mukaiyama hydroperoxidation (see Section 7.7 for further details),118,119 and have applied their findings to the synthesis of cyclic peroxides by hydroperoxidation of dienes.120,121 Nojima and coworkers have also made various peroxides, such as analogs of yingzhaosu A, using the Mukaiyama/Isayama silylhydroperoxidation procedure (see also Section 2.5).122,123,124,125,126
A wide variety of cyclic peroxides such as 1,2-dioxanes, 120,127,128,129,130 1,2-dioxolanes,118,119,120,121,129,130,131,132 and 1,2,4-trioxanes,117,133,134,135,136,137,138,139,140,141 may be obtained using the Mukaiyama/Isayama103,104 method on particular substrates. These have been reviewed elsewhere.142,143
In one example published after the aforementioned reviews, Woerpel and coworkers applied the Mukaiyama/Isayama silylhydroperoxidation103,104 to triethylsilyl enol ethers, which they then employ in Lewis acid mediated [3+2] annulations with alkenes to form 1,2-dioxolanes (Figure 26).144 For example, hydrosilylperoxidation of silyl enol ether 18 gives peroxy compound 19, and treatment of 19 with catalytic SnCl4 at −78 °C in the presence of an alkene provides 1,2-dioxolane 20. Woerpel has also made 1,2-dioxepane rings.145
Figure 26.

Woerpel’s synthesis of 1,2-dioxolanes.
In the early 1990’s, Matsushita, Sugamoto and Matsui reported that conjugated olefins are oxidized to the corresponding ketones with oxygen and triethylsilane in the presence of [5,10,15,20-tetrakis(2,6-di-chlorophenyl)porphinato]cobalt(II) complex [CoII(tdcpp)] as a catalyst followed by treatment with Ac2O and DMAP.146,147 Alternatively, treatment with trimethyl phosphite yielded alcohols.148 These reactions proceed via an unstable hydroperoxide intermediate.
In 1995, Matsushita and coworkers reported that they could isolate the unstable intermediate hydroperoxide products of α,β,γ,δ-unsaturated carbonyl compounds in this reaction (Figure 27).149,150 Remarkably, this reaction furnished only the γ-peroxy-α,β-unsaturated compounds from α,β,γ,δ-unsaturated carbonyl compounds with no other peroxide isomers. The corresponding γ-oxo-α,β-unsaturated compounds were also isolated as minor byproducts as a result of hydroperoxide decomposition. The authors later expanded the scope of the reaction to include agr;,β-unsaturated esters and styrenes, which are converted to the corresponding α-hydroxy compounds.151
Figure 27.

Matsushita and Sugamoto’s hydroperoxidation of conjugated alkenes.
The Co(tdcpp) catalyst is remarkably efficient, generally requiring only a 0.1 mol% catalyst loading, and the reaction is rapid, proceeding to completion in 30 minutes to 3 hours. In the case of the styrenes, the catalyst loading was dropped to just 0.01 mol% (Figure 27). However, at higher catalyst loadings and longer reaction times, styrenyl hydroperoxides were converted to the corresponding ketones. The reaction is run at 28–30 °C – room temperature in the authors’ laboratories. In the case of α,β,γ,δ-unsaturated carbonyl compounds, the presence of an electron withdrawing group is required to obtain a single regioisomer. One exception is symmetrical 1,3-cyclooctadiene, which reacts to give the corresponding hydroperoxide in excellent yield (92%). Unsymmetrical dienes gave inseparable mixtures of peroxide regioisomers. Acrylates and α-substituted acrylates were converted in good yield to the corresponding α-hydroperoxy esters. Unlike Isayama and Mukaiyama’s conditions,103,104 unconjugated alkenes were unreactive under Matsushita and Sugamoto’s conditions.
2.3 Ketone formation
The intermediacy of a hydroperoxide in the aforementioned aerobic hydration reactions may also be leveraged to form ketones under certain conditions. There are at least three notable cases where this has been explicitly done, and these are discussed below.
In 1989, the same year that Mukaiyama discovered his eponymous hydration reaction, Kato and Mukaiyama showed that vinyl silanes can react with molecular oxygen to give ketones directly via the intermediacy of a peroxide (Figure 28).152 The reaction employs Co(ecbo)2 as a catalyst in i-PrOH at 75 °C with an atmosphere of molecular oxygen under dehydrating conditions (4Å MS). Lower catalyst loadings resulted in longer reactions times (on the order of days) but with only minor decreases in yield. The substrate scope shows that a variety of simple vinyl silanes, including one which contains the acid sensitive THP group, are converted to the corresponding ketones in good yield. This reaction was discovered before Mukaiyama had realized that silanes bearing a Si-H bond could effect hydration and hydroperoxidation under very mild conditions,38 and Mukaiyama did not revisit this reaction with silanes or with cobalt complexes bearing simpler ligands. This reaction has seen limited use in synthesis.153
Figure 28.

Mukaiyama’s method for direct conversion of vinyl silanes to ketones with examples of substrate scope. ecbo = 2-ethoxycarbonyl-1,3-butanedionato.
As mentioned previously (Figure 27, Section 2.3), Matsushita and coworkers showed in the 1990’s that they could convert α,β,γ,δ-unsaturated esters, amides and nitriles to the corresponding γ-oxo-α,β-unsaturated esters, amides and nitriles via the intermediacy of a hydroperoxide.147 This reaction employs Co(tdcpp) in very low catalyst loadings with triethylsilane as reductant and molecular oxygen as oxidant to generate a hydroperoxide, which is subsequently converted to the corresponding γ-oxo-α,β-unsaturated complex in good yield through the action of acetic anhydride and DMAP. Figure 29 shows an example of this transformation. The authors show that this reaction occurs cleanly in the presence of unactivated alkenes. These reaction conditions may also be applied to styrenes for the synthesis of alkyl aryl ketones.146
Figure 29.

Matsushita’s synthesis of γ-oxo-α,β-unsaturated esters, amides and nitriles. Co(tdcpp) = [5,10,15,20-tetra(2,6-dichlorophenyl)porphinato]cobalt(II)
During their work on ryanodol, Inoue and coworkers found that they could convert the silylperoxides generated from alkenes with the Mukaiyama/Isayama104,105 hydrosilylperoxidation to the corresponding ketones (Figure 30).154,155 This transformation occurs upon treatment of the intermediate silyl peroxide with a sulfonyl fluoride and DBU, and proceeds through the intermediacy of a sulfonylated peroxide. The reaction conditions are mild and the authors show that a variety of sulfonyl fluorides may be employed in this reaction. Notably, nucleophilic atoms were protected with benzoyl, tosyl and tert-butyl dimethyl silyl groups in all the examples listed by the authors.
Figure 30.

Inoue’s method for the synthesis of ketones from silylperoxides.
2.4 Hydroalkoxylation
In a conceptually different approach, Shigehisa et al. reported a method for the hydroalkoxylation of unactivated olefins under oxidizing conditions via the intermediacy of a carbocation, which is subsequently trapped with an alcohol nucleophile (Figure 31).156,157 The cobalt(II) salen complex 14 proved an effective catalyst for this transformation when used in combination with the electrophilic fluorine-based oxidants (15 or 16), and silanes [PhSiH3 or (Me2SiH)2O]. The optimal combination was substrate dependent and demonstrated on an array of ~60 compounds, selected examples of which are shown in Figure 31. The scope focuses on simple linear alkenes to give the corresponding Markovnikov ethereal products, but 2,2-disubstituted and trisubstituted alkenes were also competent substrates. The reaction tolerates TBS-ethers, PMB and acetal protecting groups, esters, amides, bromo, nitro, tosylates, heterocycles, amino surrogates and some amines. These reactions were generally run with the alcohol as solvent or co-solvent, but the team showed in one case that the alcohol moiety could be used in stoichiometric amounts if an inert solvent (PhCF3) was employed.
Figure 31.

Shigehisa and coworkers’ hydroalkoxylation method.
In 2016, Shigehisa and coworkers further showed that fluorous alcoholic solvents such as trifluoroethanol (TFE) and hexafluoro-isopropanol (HFIP) are competent nucleophiles under their reaction conditions.158 A variety of styrenes and phenyl propanoids are competent substrates in this reaction. Other substrates failed to give the desired product, and the authors surmise that this is due to lack of carbocation stabilization. In the case of styrenes, the plausible benzyl cationic species is stabilized by the adjacent aromatic ring, and phenyl propanoids may be stabilized by a non-classical interaction between the carbocationic species and the aromatic ring.159
2.5 C-O Bond Formation in Natural Products and Complex Molecule Synthesis
Perhaps more effectively than any simple substrate scope, a survey of the uses of these radical C-O bond-forming reactions in natural products and complex molecule synthesis can best demonstrate their merits – mild reaction conditions, high chemoselectivity, and high functional group tolerance. Mukaiyama’s hydration conditions38,105 have acquired a privileged position among these, and most instances employ some variation of his procedure, which highlights its adaptability. Examples are briefly discussed here in chronological order.
In 1994, Matsushita et al. applied their peroxygenation/ketone formation method146 to the synthesis of (−)-pyrenophorin (26), an antifungal macrodiolide (Figure 32).160 Specifically, dienone 24 was treated with Co(tcdpp), Et3SiH and O2 to form a γ-hydroperoxide, which is cleanly converted to the 1,4-dicarbonyl (25) upon addition of acetic anhydride and DMAP.
Figure 32.

Matsushita’s synthesis of (−)-pyrenophorin (26).
In a similar fashion, Matsushita et al. applied their γ-hydroxylation method149 in 1995 to the synthesis of rac-hydroxyshogaol and related furanoids150 and in 1997 to the enantioselective syntheses of 10-oxo-11(E)-octadecen-13-olide and related fatty acids.161
In 1994, Wakamatsu and coworkers employed Mukiayama’s conditions106 for α-hydroxylation of α,β-unsaturated esters en route to the synthesis of optically pure metabolites of gomisin A (29) (Figure 33).162 Specifically, 27 was hydrated to the desired stereoisomer 28 with a diastereoselectivity of 7:1. In a later paper, the team was able to improve the yield of this reaction for a similar substrate (different protecting groups on the aromatic alcohols) to 86% by heating at 70 °C, although the diastereoselectivity dropped slightly to 5:1.163 The authors stated in the corresponding full paper on the syntheses of (+)-schizandrin, (+)-gomisin A, and (+)-isoschizandrin that Co(acac)2 gave 1:1 diastereoselectivity, while Mn(acac)2 improved the diastereomeric ratio to 5:1.164,165 However, the authors do not comment on the reason for this difference. Magnus107,221 has argued that in the case of manganese, α-hydroxylation occurs via a mechanism that involves initial 1,4-hydride addition of a manganese hydride to form a manganese enolate (cf. Figure 20 and Section 7.8). The manganese atom can coordinate molecular oxygen, which may then react with the enolate in an intramolecular fashion to form the observed α-hydroxylation products. This kind of mechanism has not been suggested for cobalt, which probably reacts via direct reaction of an α-keto radical with molecular oxygen or a peroxy-cobalt radical. We speculate that this may be the reason for the observed difference in diastereoselectivity.
Figure 33.

Wakamatsu’s synthesis of (+)-schizandrin, (+)-gomisin A (29), (+)-isoschizandrin and metabolites thereof.
Tietze and Raschke synthesized the natural norsesquiterpene, 7-desmethyl-2-methoxy-calamenene (32) with an intramolecular enantioselective Heck reaction as their key step (Figure 34).166 Subsequent functional group interconversions involved a Mukaiyama hydration.105 Note that although inconsequential for the synthesis itself, the alcohol diastereoselectivity arising from Mukaiyama’s procedure is opposite that observed with oxymercuration/reduction conditions.
Figure 34.

Tietze’s synthesis of 7-desmethyl-2-methoxy-calamenene (32).
Xu and Dong have employed Mukaiyama’s silylhydroperoxidation procedure to the synthesis of yingzhaosu C (35) (Figure 35).167 Under Mukaiyama/Isayama conditions, olefin 33 was cleanly converted into triethylsilyl peroxide 34 in 93% yield. Silyl peroxide 34 was used to make two of the four possible isomers of yingzhaosu C.
Figure 35.

Xu and Dong’s synthesis of yingzhaosu C (35).
In 2000, Enders and Ridder reported the first asymmetric synthesis of stigmolone (37), the fruiting body inducing pheromone of the myxobacterium Stigmatella aurantiaca.168 The lone stereogenic center was generated via the SAMP/RAMP hydrazone method.169 Mukaiyama’s Co(tfa)2 method99 was employed to install the tertiary alcohol by reaction with alkene 36 (Figure 36). As originally reported, a Sohxlet extractor containing activated 4Å molecular sieves was employed for azeotropic removal of water from the reaction and improved the yield. The reaction proceeded without any detectable racemization.
Figure 36.

Enders’ synthesis of stigmolone (37).
From 2000–2002, Philip Magnus and coworkers reported use of Mukaiyama’s enone α-hydroxlation method106 which Magnus further developed,107 in their synthetic work on the Kopsia alkaloids, including lahadinine B (40), 11-methoxykopsilongine,170 demethoxypauciflorine B and pauciflorine B.171,172 An example of Magnus’ use of this reaction en route to lahadinine B (40) is shown in Figure 37. The reaction proceeds in 83% yield to convert 38 to 39 in a completely diastereo- and regioselective manner. Formation of the carbamate (which is present in the natural product) at the indoline nitrogen was required, as no product was observed in the presence of the free indoline.
Figure 37.

Magnus’ synthesis of (±)-lahadinine B (40).
In 2004, Walker and Bruce employed Mukaiyama hydration conditions in their preparation of oxycodone (44) from codeine (41) (Figure 38).173 Codeine (41) may be oxidized to the ketone (42) by the NADP+-dependent morphine dehydrogenase (MDH) enzyme. In water, 42 and 43 exist as an equilibrium mixture, able to interconvert through the dienol tautomer. The authors found that when a mixture of 42 and 43 (74:26 in THF) was subjected to Mukaiyama’s hydration conditions,105 trisubstituted alkene 43 could be hydrated to oxycodone (44) selectively without hydrating the enone 42. Walker and Bruce sought to drive the reaction to completion under Le Chatelier’s principle, but interconversion of 42 and 43 was too slow in standard solvents or the solvents were incompatible with the Mukaiyama hydration (e.g. water); and the MDH oxidation was also poorly tolerant of organic solvents. The team sought to convert codeine 41 to oxycodone 44 in one pot. They found that by using the ionic liquid, 3-HOPMIm glycolate, codeine (41) could be oxidized by MDH and stoichiometric NADP to a 86:14 mixture of 42 to 43. Following removal of the MDH enzyme and cofactor from solution by dialysis, Co(acac)2 was added to the equilibrium mixture of 42 and 43 followed by gradual addition of phenylsilane with oxygen bubbling. This effected conversion of 43 to oxycodone 44 over 12 hours. Gradual addition of phenylsilane was required in 3-HOPMIm glycolate because reduction of the enone 42 to the corresponding ketone occurred when phenylsilane was added in one portion. Despite all this, equilibration of 42 to 43 was still too slow in HOPMIm glycolate to convert all of 42:43 to 44 on the timescale of the hydration reaction, but the authors state that they could repeat the Mukaiyama hydration on the same mixture after waiting for equilibration to finish. They were eventually able to convert up to 42% of the initial 42:43 mixture to oxycodone (44) via this iterative process.
Figure 38.

Walker and Bruce’ one-pot conversion of codeine (41) into oxycodone (44).
In 2005, Paquette and coworkers employed a Mukaiyama106/Magnus107 enone α-hydroxylation reaction in their synthetic work on fragments of pectenotoxin-2 (Figure 39).174,175 In particular, enone 45 was converted to alcohol 46 in 76% yield (at 75% conversion).
Figure 39.

Paquette’s synthetic work towards pectenotoxin-2.
In 2005, Shibasaki and coworkers completed the first total synthesis of (±)-garsubellin A (49), a polyprenylated phlorglucin derivative which exhibits potent neurotrophic activity by inducing choline acetyltransferase (Figure 40).176,177 Application Mukayaima hydration conditions106 to compound 47 installed the tertiary alcohol of 48, which served as a protecting group for the alkene. A later step entailed dehydrating this alcohol to regenerate the alkene (highlighted in red). Alkene protection was required because subsequent steps in this synthesis involved alkene dihydroxylation, ring closing metathesis, and Wacker oxidation.
Figure 40.

Shibasaki and coworkers synthesis of (±)-garsubellin A (49).
The Baran lab of the Scripps Research Institute has made use of the Mukaiyama hydration in many of their synthetic endeavors. The first example may be found in Baran’s 2008 semi-synthesis of (±)-cortistatin A (52) from prednisone (Figure 41).178,179,180,181 The team built orthoester 51 in a one pot procedure from alkene 50 using a Mukaiyama hydration105 followed by condensation of the formamido-diol with trimethyl orthoformate. The desired stereochemistry of the nascent alcohol arises because molecular oxygen is trapped from the bottom face of the molecule. This likely occurs because of the preference of the A-ring to adopt a half-chair conformation in which the nascent tertiary radical is sp3 hybridized and sits in the axial position with the more sterically demanding alkyl substituents occupying the equatorial position.180
Figure 41.

Baran’s synthesis of (+)-cortistatin A (52).
Also in 2008, Erick Carreira and his team reported the enantioselective synthesis of the core (55) common to banyaside, suomilide and spumigin HKVV (Figure 42).182 Banyaside B was completed in 2010.183 The team found that Mn(dpm)3 worked well in this reaction, affording the hydrate 54 from alkene-containing 53 as a single regio- and stereoisomer. This discovery arose after attempts to epoxidize and hydroborate with mCPBA, BH3•SMe2 and 9-BBN proved fruitless. Curiously, the axial alcohol in 53 was crucial to the success of the reaction; the acylated congener was unreactive.
Figure 42.

Carreira’s synthesis of the core (55) of banyaside, suomilide and spumigin HKVV.
In 2009, Harwood and coworkers reported hydroperoxidation studies towards the synthesis of mycaperoxide B (58), which possesses cytotoxic activity against various cancer cell lines and antiviral activity (Figure 43). In their work, alkene 56 was converted to silylperoxide 57 under Mukaiyama/Isayama conditions,103,104 and after removal of the TBDPS protecting group and oxidation of the primary alcohol to an aldehyde, cyclization proceeded smoothly to give a 1,2-dioxane ring analogous to that found in mycaperoxide B (58). They found that protection of the primary alcohol was necessary to obtain synthetically useful yields. A number of similar substrates were also explored.
Figure 43.

Harwood’s hydroperoxidation studies towards mycaperoxide B (58).
In 2009, Boger and coworkers reported the application of their hydration method to the synthesis of vinblastine (2) from anhydrovinblastine (1) (Figure 44; see Section 2.1, 7.11 for further details).26 The team found that use of excess iron(III) oxalate with sodium borohydride in air could effect selective oxidation of the C-20′ position of anhydrovinblastine (1) to a 5:2 mixture of vinblastine epimers. The chemoselectivity of this reaction is remarkable, given the complexity of the substrate. These conditions furnished the desired isomer as the major product, while other oxidation methods failed to do so. Notably, Mukaiyama conditions failed to give the desired hydration product at all. The Boger lab has further applied their general method for alkene functionalization to the synthesis of a wide variety of other vinblastine analogues.25,184,185,186,187,188,189,190
Figure 44.

Boger’s synthesis of vinblastine (2).
In 2010, Cassayre and coworkers published a paper in which they applied MnIII-catalyzed olefin hydration technology to the selective functionalization of avermectin B1.191 Interest in selective functionalization106,107 of avermectin B1 stems from the importance of these molecules in crop science as acaricides and insecticides and the desire to identify novel derivatives with improved biological activity. An example of this procedure applied to one avermectin B1 derivative is shown in Figure 45. Treatment of compound 59 with catalytic Mn(dpm)3, PhSiH3 and O2 in isopropanol led to a 7:3 diastereomeric mixture of compound 60 and its C-4 epimer in 67% yield. This reaction was chemoselective for the electron deficient alkene; the diene, trisubstituted alkene and cis-alkene were not reduced or hydrated to an appreciable degree. The authors remark at the end of their paper that “there is no doubt that [MnIII hydration technology] has high potential for the selective transformation of complex natural products such as avermectins and the semi-synthesis of novel derivatives of biological interest, competing in that respect with biocatalytic transformations.”
Figure 45.

Modification of avermectin B1 with manganese(III) hydration technology.
In 2011, Gang and Romo completed the total synthesis of (+)-omphadiol (63) from the chiral pool material (R)-carvone (61) (Figure 46).192 The first step of their synthesis enlisted a Mukaiyama106/Magnus107 enone α-hydroxylation reaction to obtain 62 in 63% yield. The reaction proceeds in low 2:1 diastereoselectivity, but this is inconsequential for Romo’s synthesis because the subsequent step involves oxidative cleavage of the α-hydroxy ketone C-C bond, which removes this nascent stereocenter.
Figure 46.

Gang and Romo’s synthesis of (+)-omphadiol (63).
Peng and Danishefsky reported their approach towards maoecrystal V (66) in 2011 (Figure 47).193 As part of their route, they made use of a Mukaiyama106/Magnus107 enone α-hydroxylation reaction to install the alcohol of 65 from enelactone 64, and this was effected in 80% yield with 1.5:1 diastereoselectivity for the desired epimer at 2 mol% catalyst loading.
Figure 47.

Peng and Danishefsky’s approach towards maoecrystal V (66).
In 2011, Herzon et al. employed Mukiayama’s early hydration conditions (which do not employ PhSiH3)98 in their synthesis of hasubanan alkaloids, including (+)-periglaucine B (68) (Figure 48).194,195,196 Specifically, treatment of 67 with stoichiometric Co(acac)2 in isopropanol at 75 °C hydrated the styrenyl alkene, and conjugate addition occured upon treatment with excess formic acid to yield (+)-periglaucine B (68) in 55% over 2 steps. The hydration reaction proceeded with 2.2:1 diastereoselectivity in favor of the desired isomer.
Figure 48.

Herzon et al.’s synthesis of (+)-periglaucine B (68).
Endoma-Arias and Hudlicky have also employed Mukaiyama’s early version98 of his eponymous reaction to effect the γ-hydroxylation of an α,β,γ,δ-unsaturated ester 69 as part of their synthetic work towards the kibdelones [kibdelone A (71) is shown in Figure 49].197 This hydration reaction yielded 70 in 55% yield over a two step sequence (with the first step being acetonide formation to give 69 from the corresponding diol). The authors do not report the diastereoselectivity of the reaction.
Figure 49.

Endoma-Arias and Hudlicky’s synthesis of kibdelone fragment 71.
In 2012, Farcet, Himmelbauer and Mulzer reported an approach towards the core (74) of bielschowskysin (75) in which they synthesize the tertiary alcohol in fragment 73 via a Mukaiyama hydration of 72 (Figure 50).198 The reaction proceeded cleanly in a regio- and stereoselective manner in 64% yield.
Figure 50.

Mulzer’s synthesis of the core (74) of bielschowskysin (75).
In 2012, Carreira completed the total synthesis and stereochemical reassignment of (±)-indoxamycin B (78), a polyketide (Figure 51).199 Completion of their synthesis entailed conversion of 76 to 77 en route to indoxamycin B (75). Regioselective hydration of the inductively electron deficient terminal alkene of 76 in the presence of an electron neutral trisubstituted alkene and an α,β–unsaturated ester was accomplished using Mn(dpm)3 as a catalyst and proceeded in a moderate 49% yield. This reaction gave a 1:1 mixture of epimers, which was inconsequential since the subsequent step involved Dess-Martin periodinane oxidation to the ketone.
Figure 51.

Carreira’s synthesis of (±)-indoxamycin B (78).
Barnych and Vatéle completed their syntheses of (−)-ent-plakortolide I (81) and seco-plakortolide E in 2012 (Figure 52).200,201 The Mukaiyama/Isayama hydrosilylperoxidation103,104 was enlisted to install the key peroxide moiety of (−)-ent-plakortolide I (81). Accordingly, substrate 79 was cleanly converted into the triethylsilyl peroxide 80 in 94% yield over 2 hours, though with 1:1 diastereoselectivity. In 2013, Vatele’s group also applied the Mukaiyama/Isayama silylperoxidation in the synthesis of andavadoic acid, a related natural product also isolated from plakortolide marine sponges.202 Similarly, in 2016, Wu and coworkers employed the Mukaiyama/Isayama hydrosilylperoxidation in their synthesis and structural reassignment of the related natural product, plakinidone.203
Figure 52.

Vatele’s synthesis of (−)-ent-plakortolide I (81).
Chen and Wu also employed a Mukaiyama/Isayama hydrosilylperoxidation reaction to install the requisite 1,2-dioxane functionality in their route to the chamigrane endoperoxide family, of which an example member is talaperoxide C (±)-86 (Figure 53).204 In their case, treatment of diene 82 furnished a mixture of three compounds, 83, 84 and 85. These arise through initial Markovnikov hydroperoxidation of the enone olefin, 6-exo-trig radical cyclization of the nascent peroxide radical followed by trapping with a second oxygen molecule, predominantly from the bottom face of the molecule, and silylation with Et3SiH. The alcohol 83 likely arises from cobalt mediated reductive O-O bond cleavage. This mixture of products was used to access a variety of chamigrane endoperoxide natural products, including talaperoxide C (86).
Figure 53.

Wu’s synthesis of the chamigrane endoperoxide secondary metabolites.
In 2013, Rizzacasa’s group published a formal total synthesis of the cytotoxic myxobacteria metabolite spirangien A (89) (Figure 54).205 To install the C-20 alcohol of spirangien A (89), the team employed a Mn(dpm)3 catalyzed hydration of the enone (87).106 This furnished a 1:1 epimeric mixture of 88, which was separated by HPLC. Subsequent treatment of 88 with camphorsulphonic acid removed the ketals and induced spirocyclization.
Figure 54.

Rizzacasa’s formal synthesis of spirangien A (89).
In 2013, Zahel and Metz published a concise enantioselective route to the guaiane sesquiterpene (−)-oxyphyllol (92) (Figure 55).206 The team employed a Mukaiyama hydration105 to convert the alkene of 90 to the tertiary alcohol 91 in 82% yield as a 2.4:1 diasteromeric mixture. They had initially tried to obtain 91 via a tactic entailing dihydroxylation, xanthate ester formation and Barton-McCombie deoxygenation, but were unsuccessful. Thus the discovery that a one step Mukaiyama hydration could regio- and diastereoselectively access 91 was welcome.
Figure 55.

Metz’s synthesis of (−)-oxyphyllol (92).
In 2013, Baran reported further work in the field of steroid synthesis with a publication detailing their semisynthesis of ouabagenin (95) from adrenosterone (Figure 56).207,208 Baran’s team found that Mukaiyama’s hydration105 procedure could install the C-14 tertiary alcohol 94 from the trisubstituted alkene 93 in 86% yield and 8:1 diastereoselectivity. Use of dioxane as solvent improved the diastereoselectivity of the nascent alcohol.
Figure 56.

Baran’s synthesis of ouabagenin (95).
Also in 2013, Hiroya’s group communicated their stereocontrolled synthesis of trichodermatide A (96), which is cyctotoxic against the A375-S2 human melanoma cell line (Figure 57).209,210 The final sequence of their synthesis converted 96 to 97 in a two-step, one pot hydration/deprotection sequence. Remarkably, a Mukaiyama hydration conducted in trifluoroethanol was able to effect this reaction in good yield with high chemo-, regio- and stereoselectivity. On the other hand, use of THF as a solvent barely hydrated 96. The authors rationalize the observed selectivity as follows: the nascent cobalt hydride reacts more readily with the electron rich enol ether olefin than the electron deficient vinylogous ester olefin; the axially oriented C-13 α-ether oxygen may coordinate to the cobalt hydride to induce an α-facial approach to place the hydrogen at C-8, and this would also lead to a radical on C-9 stabilized by the adjacent oxygen atom; the C-9 radical then reacts with molecular oxygen to form a peroxide, which is degraded by Co(acac)2 and PhSiH3 to give the alcohol of 97. A solvent swap and treatment with K2CO3 to removed the aryl ester furnished trichodermatide (97) in 83% yield.
Figure 57.

Hiroya’s synthesis of trichodermatide A (97).
In 2014, Tietze et al. reported their domino approach to the enantioselective total syntheses of blennolide C (100) and gonytolide C (Figure 58).211 They accessed both molecules through intermediate 99, which was obtained in a two step redox manipulation entailing a Mn(dpm)3 catalyzed hydration106,107 of 98, followed by a Ley oxidation.
Figure 58.

Tietze et al.’s synthesis of blennolide C (100) and gonytolide C.
Also in 2014, the Inoue group completed their total synthesis of ryanodol (104), an insecticidal compound of the South American plant Ryania speciosa (Figure 59). 154,212 One step of their synthesis required oxidation of the C-15 alcohol to the ketone oxidation state. After many initial attempts were rebuffed, the team developed a two-step oxidation protocol based on the Mukaiyama/Isayama hydrosilylperoxidation procedure. In the first step of this procedure, alkene 101 was converted to triethylsilyl peroxide 102 in 75% yield along with 5% of 103, which results from decomposition of the triethylsilyl peroxide 102 to the corresponding C-15 ketone followed by ketone hydration and hemi-ketal formation. The remaining silyl peroxide 102 was converted to hemi-ketal 103 by addition of nonafluoro-1-butanesulfonyl fluoride (NfF) and DBU. Inoue and coworkers have shown that this method for the conversion of alkenes to ketones is applicable to other alkenes as well.155
Figure 59.

Inoue’s synthesis of ryanodol (104).
In 2014, the Baran lab published a unified approach to the ent-atisane diterpenes and related alkaloids, including (−)-methyl atisenoate (107) and (−)-isoatisine (110).213 Their route featured creative use of Mukaiyama/Isayama hydrosilylperoxidation conditions103,104 (Figure 60). In the case of (−)-methyl atisenoate (107), commercially available (−)-steviol (105) was converted in one step to a corresponding methyl ester, which was treated with Co(acac)2, O2 and Et3SiH in DCE at 60 °C to give an intermediate triethylsilyl peroxide, which underwent cleavage of the C-13/C-16 bond concomitant with silyl peroxide O-O bond cleavage to give the diketone 106. A similar sequence was employed to convert 108 to 109, which resulted after an aldol addition was induced by the acidic resin, Amberlyst ® 15, and acetylation of the C-13 alcohol.
Figure 60.

Baran’s synthesis of ent-atisine diterpenes and related alkaloids.
Also in 2014, the Baran lab enlisted a variety of chemical and enzymatic C-H oxidation techniques to oxidize steroid skeletons in order to improve their physical/pharmokinetic properties (Figure 61).214 One aspect of their strategy involved functionalization of the C-12 and C-13 carbons via a guided desaturation approach. To accomplish oxidation of the C-12 position, Baran first employed a modification of Carreira’s procedure for C-O/C-N bond formation (see Sections 2.1 and 3.1–3). 115 Specifically, Baran’s team installed the tertiary alcohol of 112 by hydration of the alkene in 111, and acetylation furnished 112. Subjection of 112 to Suárez’s conditions215 resulted in O-centered radical formation, C-H abstraction from C-12, iodine trap of the nascent radical, and displacement to give the ether 113 in 30% yield. To access the olefin 116, the team employed a similar but distinct approach. After acetylation of the alcohols in 111, the hydroperoxide 115 was obtained under similar conditions to 112 but by using Et3SiH instead of PhSiH3 and avoiding a reductive work-up according to Mukaiyama’s hydrosilylperoxidation conditions.103 Treatment again with Suarez’s conditions furnished the olefin 116. The mechanism of this transformation purportedly involves hydrogen abstraction followed by oxidation of the nascent secondary radical to the carbocation and proton elimination.
Figure 61.

Baran’s modification of steroids by C-H oxidation.
Maimone’s 2014 four step synthesis of the antimalarial (+)-cardamom peroxide (118) presents a beautiful example of manganese-catalyzed peroxidation technology (Figure 62).216 The key step in Maimone’s transformation involves formation of the endoperoxide of 118 from 117. Maimone’s group was able to accomplish this feat by use of catalytic Mn(dpm)3 in i-PrOH/CH2Cl2 with slow addition of PhSiH3 at −10 °C, followed by mild reductive work-up with PPh3. The cascade is thought to proceed via initial formation of a tertiary α-keto radical. The more electron deficient alkene, flanked by two carbonyls, is also the most sterically accessible and so reacts the fastest with a [Mn]-H to generate the more stable tertiary α-keto radical. This in turn reacts with molecular oxygen to form a terminal peroxy radical that then cyclizes in a 7-endo-trig fashion to forge the 7-membered endoperoxide ring of 118 and concomitantly generates a second tertiary α-keto radical. This radical reacts with O2 to form a hydroperoxide radical, which is reduced to the hydroperoxide under the reaction conditions. This hydroperoxide is completely reduced to form the tertiary alcohol of 118 upon addition of PPh3 to give 118 in 52% yield.
Figure 62.

Maimone’s synthesis of (+)-cardamom peroxide (118).
Tran and Cramer published the biomimetic syntheses of (+)-ledene (119), (−)-palustrol (120), (+)-viridiflorol (121), (+)-spathulenol, and psiguadial A, C, and D from the terpene (+)-bicyclogermacrene in 2014 (Figure 63).217 The authors accessed (−)-palustrol (120) by a Mukaiyama hydration105 of (+)-ledene (119). This reaction proceeded in a moderate 45% yield, although this yield was based on a reaction run on a μmol scale, with the correct regio- and stereoselectivity. In contrast, hydroboration with BH3•THF then oxidative work-up with H2O2/NaOH gave (+)-viridiflorol (121), which has the opposite regio- and stereo-selectivity. Cramer has also employed the Mukaiyama hydration reaction to the generate cyclobutanols from cyclobutenes.218
Figure 63.

Cramer’s synthesis of (−)-palustrol (120) via Mukaiyama hydration of (+)-ledene (119).
In 2015, the Xie lab published the total synthesis of (−)-conolutine (124) in which they employed a Mukaiyama hydration105 on substrate 122 to obtain 123 (Figure 64).219 Acid mediated hydration conditions were incompatible with the functionalities present in 122. The authors explored a variety of metal salts, particularly of iron and cobalt, known to effect radical oxidation. Their efforts revealed that although Boger’s hydration conditions81 with excess Fe2(ox)3 and NaBH4 were effective, these gave a significant amount of reduction product. Although Mukaiyama’s conditions gave comparable yields, the authors preferred it because it was operationally more convenient (no need for excess metal complex and dilute conditions). Further details may be found in Xie’s paper.219
Figure 64.

Xie’s synthesis of (−)-conolutine (124).
Zhu and coworkers reported total syntheses of (−)-mersicarpine, (−)-scholarisine G, (+)-melodinine E, (−)-leuconoxine, (−)-leuconolam, (−)-leuconodine A, (+)-leuconodine F, and (−)-leuconodine C in 2015.220 The team found that (−)-scholarisine G (126) could be obtained directly from (+)-melodinine E (125) by means of a Mukaiyama106/Magnus107 enone α-hydroxylation reaction, which proceeds in this case to give the β-hydroxylated product (Figure 65). This is not entirely unexpected as Mukaiyama106 and Magnus221 both comment that regioselectivity can be reversed for β,β-disubstituted enones due to an increase in radical stability at the β-position, and in this case the enone is β, β-disubstituted, and the β position is benzylic. Zhu and coworkers also propose an alternative explanation based on the possibility that the central aminal functionality opens under the reaction conditions.
Figure 65.

Zhu and coworkers’ synthesis of (−)-scholarisine G (126) from (+)-melodinine E (125).
In 2015, the Baran lab completed syntheses of the remarkable secondary metabolites, fumitremogin A (130) and verruculogen (131), which possess an electron-deficient endoperoxide alongside an electron-rich indole (Figure 66).222 Baran employed Mukaiyama/Isayama hydrosilylperoxidation conditions103,104 to build the peroxide fragment 129 which was later incorporated into the target compounds 130 and 131. Specifically, aldehyde 127 was converted to an acetal then subjected to Co(modp)2 catalyzed hydrosilylperoxidation with Et3SiH reductant to give 128. Another 3 steps involving TES removal, TBDPS protection and acetal deprotection furnished hydrosilylperoxy aldehyde 129.
Figure 66.

Baran’s synthesis of fumitremogin A (130) and verruculogen (131).
In 2015, See, Herrmann, Aihara and Baran published an improved version of Schonecker’s Cu-mediated C-H oxidation procedure and applied it in the synthesis of several polyoxypregnanes, including utendin (134), pergularin (135), and tomentogenin (133) (Figure 67).223 Similar to the case of ouabagenin (Figure 56), subjection of substrate 132 to modified Mukaiyama hydration conditions led to alcohol 133 in 67% yield in a chemo- and stereoselective fashion. Addition of PPh3 served to decompose the intermediate peroxide.
Figure 67.

Baran’s synthesis of polyoxypregnanes.
In another example of Mukaiyama hydration105 technology in steroid synthesis, Zhu and Yu reported hydration of alkene 137 to alcohol 138 en route to linkosides A (139) and B (140), neuritogenic natural products of Linckia laevigata.224 Oxidation of the C-8 carbon was challenging but could be induced with stoichiometric Co(acac)2 at 50 °C in a moderate 46% yield. All other methods attempted by the authors were unable to oxidize 137 to 138.
In 2015, Song et al. reported the total synthesis of several atisane-type diterpenoids, including rac-crotobarin (144) and rac-crotogroudin (146), through general application of a Diels-Alder cycloaddition of Podocarpane-type unmasked ortho-benzoquinones (Figure 69).225,226 Subsequent manipulations involved use of manganese and cobalt hydration technology to install Markovnikov alcohols (Figure 69). In particular, use of Mn(dpm)3 as catalyst hydrated both dienes of 141 to give 142 en route to rac-crotobarin (143). Use of Co(acac)2 gave lower yields of 142, while Fe(acac)3 gave none of the desired product. The authors found that the northern olefin of 141 was more reactive, undergoing hydration at 0 °C, while the southern olefin underwent hydration upon warming to ambient temperature. In contrast, hydration of alkene 144 to 145 occurred solely as a single diastereomer through use of Co(acac)2, while both Mn(dpm)3 and Fe(acac)3 afforded complex mixtures. Compound 145 was made en route to rac-crotogroudin (146). Finally, Song et al. found that they could effect a remarkable rearrangement reaction of ketone 147 under Mukaiyama hydration reactions. The proximity of a ketone to the nascent carbon-centered radical generated under Mukaiyama hydration conditions enabled efficient formation of a transient cyclopropane (148) along with an O-centered radical. This can collapse to the [2.3.1] bridged ring system 149, which gives alcohol 150 upon reaction with oxygen and [Mn]. Compound 150 possesses the skeletal structure of the scopadulane-type diterpenoids and thus this radical rearrangement presents a remarkable way to access this family of secondary metabolites.
Figure 69.

Song et al.’s used of [Co] and [Mn] radical chemistry in the syntheses of a) rac-crotobarin (143), b) rac-crotogroudin (146), and c) the scopadulane-type diterpenoid skeleton.
In 2016, Keßberg and Metz reported enantioselective syntheses of brosimine A, brosimine B, and brosimacutin L (152) (Figure 70).227 Use of Mukaiyama’s hydration105 in the last step followed by a TBAF deprotection enabled conversion of 151 to brosimacutin L (152).
Figure 70.

Metz’s synthesis of brosimacutin L (152).
Recently in 2016, the Baran lab completed a remarkable 19-step total synthesis of (+)-phorbol (156), of which two steps involved a modified Mukaiyama hydration (Figure 71).228 The first sequence installed the TMS-protected alcohol 154 from substrate 153 in good yield, albeit with stoichiometric Mn(acac)2. After a TFDO oxidation of the C-12 methylene and Zn2+/Mg2+ mediated dehydration with cyclopropane ring opening to give an intermediate diene, a second Mukayaima hydration was employed to selectively hydrate the 2,2-disubstituted alkene to give tertiary carbinol 155 in 34% yield over 2 steps alongside 62% of recovered 154. Addition of PPh3 was an essential additive in this second step as it reduced the intermediate peroxide adduct to an alcohol, preventing unwanted by-products.
Figure 71.

Baran’s synthesis of (+)-phorbol (156).
Also in 2016, Ang Li’s group completed the total synthesis of rubriflordilactone B (159) (Figure 72).229 Their convergent synthesis forged the left and right fragments of rubriflordilactone B (159) separately before joining them together at the aromatic ring. Construction of the left hand fragment involved Mukaiyama hydration105 of alkene 157, followed by sodium hydroxide mediated lactonization to afford 158. Treatment of 157 with strong aqueous acid could also effect the transformation directly in one step, but occurs with racemization.
Figure 72.

Li and coworker’s synthesis of rubriflordilactone B (159).
In addition to the positive examples listed above, several examples (not exhaustive) from synthesis in which these types of reactions did not work as desired may be found in these references.230,231,232
3. C-N bonds
3.1 Nitrosation and Oximation
The ability to capture molecular oxygen with carbon-centered radicals generated from alkenes by transition metal catalysts in the presence of a reductant prompted researchers to consider what other sorts of gases might be used. Chief among these was nitric oxide (NO), which like molecular oxygen, has the ability to engage in radical chemistry from its ground state electronic configuration. The topic of “organic radical reactions associated with nitrogen monoxide” has been reviewed.233
After demonstrating the viability of hydration with molecular oxygen 1981,74,75 Okamoto and coworkers reported in 1987 a method for the hydronitrosation of styrenyl alkenes with nitric oxide through the action of a cobalt catalyst and BH4− to form oximes (Figure 73).234
Figure 73.

Nitrosation of styrenyl alkenes from Okamoto et al.
This reaction employed Co(dmg)2(pyr)Cl as a catalyst with tetraethyl ammonium borohydride as reductant in a 1:1 mixture of DME and i-PrOH. The scope of the reaction extended to a variety of substituted alkyl aryl alkenes and polyaromatic compounds. Styrenes with either electron withdrawing substituents (Cl, Br, NO2) or electron donating substituents (OMe, vinyl, O-allyl) underwent hydronitrosation, although the latter proceeded with 10–30% lower yields in general. The highest yield reported for this reaction was 69% for p-Cl styrene. Whereas Co(TPP) and Co(Pc) were known to engage in the hydration reaction with molecular oxygen,74 both complexes were ineffective in the presence of nitric oxide. The authors speculated that this difference may result from product inhibition and/or from NO poisoning of the catalysts. Okamoto et al. proposed a mechanism which involved nitrosation by radical recombination of a benzylic radical with NO, followed by tautomerization. The benzylic radical was thought to be generated by homolysis of a Co-C bond. Generation of the Co-C bond was not thought to occur via a coordination-insertion mechanism, but a HAT-type mechanism was not explicitly proposed. Coordination of nitric oxide to cobalt in the catalytic cycle is not proposed by the authors, who state that although stoichiometric reactions were known, prior to their work “no catalytic reaction involving NO has been reported probably because the coordination abilities of insertion products are too high for replacing them by either NO or the organic substrates, a process necessary for ensuring the catalytic cycle.”
It is perhaps for this reason – nitric oxide’s tendency to form stable complexes with many metals and thereby inhibit catalysis – that Okamoto et al. replaced nitric oxide with ethyl nitrite (EtONO) in their further study of this reaction in 1988 (Figure 74).235 Their substrate scope was slightly expanded, and included substituted styrenes, 1-phenyl-1,3-butadiene, and some cyclic aryl-conjugated ethylenes. Use of EtONO improved the yield of this reaction considerably (> 90% for styrenes) while halving the catalyst loading and permitting use of NaBH4 in a few cases. Benzene proved to be the best solvent under these conditions.
Figure 74.

2nd generation conditions for hydronitrosation from Okamoto et al.
In 1990, Kato and Mukaiyama reported the hydronitrosation of α,β-unsaturated carboxamides catalyzed by the cobalt complex, Co(eobe), with nitric oxide as nitrogen source and triethylsilane as reductant (Figure 75).236 The reaction proceeded in 67–96% yield, giving isomeric mixtures of nitroso and oxime products, with reaction times of 16 to 66 hours. Catalysts other than Co(oebe) exhibited poor conversion or yielded primarily reduction products. The authors found that partial pressure of nitric oxide improved the reaction yield, so a 1:1 mixture of NO:N2 was used at atmospheric pressure. The team also found that 3 equivalents of triethylsilane were optimal, with yields dropping when either 2 or 4 equivalents were used instead. With respect to the mechanism of the transformation, the authors stated that “it is assumed that 2-cobalt-carboxamide complex is initially generated as an intermediate by the reaction of cobalt(II) complex and Et3SiH, and successive substitution of cobalt with NO takes place to produce the 2-nitrosocarboxamide.”
Figure 75.

Kato and Mukaiyama’s hydronitrosation of α,β-unsaturated carboxamides with substrate scope.
Kato and Mukaiyama soon after extended the scope of their reaction to α,β-unsaturated esters by using n-butyl nitrite instead of nitric oxide (Figure 76).237 Additionally, use of phenylsilane as reductant and THF as solvent provided the best yields, although diethylsilane in THF also worked very well. The reaction was generally run for one to two days and its utility was demonstrated by synthesis of several products in good yield. Oxime formation occurred exclusively; the tautomeric hydronitrosation products were not observed.
Figure 76.

Kato and Mukaiyama’s hydronitrosation of α,β-unsaturated esters.
Kato and Mukaiyama also expanded the scope of this reaction to α,β-unsaturated ketones, nitriles, and amides alongside discussion concerning the development of this reaction.238
In 1992, Kato and Mukaiyama extended the preceding work to convert unactivated terminal and 1,2-disubstituted alkenes with an iron catalyst to nitroso alkane dimers (Figure 77).239 In particular, the team found that treatment of alkyl substituted alkenes with Fe(acac)3 as a catalyst in the presence of n-butyl nitrite and phenylsilane proceeded smoothly to provide the corresponding nitroso dimers. While Fe(acac)3 provided the best yields and was the most readily available iron(III) complex, Fe(dpm)3 (dpm = 2,2,6,6,-tetramethyl-3,5-heptanedionato) and Fe(acp)3 (acp = 2-acetylcyclopentanato) also worked well. Use of iron complexes bearing electron deficient ligands resulted in significantly worse yields, however. Bubbling nitrogen gas through the reaction improved the yield of the reaction, purportedly by removing nitric oxide. Nitric oxide is thought to arise from gradual decomposition of butyl nitrite and is pernicious because it binds the catalyst, deactivating it. Several terminal and 1,2-disubstituted alkene substrates successfully reacted to give the corresponding alkyl nitroso dimers in moderate to good yields, but trisubstituted alkenes did not react. In the case of allyl benzoate 160, the oxime compound 162 was also obtained in a 2:1 ratio with the dimer 161. It is known that the formation of nitrosoalkane dimers occurs with great facility from the individual alkyl nitroso monomers and are remarkably stable to retro-dimerization.240,241 Aryl and keto-substituted nitroso compounds tautomerize to form the oxime instead.
Figure 77.

Mukaiyama’s and Kato’s synthesis of nitroso alkanes from alkenes.
Mukaiyama has reviewed his group’s work on the nitrosation and nitration of olefins with nitrogen monoxide.242
In 1998, Sugamoto et al. showed that a cobalt porphyrin [Co(TPP)] system may also effect the oximation of styrenes (Figure 78), α,β-unsaturated carbonyls (Figure 79), and α,β,γ,δ-unsaturated carbonyls (Figure 80).243 With use of t-BuONO as nitrite equivalent and triethylsilane as reductant, several para-substituted styrenes were cleanly converted to the corresponding oximes. Yields were highest for styrenes bearing electron-withdrawing groups. Ketone formation also occurred as a minor byproduct in some cases. The high activity of Co(TPP) in this reaction stands in stark contrast to Okamoto’s system234 where it showed no activity with ethyl nitrite and tetrabutyl ammonium borohydride. MnIII(TPP)Cl, FeIII(TPP)Cl, Co(acac)2, Co(salen) showed little or no reactivity under the reaction conditions. Other cobalt(II) porphyrin complexes, especially those bearing electron donating substituents, also worked well, but Co(TPP) was chosen for substrate scope exploration because of its commercial availability.
Figure 78.

Oximation of p-styrenes from Sugamoto et al.
Figure 79.

Oximation of α,β-unsaturated esters, ketones and aldehydes from Sugamoto et al.
Figure 80.

Oximation of α,β,γ,δ-unsaturated carbonyls from Sugamoto et al.
Under the same conditions, α,β-unsaturated esters, ketones and aldehydes furnished the corresponding α-oximes (Figure 79). Notably, β,β-disubstituted α,β-unsaturated ester 163 did not react at all.243
Furthermore, Sugamoto et al. demonstrated the capacity for this reaction to convert a number of α,β,γ,δ-unsaturated carbonyls to the corresponding γ-oxime-α,β-unsaturated carbonyls in moderate to high yields (Figure 80).243
Boger and coworkers have demonstrated that a wide variety of radical traps can be employed with their iron(III) oxalate-mediated method for hydrofunctionalization of unactivated alkenes.26,81 Among these, Boger has shown that excess NaNO2 may be used to obtain a nitrosoalkane from an alkene (Figure 81).This method was also used by Boger and coworkers to provide the C-20′ nitroso derivative of vinblastine with the leurosidine stereochemistry.26
Figure 81.

Boger’s nitrosation of unactivated alkenes.
In 2009, Matthias Beller and coworkers showed that FeIIPc can also effect the oximation of styrenes and aryl substituted alkenes in ethanol using sodium borohydride as reductant and t-BuONO as nitric oxide equivalent (Figure 82).244 Beller’s team found that hydrogen was evolved during the initial course of the reaction and B(OEt)3 was observed by gas chromatography. The authors discovered that running the reaction under 10 bar of hydrogen gas increased yields. Various metal complexes were tested in this reaction, and although FePc worked best, vitamin B12 and Co(dmgH2)(py)Cl also worked well. FeCl3 could also catalyze this reaction when mixed with 2–4 equivalents of di- or tri-dentate pyridine type ligands. A variety of substituted styrenes reacted to give the corresponding oximes in useful yields, with electron deficient styrenes giving the best yields. Additionally, oximes of a 2-vinyl pyridine (164), alkyl aryl alkenes (165 and 167), and a di-aryl alkene (166) could be obtained. The authors propose the intermediacy of a σ-alkyl Fe(III)(Pc) complex in this reaction, which reacts with t-BuONO to afford an oxime. Interestingly, in contrast to the Co(TPP) system of Sugamoto et al.,243 when α,β-unsaturated esters were subjected to the reaction conditions, the corresponding saturated esters were instead obtained. This difference may be due to a radical-polar crossover as observed elsewhere with iron complexes in related chemistry (cf. Section 4.4, 4.8 and 4.9), although the authors do not comment.
Figure 82.

Beller’s oximation of aryl substituted alkenes.
In 2014, Lahiri et al. published a paper on a second method for iron catalyzed aryl alkene oximation (Figure 83).245 An iron(II) bis tetrafluoroborate hexahydrate/dipicolinic acid combination served as the metal salt/ligand system. The authors found that a 5:1 mixture of MeOH-H2O to be best for this transformation. Use of a 5:1 mixture of EtOH-H2O under otherwise identical conditions, curiously, gave the ketone rather than the oxime as the major product. The authors observed that reaction yields decreased with longer reaction times and higher catalyst loadings, presumably due to reduction of the oxime products. As Beller had previously noticed, Lahiri et al. also observed that their yields improved when the reaction was run under 10 bar of hydrogen gas. A variety of styrene substrates were converted to the corresponding oximes in good yields. Notably, no dehalogenation occurred with halogenated styrenes.
Figure 83.

Lahiri et al.’s method for oximation of styrenes.
3.2 Hydrohydrazination
In the absence of other reagents, alkenes and azodicarboxylates will react via an ene reaction to form a C-N bond. A trisubstituted alkene exhibits strong preference for bond formation at the less-substituted carbon in accord with stabilization of developing positive charge in the transition state,246 that is, anti-Markovnikov selectivity. The alternative Markovnikov C-N bond formation can arise when the nitrogen enophile is tethered and geometrical constraints force this alternative regioselectivity. On the other hand, Markovnikov C-N bond formation can be achieved classically via a Ritter reaction using strong and concentrated Brønsted acid. Alternatively, carbon radicals can add to azodicarboxylates,247,248,249 as can organometallic nucleophiles,250,251 offering an alternative to classical reactivity if a radical or organometallic can be generated from an alkene.
In 2004, as one of the earliest expansions of Mukaiyama’s reactivity engine,99,103,104,105,118 Waser and Carreira demonstrated that a cobalt(III)252 complex could catalyze the Markovnikov hydrohydrazination of diverse electron neutral alkenes (Figure 84).115 This reductive coupling allows the normally electrophilic diazodicarboxylates253 to occupy a position previously occupied by strictly nucleophilic nitrogen sources. Furthermore, exquisite chemoselectivity is observed in the reaction: alcohols are unreactive, benzyl groups are not affected, esters, ketones and acetals (not shown) remain intact and even primary alkyl halides are untouched (22 substrates are included). Yields ranges from 62–94% and a variety of cyclic and acyclic 1,1- and 1,2-disubstituted and trisubstituted alkenes may undergo hydrohydrazination. During their development of this reaction, the authors found that previously reported cobalt complexes were unable to effect the desired addition and so screened a variety of ligands known to mediate epoxidation or peroxidation reaction of alkenes or alkanes.254 They eventually found that the Schiff base cobalt(III) complex 168 provided the best results.
Figure 84.

(a) Carreira’s cobalt catalyzed hydrohydrazination procedure (b) with selected substrates (c) and the cobalt precatalyst (168).
Waser and Carreira soon after showed that the simple manganese complex, Mn(dpm)3, could effect this transformation as well, but with enhanced reactivity – proceeding with lower catalyst loading (2% vs. 5%), shorter reaction times (2–3 hrs. vs. 5–20 hrs.), and expanded substrate scope compared to the cobalt catalyst (Figure 85).255 With particular regard to the expanded scope, the authors were able to hydrohydrazinate 1,2-disubstituted alkenes, such as cyclohexene and crotyl alcohol, and tetrasubstituted alkenes such as dimethylbutene, which had previously exhibited poor yields under cobalt catalysis. Use of Mn(dpm)3 as catalyst was also compatible with different silanes, including the much cheaper PMHS [poly(methylhydrosiloxane)].
Figure 85.

(a) Carreira’s manganese catalyzed hydrohydrazination procedure (b) with selected substrates. The conditions for the [Co] reactions are those shown in Figure 84.
Further work by Waser et al. on this hydrohydrazination system showed that allylic and propargylic hydrazines could be accessed by a cobalt-catalyzed hydrohydrazination of dienes and enynes (Figure 86).256 The authors had earlier observed that olefins in conjugation with an aromatic ring (styrene derivatives) reacted more quickly in cobalt catalyzed hydrazination of olefins and found that they were able to harness this rate difference to access allylic and propargylic hydrazides. Notably, the authors had to suppress competitive semi-reduction, di-hydrohydrazination and Diels-Alder cycloaddition pathways. Thus the authors found that use of the less-active cobalt catalyst 169, gave the best results for both diene and enyne substrates. In the case of the more reactive dienes, 2.5–5 mol% of 169 alongside tetramethyldisiloxane (TMDSO) proved optimal, with the 10 substrates tested furnishing the corresponding allylic hydrazines in 45–90% yield. Isomerization occurred in cases where a more substituted olefin resulted (170 and 171), whereas isomerization did not otherwise occur for other examples (172). Both cyclic and acyclic substrates worked well. In the case of enyne substrates, the alkene portion always reacted preferentially and no isomerization was observed. PhSiH3 rather than TMDSO gave the best results for the 12 enyne substrates tested, providing the desired propargyl hydrazines in 42–83% yield.
Figure 86.

(a) Carreira’s cobalt catalyzed hydrohydrazination procedure for dienes and enynes (b) with selected diene substrates (c) with selected enyne substrates (d) the cobalt precatalyst (169) and TMDSO.
In their 2006 full paper, Carreira and coworkers showed that this hydrohydrazination reaction also works well (12 substrates, 60–98% yield) for a variety of aryl- and heteroaryl-substituted alkenes, including vinyl-furans, thiophenes, pyrroles, imidazoles, pyridine, pyrazines and indoles.115 Notably, two substrates, 4-vinyl aniline and 2-vinyl pyrrole gave low yields (<40%) when unprotected, but were fine when protected with tosyl and f-moc moieties. The authors also discuss in details the effects of the metal and ligand identity on the aforementioned reactions as well as mechanistic studies (see Section 7.11).
In 2005, Sato et al. showed that it was possible to effect the asymmetric α-hydrazination of α,β-unsaturated carboxylates by use of alkenoates bearing a camphorsultam chiral auxiliary (χc*) alongside Mn(dpm)3, PhSiH3 and di-tert-butyl azodicarboxylate (DBAD) (Figure 87).257 The reaction proceeds in moderate to good yield with good diastereoselectivity for a variety of alkyl, aryl, and thioether-containing products. The authors propose the intermediacy of a Mn-C bond, but restrict their speculation regarding the mechanism to this.
Figure 87.

Yamada and coworkers’ asymmetric hydrohydrazination.
Although it involves cyclopropanes rather than alkenes, Bunker et al. of Pfizer took note of Waser and Carreira’s work115,255,252 by showing in 2011 that [1.1.1]propellane (173) also reacts with di-tert-butyl azodicarboxylate and phenylsilane in the presence of Mn(dpm)3 to give 174, which may be converted to the corresponding amine hydrochloride salt by a deprotection/hydrogenation sequence (Figure 88).258 Propellamines are of interest in drug discovery.259 The reaction likely proceeds via a radical mechanism.
Figure 88.

Bunker et al.’s synthesis of propellamine.
In 2016, Cui and coworkers developed a method for coupling unactivated olefins with α-diazocarbonyl compounds to generate terminally N-substituted stabilized hydrazones (Figure 89).260 This approach complements the hydrohydrazination reactions discussed above as another method for reductive Markovnikov installation of an N-N unit across an alkene under fairly mild conditions, albeit this method results in pendant hydrazones instead of hydrazines by utilizing an alternative hydrazine-equivalent source. As noted in two recent entries to the field of diazocarbonyl chemistry,261,262 nucleophilic addition into the terminal nitrogen represents an uncommon mode of reactivity for α-diazocarbonyl compounds, although the reactivity of such compounds has been well studied and recently reviewed.263
Figure 89.

(a) Cui and coworkers’ radical addition into diazo compounds (b) with selected examples of olefin and diazo variation, and intramolecular cyclization.
Typical conditions in this reaction utilize one equivalent of diazo compound and two equivalents of olefin in presence of 10 mol % Fe(acac)3 and two equivalents phenylsilane with ethanol or THF/EtOH as the solvent. These conditions are analogous to those employed by Baran and coworkers in their various hydrofunctionalization reactions20,21,264,265 (discussed in Sections 4.4 and 4.5), as well as those used by Cui and coworkers in their hydrostyrenylation266 (Section 4.6). The authors showed a variety of olefins were competent to undergo this reaction, including monosubstituted, symmetrical 1,2-disubstituted, and 2,2-disubstituted olefins. One example of selective heteroatom-directed radical generation21 was also demonstrated.
Additionally, a range of stabilized diazo compounds were investigated as utilizing β-citronellol as the olefin partner. These necessarily included at least one electron-withdrawing substituent, typically a substituted carboxylate moiety, but in two cases substituted amide-type moieties were demonstrated (diazo derivatives of N-methyl oxindole and N,N′-dimethyl barbituric acid, respectively, not shown). All examples demonstrated also included a second substituent proximal to the diazo group, which could be electronically varied. Electron-deficient secondary substituents such as carboxylate, phenylsulfone, and phosphonate ester were shown to be tolerated, as well as an electronically neutral benzyl moiety, and electron rich arenes such as 2-naphthalene. The authors also demonstrated that this reaction could be applied in an intramolecular sense, generating six, seven, and even eight membered rings successfully.
3.3 Hydroazidation
In 2005, Carreira reported another variant of the Mukaiyama reaction manifold which is capable of introducing an azide at the Markovnikov position of an olefin through the use of p-toluenesulfonyl azide as electrophilic nitrogen source (Figure 90).115,267,268 Organoazides are convenient intermediates to amines, amides, anilines and heterocycles, as well as end-products in themselves or substrates for the highly useful cycloaddition with alkynes.269 Carreira’s hydroazidation, in contrast to the hydrohyrazidation, benefits from formation of cobalt complex in situ from cobalt tetrafluoroborate tetrahydrate [Co(BF4)2•H2O] and ligand 172. In some cases, tetramethyldisiloxane (TMDSO) outperforms phenylsilane, which gives larger amounts of reduction by-products. Addition of TBHP (30 mol%) as an additive was found to accelerate initiation of the reaction. Unlike the examples in Figure 84 (Section 3.2), alcohol substrates are not tolerated, even though the reaction is run in ethanol. Instead, proximal alcohol functions must be protected with sterically large silyl groups for efficient reaction. Even use of a benzyl ether leads to diminished yields, which appears to implicate an organometallic intermediate with a vacant orbital as a necessary component of the catalytic cycle. A total of 13 substrates, including terminal, geminally disubstituted, and trisubstituted alkenes, successfully underwent hydroazidation with yields ranging from 35–90%. Subsequent work further expanded the scope of this reaction to a variety of other geminally disubstituted alkenes268 and investigated the mechanism115 of the reaction (see Section 7.11).
Figure 90.

(a) Carreira’s cobalt catalyzed hydrohydrazidation procedure for alkenes (b) with selected substrates (c) the cobalt ligand (175) and TMDSO.
In 2012, the Boger lab also reported a procedure for hydroazidation of unactivated alkenes as part of a widely general method for the hydrofunctionalization of unactivated alkenes with Fe2ox3 in the presence of sodium borohydride and a radical trapping agent (Figure 91).81 Following their initial reports of this reactivity in 2009,26 Boger’s team demonstrated that use of stoichiometric sodium azide allowed for hydroazidation of unactivated alkenes in the presence of an impressive array of other functional groups, including an epoxide, a carboxylic acid, alcohols, an aniline, an ether and a primary alkyl bromide (not shown). Aryl substituted alkenes were also competent substrates. Other azide salts (LiN3 and CsN3) were also effective azide sources. Mechanistic studies indicate a radical mechanism for hydrofunctionalization (see Section 7.12 for further details).
Figure 91.

Boger’s method for hydroazidation using Fe2ox3/NaBH4 with selected substrate scope.
3.4 Hydroamination
In 2014, Shigehisa and coworkers reported a method for the Markovnikov-selective intramolecular hydroamination of unactivated olefins (Figure 92).270 This work expanded upon a series of cobalt-Shiff-base catalyzed hydrofunctionalizations by the same authors156,157,271 and utilized similar conditions to these previous methods (see Sections 2.4 and 5.1). Reaction conditions for this protocol typically employed two equivalents of tetramethyldisiloxane (TMDSO) as the hydride source, 3 to 6 mol% of cobalt complex 21 as catalyst, and fluoropyridinium tetrafluoroborate 23 as an oxidant. In cases with slower reaction times, use of the tosylate counterion (22) instead of tetrafluoroborate could help suppress side reactions, particularly hydrofluorination. Substrates for this hydroamination were typically terminal monosubstituted olefins, although two 2,2-disubstituted olefins demonstrated competency (not shown). Exclusively non-basic amides or sulfonamides were employed, with a range of protecting groups or otherwise functionalized (sulfon)amide substituents tolerated.
Figure 92.

(a) Shigehisa et al.’s cobalt catalyzed intramolecular hydroamination (b) with cobalt (II) precatalyst, oxidant, (c) selected examples of scope and (d) examples of heteroatom-selectivity reversal.
Selected examples of amide substituents investigated include trifluoroacetamides, benzamides, Boc-protected amino acid derivatives, and heteroaryl amides. A range of ring-sizes could be successfully constructed, as exemplified by tosyl aziridine 176, carboxybenzyl indoline 177, nosyl tetrahydroisoquinoline 178, and tosyl benzazepine 179. Other tolerated substituents included a distal primary alcohol, distal free and protected phenols, an aryl bromide, and strained cyclopropane and epoxide functionalities (not shown). Interestingly, depending on substrate functionalization and sterics proximal to the reacting olefin, N to O heteroatom selectivity-reversal products could be isolated. For example, although 176 and 180 are both derived from an allylamine fragment, the expected hydroamination product, namely aziridine 176, was isolated when tosylated, whereas oxazoline 180 was isolated instead when a benzamide group was utilized. Similarly, sulfonimidate 181 was obtained instead of the corresponding hydroamination product, which was obtained when a truncated version of the same sulfonamide was used (5 membered ring formation instead, not shown).
Baran and coworkers reported in 2015 a method for formal olefin hydroamination utilizing nitroarenes and nitro-heteroarenes (Figure 93). 264 Extending observations made in their related radical conjugate addition reaction (see Section 4.4 below),20,21 they sought to use readily available nitroarenes directly as the nitrogenous partner for the formation of C-N bonds. Previous reports272,273,274,275,276 have demonstrated that the nitroso functional group can serve as a competent radical acceptor. Using Mukaiyama-type conditions, nitroso compounds are generated in situ concomitant with carbon-centered radical generation, which results in net C-N bond formation via radical addition. As discussed in Section 3.1, although conceptually similar hydronitrosation/hydrooximation had been previously reported,236,237,238,239 previous iterations of this strategy were limited to simple nitrogen sources such as nitric oxide or alkyl nitrites.
Figure 93.

(a) Baran’s Fe(acac)3 catalyzed hydroamination of olefins (b) with selected substrate scope.
In a typical reaction sequence, nitroarenes are treated with excess olefin and PhSiH3 in presence of catalytic Fe(acac)3 to effect the desired Markovnikov hydroamination via hydroxylamine intermediates (Figure 93). Although these intermediates in some cases are reduced under the reaction conditions, a reductive workup with zinc metal is usually employed to increase yield. This reductive work-up also cleaves the N-O bond of O-alkylated hydroxylamine byproducts, which in turn probably result from the nascent nitroxyl radicals reacting with a second equivalent of carbon radical. These hindered hydroxylamines may also be of some value.
Beyond application of this transformation towards the synthesis of hindered secondary amines, the broad scope of nitroheteroaryl compounds tolerated enabled orthogonal functionalization strategies to be employed. In addition to electron rich and highly electron deficient arenes, electronically and structurally diverse nitro-heteroarenes were found to participate. Halogens, psuedohalogens, boronic acids, and free alkyl amines were all tolerated, any of which could find use in sequential coupling operations, and all of which provide orthogonal and complementary reactivity to established arylamine cross-coupling strategies. Furthermore, this method complements traditional alkylation and reductive amination strategies; 182 and 183, which could be retrosynthetically disconnected in either of these transforms would likely find chemoselectivity challenging to achieve in a forward sense due to sensitive alcohol, cyano or ketone moieties. Hindered Michael-type adducts such as electron-deficient aniline 184 are accessible, but might be otherwise difficult to access.
Recently, the groups of Shenvi116 and Thomas277 have independently reported modifications to the conditions developed by Baran and coworkers,264 both of which demonstrated the reaction at room temperature with reduced catalyst loading (1–2 mol% iron catalyst).
Shenvi and coworkers116 demonstrated that using isopropoxy(phenyl)silane [Ph(i-PrO)SiH2] as the reductant in place of phenylsilane allowed the hydroamination reaction to be run at room temperature in a mixture of isopropanol and ethyl acetate with 1 mol% of the original iron(III)acetoacetonate catalyst (entry B, 75% yield, Figure 94) whereas under identical conditions with phenylsilane (entry A) a much reduced yield was obtained (37%). It was additionally demonstrated that increased yields (88%) could be obtained as well when the original 60 °C reaction temperature was used with this silane and the same 1 mol% catalyst loading (entry C). Evidence within this report116 suggests the origin of this improvement is enhanced precatalyst activation by the silane examined (see Section 7.11).
Figure 94.

Alternative conditions for hydroamination. aSee ref 116. bSee ref 277.
In an orthogonal modification on Baran’s264 reaction conditions, Thomas and coworkers277 reported that replacing iron(III)acetoacetonate with iron phenolate complex 185 could effect the desired hydroamination at 2 mol % catalyst loading and room temperature in 1 hour (entry D, Figure 94), with other conditions being unchanged (ethanol solvent, 2 equivalents phenylsilane, 3 equivalents olefin). A subset of the substrates present in Baran’s report were shown to have similar or elevated yields under these modified conditions. In this study, the authors also noted the greatly reduced product yield from the room temperature reaction utilizing iron(III)acetoacetonate at 2 mol%. Identical conditions were also reported by these authors in a separate study focused on this catalyst’s ability to effect both this reaction and the reduction of aryl nitro groups to anilines.278
3.5 C-N Bond Formation in Natural Products and Complex Molecule Synthesis
Although there are currently few examples of radical hydrofunctionalization in complex molecule synthesis, Boger’s 2012 report of anhydrovinblastine hydrofunctionalization81 puts forward a powerful testament to its value. Along with the many radical traps shown capable of engaging the putative carbon radicals generated in this report, they demonstrated hydroazidation of in-situ generated anhydrovinblastine (1) with Fe2ox3 in the presence of sodium borohydride and an azide source (Figure 95; see Sections 2.1, 7.6, and 7.12 for more details). Boger’s team found that the inorganic azide source played an important role in the determination of the stereochemistry of the resulting analogs. Whereas lithium and sodium azides resulted in only production of azidoleurosidine (187), cesium azide proved capable of providing usable quantities of the desired epimer azidovinblastine (186). Notably, these conditions could deliver these complex azido-analogs in a combined yield of 74%. This achievement of selective late-stage hydrofunctionalization has since enabled the study of a plethora of 20′-N-substituted vinblastine and leurosidine analogs.25,81,184,189
Figure 95.

Boger’s hydroazidation of anhydrovinblastine (1) to azidovinblastine (186) and azidoleurosidine (187).
4. C-C bonds
Although early chemists in this field may not have understood or considered many of the mechanistic nuances of the preceding chemistry, they did realize that the ability to generate a carbon-centered radical from an alkene is powerful. Many useful transformations have and continue to be discovered by trapping these nascent carbon-centered radicals with appropriate molecular partners. It is unsurprising then, that these discoveries also sparked interest and research into using HAT chemistry to forge the all-important C-C bond. The following Sections aim to discuss general classes of these reactions and to list relevant synthetically useful reactions. The reader is directed to Section 7.10 for further mechanistic discussion.
4.1 Reductive Carbocyclization
Hydrogen atom transfer from a transition metal hydride to an alkene in the presence of other unsaturation can result in C-C bond formation via cyclization. A number of groups have studied these reactions, although the most work has been done from a mechanistic point of view rather than a preparative one. This section enumerates these various reactions, the substrates that have been studied and may be synthetically useful, alongside brief historical commentary. The interested reader is referred to the Section 7.4, 7.8, and 7.10 for further mechanistic discussion and details concerning these reactions.
As discussed in Section 2 above, the discovery of the alkyl cobalt bond in vitamin B12 led to the exploration of Schrauzer’s cobaloxime69,70,71,72 complex (Section 2.1, Figure 3). Following these discoveries in the 1960’s were a number of reports in the 1980’s and 1990’s that employed related cobalt complexes in C-C bond coupling between alkyl halides and alkenes,279,280,281,282,283,284,285 and to effect radical polymerization286 of acrylates.287
Building on this remarkable precedence, van der Donk and coworkers reported in 2002 a method for aryl alkene dimerization using vitamin B12 (see Section 4.2).288 As part of their investigation of the mechanism of this reaction, they studied the reactivity of pyrroles 188 and 191 (Figure 96). They found that treatment of pyrrole 188 with catalytic cyanocobalamin, a titanium(III) citrate solution containing tetrabutylammonium hydroxide as a phase transfer catalyst, an aqueous Tris buffer (pH 8), and ethanol under a nitrogen atmosphere gave 42% of a cycloisomerization product (189) and 28% of a reductive cyclization product (190). Use of substrate 191 gave an 80% yield of a reductive cyclization product 192 only. These reactions are thought to proceed via a radical 5-exo-trig cyclization to give intermediate 193, which either abstracts a proton to give a reduced product (190 and 192) or loses a hydrogen atom to yield the cycloisomer 189. This reaction may proceed via a HAT from a cobalt(III) hydride, which is initially generated by protonation of a cobalt(I) complex [resulting from Ti(III)citrate reduction of cyanocobalamin].289,290 A subsequent paper from van der Donk in 2006 applied this method to various dienes to make substituted tetrahydrofurans and pyrrole-containing products via reductive cyclization and cycloisomerization pathways.289
Figure 96.

Van der Donk’s reductive cyclization of dienes catalyzed by vitamin B12.
Jack Norton’s group has conducted pioneering studies on reductive cyclizations of dienes and polyenes initiated by hydrogen atom transfer from a transition metal hydride.290,291,292 This section will only briefly discuss reductive carbocyclization applications of Norton’s work. The interested reader is directed to Section 6.2 and 7.4 for further discussion of related content.
In 2007, the Norton group demonstrated reductive cyclization of diene 194 in a 5-exo-trig fashion to obtain the substituted cyclopentane 195 as a mixture of diastereomers in excellent yield by using the catalytically generated chromium hydride, CpCr(CO)3H, which the group had studied extensively290,291,293,294,295,296,297,298,299,300,301 (Figure 97; see also Section 6.2). The geminal diesters of 195 were required to obtain this high yield by virtue of the accelerating Thorpe-Ingold effect, but reductive cyclization products could still be obtained as the major products, though in lower yield, without these present.
Figure 97.

Norton’s reductive cyclization of dienes using CpCr(CO)3H.
Subsequent work in 2008 showed that decalin 197 could also be obtained from polyene 196 via a HAT-initiated radical cyclization from catalytic CpCr(CO)3H, albeit in 23% yield (Figure 98).290 Use of the vanadium hydride, (dppe)(CO)4VH, in a stoichiometric fashion also gave decalin 197 in a moderately improved yield of 32%.290 Although not yet fully realized, this reaction demonstrates the potential for HAT radical cyclizations to effect polycyclization reactions. The low yield may be attributed to the many other pathways, especially premature termination of the cascade cyclization by C-H bond formation.
Figure 98.

Norton’s demonstration of a polyene cyclization to obtain a decalin 197 initiated by chromium and vanadium hydrides.
In 2015, the Norton group reported their studies on the reactivity of the three different kinds of metal hydrides towards radical cyclization of enol ethers into aryl substituted alkenes to make substituted tetrahydrofurans (Figure 99).291 Of these, the group found that the vanadium hydride was the only one which could effect cyclization of 198 to substituted tetrahydrofuran 201.
Figure 99.
Norton’s comparison of three kinds of metal hydrides in the reductive cyclization of diene 194.
The chromium hydride does not react productively with the vinyl ether group in the reversible HAT step, while the in situ generated cobalt hydride removed the vinyl group to give alcohol 200. An acidic species is known to be generated by treatment of Co(dmgBF2)2(THF)2 under hydrogen gas, which may catalyze hydrolysis of the vinyl ether. 302 The authors attribute the effectiveness of HV(CO)4dppe over the chromium hydride to the weakness of its V-H bond (57.5 cal/mol). HAT to the vinyl ether favors α-ether radical formation in the case of the weak vanadium hydride, so cyclization can occur. In the case of the chromium hydride, this equilibrium lies too far on the side of the metal hydride/olefin for a productive reaction to proceed. The authors composed a short substrate table to demonstrate the scope of this vanadium hydride reductive cyclization, of which selected examples are shown in Figure 100. The substituted tetrahydrofurans were obtained in moderate to good yield (47–91%) and diastereoselectivity (3:2 to 12:1), in all cases favoring the cis diastereomer due to a conformational preference303,304 for a pseudoequitorial positions of the substitutents in the transition state.
Figure 100.

Selected scope of Norton’s vanadium catalyzed reductive cyclization of enol ethers with substituted alkenes.
In 2012, as part of radical clock experiments to probe the mechanism of their reaction, the Boger lab has also shown that their Fe2ox3/NaBH4 hydrofunctionalization method could effect reductive radical cyclization of 1,6-dienes to form substituted cyclopentanes.81 Likewise, the Carreira and Shenvi groups have performed similar experiments.24,115
4.2 Intermolecular Reductive Dimerization
As mentioned above (Section 4.1), van der Donk and coworkers have reported reductive dimerization of styrenes using a vitamin B12/Ti(III) citrate/pH 8 aqueous buffer system (Figure 101).288 Treatment of styrenes with vitamin B12 (cyanocobalamin), titanium(III) citrate as an aqueous solution with tetraethyl ammonium hydroxide as a phase transfer agent, an aqueous pH 8 Tris buffer in ethanol under a nitrogen atmosphere resulted in a mixture of dimeric and reduction products for 7 different styrenes. The reaction conditions likely generate a Co(III) hydride which generates benzylic radicals via HAT to the styrenes. These persistent radicals may then either dimerize, abstract a hydrogen atom from solvent, or polymerize (not shown). Shenvi and coworkers have noted that styrenes tend to dimerize under the conditions of alkene reductions using Mn(dpm)3/PhSiH3.24,116
Figure 101.

Van der Donk’s procedure for styrene dimerization.
4.3 Hydrocyanation
Alkene hydrocyanation can convert relatively inert alkenes into useful intermediates for further elaboration by nitrile hydrolysis, reduction, or alkylation. However, unactivated alkenes are particularly challenging substrates for addition of HCN, which itself presents severe restrictions on handling. In 2007, building from his work on hydroazidation and hydrohydrazidation (see Sections 3.2 and 3.3), Carreira reported the hydrocyanation of a broad range of alkenes, highlighting the remarkable functional group tolerance of this method.22 The addition of carbon-centered radicals to the cyanide donor, p-toluenesulfonyl cyanide (TsCN) finds some precedence in the literature,305,306,307,308 but the mechanism of the Mukaiyama-type reactions was even less clear at the time, so a direct connection was not obvious. After observation that prior catalysts used in hydrohydrazidation fared poorly in hydrocyanation, it was found that Co(salen) catalysts 21 and 202 performed well. Selected examples in Figure 102 illustrate the use of electron neutral alkenes and the establishment of consonant (204), dissonant (205) and high value (206) compounds. (Note: “consonant” and “dissonant” refer to the ‘matched’ and ‘mismatched’ polarization, respectively, of a molecule. For example, 205 is dissonant because a 1,4-relationship exists between the two carbon atoms which bear heteroatoms.)9,19
Figure 102.

(a) Carreira’s method for hydrocyanation of alkenes (b) with selected examples and (c) cobalt precatalysts.
Boger and coworkers have similarly shown that use of tosyl cyanide as a radical trap can be used in concert with their iron(III)oxalate-mediated hydrofunctionalization method to generate a hydrocyanide from an unactivated alkene, albeit in low yield (Figure 103).81
Figure 103.

Boger’s method for hydrocyanation of alkenes.
4.4 Conjugate Addition
The conjugate addition of carbon-centered radicals into unsaturated electrophiles, known as the Giese reaction,309 is a powerful tool for carbon-carbon bond construction, both in an intra- and intermolecular sense.310,311 In light of this, the recognition that olefins may be utilized as radical synthetic equivalents (“radical donors”) under metal-mediated reductive conditions led Baran and coworkers to pair them with electron-withdrawn (“radical acceptor”) olefins to effect such radical conjugate addition (Figure 105).20,21 Conceptually following the work by Boger81 and Carreira22,23 in the utilization of carbon electrophiles to capture nascent radicals, this approach, which may also be thought of as a reductive olefin cross-coupling,312 allows rapid construction of carbon-carbon bonds and is capable of generating strained systems and contiguous quaternary centers. After the first20 communication of this work, which centered on intra- and intermolecular radical conjugate addition using all-carbon/hydrogen substituted olefins as donor substrates, Baran and coworkers reported a related second-generation protocol, which expanded this methodology to a wide range of heteroatom-substituted olefin donor substrates.21
Figure 105.

Baran’s first-generation conjugate addition.
Baran’s first-generation conditions20 (Figure 105) utilized phenylsilane as a hydride source in presence of catalytic or stoichiometric iron (III) acetylacetonate to perform this radical conjugate addition. These reactions were typically run in ethanol, although the authors indicate that the use of a 5:1 mixture 1,2-dichloroethane/ethylene glycol as solvent was beneficial in their catalytic protocol due to occasional difficulty removing triethoxy(phenyl)silane. As reported, this protocol could be run under ambient atmosphere, was tolerant of water, and was demonstrated on gram scale. As the stated intent of developing this reaction was to form carbon-carbon bonds within terpenoid scaffolds, several examples of such an intramolecular transformation are demonstrated, with examples such as α-ionone derivative 207 and citral derivative 208, and these generally proceeded in good to excellent yields. In addition to intramolecular cyclizations, Baran and coworkers reported the transformation was competent in an intermolecular sense, even tolerating vinyl-pyridine or heteroaryl donors or acceptors, respectively. A number of donor olefins were shown competent to engage in this reactivity, including monosubstituted, 2,2-disubstituted, and trisubstituted olefins, and a number of acceptor olefins as well, including acrylates, cyanoacrylate, acrylamides, fumarates, vinyl sulfones, and cyclic unsaturated lactones and ketones (not shown).
Baran’s second-generation conditions21 (Figure 106) mainly differed from the first generation in implementing the diisobutyrlymethane iron ligand (dibm, instead of acetoacetonate, acac), as well as by the observation that addition of Na2HPO4 improved yields (except in substrates containing carboxylic acids, in which case it was left out). This protocol, generally run in EtOH with 2 equivalents of PhSiH3, proved capable of expanding the substituent pattern of donor olefins tolerated to include a wide array of heteroatoms, including oxygen, nitrogen, sulfur, silicon and halogen substituents, and in most cases dropped the catalyst loading required to 5 mol %. As a result of heteroatom stabilization of the nascent radical, the products obtained are formally umpolung Michael-type adducts. Examples of the catalytic protocol using Fe(dibm)3 and 2 equivalents of PhSiH3 included silyl- and regular enol ethers, enecarbamates and enamides and thio-enol ethers. Alkenyl silanes, boronate esters and haloalkenes all utilized variants of the first generation protocol, with alkenyl silanes requiring higher catalyst loading and temperatures (n-PrOH at 50 mol % catalyst and 80 °C), boronate esters being most effective with 3 equivalents PhSiH3 and Fe(acac)3 instead of Fe(dibm)3, and alkyl halides also using 3 equivalents PhSiH3 as well as requiring stoichiometric Fe(acac)3.
Figure 106.

Heteroatom-bearing olefins in Baran’s second-generation conjugate addition.
Shenvi and coworkers116 recently demonstrated that using isopropoxy(phenyl)silane [Ph(i-PrO)SiH2] as the reductant in place of phenylsilane accelerated Baran’s first-generation conjugate addition reaction20 such that it could be run at room temperature in a mixture of isopropanol and another solvent (Figure 107, entries 2 and 4) with a reduction of catalyst loading to 5 mol % iron(III)acetoacetonate. Under similar conditions with phenylsilane (entries 1 and 3) a much reduced yield was obtained. Isopropanol was used in this comparison so as to control the silane ligand sphere, but similar reactivity was observed with ethanol as the solvent. Comparable yields were reported under Baran’s first-generation conditions, although the second-generation conditions21 were not examined.
Figure 107.

Comparison of PhSiH3 and Ph(i-PrO)SiH2 in Baran’s conjugate addition.
In 2016, Cui and coworkers reported that para-quinone methides act as excellent electrophiles for carbon-centered radicals generated by the action of Fe(acac)3/PhSiH3 on olefins (Figure 108).313 The reaction is useful for the synthesis of para-substituted 2,6-di-tert-butyl phenols via a 1,6-conjugate radical addition. A variety of substituted phenols (R4 = Me, Ar) function as competent electrophiles, and the alkene moiety may be substituted with hydrogen, alkyl, alcohol, ketone, ester and amide functional groups. All quinone methide electrophiles possess tert-butyl groups at the 2 and 6 positions, except for one case. In this case, methyl groups replaced the tert-butyl groups and the yield of the conjugate addition dropped to 30%. This may be due to competitive 1,4-addition and/or hydrogenation pathways, although the authors do not comment. In cases where the alkene is unsymmetrical, the authors observed modest diastereoselectivity in the resulting coupled products. The addition reaction generally proceeds in good to excellent yield (40–94%).
Figure 108.

Phenol synthesis from Cui and coworkers.
4.5 Hydromethylation
Following their reports of conjugate addition,20,21 Baran and coworkers sought to solve the conceptually simple yet practically difficult problem of the formal addition of methane across an unactivated olefin. 265 Although limited precedent existed in the literature for variants on such a transformation, these typically employed conditions which would likely prove intolerant to many reactive moieties or otherwise be difficult to apply broadly, as the conditions required utilized, for example, strong Lewis acid activation,314 highly nucleophilic and pyrophoric organometallic species, 315,316 or long reaction times.316 Baran accomplished this transformation instead by applying HAT radical generation followed by addition into a methyl-equivalent ablative functional group, specifically the formaldehyde hydrazone (Figure 110), as inspired by Kim and Cho who demonstrated hydrazone addition-ablation in the context of classical radical generation and intramolecular cyclization.317
Figure 110.

Hydromethylation reaction conditions and representative examples.
Typical reaction conditions for Baran’s hydromethylation require in-situ formation of intermediate formaldehyde hydrazone via condensation of commercial formaldehyde solutions with n-octylsulfoyl hydrazide. It is noted that by applying isotopically-labeled formaldehyde, one may obtain radiolabeled or otherwise isotopically enriched methylation. This reagent is then applied to the substrate olefin in presence of Fe(acac)3, methanol and PhSiH3 followed by freeze-pump-thaw cycles to degas the resulting reaction mixtures. It should be noted that equivalents of each reagent are optimized depending on several conditions including substrate and formaldehyde source, and that the Baran group found that the reaction could be rendered catalytic in iron by the addition of superstoichiometric trimethylborate. Following reaction completion and solvent removal, the resulting (unstable) sulfonyl hydrazides are heated in methanol in presence of sodium acetate to effect sulfonyl hydrazide removal, presumably via loss of sulfinic acid and nitrogen extrusion.
This transformation was demonstrated with a range of mono-, 2,2-disubstituted and trisubstituted olefin substrates bearing distal functionality such as azide, boronate ester, TMS-capped aryl acetylene, and benzyl alcohol. Other examples of functional group tolerance include aryl and alkyl bromides and iodides; ureas, amides and carbamates; a free phenol and phenolic triflate; a silyl ether, acetals, and esters (not shown). Noteworthy examples of late-stage functionalization on complex substrates bearing Lewis-basic and potentially sensitive functionality include 209, derived from picrotoxinin, and 210, derived from quinine, although in these cases yields were modest. As mentioned above, isotopic labeling was demonstrated to generate deuterium- and 14C-labeled products from their respective labeled precursors (formaldehyde-d2 and 14C-formaldehyde, respectively, not shown).
4.6 Hydrostyrenylation
Following the HAT-initiated radical addition reactions developed in the Baran lab (see Sections 4.4, 4.5), 20,21,264 Cui and coworkers demonstrated that similar radical-generating conditions could be applied to effect a radical conjugate addition-elimination on β-nitrostyrenes (Figure 131), yielding β-alkylated styrenes. 266 The nitrostyrene radical-addition/elimination paradigm is known in the literature to proceed under a variety of radical-generation methods including diazo thermolysis,318 the action of alkyl-gallium,319 -borane320 and -aluminum321 species, and peroxide homolysis.322 It also should be noted that similar radical-addition-elimination sequences have been shown to take place utilizing a number of other β-styrene substituents as single electron nucleofuges.323
Figure 131.

An example of cobalt-mediated hydroselenation of alkenes.
As shown in Figure 112, Cui and coworkers’ report266 also utilized Fe(acac)3 and PhSiH3 to effect Markovnikov-selective radical generation and reductively add unactivated olefins into the beta position of the acceptor styrene in the above discussed addition-elimination fashion. Typical reaction conditions utilized 10 mol % Fe(acac)3, 2 equivalents of donor olefin and silane, and a reaction temperature of 60 °C with ethanol as the solvent under an atmosphere of argon. The scope of donor olefin was examined and representative examples include 211 and 212, derived from allyl trimethylsilane and allyl cyanide respectively, as well as 213, derived from methylene N-boc-piperidine. Additional donor olefins included unfunctionalized cyclic alkenes, allylic and homoallylic alcohols, and phenoxy ethers (not shown). The scope of acceptor nitrostyrene arene was also examined and representative examples include phenol 214 and 3-pyridine 215, as well as methyl, dimethylamino, methoxy, and halogen (F, Cl, and Br) substituted styrenes (not shown). Other heteroaryl nitrostyrene analogs included furan, thiophene, and N-tosyl indole (not shown).
Figure 112.

Cui’s Fe(acac)3 catalyzed hydrostyrenylation protocol.
4.7 Hydroarylation
In 2015, Gui et al. reported a method for the synthesis of oxindoles from α,β-unsaturated N-aryl amides in what may be considered a formal intramolecular hydroarylation reaction (Figure 113).324 The authors found that use of stoichiometric iron(III) chloride with sodium borohydride as reductant in DME with heating at 70 ºC could effect cyclization in good yield (58–87%) to make a variety of oxindoles. Deuterium labelling experiments indicated that a deuterium atom from NaBD4 is incorporated at the terminal position of the olefin. The authors propose a mechanism that involves Markovnikov hydrometallation of a iron(III) hydride across the the olefin to form a Fe-C bond α to the amide carbonyl. This Fe-C bond would homolytically cleave upon binding O2 to generate a carbon-centered radical which undergoes a 5-exo-trig radical cyclization. Oxidation of the resultant radical with loss of a proton gives the rearomatized product oxindole.
Figure 113.

Gui et al.’s synthesis of oxindoles via radical cyclization onto an aryl ring.
In 2016, Shenvi and coworkers applied a modification of their hydrogenation conditions to synthesize the 8-aryl menthol class of chiral controllers from aryl-sulfonylated isopulegol (Figure 114).325 The reaction proceeds via an ipso-radical Smiles-Truce rearrangement to effect an aryl transfer in a Markovnikov manner across the olefin. Initiation of the reaction proceeds via HAT across the alkene. The reaction occurs with loss of SO2, which appears to irreversibly bind manganese. Consequently, the reaction is stoichiometric in manganese. The authors report than the reaction can be run with catalytic Mn(dpm)3 if Mn(OAc)3 is used as a stoichiometric manganese donor. Through this two-step protocol, the team was able to append a variety of electron deficient, electron neutral, and electron rich carboaromatics and heteroaromatics in a regioselective fashion, thereby enabling easy access to these priviledged chiral building blocks.
Figure 114.

Synthesis of chiral 8-aryl menthols via a HAT initiated Smiles-Truce rearrangement.
4.8 Reductive Coupling of α,β-Unsaturated Compounds with Carbonyls
Along with seminal reports on alkene hydration38,97,98,99,100,103,104,105,106 and oximation/nitrosation236,237,238,239 in 1989 and the early 1990’s, Mukaiyama described a cobalt-catalyzed addition reaction of α,β-unsaturated substrates into aldehydes in the presence of a silane (Figure 115).326 This transformation forms a new C-C bond in good yield, providing β-silyloxy nitriles, amides, or esters in a process which may be described as a reductive aldol-like addition reaction. Prior to Mukaiyama, there were no examples of catalytic methods to effect this transformation.327,328 Since his publication, however, numerous catalytic enone hydrometallation processes effected via copper,329 palladium,330,331 iridium332 and rhodium catalysis333,334,335 in the presence of a hydride donor have been developed for catalysis of reductive aldol-like reactions.
Figure 115.

Cobalt-catalyzed reductive coupling with aldehydes.
As an example of Mukaiyama’s reductive aldol-type reaction,326 acrylonitrile and benzaldehyde in 1,2-dichloroethane afforded β-hydroxy nitrile 216 in 70% yield along with traces of benzylalcohol when treated with phenylsilane and catalytic amounts (down to 5 mol%) of Co(acac)2 at 70 ºC under an argon atmosphere (Figure 115). Interestingly, Co(dpm)2 catalyzed the process more efficiently than Co(acac)2, leading to a 93% yield at 20 ºC. Consequently, this precatalyst was chosen for further substrate scope exploration. The transformation tolerated α- and β-substitution (217 and 219, respectively) but showed no diastereoselectivity in any of these examples. Coupling between acrylate and cinnamaldehyde led to an equimolar mixture of syn and anti products 219 in 80% isolated yield. In the case of amides, the coupling showed some diastereoselectivity towards 220 (95% after 2 hours, syn:anti = 80:20). Finally, the less activated 3-phenylpropanal reacted slower but afforded product 221 in 90% after 6 hours, syn:anti = 70:30. Acidic quenching with 10% HCl in methanol afforded smoothly the β-hydroxy products quantitatively.
Using a similar approach, Yamada reported the cobalt-catalyzed carboxylation of substituted acrylonitriles with carbon dioxide (Figure 116).336 When the team employed the same conditions as Mukaiyama,326 they predominately observed conjugate reduction of the enone (222), and initial attempts to capture CO2 under these conditions were unsuccessful. However, a screen of reducing agents revealed that use of diethylzinc instead of a silane or borane enabled quantitative conversion to the desired product after methylation with TMSCHN2. These reactions are conducted with catalytic cobalt(II) acetylacetonate loading under atmospheric pressure of CO2. In the absence of a cobalt catalyst, ethyl-substituted products 223 were obtained, which result from conjugate addition into the enone moiety by diethylzinc. Both Z- and E-alkenes were competent substrates; alkyl, ether, ester, and halogen substitutions on pendant aromatic rings were all well tolerated and led to the corresponding products in excellent yields. This transformation proceeded on both α- and β-monoaryl-substituted acrylonitriles (224 and 225, respectively). Interestingly, the efficiency of the reaction dropped to 38% in the presence of an α-methyl substituent (226) and only trace product was observed with β,β’-disubstitution (227, 9%, 60:40 dr). Finally, tetrasubstituted alkenes (228) showed no reactivity. The authors propose a mechanism that involves formation of a cobalt hydride (via reaction between Et2Zn and Co(acac)2), which undergoes conjugate hydrometallation of the α,β-unsaturated nitrile. The resultant enolate then traps CO2.
Figure 116.

Cobalt-catalyzed reductive carboxylation of acrylonitirles.
In 2001, Krische expanded this kind of reductive coupling reaction to the intramolecular cyclization α,β-unsaturated ketones (Figure 117).337 Although enormous advances had been made in the performance of aldol reactions, most systems exhibited suboptimal stereocontrol with symmetrical substrates. For these substrates, Krische’s methodology permitted formation of cyclization products with nearly exclusive selectivity for a syn configuration due to geometrical requirements of the transition state. Krische’s conditions proved quite general for five-, six- and seven-membered ring formation, albeit reduced yields are observed in the latter case. Nonetheless, stereocontrol remains high in all cases, with a syn:anti ratio of 99:1 determined for 229, 230 and 233. Heteroaromatic enones also underwent cycloreduction in good yields as in the formation of 231, while aliphatic enone partners gave diminished yields of the desired products, for example, 232.
Figure 117.

Syn-diastereoselective cobalt-catalyzed aldol cycloreduction.
When bis-enones were subjected to these same conditions, an analogous Michael cycloreduction occurred (Figure 118). Curiously, anti-stereochemistry was observed exclusively for this transformation, affording diastereoselectivity opposite to that observed for the aldol addition reaction. In the case of symmetrical enones, the formation of five- and six-membered rings proceeded efficiently at 50 ºC (62% of 234 and 73% of 235, respectively). Furthermore, cycloreduction of unsymmetrical substrates revealed that electronic differences in the enone could be exploited to favor one regioisomer over another. Thus, isomeric products containing phenyl- and methyl-ketone substituents (237:238) were obtained in 62% yield as a 3:1 mixture at 70 ºC. However, use of phenyl- versus 2-furyl-substituted enone moieties led to a 1:1 mixture of isomeric products (239:240) in 54% yield. Finally, the authors developed a formal [2+2] cycloaddition of bis-enones by changing only the reducing agent.338 In particular, use of PhMeSiH2 instead of phenylsilane provided the substituted bicyclo[3.2.0] ring system 236 completely diastereoselectively in 72% yield. These transformations are thought to proceed through a coordination/cyclometallation-type mechanism rather than a TM HAT mechanism.
Figure 118.

Anti-diastereoselective cobalt-catalyzed Michael cycloreduction and formal [2+2].
4.9 Reductive Addition of Alkenes to Sulfonyl Oximes
Following their work on the hydrocyanation of alkenes (Section 4.3),22 Carreira’s group reported a cobalt-catalyzed method for the synthesis of oxime ethers from sulfonyl oximes and unactivated alkenes (Figure 119).23 The authors found precedence for this reaction in the work of Kim and Kim, who had previously shown that phenyl sulfonyl oximes can participate in radical reactions.339 This olefin functionalization reaction was proposed to proceed via hydrocobaltation of the olefin with a cobalt hydride, followed by nucleophilic attack at the carbon of the sulfonyl oxime. Subsequent elimination of the p-toluenesulfonyl group restores the oxidation state (Section 7.12, Figure 204).340 Although many methods have been reported for the synthesis of oximes, Carreira’s protocol allows for the direct, chemoselective conversion of unactivated alkenes to aldoximes and oximonitriles in excellent yields. For example, treatment of 4-phenylbutene with cobalt complex 21, phenylsilane, and a sulfonyl oxime in ethanol furnished the desired product 241 (where X = H) in quantitative yield. In contrast, no product was observed when a methyl or a trifluoromethyl ketoxime was used, presumably as a consequence of steric congestion. However, ketoximes with resonance-stabilized electron-withdrawing groups such as esters or nitriles performed well, with nitriles being significantly more efficient.
Figure 119.

Cobalt-catalyzed direct synthesis of oxime ethers from alkenes.
Figure 204.

Mechanistic tests for the hydrochlorination of alkenes.
During the study of the scope of the transformation, N-(benzyloxy)-1-(phenylsulfonyl)-methanimidoyl cyanide (X = CN) gave much better yields than phenylsulfonylmethanal O-benzyloxime (X = H). All examples showed excellent Markonikov selectivity with both reagents, as only branched products were detected. Ethers and silyl ethers were tolerated, as were aldehydes and furans. Furthermore, 2,2′-disubstituted and trisubstituted alkenes afforded the desired products in excellent yields. Finally, styrenes and indenes were also competent substrates.
4.10 C-C Bond Formation in Natural Products and Complex Molecule Synthesis
There are only a few examples of C-C bond formation in natural products and complex molecule synthesis that employ the preceding reactions, especially in comparison with the C-O bond forming reactions. One reason for this may be that of time: these reactions are new relative to hydration or peroxidation. A more significant reason is probably the fact that only relatively small carbon units (mostly C1) can be appended with these methods. However, Baran’s recent expansion of this reaction paradigm to Michael acceptors20,21 has expanded the size of carbon units which may be appended to alkenes and several of the examples below employ his methods. Undoubtedly, further application of these methods will occur with time.
In 2015, Carreira and coworkers completed a total synthesis of (±)-hippolachnin A (245) (Figure 120)341 During this endeavor, the team explored several strategies to set the desired cis-ethyl stereochemistry. One of these involved applying Baran’s conjugate addition reaction.20 Indeed, subjection of 242 to Baran’s conditions did effect cyclization to the hippolachnin A skeleton epimer 243 in 24% yield, and also gave 244, which results from opposite regioselectivity of the initial HAT. Unfortunately, 243 possesses the undesired trans stereochemistry at the nascent C-C bond, and so this route was not pursued further. Nonetheless, Carreira’s example highlights the potential utility of this reaction in total synthesis.
Figure 120.

Carreira’s application of Baran’s conjugate addition reaction during Carreira’s work on hippolachnin A (245).
Also in 2015, the Pronin lab completed a remarkable and concise (11-step) stereocontrolled synthesis of emindole SB (251) (Figure 121).342 A key step in Pronin’s synthesis features a polycyclization cascade which expands upon precedence laid down in Baran’s work on radical conjugate additions (Figure 4.4).20,21 The authors had envisioned that formation of the trans-junction of the natural product could be forged via a radical-polar crossover reaction, initiated by a chemoselective hydrogen atom transfer to the 2,2-disubstituted alkene in 246, and followed by an aldol addition (Figure 121). Exploration of various HAT conditions revealed that use of Fe(acac)3 and Ph(i-PrO)SiH2 in a mixture of 1,2-dichloroethane and ethylene glycol at 0 ºC could enable the formation of a nearly equimolar mixture of isomers 247 and 248 in 58% yield in a highly diastereoselective manner. (Identification of Ph(i-PrO)SiH2 was facilitated by private correspondence with the Shenvi lab, who were studying the beneficial effects of Ph(i-PrO)SiH2 on these Mukaiyama-type reactions at the same time.)116 Curiously, the saturated aldehyde in 246 was observed to exist predominantly as a hemiacetal in the presence of alcoholic solvents. The authors reasoned that replacement of the ketone in 246 with a hydroxyl group (i.e. 249) could allow reversible formation of a cyclic hemiacetal intermediate (250), which might in turn promote the inverse stereocontrol during the tandem reaction. Indeed, substrate 249 cyclized with 5:1 diastereoselectivity to the analogous product. In this case, the diastereomeric ratio improved upon lowering the temperature whereas replacement of the solvent with ethanol or methylation of alcohol 249 diminished formation of the desired isomer.
Figure 121.

Pronin’s synthesis of emindole SB (251).
As discussed above (Section 4.5; Figure 110), the Baran lab has demonstrated that their radical hydromethylation method265 may also be used to modify natural products such as picrotoxinin, gibberellic acid, rotenone and quinine.
5. C-X bonds (X = Halogen, S, Se)
5.1 Hydrofluorination
The Markovnikov addition of hydrogen fluoride across an olefin is a simple strategy to access monofluorinated compounds, which have become increasingly important in the pharmaceutical industry.343 While classical methods, such as Olah’s hydrofluorination with HF-pyridine,344 are effective for C-F bond formation in simple alkenes, the acidic and ionic conditions required for these procedures limit functional group compatibility. However, radical functionalization methods display functional group compatibility orthogonal to polar methods. Reports by Sammis345,346 and Li347 have demonstrated the viability of radical fluorination using electrophilic N-fluoro reagents, which are synthetically attractive because they are well explored and easy to handle.348 The use of these reagents as a source of F• in hydrofluorinations was recognized by Boger349 and then Shigehisa,271 who have developed methods for Markovnikov hydrofluorination of alkenes under effectively neutral conditions (Figure 122).
Figure 122.

Radical hydrofluorination of olefins.
Boger’s method for radical olefin hydrofluorination349 followed his previous work with olefin radical hydrofunctionalization26,81 and also uses sodium borohydride and iron(III)oxalate to effect Markovnikov hydrofunctionalization via carbon-centered radicals (Figure 123). The radical is then intercepted by the fluorinating reagent F-TEDA (N-Chloromethyl-N-fluorotriethylenediammonium ditetrafluoroborate, also known by its trade name, SelectFluor®) to provide mono-fluorinated products (23 examples reported).
Figure 123.

Boger’s iron-mediated radical hydrofluorination. (a) Typical reaction conditions employed. (b) selected examples of substrate scope.
Although superstoichiometric in reagents, this transformation exhibits remarkable functional group compatibility. Tolerated functional groups include acid sensitive moieties such as acetonide-protected sugar 252 and Boc-protected amine 253; unprotected alcohols (259) and free amines (258); potential intra- or intermolecular nucleophiles such as unprotected carboxylic acid 254 and unprotected phenols 256 and 257. Phenol 257, derived from eugenol, represents an instructive case in selectivity. Even though para-substituted phenols have previously been shown to undergo oxidative fluorination to 4-fluorocyclohexa-2,5-dieneones under similar conditions350 (F-TEDA, MeCN, 22 °C), they do not react in this case. The high chemoselectivity is presumably due to the extremely rapid rate at which the desired reaction takes place: as short as 5 minutes at 0 °C, which is striking considering that these reactions are run at fairly high dilution (0.0125 M). Similarly, this reaction outcompetes radical capture by adventitious O2 – these reactions were all run under an atmosphere of air. It is noted that substrate-controlled diastereoselectivity is possible under the reaction conditions, as evidenced by a 5:1 diastereomeric ratio (favoring axial fluoride delivery) observed in product 255.
Another radical hydrofluorination protocol was developed by Shigehisa, Hiroya and coworkers in 2013 (Figure 124).271 Inspired by Carreira’s cobalt catalyzed hydrofunctionalization methodology, 22,23,115,252,255,256,267,268,340 this protocol bears a good deal of similarity to the hydroalkoxylation conditions (see Section 2.4) that directly preceded it,156 (which, according to the authors, was incidentally discovered while pursuing this hydrofluorination). As shown in Figure 124, a typical reaction setup is run under argon in degassed α,α,α-trifluorotoluene, where the substrate olefin is treated with 1,1,3,3-tetramethyldisiloxane (TMDSO) and N-fluoropyridinium salt (23) in presence of catalytic salen cobalt catalyst 21. The authors remark that PhCF3 as solvent is crucial to the success of this reaction, and that degassing is required to suppress the undesired side reaction of olefin hydration. Notable examples of olefin scope include fluoride-sensitive silyl protecting groups such as 260, acid-labile acetal 261, primary tosylate and aryl halide 262 and 263. One example of a potential nucleophilic moiety (alcohol 264) was demonstrated; however, the authors note that double bond isomerization is a problematic side-reaction in this case. It is unclear whether this isomerization is due to radical28 or cationic156 side-reactivity. A potentially reduced nitro group was also tolerated under the reaction conditions (265).
Figure 124.

Cobalt-catalyzed radical hydrofluorination. (a) Typical reaction conditions employed. (b) Selected examples of substrate scope. (c) Catalyst and hydride source.
5.2 Hydrochlorination/Bromination/Iodination
As noted in the introduction (Section 1), a significant benefit of the putative MH HAT reaction is its selectivity for functionalization of electron neutral alkenes in the presence of multiple other functional groups. In stark contrast, other hydrofunctionalization conditions like hydrochlorination with Brønsted acids react preferentially with more Lewis basic functional groups and can therefore lead to undesired byproducts or exclusively the wrong product. Hydrochlorination of alkenes is a particularly compelling example because, as Carreira points out, it is a reaction frequently taught in introductory organic chemistry, but seldom applied outside of the most basic industrial processes.340
In light of this restriction and as a continuation of their preceding work22,23,115,252,255,256,267,268 on olefin functionalization, Carreira and coworkers developed a Markovnikov-selective hydrochlorination procedure for the synthesis of alkyl chlorides (Figure 125). A screen of conditions based on their prior work revealed two conditions that could be used for the mono-, 2,2-di- and tri-substituted olefins that compose their substrate table. Use of the catalyst 21 (2–8 mol%) with phenylsilane as reductant in ethanol at room temperature under an argon atmosphere effected the desired transformation for all substrates, but gave slightly better yields at lower catalyst loading for mono-substituted alkenes. Otherwise, Co(BF4)2•6H2O could be mixed with ligand 175 to form an active catalyst in situ. The addition of catalytic t-BuOOH (30 mol%) provided an accelerating effect. Carreira proposes a mechanism involving hydrocobaltation of the olefin with a cobalt hydride, Co-C bond homolysis, and trapping of the radical with tosyl chloride.
Figure 125.

(a) Carreira’s hydrochlorination reaction. (b) Selected substrates. (c) Cobalt(III) ligand (175) and cobalt salen complex (21).
The Boger lab has demonstrated that their iron(III) mediated hydrofunctionalization methodology (see Section 2.1) can also effect hydrochlorination of unactivated alkenes when 4-AcNHC6H4SO2Cl is employed as the chlorine atom source (Figure 126).81 Treatment of β-citronellol with 5 equivalents of iron(III) oxalate hexahydrate, 6.4 equivalents of sodium borohydride, 5 equivalents of 4-AcNHC6H4SO2Cl at 0 ºC for 30 minutes provided the corresponding alkyl chloride in 62% yield.
Figure 126.

Boger’s hydrochlorination method.
In 2010, Ishibashi and coworkers reported iron-catalyzed and iron-mediated methods for radical cyclizations of 1,6-dienes and enynes.87 Use of catalytic FePc could generate alcohols (see Section 2.1), whereas use of stoichiometric FeCl3 instead gave alkyl halides (Figure 127) when employed alongside NaBH4 as reductant with a series of linear dienes. In the case of unsymmetrical substrates 266 and 269, 266 gave an equal mixture of products 267 and 268, while 269 gave predominately 270, in which radical cyclization is initiated from the 2,2-disubstituted alkene of 269. Several enynes also underwent cyclization to the corresponding vinyl halides. A mixture of diastereomers was obtained in most cases. Use of FeBr3 instead of FeCl3 furnished the alkyl bromide 272, while use of ICH2CH2I as an iodine atom source gave the analogous alkyl iodide 273. The authors propose formation of an intermediate Fe-C bond, which undergoes homolysis prior to cyclization. The manner in which the initial Fe-C bond is formed from a putative iron hydride is not discussed. Notably, the use of an FeIII salt in combination with a reductant distinguished the reactivity of this system from simple Lewis acidic behavior.351,352 Use of FeII complexes rather than FeIII complexes with NaBH4 as reductant appears to generate hydrido iron(I) complexes which can reduce alkyl bromides and iodides to the corresponding carbon-centered radical. These high-energy radicals can undergo 5-exo-trig radical cyclizations.353
Figure 127.

Ishibashi and coworkers’ halocyclization reaction.
In 2015, Ma and Herzon reported that they could effect the hydrobromination and hydroiodination of alkenes and alkenyl bromides/iodides (Figure 128), along with other transformations (see Sections 5.2 and 6.1 for hydroselenation and hydrogenation findings).354 They accomplish these transformations of alkenes to halides with stoichiometric Co(acac)2, alongside triethylsilane and 1,4-dihydrobenzene as reductants, tert-butyl hydroperoxide as an additive/oxidant, and tosyl bromide (TsBr) or diiodomethane as sources of halogen. Bromides 274 or 276 as well as iodides 275 or 277 could be generated from the corresponding methallyl- or prenyl-esters. Similarly, bromide 278 and iodide 279 could be formed from a corresponding terminal alkene. Remarkably, dihalogenated compounds 280 and 281 could also be formed from the precursor vinyl chloride and bromide, respectively. These reactions are thought to proceed through carbon-centered radicals generated through HAT from a cobalt hydride.
Figure 128.

Herzon’s method for hydrobromination and hydroiodination of alkenes.
5.3 Hydrochalcogenation
One of the most important methods for the synthesis of alkyl and vinyl sulfides and selenides is the addition of sulfides/selenides to carbon-carbon unsaturated bonds via ionic or radical processes.355 While there are numerous examples in the literature of transition-metal catalyzed addition of heteroatom compounds to alkynes, alkenes and allenes, these transformations tend to proceed rather inefficiently when sulphur or selenium atoms are involved. This is commonly attributed to catalyst poisoning via metal binding, or to formation of aggregates.356
In 1990 Kano and coworkers reported the hydrothiolation of styrenes using a porphinatoiron(III) catalyst with diphenyl disulfide and sodium borohydride (Figure 129).357 The reaction led to the desired product 282 in moderate yield after 24 hours at room temperature in a 1:1 mixture of benzene and ethanol. Although indenes and stilbenes showed some reactivity the scope of the transformation was rather limited for these cases.358
Figure 129.

Iron-catalyzed hydrothiolation of styrenes.
In 2011, Girijavallabhan took inspiration from Carreira’s work and reported a cobalt-catalyzed hydrothioetherification of unactivated alkenes (Figure 130).359 The protocol employs phenyl silane as reductant in combination with an electrophilic sulfur source, and is run in ethanol. The reaction proceeds efficiently under mild conditions and displays good functional group compatibility. Thus, complex 21 (2 mol%) catalyzed hydrothiolation of substrate 283 using either tosyl sulfide or S-phenyl benzenethiosulfonate. That either electrophilic sulfur source may be used suggests that the electronics of the sulfide do not have a great impact on the yield of the reaction. However, hydrogenation of the double bond was observed as a single product when thiophenol or diphenyl disulfide were used as the sulphur source. Minor amounts of hydrogenated byproduct were also detected when using less than 3 equivalents of radical trap. The transformation was competent for 2,2-disubstituted and trisubstituted alkenes as well as styrenes (284, 286 and 285, respectively).
Figure 130.

Cobalt-catalyzed hydrothiolation of alkenes.
As discussed above (see Section 5.2), Ma and Herzon354 reported in 2015 the radical hydrobromination and hydroiodination of alkenes using tosyl bromide and diiodomethane as the halogen sources, respectively, in analogy to Carreira’s tosyl chloride.340 The same method was also applied to the hydroselenation of olefins (Figure 131). Thus, treatment of a substrate with stoichiometric amounts of Co(acac)2/TBHP and an excess of Et3SiH and 1,4-dihydrobenzene as reductants in n-propanol gave Markovnikov like products in good yields. In this reaction, Se-phenyl 4-methylbenzenesulfonoselenoate was used as the radical trap to functionalize mono-, di-, and trisubstituted alkenes.
5.4 C-X Bond Formation in Natural Products and Complex Molecule Synthesis
Application of metal-catalyzed radical hydrofunctionalization technology to C-X bond formation has not yet occurred in natural products total synthesis, to the best of our knowledge. Nevertheless, some of these reactions have been used for the synthesis of natural product analogs, similar to those seen in the C-N bond Section (Section 3.5). Indeed, all the transformations explained in this section could lead to a library of products with different heteroatoms. For instance, Boger prepared the thiocyanate analog of vinblastine utilizing his Fe2ox3 technology to install the C-S bond in the natural product analog 287 (Figure 132).81 Similarly, Baran applied Boger’s hydrofluorination protocol to an ouabagenin precursor208 to afford the fluorinated analog 288.
Figure 132.

Synthesis of analogs containing C-X bonds.
6. Hydrogenation and Isomerization
In the absence of an external radical trap, a simple alkene subjected to transition metal-catalyzed or -mediated HAT can take two main pathways leading either to isomerization or hydrogenation (Figure 133). After initial HAT to a radical pair, the nascent carbon-centered radical may abstract a second hydrogen atom to give a hydrogenated compound. Alternatively, the metallo-radical could abstract a hydrogen atom from the adjacent methylene to form an isomeric olefin.
Figure 133.

Hydrogenation versus isomerization pathways for a simple alkene.
Additional pathways–cyclization, cycloisomerization, retro-cycloisomerization–and mixtures of products–positional and E/Z geometrical isomers can– result for substrates that contain multiple olefins or varying substitution patterns. The challenge lies in obtaining selectivity for a desired pathway. The following discussion details relevant synthetic efforts with brief historical commentary.
6.1 Hydrogenation
The first hydrogenation reactions which operate via a transition metal catalyzed (or mediated) HAT were discovered in the mid-20th century, but were not recognized as HAT reactions until 1975.360,361 In 1949, Adkins and Krsek were studying the hydroformylation362 of various alkenes with Co2(CO)10 in the presence of H2 (g) and CO (g) (100–150 atm, 1:1) at 120–125 ºC in benzene.363 They reported that α,β-unsaturated aldehydes, ketones and esters (crotonaldehyde, acrolein, methyl vinyl ketone, mesityl oxide, ethyl cinnamate and ethyl β-(2-furan)-acrylate) did not undergo hydroformylation. Rather, they were reduced to the corresponding saturated carbonyl compounds in 40–90% yields (Figure 134).
Figure 134.

α,β-unsaturated carbonyl compounds which undergo reduction rather than hydroformylation.
This unusual result was noted by Irving Wender and coworkers who proceeded to study this reaction further by treating olefins with 150–300 atmospheres of H2 (g) and CO (g) at 180–185 °C in the presence of cobalt.364 Under these conditions, alkenes were hydroformylated and reduced to the corresponding alcohol. In some cases, the olefin was only reduced. The authors found that whether a given olefin underwent reduction or hydroformylation and reduction depended on the amount of olefinic character it possessed – hydrogenation became dominant as the degree of double-bond character of an olefin diminished as a result of conjugation. Highly aromatic rings were unreactive. For example, 2,5-dimethylfuran was hydroformylated and reduced to 2,5-dimethyl-3-tetrahydrofurfural; thiophene was hydrogenated to thiolane; phenanthrene was hydrogenated to 9,10-dihydrophenanthrene and 1,2,3,4-tetrahydrophenanthrene; and benzene was inert. Wender proposed a free radical mechanism to account for the observed reactivity: initial addition of a hydrogen radical (•H) to an unsaturated moiety (aldehyde, alkene or polyaromatic compound) results in a stabilized radical with the spin centered so as to maximize stabilization (especially for polyaromatic compounds), and is followed by abstraction of a hydrogen atom from H-Co(CO)4 to give the saturated product (Figure 135).364 Halpern later amended this proposal, arguing that it is not a free hydrogen atom (H•) but H-Co(CO)4 which transfers the initial hydrogen atom.360,361
Figure 135.

Wender’s mechanistic proposal for the radical reduction of an aldehyde.
Subsequent work from Wender elaborated the scope of this reaction in the selective partial reduction of polyaromatic compounds (Figure 136).365,366 In 1959, Wender showed that this reaction reduced double-bonds of lesser aromatic character to give di- or tetra-hydro-aromatic compounds exclusively.366 Highly stabilized aromatic compounds like fluorene and triphenylene were not reduced. Thus, this method enabled access to partially reduced polyaromatic compounds which had previously been difficult or impossible to access with other hydrogenation catalysts, albeit at high temperature and pressure. As observed in earlier work,363,364 Wender notes that these reactions are not adversely affected by sulfur-containing compounds, which is remarkable because other homogeneous hydrogenation catalysts are normally poisoned. This early observation is prescient of the chemoselectivity that later transition metal HAT reactions would exhibit.
Figure 136.

Partial reduction of polyaromatic compounds under the action of (CO)4Co-H.
In 1975, Halpern argued that HAT from transition metal hydrides was the primary mechanism operative in the aforementioned364,365,366,367 (CO)4Co-H reductions of polyaromatic compounds.360
In a similar vein, Iguchi discovered in 1942 that aqueous cobalt cyanide solutions could rapidly absorb hydrogen and that increased quantities of hydrogen were absorbed in the presence of sodium cinnamate or isatin.368 A number of groups subsequently studied this system and found that [(CN)5CoH]3− was formed under the reaction conditions.367 In 1962, Kwiatek and coworkers reported that [(CN)5CoIIIH]3− was able to catalytically reduce a wide array of conjugated functional groups such as aliphatic dienes, conjugated aromatic olefins, α,β-unsaturated carbonyls (acids, esters and aldehydes), 1,2-diketones, epoxides and azoxy compounds under an atmosphere of hydrogen gas (1 atm).369,370 These substrates include styrene, isoprene, 1,3-cyclohexadiene, tiglic aldehyde, methacrylic acid, sorbic acid, cinnamic acid, methyl methacrylate, benzil, cyclohexene oxide, styrene oxide, and azoxybenzene, among others. Each was reduced by one degree of unsaturation. Alkyl substituted alkenes and aromatic compounds (diphenylethylene, indene and propenylbenzene) were not reduced. These reactions were conducted in water with an organic co-solvent added for water insoluble substrates. [(CN)5CoH]3− was generated in situ from K3Co(CN)5 by the action of hydrogen gas. This system has been intensely studied and radical intermediates have been implicated in the reaction.367,371,372,373 Halpern proposed in 1968 that reductions of α,β-unsaturated carbonyls with [(CN)5CoH]3− might proceed through a HAT type-mechanism.372,360
In 1977, Halpern’s team convincingly showed that HAT was operative in the reduction of styrenes with (OC)5Mn-H complexes.361 They observed chemically induced dynamic nuclear polarization (CIDNP), which is evidence of the radical character of this reaction, and an inverse isotope effect for HMn(CO)5/DMn(CO)5, which suggests that the first hydrogen atom transfer (HAT) is reversible (see Section 7.3 for further discussion).374,375
Subsequent to Halpern’s seminal361 unambiguous demonstration that HAT is the primary pathway operative in the H-Mn(CO)5 reduction of styrenes, an extensive number of studies were published on the subject of metal-mediated hydrogen atom transfer.29,375 Like those of Halpern, these early studies focused on reductions of activated alkenes such as polyaromatic compounds, styrenes, acrylates, 1,3-dienes and allenes with transition metal hydrides bearing strongly π-acidic ligands like :C≡O along with a phosphine or Cp/Cp* ligand.375 However, addition across unactivated alkenes was not observed. These studies were primarily concerned with understanding kinetic and thermodynamic parameters governing these HAT reactions rather than leveraging their reactivity in organic synthesis.375 Thus the value of these studies lies primarily in their elucidation of physical organic chemistry principles and mechanistic details surrounding transition metal hydrogen atom transfer chemistry and so are discussed and/or referenced in Section 7. The following discussion below shall focus rather on reactions developed within the synthetic organic chemistry community.
In the synthetic organic chemistry literature which arose from and around Mukaiyama’s research,38 hydrogenated products have been observed in reactions (which retrospectively may involve transition metal HAT) as undesired side products. For example, in Mukaiyama’s seminal 1989 contribution detailing the oxidation of unactivated alkenes with molecular oxygen using Co(acac)2 in isopropanol, he observed butylbenzene as a minor product in up to 17% yield in attempts to oxidize 4-phenyl-1-butene (Figure 137).97 However, since Mukaiyama’s team was interested in obtaining oxidation products, this side product was not pursued further.
Figure 137.

A reduced side product was observed by Mukaiyama’s team during development of their hydration reaction.
Other researchers observed in control experiments that if O2 was excluded from their systems, formation of hydrogenated products increase (see Section 2). Indeed, Magnus and coworkers noticed that reduction occurred in the absence of O2 during their control studies involving conjugate reduction of α,β-unsaturated ketones with a putative Mn(III) hydride107 and developed this into a method for selective conjugate reduction of enones, which they reported in 2000.221 This reaction selectively reduces the α,β-unsaturated moiety of enones in the presence of other alkenes (Figure 138). However, rather than a HAT mechanism, this reaction is thought to proceed via a polar 1,4-addition of a metal hydride into an enone to form a [Mn] enolate, which is protonated to turn over the catalyst. β,β-disubstituted α,β-unsaturated ketones (and esters) are not reduced under these conditions. However, the authors did observe H-addition to the β-position of β,β-disubstituted α,β-unsaturated ketones if oxygen was present (see Section 7.6 for further discussion of this observation).107,221 Magnus’ system was modified from Mukaiyama’s work,106 and the Shenvi group24 later adapted the same system. Both Mukaiyama and Shenvi propose the intermediacy of carbon centered radicals rather than the closed shell system Magnus proposes, although the dominant pathway probably depends on the substrate and presence of additives. In the case of Shenvi, tert-butyl hydroperoxide (TBHP) is added and expands the reactivity to unactivated alkenes, as well as enabling catalyst turnover by oxidation of [Mn]+2 to [Mn]+3 (vide infra).24
Figure 138.

Magnus’ conjugate reduction method with substrate scope.
In Halpern’s 1975 paper,360 the observation that reduction of 9,10-dimethylanthracene by H-Co(CO)4 resulted in an epimeric mixture of the cis- and trans- di-hydro-products was considered one indication that the reaction proceeds through a radical intermediate (Figure 139). If one only wants one of the epimers, then the intermediacy of radicals is a disadvantage, since both result. On the other hand, the advantage of HAT could be counted two-fold. First, it shows interesting chemoselectivity – only the two reduced products shown in Figure 139 result while the remaining unsaturation is not affected. Second, the trans-isomer is obtained in one step, a stereochemically interesting result that would be difficult to obtain if the reaction proceeded by simultaneous hydrogen delivery from a metal center, which one would expect to give the cis-product.
Figure 139.

Halpern’s examination of [Co]-H reduction of polyaromatics prompted him to posit transition metal HAT as a mechanistic pathway.
In 2014, the Shenvi24 and Herzon27 labs found ways to use transition metal HAT to solve unaddressed problems related to the hydrogenation of alkenes. In so doing they demonstrated chemoselectivity and stereochemical benefits of transition metal HAT reactions. The Shenvi lab showed how the intermediacy of a carbon-centered radical could be exploited to obtain hydrogenated products with thermodynamic stereocontrol.24 The Herzon group, and the Shenvi group to a lesser degree, employed transition metal HAT for the chemoselective reduction of haloalkenes to haloalkanes without reduction of the carbon-halide bonds.27 The Herzon lab has also shown that the alkene-selectivity of a transition metal HAT pathway can be complementary to classical hydrogenation techniques.354 It is worth noting that identification of the general problems solved by both groups arose in the context of natural products synthesis. We will discuss both contributions in turn.
The Shenvi lab’s interest in hydrogenation arose during their synthetic studies of the asmarine alkaloids.376,377 During the course of the project, it became necessary to reduce an exocyclic methylene of 289 to the thermodynamically-favored product 291 (Figure 140). However, attempts to do so with a variety of homogeneous, heterogeneous and pericyclic methods resulted primarily in formation of the kinetic product 290 or in nearly equal mixtures of the two products 290 and 291.
Figure 140.

The Shenvi group developed a HAT hydrogenation method when they could not achieve chemo- and stereoselective reduction to the thermodynamic product 291 with any known method.
Their solution led to transition metal HAT via examination of the reduction of 4-tert-butylmethylenecyclohexane 292 (Figure 141).
Figure 141.

HAT bypasses high energy intermediates in dissolving metal pathway.
It was known that dissolving metal conditions (Liº, EDA, 35 ºC) alone were able to access the thermodynamic product 297.378 This selectivity occurs through a mechanism that involves initial population of the high-energy LUMO π* to form a high energy radical anion 293, protonation of the anion by the solvent (NH3) to give tertiary radical 297, a second electron transfer to form a tertiary anion 294, followed by a final proton transfer to yield the reduced products 296 and 297 (Figure 141). The thermodynamic hydrogenation product 297 results as the major product because the tertiary radicals 294 and anion 295 are mainly sp3 hybridized379 and predominantly occupy the lower energy equatorial conformation. Thus when protonation occurs, it does so from the more sterically hindered face to give the equatorially oriented methyl group (297). However, dissolving metal reductions are infrequently used because they exhibit poor chemoselectivity. The π* orbital of an alkyl substituted alkene is infrequently the LUMO of a molecule, and this fact in combination with the high reducing potential of the alkali and alkali-earth metals used in dissolving metal reductions means that almost all other functionality (e.g. halides, carbonyls, heteroaromatic rings, etc.) are reduced before alkyl-substituted alkenes. A direct hydrogen atom transfer (HAT) circumvents this problem of requiring a large reduction potential. By bundling the electron (e−) and proton (H+) to form a hydrogen atom (H•) and delivering this directly to an alkene to form an intermediate tertiary radical 294, HAT bypasses the high energy radical anion 293 and therefore shows much improved chemoselectivity for alkyl substituted alkenes over other reduction labile functionality.
While this strategy was attractive, conditions for transition metal mediated/catalyzed HAT reduction of unactivated (i.e. alkyl-substituted) alkenes were “unknown” at the time. More correctly, conditions for TM HAT reduction were not widely recognized under the terms of hydrogen atom transfer chemistry. Mukaiyama’s hydration38 and related reactions had long demonstrated that carbon centered radicals can be generated from unactivated alkenes under mild conditions by the action of Co, Fe, and Mn complexes bearing β-diketonate ligands in the presence of a silane.38 However, these and related reactions were thought to proceed through a hydrometallation/M-C bond homolysis mechanism. Notably, prior to Shenvi and Herzon, Norton298 had noted in passing that these reactions probably298 proceed through a HAT mechanism. Nojima118,119,120 indicated similar possibilities, but depicted radical character at the transition state, not as a discrete step. Boger’s iron(III)-borohydride system26,81 was described as distinct from the reactions of carbonyliron hydrides, and characterized as a radical hydrogen atom addition. For unknown reasons, these mechanistic intimations were not adapted until after 2014, at which point most subsequent papers adopted HAT as the accepted mechanism. While both a TM HAT mechanism and a hydrometallation/homolytic M-C bond cleavage mechanism would generate carbon centered radicals, distinguishing between these two is significant for at least two synthetically relevant reasons. First, invoking a HAT mechanism is better able to explain the exclusive Markovnikov selectivity observed for these reactions. This selectivity is especially meaningful for sterically encumbered alkenes, for which one would expect inverted regiochemistry especially for highly sterically differentiated alkenes. Instead, regiochemical inversion is only observed for cases where radical stability differs. Second, the unusual tolerance of these reactions for Lewis basic functionality is also more reasonable for a mechanism that does not require alkene coordination to Lewis acidic metal center cis to a metal hydride.
With these considerations in mind, Shenvi and co-workers explored Mukaiyama’s conditions and those of related reactions and eventually found that use of Co(dpm)2 or Mn(dpm)3 (10 mol%) in the presence of PhSiH3 and tert-butyl hydroperoxide in i-PrOH under an inert atmosphere were able to catalytically hydrogenate unactivated alkenes with thermodynamic selectivity (Figure 142). These conditions showed high chemoselectivity, reducing alkenes even in the presence of a sulfide 298 and Lewis basic heterocycles (299, 306, 307). The high chemoselectivity and Lewis basic functional group tolerance was taken to suggest that a non-coordinative hydrogenation mechanism is operative. Furthermore, alkenes are reduced in the presence of aryl halides (F, Cl, Br, and I) to their thermodynamic products with 5–6:1 diastereoselectivity (301 and 307). A chloro- (302) and bromo-alkene (285), silyl enol ether (304) and enamide (305) were similarly reduced, although the former three show lower diastereoselectivity (3.7–3.9:1) than alkyl substituted alkenes. Presumably this is due to diminished sp3 character of the intermediate carbon-centered radical due to hyperconjugative stabilization of the heteroatom lone pairs. The enamide (305) was reduced with 6.7:1 diastereoselectivity. Alkenes are reduced even in the presence of aldehydes 309 and free thiols 308, although the latter undergoes disulfide bond formation and the former is highly sensitive to carboxylic acid impurities.
Figure 142.

Selected examples of Shenvi’s HAT reduction method.
The divergent stereocontrol of Shenvi’s hydrogenation method was demonstrated with some compounds known in the literature. The reduction of drimene (311) to drimane (312) is one such example (Figure 143). The point here is to show the stereodivergent route that this hydrogenation pathway takes compared with non-radical hydrogenation pathways (cf. 310).380
Figure 143.

Example of divergent stereocontrol.
In 2016, the Shenvi lab published further details regarding their development of this reaction. Their chief finding was the role of Ph(i-PrO)SiH2 in the reaction, which appears to be a superior reductant to PhSiH3. Use of this silane improves the yields of their hydrogenation reaction with Mn(dpm)3 and enables many other HAT type-reactions which employ manganese and iron complexes.116 In addition, the use of this silane allows the hydrogenation reaction to be run in a wide variety of different solvents, whereas previously the Mn(dpm)3 reaction required an alcoholic solvent. Reactions with cobalt complexes are not significantly affected by this silane, presumably because initial formation of a metal hydride is not rate determining.
Meanwhile, the Herzon lab was interested in accessing the natural product (−)-acutumine (314) from dehydroacutumine (313) via hydrogenation of a vinyl chloride (Figure 144).194,195,381 However, they had found only one method (using a [Rh] catalyst under a hydrogen gas atmosphere) which could give them their desired product, and this in only 17% yield (56% based on unreacted starting material). This underwhelming result prompted them to develop a better method.27
Figure 144.

Herzon’s synthesis of (−)-acutumine (314).
Based on their extensive survey of reduction methods, Herzon concluded that the challenge of haloalkene reduction lies with suppressing hydrodehalogenation (Figure 145). Presumably, the hydrodehalogenation product 317 occurs via β-elimination of a β-haloalkylmetal species 315 followed by hydrogenation of an intermediate alkene 316. Alternatively, a C-X oxidative addition, reductive elimination sequence could lead to alkene 316. Herzon reasoned that since halogen atoms stabilize adjacent (α) radicals, an initial transition metal catalyzed HAT to a vinyl halide should occur to give a stabilized α-haloradical 318, followed by a second HAT to obtain the desired alkyl halide 301. This would avoid formation of an alkyl-metal intermediate 315, which leads to dehalogenation. Indeed, they found that transition metal catalyzed HAT was a general solution for the reduction of fluoro, chloro, bromo, and iodoalkenes, cyclic and acyclic alkenyl halides, and gem-dihaloalkenes (Figure 146).
Figure 145.

Herzon showed that HAT bypasses dehalogenation pathways.
Figure 146.

Herzon’s method for alkyl halide synthesis by reduction of vinyl halides.
Specifically, Herzon explored a variety of manganese and cobalt-based catalysts and terminal reductants and uncovered two ways to effect this reduction. The first (a in Figure 146) is selective for 2,2-disubstituted alkenes and comprises of Co(acac)2 (25 mol %), t-butylhydroperoxide (TBHP, 25 mol%), tricyclohexylphosphine (25 mol%), 2,6-di-t-butyl-4-methylpyridine (DTBMP), 1,4-cyclohexadiene (1,4-CHD, 5 equiv.) and triethylsilane (5 equiv.) in isopropanol ([0.3 M]) at 50 ºC under an Argon atmosphere. Alternatively, tricyclohexylphosphine could be excluded from the reaction if Co(acac)2 and TBHP are added equimolar with substrate (1 equiv. each) (method b Figure 146). This latter reaction could also be run at room temperature under air and could additionally reduce tri- and tetra-substituted alkenes.
Following this work, Herzon showed in 2015 how transition metal HAT reduction demonstrates selectivity that is complementary to classical methods.354 While classical methods obtain selectivity for a given olefin based on strain, steric environment, or proximity to a directing group, Herzon demonstrated that selectivity in his HAT hydrogenation method depends on intermediate radical stability. In particular, Herzon’s team demonstrated with a series of competition experiments that useful selectivity could be obtained for 8 pairs of substrates. These include hydrogenation of 2,2-disubstituted olefins over α-olefins; bromo- and chloro- alkenes over α-olefins; 2,2-disubstituted alkenes over 1,2-disubstituted alkenes; 2,2-disubstituted alkenes over internal alkynes; trisubstituted alkenes over α-olefins; and trisubstituted olefins over cis- and trans-1,2-disubstituted alkenes.
Taken together, Shenvi24 and Herzon’s27 hydrogenation methods re-contextualized the Mukaiyama hydrofunctionalization methodology in light of transition metal hydrogen atom transfer chemistry and suggest that TM HAT may in many cases better fit the experimental data (see Section 6.2, 7.8 for further discussion).
One other paper should be noted. Prior to any of Mukaiyama’s work, Chung and coworkers published in 1979 a method detailing the reduction of mono-, and disubstituted olefins by a CoCl2/NaBH4 system that bears resemblance to the aforementioned reactions.382,383 This work stems from an interest in tempering the reactivity of borohydride reductants by use of metal salts. However, later work384 revealed that these conditions generate cobalt boride and hydrogen gas in situ,385 although the mechanism of alkene/alkyne reduction with this heterogeneration mixture remains unclear. Consequently, we do not discuss these numerous reactions further except to state that a number of groups have found uses for this reaction in complex molecule and natural product syntheses.207,208,386,387,388,389,390,391
In addition to the aforementioned work on cobalt and manganese complexes based on Mukaiyama’s work, a few methods have been developed from the iron-based chemistry initially employed in aerobic oxidation chemistry (see Section 2.1).
In 1991, Kano and coworkers reported the reduction of styrene to ethyl benzene with iron porphyrin complexes (Figure 147).392 Kano and coworkers were not interested in developing a hydrogenation method. Rather, they were interested in assessing the active species involved in the aerobic oxidation of olefins to alcohols with Fe(TPP) complexes in benzene-ethanol as solvent with NaBH4 as reductant (see Section 2.1). Santa et al.62 had assumed that an FeV-oxo porphyrin intermediate first epoxidizes an olefin, which is subsequently reduced with NaBH4. Kano doubted that the mechanism involved an FeV-oxo porphyrin intermediate because no epoxidation products had ever been observed and reduction of epoxides with BH4− is slow. Kano’s paper demonstrates that in the absence of oxygen, an FeIII porphyrin chloride complex in the presence of sodium borohydride can reduce styrene, which implies that an FeV-oxo porphyrin complex need not be involved. The authors found that different electronic substitution on the porphyrin ligand and solvents resulted in different product distribution and conversion. Use of 5,10,15,20-tetrakis(para-Cl phenyl)-porphyrinatoiron(III) chloride gave the highest yield of hydrogenation product. The byproducts in this reaction are meso and racemic 2,3-diphenylbutane, which likely result from benzylic radical dimerization. The authors propose the intermediacy of a σ-alkyliron(III) porphyrin complex.
Figure 147.

Kano’s hydrogenation of styrene.
Thomas and coworkers reported in 2014 a general and operationally simple formal hydrogenation method using Fe(OTf)3 and NaBH4 (Figure 148).393 This work was developed based on the precedent of Kano64,392 and Boger,25,26,81, who reported methods for the hydrofunctionalization of olefins with iron complexes and sodium borohydride reductants. The reaction converts terminal alkenes to the corresponding alkane. Other alkene substitution patterns either do not react, or give only trace products. The authors propose that the reaction proceeds through a polar mechanism, not through radicals. Thus, this transformation is included for the sake of completeness.
Figure 148.

Thomas’ alkene reduction method.
6.2 Isomerization
As transition metal hydrogen atom transfer (TM HAT) isomerization reactions are mechanistically distinct from those that proceed through metal-hydride insertion-elimination and π-allyl pathways, they can be used to access structural motifs that may otherwise be difficult to access.394
An isomerization reaction that proceeds through a TM HAT mechanism leverages the known reactivity of carbon-centered radicals to form and/or break bonds. The metal complex regulates the reactivity of the intermediate radical only in so far as it mediates the kinetics of competive reactions (H-atom abstraction, radical collapse, radical reduction). In contrast, canonical metal mediated/catalyzed isomerization reactions require coordination of the alkenes/alkynes to the metal center and operate via hydrometallation, insertion, and reductive elimination pathways. Consequently, these reactions are often inhibited by Lewis basic functionalities, which are normally superior to alkenes/alkynes as ligands for a given metal complex. Thus a TM HAT isomerization can exhibit unusual chemoselectivity compared to other transition metal isomerization reactions. Examples of isomerization, cycloisomerization and retrocycloisomerization of alkenes via transition metal mediated/catalyzed hydrogen atom transfer (TM HAT) are known in the literature and are discussed below.
In the 2000’s, van der Donk and coworkers observed cycloisomerization in their study of vitamin B12 catalyzed cyclizations of aryl alkenes (Figure 150).288,289 The authors found that use of vitamin B12, titanium(III) citrate in an ethanol/water solvent mixture with Et4NOH as a phase transfer agent could effect radical cyclizations of dienes. They further noted that the product distribution is affected by pH, with a lower pH favoring cycloisomerization over reductive cyclization (see also Section 4.1).289 Use of t-BuOH instead of ethanol also increased formation of the cycloisomerized product, presumably because ethanol has more homolytically labile C-H bonds. The authors speculate that initiation of the radical cyclization proceeds via a hydrogen atom transfer from a cobalt(III) hydride to the aryl alkene.
Figure 150.

Van der Donk and coworkers demonstrated the cycloisomerization of dienes with vitamin B12.
The Norton group has observed isomerization, hydrogenation and reductive cyclization (Section 4.1) in their pioneering studies on transition metal hydrogen atom transfer chemistry.2,375 These studies have focused primarily on elucidation of physical organic chemistry parameters (rate constants, bond strengths, structural parameters which affect reactivity, etc.) and have yielded many synthetically useful insights. Observations regarding the mechanism of TM HAT are discussed in Section 7.4, while discussion here focuses on isomerization applications in the synthesis of organic molecules.
Of particular relevance to this review, Norton has studied the reactivity of the chromium hydride, CpCr(CO)3H,290,291,293,294,295,296,297,298,299,300,301 vanadium hydrides of the type, HV(CO)4(P-P) [where (P-P) is a bidendate di-phosphine ligand],291,293,395 and a putative396 cobalt hydride generated from (H2O)2Co(dmgBF2)2 under H2(g) pressure,291,302,397,398,399 with dienes such as 320 (Figure 151).292 The chromium hydride and cobalt hydride systems are particularly notable because these metal hydrides can be generated catalytically under hydrogen pressure. The vanadium hydride, (P-P)(CO)4V-H undergoes the most rapid HAT to olefins due to a very weak V-H bond (56 kcal/mol) and so can initiate radical cyclizations of dienes more rapidly and under milder conditions than can CpCr(CO)3H (62 kcal/mol).395 By the same virtue, however, these vanadium hydrides cannot be generated catalytically under H2(g) because •V(CO)4(P-P) cannot cleave the H-H bond of molecular hydrogen. Norton has also demonstrated, however, that HAT to olefins is not just dependent on M-H and olefin bond strengths, a thermodynamic consideration, but also on on steric and electronic properties of the metal hydride and acceptor olefin, which are kinetic factors.395
Figure 151.

Several competing pathways are available to a diene 320 reacting with a metal hydride via TM HAT.
The Norton group has articulated the challenge of obtaining selectivity in TM HAT cyclizations (Figure 151).299,292 For example, consider the case of diene 320. Norton has shown that initial HAT generally proceeds more rapidly with the less sterically hindered 2,2-disubstituted enone alkene to give radical 321.298 For this stabilized radical, there are a variety of different pathways available (a few of which are shown in Figure 151). Reductive cyclization (323) and cycloisomerization products, both (via intermediate radical 322), hydrogenation (324) isomerization (325) products may all result. The course which a given reaction takes is a function of the concentration of reactive species in solution (such as M-H and M•) and the relative magnitude of the the rate constants for the individual cyclization, hydrogenation and isomerization pathways (kcyc, kH, kiso, and k′iso). These rate constants are in turn dependent of the steric accessibility of the olefins, stability of intermediate radicals, and metal hydride bond dissociation energy, among others. Norton has been largely responsible for measuring and parsing these relationships.
With respect to obtaining selectivity for isomerization pathways, when Norton’s group first began looking at radical cyclization of dienes like 327 with catalytically generated CpCr(CO)3H, (a system which the Norton group had first studied in the context of chain transfer radical polymerization of methyl methacrylate293,294,295,296,297,298), they observed linear isomerization product 330 as a minor product in a maximum yield of 15% (Figure 152).299 The relative ratios of reductive cyclization product (328), linear hydrogenation product (329) and linear isomerization product (330) were were observed with varying reaction temperature, concentration or reaction length; dilute conditions provided the best yield of 330.
Figure 152.

Norton observed isomerization (330) in the reaction of diene 327 with CpCr(CO)3H.
However, Norton showed that incorporation of a quaternary center into a similar substrate, 194, strongly favored reductive cyclization to give 195 (>95% yield), even at a higher reaction concentration (0.30 M) and shorter reaction times (Figure 153) by virtue of the Thorpe-Ingold effect (see also Section 4.1).299
Figure 153.

Thorpe-Ingold effect favors reductive cyclization of 194 under [Cr] catalysis.
The Norton group subsequently demonstrated that a putative metal hydride generated from (H2O)2Co(dmgBF2)2 under 3 atmospheres of hydrogen gas is able to effect the isomerization of compound 327 to the linear isomer 330 (Figure 154).397 The difference in reactivity between a [Cr]-H (Figure 152) and this putative [Co]-H is remarkable (Figure 154).
Figure 154.

Isomerization product 330 is favored by a putative [Co]-H.
The Norton lab’s careful measurements of the physical constants governing these reactions (rate constants and bond strengths) allowed them to explain this difference.397 Consider the case of diene 332 (Figure 155).397 In the case of the chromium complex, activation of H2(g) to form the chromium hydride, CpCr(CO)3H, is faster than HAT to the olefin, so metal hydride is readily available to a nascent carbon-centered radical whenever it is formed, leading predominately to the hydrogenated product (331). In the case of the cobalt hydride, activation of H2(g) is slower than HAT to the olefin, so the cobalt(III) hydride population is low while the [Co]II metallo-radical population is high. Therefore, the [Co]II complex is readily available to abstract a hydrogen atom from a C-H bond adjacent to the nascent carbon-centered radical (334). Since linear isomerization is not possible in the case of 331, the initial nascent carbon-centered radical (334) either reverts back to the starting material in an unproductive pathway, abstracts a hydrogen atom from an available donor to give hydrogenated product (331), or cyclizes in a 5-exo-trig fashion to give a stabilized benzylic radical (335), which gives cycloisomerization product 333 upon HAT back to a [Co]II species. Curiously, no reductive cyclization product is observed.
Figure 155.

A second example of distinct behavior of a chromium hydride versus a cobalt hydride.
Norton’s group also observed cycloisomerization during the course of their 2014 study on the effect of double-bond substituents on the rate of cyclization of α-carbomethoxyhex-5-enyl radicals generated via HAT from a transition metal hydride to an olefin (Figure 156).292 In particular, diene 336 gives cycloisomer 337 in 41% yield along with hydrogenated product 338 and linear isomer 339.
Figure 156.

Examples of isomerization and cycloisomerization from Norton and coworkers.
In 2016, the Norton group published a detailed account of alkene isomerization and diene cycloisomerization using Co(dmgBF2)(THF)2 as a catalyst under hydrogen gas.399 While paying particular attention to factors which affect the outcome of a given TM HAT reaction, the authors also demonstrate the synthetic utility of this catalytic system (Figure 157).
Figure 157.

Selected examples of HAT isomerization and cycloisomerization from Norton and coworkers.
They show that 2,2-aryl, alkyl-substituted alkenes selectively isomerize by one position (10 substrates). In the case of 340, isomerization generates an enol, which tautomers to its aldehyde form (341). The reaction is selective for 2,2-aryl, alkyl-substituted alkenes, so skipped diene 343 is obtained nearly quantitatively from isomerization of 342; the terminal alkene is not reactive under these conditions. Similarly, pyridine-substituted diene 344 is also cleanly isomerized to 345. Cycloisomerization is also possible when the HAT acceptor alkene is a terminal or 2,2-disubstituted alkene and bears an electron-withdrawing group, and the other alkene is aryl substituted. Consequently, cyclopentenone 347 is obtained directly from enone 346; indole derivative 349 arises from cycloisomerization of 348, and indane 351 from diene 350. Cyclopentenone 347 is particularly interesting because the isomerized olefin ends up within the nascent pentacycle; in all other examples it becomes exocyclic. Notably, this isomerization reaction employs hydrogen gas directly. Norton notes that no hydrogenation byproducts are observed in these isomerization reactions because the concentration of cobalt hydride is very low relative to [Co]II at any given time. Consequently, HAT back to [Co]II to give isomerization/cycloisomerization products is the only pathway observed.
The Shenvi group has approached TM HAT isomerization from the purview of organic chemists interested in synthetic utility. After publishing their HAT hydrogenation paper in 2014,24 Shenvi and coworkers reasoned that they might be able to minimize hydrogenation pathways in favor of isomerization pathways by ligand modification. They found that this was true and reported in 2014 a cobalt catalyzed HAT isomerization, cycloisomerization and retrocycloisomerization of a variety of alkyl substituted alkenes and dienes.28 This transformation is effected by racemic Co(SalentBu,tBu)Cl (1–10 mol%; Figure 158) with PhSiH3 (1–50 mol%) in benzene under an Argon atmosphere.
Figure 158.

The Co(SalentBu,tBu)Cl precatalyst employed in Shenvi’s HAT isomerization reaction.
The Shenvi group found that use of the privileged tetradentate salentBu,tBu ligand suppresses a hydrogenation pathway in preference to an isomerization pathway (Figure 159). The active catalyst in this reaction is selective for terminal alkenes and so can selectively isomerize terminal alkenes internally by one position. Thus tri-substituted alkene 353 is obtained in good yield from 352, and skipped dienes like 355 can be accessed (from 354 for 355). This reactivity is complementary to the isomerization/hydroboration catalyst of Chirik, which isomerizes internal alkenes to the terminal position; the terminal alkene is then hydroborated.400 In the absence of steric bias, the cobalt catalyst appears to abstract the weaker of two adjacent C-H bonds, so tetra-substituted alkene 357 is obtained selectively by isomerization of 356.
Figure 159.

Selected substrate scope of Shenvi’s HAT isomerization reaction.
Since the reaction proceeds through a transition metal catalyzed HAT pathway, the intermediate carbon centered radical can be leveraged to effect bond formation or cleavage events (Figure 159). This behavior is not reported in cobalt isomerization catalysts which proceed through coordination/insertion/β-H elimination pathways.401,402 The nascent carbon centered radical is capable of adding into a variety of unsaturated C-C bonds, including those of aromatic rings (359 and 361). It is also able to cleave strained rings. In the case of substrate 360, an adjacent cyclopropane ring is homolytically cleaved following the initial HAT, and the resultant secondary radical can add into the pendant aromatic ring to give 361 after rearomatization. In another example of retrocycloisomerization, the strained cyclobutane ring of caryophyllene oxide 362 is cleaved to give (−)-humulene II oxide 363 in excellent (95%) yield.
The team further found that by modulating the electronic properties of the salen ligand, they could alter the ratio of linear isomerization to cycloisomerization products, which suggests that the electronics of the metallo-radical affects the persistence of the nascent carbon-centered radical.
While this reaction is relevant to synthetic organic chemistry, the link it forges between the Mukiayama hydrofunctionalization literature and the TM HAT work pioneered by Halpern, Norton, and others is perhaps more significant. The putative cobalt(III) hydride, Co(SalentBu,tBu)H, that is generated in this reaction bears the planar tetradentate salen ligand. The metal hydride is formed through the action of phenylsilane on the Co(SalentBu,tBu)Cl pre-catalyst. A defining feature of Mukaiyama’s work is reduction of metal complexes with silanes. Significantly, this planar tetradentate ligand occupies all coordination sites cis- to a putative metal hydride. Therefore, barring ligand dissociation or a highly distorted ligand binding mode, alkene insertion/β-hydride elimination is not possible for lack of an open cis-coordination site. Moreover, the authors observed inhibition of catalysis by formation of a cobalt-alkyl (2º) bond, which results from radical cage pair collapse following the initial HAT. This organometallic bond could be cleaved homolytically upon heating. Support for a TM HAT mechanism over a coordination mechanism in this case comes from the observation that approximately two turnovers of the catalytic cycle were observed before the reaction stalled completely. No turnovers would be expected if the mechanism proceeded through a hydrometallation mechanism.
Further study of this reaction, particularly with regard to physical organic and inorganic chemistry aspects are an exciting field of research that is sure to yield further fruitful insights into how and why these reactions work.
6.3 Hydrogenation and Isomerization in Natural Products and Complex Molecule Synthesis
Use of HAT hydrogenation and isomerization methods in complex molecule and natural products synthesis is less widespread than C-O bond forming reactions, but is still present.
As mentioned previously (Figures 143 and 159), Shenvi and coworkers demonstrated in 2014 that their hydrogenation reaction could access drimane (312) in one step from an alkene precursor24 Similarly, Shenvi has also shown that their isomerization conditions can efficiently convert caryophyllene 362 to humulene II oxide 363 via a retro-cycloisomerization (Figure 159) and can convert β-cedrene to α-cedrene via a linear isomerization.28
In 2015, during their recent study and total synthesis of (±)-hippolachnin A (245), Carreira and coworkers investigated use of radical reduction conditions on substrate 364 (Figure 160).341 They found that use of Mn(dpm)3 (75% yield), Co(acac)2 (63% yield) and Fe(acac)3 (46% yield) all provided the undesired isomer 365 in >99:1 diastereoselectivity. Density function theory calculations at the B3LYP/6–311 + G(d,p) level of theory indicated that isomer 365 is more stable than the alternative epimer by ΔΔGº=1.0 kcal mol−1. Thus, although these radical hydrogenation reactions provided the wrong isomer required to access hippolachnin A (245), they did provide the more thermodynamically stable isomer with excellent diastereoselectivity. Other heterogeneous and homogeneous catalysts provided nearly equivalent amounts of both diastereomers or formed predominately the opposite and desired epimer of 365, which corresponds to hippolachnin (245).
Figure 160.

Carreira’s synthesis of hippolachnin A (245)
In a similar example, Bosch et al. have employed radical hydrogenation conditions to access the desired thermodynamic isomer 367 from alkene 366 en route to Serratezomine E (369) (Figure 161).403 Use of Mn and Fe complexes provided 367 in ~3:1 ratio with its isomer (368). The best yields were obtained with Mn(dpm)3, although purification was difficult and ~30% of unidentified by-products were obtained. Further exploration of reaction conditions revealed that use of H2 (g) and [RhCl(PPh)3] gave a 96:4 ratio of 367:368 in quantitative yield, so these conditions were employed instead.
Figure 161.

Bosch et al. synthesis of serratezomine E (369).
In their methodology paper on the construction of bicycle[6:4:0] and -[7:4:0] frameworks, Mukai et al. found in one case that treatment of oxabicyclo[7:4:0] compound 370 with Co(acac)2, TBHP and PhSiH3 efficiently removed the vinyl sulfonyl group to give 371 without further reduction.404 This constitutes a unique example of sulphonyl reduction under Co(acac)3 mediation. The authors do not speculate on the mechanism of this reaction.
In 2016, Shenvi and coworkers completed the enantioselective total synthesis of (+)-7,20-diisocyanoadociane (374) (Figure 163).405 One step of their synthesis employed the group’s radical hydrogenation methodology24 to set two stereocenters in (+)-7,20-diisocyanoadociane (374). After an initial addition of methyl magnesium bromide to install the southern tertiary alcohol of 373 from enone 372, treatment with Mn(dpm)3/PhSiH3 reduced the enone double bond to give the trans-configuration of 373 with 3:1 diastereoselectivity in 51% yield.
Figure 163.

Lu et al.’s synthesis of (+)-7,20-diisocanoadociane (374).
7. Mechanistic Overview
7.1 Introduction
The preceding synopsis presents a summary of synthetically useful methods for the Markovnikov-selective radical hydrofunctionalization of alkenes that proceed by reaction with Mn, Fe and Co complexes and reductants. We include reactions based on the possibility that carbon radical generation arises via hydrogen atom transfer from a metal hydride to an alkene. The difficulty with grouping these reactions by a specific mechanism (namely, hydrogen atom transfer) is that there is a paucity of experimental evidence to support definitive assertions, especially within the synthetic organic chemistry literature. Indeed, many early developments in the field of alkene functionalization precede Halpern’s articulation361,374,375 of transition metal hydrogen atom transfer (TM HAT) as a viable mechanistic pathway, and so TM HAT was not even considered as a possibility in these cases. Even after Halpern’s work, later synthetic literature only rarely mentions HAT as a mechanistic possibility, despite its invocation in the inorganic, physical organic, and polymer literature.5 The idea that the reactions in Sections 2–6 instead proceed through initial hydrometallation remains an alternative hypothesis, but we suggest that HAT be considered a reasonable and general mechanism for initial radical generation. Post 2014, this MH HAT hypothesis was suddenly and widely adapted in research articles and literature reviews to characterize the field.5,6,7,24,27,397
The reactions discussed in this review have been studied by various kinds of chemists: physical organic, inorganic, polymer, and synthetic organic. This diversity of scientific interest and expertise, in combination with the breadth of conditions [metals (V, Cr, Mn, Fe, Co), ligands, reducing agents (silanes, boranes, etc.), solvents, etc.] employed for these radical hydrofunctionalization reactions makes synopsis and discussion of all mechanistic studies difficult. Therefore, this section aims to present a critical discussion of the main mechanistic studies conducted for the preceding reactions, and draws some common conclusions from this work.
Some general comments may be made about the following studies. Although coherent mechanistic proposals have been made on the basis of kinetic studies and labeling experiments, isolation of intermediates has proven challenging. Regardless, most studies consistently propose formation of a metal hydride followed by reaction with a double bond, independent of the metal, the reductant, the substrate, or even the particular transformation. Retrospective application of a MH HAT mechanism to net redox-neutral catalytic transformations would resemble a cycle like that in Figure 164,28 variants of which have now appeared in literature reviews.5,6,7 Such metal hydride reactivity is based on mechanistic investigations that began in the latter half of the last century.
Figure 164.

Reductive-oxidative cycle towards alkene hydrofunctionalization.
Study of the properties and reactivity of transition metal hydrides was of great interest in the early 1960s because of their roles in the hydrogenation and hydroformylation of alkenes.406,407 The metal-hydrogen bond energies have been extensively measured and compared using different techniques. As Pearson has discussed, transition metal hydrides typically do not possess a large dipole, so three different types of behavior may be observed upon M-H bond cleavage (Figure 165).408,409 One may observe release of H−, H· or H+ depending upon the relative stability of the resulting cationic, radical or anionic metal species, as well as the accessible kinetic pathways for bond cleavage. Analysis of the pKa’s of a set of transition metal hydrides (V vs. Re) indicated that the 1st row metals were generally more acidic than 3rd row metals. Comparison within a row between Mn, Fe, and Co complexes bearing carbon monoxide ligands showed a clear decrease in pKa from left to right in the periodic table, which trends with the corresponding increase in electron affinity (Figure 165). Metal hydride acidity is also affected by the ability of a ligand to accept π back donation from the metal center, the oxidation state of the metal, and the geometry of the ligand coordination sphere.410 For example, replacement of a carbonyl ligand with a triphenylphosphine ligand, a worse π-accepting ligand, causes the pKa of a metal hydride to increase. Since homolytic cleavage is also a reductive process (with respect to the metal), a general correlation between bond strengths and Brønsted acidities was established.
Figure 165.

Acidities and bond strengths of metal hydrides.
7.2 Reaction of a Metal Hydride with an Alkene
The nature of the between metal hydride and alkene has also been well studied, and perspectives have evolved alongside the development of novel catalysts and transformations. Early UV studies on the addition of [HCo(CN)5]−3 to a set of activated alkenes revealed a significant correlation between the electron density of the alkene and the reaction rate (Figure 166).372 More electron-donating groups and/or α-substitution increased the rate of formation of an organocobalt adduct like 375. Kinetic measurements suggested participation of the alkene as a nucleophile and the metal hydride as an electrophile.
Figure 166.

Interaction between a metal hydride and an alkene.
Several hypotheses were articulated for the initial interaction between Co-H and the substrate as explained below. First, a concerted addition of the metal hydride to the double bond through a four-center transition state 376 was considered. This was regarded as rather unlikely since it implies simultaneous Co-C bond formation, and would have to accommodate a highly sterically disfavored configuration in the transition state. Only Markovnikov products were ever observed with this reactivity, so this putative concerted addition would have to proceed through a sterically disfavored transition state completely regioselectively. Secondly, the reaction may proceed via initial protonation 377 of an alkene by the metal hydride. This idea was discarded though because of the high pKa of HCo(CN)5−3 (20.0). Thirdly, the reaction may proceed via a direct hydrogen atom transfer from the metal hydride to the alkene via 378 (BDE (Co-H) = 57 kcal/mol). This mechanism seemed the more probable. Moreover, the reaction rate showed no [CN−] dependence, which ruled out a pre-equilibrium involving the coordination of the double bond and the metal.
Interestingly, a later study on the addition of HCo(CO)4 to styrene (379) showed competitive formation of the organocobalt adduct 382 and hydrogenation of the substrate to ethylbenzene (381) (Figure 167).411 Monitoring the transformation by IR revealed that both pathways were first order in metal hydride and in styrene. More importantly, the relative rates [d/dt(k2/k3)=0] remained constant during the course of the reaction. The reversible formation of a common radical pair was suggested and later confirmed by CIDNP effects (chemically induced dynamic nuclear polarization). This NMR technique detects enhanced absorption or emission of signals, usually using 1H NMR, when unpaired electrons are generated under the reaction conditions. Thus, the magnetic moment of the electron polarizes the spin of many protons changing the usual thermal Boltzmann distribution. This experiment indicated the presence of a radical pair collapse competing with either the back-reaction to reform the reactants or separation of the radicals (cage escape) to ultimately form the products. The solvent-separated free radical 380 could react further through fast hydrogenation. Alternatively, radical collapse could yield the organocobalt complex 382,412,413,414 which had been previously trapped with phosphines and further characterized.415
Figure 167.

Competition between cage escape and radical collapse towards hydrogenation/hydroformylation.
In a secondary process, formation of 2-phenyl-propanal (384) was observed, especially at low styrene/HCo(CO)4 ratios. In this case, insertion of the organocobalt species 382 in a carbon monoxide moiety occurs prior to reaction with another equivalent of the metal hydride. However, at longer reaction times or higher temperatures, slow evolution of CO led to the formation of (phenylethyl)cobalt complex 383 (1699 to 1715 cm−1). This intermediate further led to mixtures of branched- and straight-chain isomers, the ratio of which was highly dependent on the substrate, according to previous studies.416 In the case of styrene (379), the amount of linear isomer (383) was less than 5% immediately after the reaction was completed, but increased over time to favor the straight-chain isomer 383 at equilibrium.
In summary, different studies on the addition of metal hydrides to alkenes have suggested the formation of radical intermediates as the kinetically preferred reaction pathway. Controlling a competitive radical collapse towards an organometallic species could ultimately tune the selectivity of the transformation.
7.3 Evidence for the Formation of a Radical Pair
The recurring proposal of a free radical mechanism for the reaction between a metal hydride and an unsaturated C-C bond spurred several studies seeking to identify these intermediates. Sweany and Halpern studied the hydrogenation of α-methyl styrene (385) with HMn(CO)5 and found that this reaction obeys a second-order rate law (Figure 168).361 He suggested that a mechanism involving hydrogen atom transfer to the alkene to generate a carbo-radical metallo-radical cage pair is consistent with this data. Evidence for radical formation was provided by the observation of CIDNP effects, which also indicate reversible formation of a radical pair prior to cage escape towards 386.
Figure 168.

Evidence for the formation of a radical pair during the hydrogenation of alkenes.
No detectable amount of the radical combination product, Ph(Me)2CMn(CO)5 (388), was observed. Ph(Me)2CMn(CO)5 (388) is expected to be unstable to decomposition via Mn-C bond homolysis. However, in the case of the reaction with styrene, transient formation of low concentrations of the organometallic coupling product, Ph(Me)CHMn(CO)5, were observed. Thus, both reduction of the radical intermediate 386 to give 387 and recombination of the [Mn] metallo-radicals are faster than cage escape. Other studies have shown that the formation of the alkyl-metal complex is highly dependent on the viscosity of the solvent used.417
Further support for a HAT mechanism was provided by use of DMn(CO)5. Hydrogenation was accompanied by isotopic exchange in the substrate, reflected in the accumulation of HMn(CO)5. This behavior was in accord with the reversibility of the formation of the radical pair. Moreover, the overall rate constant of the hydrogenation with DMn(CO)5 was larger than with HMn(CO)5 (kH/kD = 0.4 at 65 ºC). This inverse kinetic isotope effect is common in reversible hydrogen atom transfers from metal hydrides due to the very low initial frequency of the M-H/M-D bond (1800 cm−1) relative to that of the C-H/C-D bond (3000 cm−1). Similarly, allenes and dienes could be also reduced via hydrogen atom transfer via a stabilized allylic radical, which could be further reduced to the corresponding alkene.418,419 However, a second alkene reduction of the resultant alkene to the alkane was never observed, indicating the importance of radical stabilization.
An analogous study was performed for the reduction of polycyclic aromatic hydrocarbons (Figure 169).360 The aromatic polycycle, 9,10-dimethylanthracene (389), was efficiently hydrogenated with HCo(CO)4, generated from Co2(CO)8 and synthesis gas (H2 + CO), through free radical intermediates (390) instead of organocobalt complexes. An equimolar mixture of cis- and trans-products (391) resulted and 9,10-dimethylanthracene (389) was reduced more rapidly than anthracene itself, suggesting low sensitivity of hydrogen atom transfer to steric influences. Instead, the higher rate observed for 9,10-dimethylanthracene (389) over anthracene probably reflects increased stabilization of the radical intermediate 390 by the two methyl groups relative to the analogous anthracene intermediate. Moreover, no competing hydroformylation was observed, which would presumably require the formation of an organocobalt adduct for the CO insertion to occur.
Figure 169.

Proposal for the reduction of polycyclic aromatic hydrocarbons.
Reaction of anthracene with DCo(CO)4 resulted in rapid isotopic exchange at the site of addition and is a consequence of the reversibility of the initial hydrogen atom transfer. Interestingly, no scrambling was observed when the reduction was performed with DMn(CO)5.420 An inverse KIE was still measured but no CIDNP effects could be detected, which was taken to mean that that reversible formation of a radical pair does not occur. Similarly, the group of Orchin studied the analogous hydrogenation of ethylidenefluorene and bifluorenylidene comparing cobalt and manganese: both followed second order kinetics.421 This case deserves special attention since the kinetic isotope effects were highly dependent on the substrate.
Hydrogen atom transfer to ethylidenefluorene (392) and bifluorenylidene (393) was endothermic (15.7 kcal/mol and 12.1 kcal/mol, respectively) (Figure 169). Ethylidenefluorene (392) showed an inverse KIE (0.4), but the latter example, bifluorenylidene (393), showed a normal KIE: 2.0. The authors suggested that the ground state for bifluorenylidene is higher in energy than ethylidenefluorene and therefore the transition state would more resemble the reactants than previous examples of TM HAT. Therefore, it was assumed that the normal KIE arose from an earlier transition state (Figure 170).
Figure 170.

Orchin’s explanation for the origin of the inverse kinetic isotope effect.
This reasoning was later discussed by Norton, who suggested irreversible hydrogen atom transfer to bifluorenylidene (393) as a more likely explanation.375 That is, return of the radical cage pair collapse to starting reagents is out-competed radical cage escape. Thus, although a single-step inverse KIE is in principle possible if it has a sufficiently late transition state, there are few if any cases where those cannot be attributed to operation of a multistep mechanism containing one or more reversible steps.422
Instead, in MH HAT the short lifetime (impersistence) of the metal radical/carbon radical cage pair and its rapid return to starting materials establish a preequilibrium prior to the rate-determining step, which is usually cage escape. The inverse isotope effect observed in MH HAT is attributable to the lower stretching frequencies of M-H/D bonds in the starting material compared to higher stretching frequencies of C-H/D bonds in the cage pair intermediate; i.e. the ratio of cage pair to starting materials is higher for deuterium and determines the overall reaction rate.
In summary, studies on the formation of a radical pair were based on the observation of CIDNP effects as well as related intermediates using different spectroscopic techniques, for example, NMR or IR. Kinetic studies also supported the proposal along with isotope exchange experiments, which generated an intensive discussion. Analogous behaviour was observed in the hydrogenation of polycyclic aromatic hydrocarbons.
7.4 Behavior of the Carbon-Centered Radical
Eisenberg and Norton reviewed early work on transition metal HAT in 1990, including the group’s study of the rates of hydrogen atom transfer from metal hydrides to a trityl radical 394 (Figure 171).375,423 These prior studies had focused on the generation of radical pairs, but little was known about the relative rates of reaction between a metal hydride and a carbon-centered radical.423 Norton found that trityl radical (394) abstraction of a hydrogen atom from a transition metal hydride (to give 395) is highly sensitive to steric factors.423 For example, changing HFeCp(CO)2 for HFeCp*(CO)2 resulted in a 40 fold decrease in rate independent of the M-H bond strength. In this case, Norton’s team determined that trityl abstraction of the hydrogen atom from the metal hydrides is an exothermic single step, is rate-determining, and exhibits a normal kinetic isotope effect [2.8 for HMn(CO)5]. Subsequent studies on the relationship between the rate of HAT to alkenes for a series of bidentate phosphine vanadium hydrides290 confirmed this conclusion: Norton found that although bidentate phosphines with larger bite angles had lower BDEs, they observed an inverse dependence in the rate of HAT to alkenes for these complexes.395,424 This suggests that steric encumberance (larger phosphine bite angles) on the metal hydride significantly affects rate of HAT to alkenes.24,354
Figure 171.

Reduction of a carbon-centered trityl radical.
At the same time, Bullock and Samsel approached this same problem of assessing relative rates of transition metal HAT using a radical clock experiment (Figure 172).425,426 Hydrogen atom transfer from a metal hydride to a vinyl cyclopropane 396 results in a mixture of rearranged and unrearranged product (398 and 400, respectively). Based on distribution and kinetic measurements, quantitative data on relative rates could be obtained. Thus, substrate 396 was reduced with several metal hydrides (Mn, Mo, Fe, W and Cr) to form a radical pair intermediate (as defined by CIDNP) during the course of the reaction. Reversible hydrogen atom transfer is suggested as the first step of the mechanism in light of observed isotope exchange when using DCrCp(CO)3 or DWCp(CO)3. Consistent with this exchange, reversible TM HAT to 2-cyclopropylpropene with metal deuterides resulted in deuterium incorporation at the methyl position of recovered 2-cyclopropylpropene. This first forward step is endothermic and shows an inverse KIE (0.55 for DWCp(CO)3).
Figure 172.

Competition between reduction and rearrangement of substrate 396.
Upon radical pair separation, the cyclopropyl radical 397 is either directly reduced to 398 or rearranges to intermediate 399 prior to a second hydrogen atom transfer to yield 400. Finally, the nascent metallo-radicals are known to rapidly homodimerize (k6 = (2 – 4)·109 M−1 s−1 at 22 ºC). Kinetic studies revealed a second order rate law for the consumption of 396 with all the metals except HCrCp(CO)3. In the case of •CrCp(CO)3, a higher concentration of this 17-electron metallo-radical species coexists at equilibrium with the Cr-dimer [Keq (k6/k−6)= 2.5·10−4 M].301 Thus, the ratio between rearranged and unrearranged products for a specific concentration of metal hydride is constant because the formation of 397 is the rate-determining step in this process. If this mechanism was correct, the ratio between rearranged and unrearranged products for a specific concentration of metal hydride should be constant. Indeed, 398/400 did not change with time and 398/400 vs. [MH] was linear, as there was more reduction of 397 before it could rearrange to 399. The equilibrium constant for the ring-opening rearrangement of cyclopropylmethyl radical is ≈ 104 (k4 = 1·108 s−1 at 25 ºC); however, the substituents on intermediate 397 decreased the equilibrium constant (Keq) because the rearrangement results in the formation of a primary radical from a highly stabilized tertiary benzylic radical. Finally, the authors determined that the second hydrogen transfer was exothermic and showed a normal KIE (k3H/k3D = 2.2 and k5H/k5D = 1.8 for DWCp(CO)3), which they attributed to a relatively early transition state.
Most of these early studies were focused on the metal hydride and explored the effects of the bond strengths and the ligand steric factors in the hydrogen atom transfer. However, reactivity is also controlled by the relative stability of the newly formed carbon-centered radical, which explains the Markonikov selectivity observed for these hydrofunctionalizations.427 In 2003, Norton undertook a kinetic study to disclose the influence of the olefin structure in the hydrogen atom transfer from HCrCp(CO)3 (Figure 173).295,298,291 Comparison between the relative rates of H/D-exchange and hydrogenation of different substituted alkenes revealed a gradually changing behavior; the rate of hydrogen atom transfer increased with increasing stabilization of the resulting carbon-centered radical and with reduced steric congestion around the double bond.
Figure 173.

Reactivity of alkenes depending on their steric and electronic properties.
Reaction with substrate 405 showed only traces of hydrogenation along with slow isotope exchange (k1 = 9.8·10−4 M−1 s−1 at 50 ºC). Otherwise 385 and 407 did not produce reduced product but deuteration proved too rapid to follow by 1H NMR at this temperature. In the case of styrene (379), the initial hydrogen atom transfer was slower because a secondary radical was formed but as this was less sterically hindered, hydrogenation of the substrate occurred exclusively. The first hydrogen atom transfer to methyl acrylate (406) occurred as rapidly as to styrene (379), but the second hydrogen atom transfer was significantly more sensitive to steric factors. The presence of a β-methyl in 404 led to a mixture 1:1 of deuterium incorporation and hydrogenation whereas in the absence of the α-methyl in 402 showed almost no exchange along with extremely slow hydrogenation. Finally, hydrogen atom transfer to unactivated alkenes was less favoured; 401 showed almost no reactivity (k1 = 2.4·10−7 M−1 s−1 at 50 ºC) and 357 consumed only 23% of metal hydride leading to a mixture 7:3 of deuterium incorporation and hydrogenation (no internal alkene was observed). The same trend was observed in the peroxidation of alkenes using CoII/Et3SiH/O2 when Nojima performed competing experiments with different alkenes: the reactivity of the substrate increased with the stability of the radical formed with more electron-rich olefins.120 Less steric congestion also accelerated this reaction.
Shenvi also performed competitive hydrogenation experiments with different alkenes using catalytic Mn(dpm)3 and PhSiH3/TBHP, which presumably occurs via a manganese hydride (Figure 173).24 In this case, various substitution patterns were readily reduced with little influence of the direct attached functionality; increased substitution slightly decreased the rate of consumption and electron-withdrawing groups had a minor accelerating effect on the reaction rate. Otherwise, Herzon found that 2,2-disubstituted alkenes reacted faster than monosubstituted alkenes when using stoichiometric cobalt hydride complexes, as did substrates containing heteroatoms. Alkynes and 1,2-disubstituted olefins reacted more slowly whereas trisubstituted alkenes reacted similarly.354
Based on such rate differences between differently substituted olefins, 1,5-dienes can undergo site-selective radical generation and partitioning between reduction, isomerization and radical cyclization can be studied (Figure 174).296,299 Since the metal hydride could be regenerated under elevated H2 pressure, the reaction could be performed with catalytic amounts of chromium (7 mol%).397 In the case of substrate 327, double bond A undergoes hydrogen atom transfer more rapidly than B, leading to the formation of intermediate 408. Subsequent radical cyclization occurred via a chair-equatorial transition state followed by reduction of the new radical cis-409 (trans-409 was minor).292 Therefore, it was determined that acceptance of a H• from the metal hydride to cis-409 was more favored than donating it to the metal radical via a reverse hydrogen atom transfer to form a new double bond. Simultaneously, direct reduction of 408 to 329 and isomerization to 330 were observed as secondary pathways.
Figure 174.

Competition between direct reduction, isomerization and radical cyclization.
In summary, the reaction between a metal hydride and a carbon-centred radical has been studied by means of trityl radicals as well as radical clock experiments. The results revealed an exothermic process highly dependent on the steric congestion of the ligand. The reactivity was also controlled by the olefin structure, which has been extensively illustrated by different groups, and allowed the development of cascade cyclizations of polyalkenes.
7.5 Chasing the in situ Formation of Metal Hydrides
Pursuit of improved alkene hydrofunctionalization methods was accompanied by the development of new metal complexes with lower air-sensitivity and higher thermally stability. Some complexes could catalyze the generation of radicals from H2: for example, (H2O)2Co(dmgBF2)2 (410) (Figure 175).397 Such “cobaloximes” were accepted early as models for vitamin B12 (containing a crucial Co-alkyl bond sensitive to homolysis)288,428 and were active towards the polymerization of acrylates or the photolytic cyclization of unsaturated alkyl iodides.429,430 The formation of a cobalt (III) hydride as intermediate was suggested in these reports and in many subsequent transformations. However, identification and characterization of specific cobalt species had proven challenging. Norton’s group focused on the study of complex 410, which reacted with H2 and then reduced trityl radical 394 to 395. This behavior suggested the formation of a metal hydride followed by a hydrogen atom transfer, analogous to Figure 171. However, in this case the rate of reduction did not change with time, thus the reaction was zero order in 394. Moreover, the reaction was first order in H2 and second order in cobalt; therefore, the activation of H2 by 410 to form the metal hydride appeared to be the rate-determining step under these conditions. In this scenario, the rate constant would be independent of the concentration of the trapping radical, which was confirmed by reaction with TEMPO.
Figure 175.

Hydrogen atom transfer towards a trityl radical under catalytic conditions.
These results indicated that the mechanisms of catalyzed alkene hydrofunctionalization methods could have completely different kinetics than the reduction of double bonds with stoichiometric amounts of metal hydrides, in spite of the chemical similarities. Moreover, different rates in the hydrogen atom transfer would also imply a different [MH]/[M•] ratio and therefore a different distribution of products, for example, in a radical cyclization like in Figure 174. These studies generated indirect evidence for the formation of a metal hydride but no spectroscopic observation was reported.397 These cobaloxime hydrides were often thermodynamically unstable and the isolation and characterization of analogous structures have created significant controversy.431,432,433 During the photochemical protonation of [Co(dmgBF2)2(CH3CN)]−, Dempsey and Gray observed an absorbance at 405 nm that was attributed to hydride 412 (Figure 175).434 Norton has observed that complex 411 under high pressure of H2 (70 atm) changed slowly but smoothly towards a new intermediate at 556 nm (pKa 13.4; ΔGH = 50.5 kcal/mol) in a reversible process.302 1H NMR studies showed no signal that could be attributed to Co-H but residual coupling to 59Co could broaden the hydride signal and make it difficult to observe. Nevertheless, formation of a new species containing an exchangeable proton (reaction with CD3OD or D2) was observed and assigned to the new O-H bond in complex 413. The fact that no desymmetrization of the ligand occurred was explained with a proton-coupled electron transfer from 411 to 413 on the NMR time scale. Interestingly, when another axial ligand was used (414), the UV-vis spectrum showed a new intermediate absorption band at 354 nm. This was assigned to the metal hydride 415, although the data was inconclusive. Swapping CH3CN for THF could modify the pKa of the hydride and slow its tautomerization. Therefore, this report proved the importance of the ligand in determining the structure of the metal hydride and questioned its involvement in the hydrogen atom transfer towards an unsaturated substrate; thus, cobaloximes were defined as cobalt complexes with non-innocent ligands.
As mentioned, radical hydrofunctionalizations have been expanded to cobalt, iron and manganese catalysis using a wide variety of reductants to form the corresponding metal hydrides. Likely the mechanism of putative metal hydride generation differs depending on whether a reaction uses a silane, a borohydride or an alcohol. In the early protocols for the hydration of alkenes reported by Mukaiyama, a secondary alcoholic solvent was used as the reductant in the presence of a β-diketonate cobalt complex under oxygen atmosphere at 75 ºC (Figure 177; see also Section 2.1, Figure 15).98,435 At this stage, only peroxy-metal radical species like 416 were suggested as the key intermediates by reaction with molecular oxygen.38 Interestingly, no reaction proceeded with primary and tertiary alcohols but isopropanol or cyclopentanol led to olefin hydration along with competing over-oxidation and reduction. The reaction was competent only when using complexes ranging from 0 V to +0.5 V in their redox potentials depending on the ligand, for example, acetylacetonate. Yamada later performed a careful study of the transformation and determined that 3 equiv. of acetone and 2 equiv. of water were produced for every equivalent of alcohol produced when the reaction was performed in isopropanol.99 Reaction in d8-i-PrOD led to partial deuterium incorporation in the final product, confirming that the secondary alcohol acts as a reductant and explaining the formation of acetone. More significantly, the relative distribution of products could be modified by the addition of an external ligand; for example, performing the reaction in the presence of pyridine could modestly affect the product distribution and decrease the amount of alkene reduction.97 Later, silanes replaced alcohols as reducing agents, which allowed the transformations to occur much more efficiently at room temperature.100
Figure 177.

Use of an alcohol as reductant in the hydration of alkenes.
Salen-type ligands also provided cobalt complexes active towards the hydration of styrenes (Figure 178).92 In this case, a chiral non-racemic catalyst (417) could catalyze the formation of enantioenriched products such as (R)-418 (82% ee), a result that was used to question the involvement of a free radical when the C-O bond was formed.436 However, this result was not reproducible in 250 batches. More disturbingly, when the reductant was changed to sodium borohydride, the opposite sense in enantioselection was obtained leading to (S)-418. This result suggested that the mechanism under those conditions and the original ones were different, but neither was further elucidated. Nevertheless, FAB-MS analysis of cobalt complex 417 treated with a modified borohydride revealed the formation of the metal hydride [M+1]; treatment with deuterated borohydride led to [M+2] detection. The formation of a cobalt hydride intermediate was also confirmed with time-resolved FT-IR method (new peak at 2250 cm−1). However, this same system proved to be efficient for the enantioselective reduction of aryl ketones, which does not typically occur by a stepwise radical process.437 This methodology was restricted to activated substrates and showed high dependence on solvent and on the borohydride’s counter-cation.
Figure 178.

Detection of a metal hydride during the hydration of styrenes.
Finally, the generation of metal hydrides has also been proposed in many reactions which employ silanes as the reducing agent. Although cobalt catalysts performed efficiently in THF, iron- and manganese- complexes required an alcoholic solvent in order to obtain competent reactivity.116 Reversible reaction between silanes and alcohols has been studied and shown to increase the hydritic character of the silane through the formation of pentavalent species (Figure 179).438 Very recently, this intermediate was observed by NMR at − 70 °C, revealing an associative exchange mechanism.439
Figure 179.

Detection of pentavalent silane/alcohol species.
In summary, obtaining direct experimental evidence for the formation of metal hydrides in the catalyzed alkene hydrofunctionalization methods has been a long sought challenge. The generation has proven dependent on the reductant used and could affect the kinetics of the whole transformation. Norton showed the complexity of those species by studying the evolution of cobaloximes in different solvents, which are complexes bearing non-innocent redox ligands.
7.6 Studies on Alkene Hydration
Mukaiyama’s discovery that silanes are mild, competent reductants in these radical hydrofunctionalizations lay the grounds for further development by many groups. Cobalt complexes with β-diketonates or salen-type ligands and with silanes were widely applied to the development of a variety of useful transformations. In 1995, Mukaiyama proposed that metal hydride (419) formation occurs by reduction of a cobalt(III) complex with the silane (or the alcohol in a few cases) (Figure 180).38 A cobalt(III) complex results from initial oxidation of the cobalt(II) complex with O2. Subsequent formation of Co-alkyl complex 420 was proposed, but the elementary steps involved in this transformation were not discussed and hydrogen atom transfer was not mentioned by name or in concept. To date, the involvement and abundance of Co-alkyl intermediates like 420 is still unclear in these reactions. Intermediate 420 could then further react with oxygen leading to a peroxy-metal radical 416 and the final product after homolysis. This proposal was supported by the generation of silyl peroxides depending on the reaction conditions.105 However, regeneration of the metal species was not discussed in detail.
Figure 180.

Early proposal for the hydration of alkenes in which H and C are counted as cations and electrons are assigned to Co as CoI.
Prior to Mukaiyama, Okamoto and Oka had effected the hydration of styrenes using (dmg)2Co(Py)Cl and sodium borohyride, which revealed complete deuterium incorporation at the terminal position when NaBD4 was used.75 Moreover, isolation of the corresponding cobalt-alkyl (420) and cobalt-peroxy (416) complexes was possible and they were tested towards the formation of the hydration product; both of them afforded 1-phenylethanol in excellent yield, which suggested that these are viable reaction intermediates. Other early examples of metal-alkyl complexes were reported in the literature in which cobalt, manganese or iron catalysts bearing electron-rich ligands were used. 61,67,74,244,392,28,411,417,426 However, no evidence has been found for most of the upcoming mechanistic studies of alkene hydrofunctionalizations.
Similarly, Kasuga conducted mechanistic studies of their styrene hydration, which uses NaBH4 as a reductant under an oxygen atmosphere, by comparing cobalt (II), iron (III) and manganese (III) complexes bearing phthalocyanine, a porphyrin-like ligand with an extended π-system (Figure 181).78 While all of these catalysts were effective for the formation of the alcohol, only the cobalt was active under anaerobic conditions, yielding ethylbenzene in the absence of oxygen. Moreover, addition of TEMPO to the reaction inhibited completely the reactivity of the iron and manganese complexes but had only minor effects on the cobalt (II) complex. The authors suggested the formation of the metal-alkyl complex in the case of cobalt and the generation of free radicals with manganese and iron in order to explain the differences in reactivity. More interestingly, the kinetic profiles were very similar for Mn and Fe but an induction period of 30 minutes was observed for cobalt.
Figure 181.

Peculiarities of cobalt complexes.
In 2009, Boger and coworkers reported a detailed study on the total synthesis of vinblastine (2) and related natural products, which involves an iron-catalyzed hydration of a double bond (Figure 182).26 The authors provide full details of the development of the direct coupling of catharanthine with vindoline, presumably initiated by generation of a radical cation with FeCl3 that undergoes oxidative fragmentation and diastereoselective coupling.26,440 Reduction of the resulting iminium ion 421 with NaBH4 leads to formation of anhydrovinblastine (1). Treatment with Fe2(ox)3/NaBH4/air effects oxidation of the desired alkene to the hydration product with a 2:1 diastereoselectivity [vinblastine (2):leurosidine (3)]. Although oxidation of anhydrovinblastine (1) had been previously observed and explored with a full range of oxidants, including O2, none had done so preferentially from the β-face. Moreover, both steps could be performed sequentially in one pot and the hydration reaction proceeded without the formation of an isomerized enamine intermediate or an oxidized iminium ion (421) according to the labeling experiments. Boger characterized the reaction as an “Fe-mediated hydrogen atom radical addition” to the trisubstituted olefin of anhydrovinblastine initiated by treatment with Fe2ox3 and NaBH4. The resulting carbon centered radical then reacts with O2 to form a hydroperoxide, which is reduced to give the final alcohol. Interestingly, Mukaiyama’s conditions for the hydration of alkenes failed to provide the desired product (Co(acac)2, O2, PhSiH3).
Figure 182.

Mechanistic tests for the hydration of anhydrovinblastine.
Whereas reactions performed in D2O led to no deuterium incorporation, 18O2 labeling studies confirmed that the alcohol oxygen originates from O2. In agreement with the proposal, complete incorporation of one deuterium was observed when the hydration was performed with NaBD4 and incorporation of two deuteriums occurred stereoselectively from the one pot coupling/oxidation. Just as interestingly, reaction of anhydrovinblastine with NaBD4 in the absence of oxygen led to the reduction of the alkene with surprising 1:2 diastereoselectivity (deoxyvinblastine:deoxyleurosidine). Finally, the involvement of radical intermediates in the mechanism was confirmed by trapping the carbon-centered radical with TEMPO, among other radical traps.
In summary, the early proposals towards the hydration of alkenes initiated by Mukaiyama consisted on the generation of metal-alkyl complexes, which evolved to metal-peroxy species that underwent homolytic cleavage. The studies were based on deuterium labelling experiments, kinetic data and use of radical traps. Boger later reported a complete work on the coupling of catharanthine with vindoline followed by oxidation towards vinblastine.
7.7 Studies on Alkene Hydroperoxidation
As shown in Figure 25, Mukaiyama also reported the peroxidation of alkenes with Et3SiH/O2 catalyzed by Co(acac)2 when using DCE or benzene as solvent (Section 2.2).105 Considering the lack of mechanistic insights accompanying the growth of alkene hydrofunctionalizations, Nojima was a pioneer in studying the intermediates involved in the cobalt-catalyzed peroxidation (Figure 183).118 The reaction was very efficient with unactivated alkenes such as 422 to form 423 by following the original Mukaiyama’s conditions; however, Isayama showed that slower substrates such as styrenes required the use of TBHP to decrease the induction period and obtain synthetically useful yields.104
Figure 183.

Proposed catalytic cycle for the peroxidation of alkenes.
Based on the previous literature, Nojima suggested the generation of Co(III)-alkylperoxo complexes to promote the peroxidation transformation. The authors decided to prepare cumylperoxo-Co(acac)2Py complex 424, which was also shown to catalyze the reaction between substrate 422 and Et3SiH/O2 affording product 423 along with cumylsilyl peroxide (Figure 183). This result implied that a metal exchange between cumylperoxide and Et3SiH occurs very rapidly and provides a metal hydride complex (new signal observed by 1H NMR at −2.8 ppm). Moreover, Co(III)-alkyl complex 425 was also catalytically active and simultaneously formed PhCH2CH2CH2OOSiEt3. Analogous to Mukaiyama’s hydration proposal, insertion of the double bond into the metal hydride was suggested to form complex 426, which would undergo homolytic cleavage of the Co-C bond.38,441 This nascent radical would react with molecular oxygen leading to complex 416,442 which would undergo transmetallation with Et3SiH to give product 423 and regenerate the metal hydride. The presence of radical species was confirmed by means of radical clock experiments: thus, vinylcyclopropane 427 led to the major formation of peroxide 428 under the reaction conditions, by ring opening. Similar behavior was observed in the peroxidation of 1,5-dienes leading to mixtures of linear peroxides and 5- or 6-membered cyclic peroxides.120 Moreover, the peroxidation performed with Et3SiD led irreversibly to non-stereoselective deuterium incorporation (d-429). However, the initiation step towards the formation of the metal hydride was not completely clear; thus, reaction of Co(modp)2 with no substrate led to the formation of Et3SiOOSiEt3 and Et3SiOH suggesting oxidation of the cobalt complex with oxygen followed by transmetallation with Et3SiH. Importantly and despite citation of Halpern’s work, Nojima still characterizes the hydrofunctionalization as a hydrometallation, not HAT, i.e. the transition state is depicted to possess radical character instead of a discrete radical intermediate being formed.
Sugamoto reported that cobalt-porphyrins also catalyze the peroxidation of substrates as α,β,γ,δ-unsaturated alkenes or styrenes (Figure 184).151 However, this system was not efficient towards unactivated alkenes, highlighting again the importance of the redox potential of the metal complex. These examples are interesting because of their regioselectivity: the more electron-rich alkene reacted first as confirmed by deuterium incorporation. EPR spectroscopy showed the formation of a five-coordinated metal species, which the authors took to suggest coordination of the alcohol or the alkene.372,411 Thus, the proposed mechanism involves coordination of the alkene with the catalyst prior to the attack of the silane to form a Co-alkyl intermediate like in Figure 183. This organometallic would react with oxygen and finally with isopropanol in order to release the product and LCo(i-PrO) to regenerate the catalyst with another equivalent of silane.
Figure 184.

Regioselective peroxidation of α,β,γ,δ-unsaturated alkenes.
Concurrently, Magnus studied the oxidation of α,β-unsaturated substrates with catalytic Mn(dpm)3 and PhSiH3/O2 in DCM/isopropanol (Figure 185).107 The reaction led to the α-peroxide (431) and then the α-hydroxy product after reductive work-up with P(OEt)3. Mukaiyama had shown that this transformation was possible with Mn(dpm)2 but Magnus suggested the in situ oxidation of MnII to a MnIII adduct as the active species (dark green-brown), which required alcohol to function.106 Thus, a solution of Mn(dpm)3 with stoichiometric amounts of silane showed no change by IR until the addition of isopropanol led to disappearance of the phenylsilane IR absorption (2152 cm−1), production of a putative metal hydride absorption (2168 cm−1) and decolorization of the dark-green solution to pale yellow. Diphenyldisiloxane was detected as a by-product in small amounts, possibly through formation of Ph(i-PrO)SiH2. Similar MnII alkoxide adducts, Mn4(OEt)4(EtOH)2(dpm)4,443 also catalyzed enone hydroxylations under identical conditions.
Figure 185.

Mechanistic tests using a manganese catalyst.
The authors also propose that the hydridic character of the putative metal hydride HMn(dpm)2 was substantially increased in the presence of oxygen since the reactivity was greatly accelerated. A significant example was the reaction of β-ionone with and without oxygen atmosphere: whereas oxidation of the conjugated alkene occurred under O2, reduction of the α,β-unsaturated olefin was observed under argon. These results suggested the formation of two distinct reducing agents with different regioselectivities: HMn(dpm)2 versus HMnO2(dpm)2.444 Interestingly, some examples showed significant reduction of the alkene depending on the way in which the reaction flask was washed since the protic surface of the glass could interfere in the reaction. More significantly, reaction with a β,β′-disubstituted enone 434 under oxygen showed the formation of product 435 and no reactivity was observed under argon, suggesting a different hydritic character between the different complexes. In this proposal, HMn(dpm)2 would react with the substrate forming intermediate 432, which would be protonated with isopropanol to 433, and HMnO2(dpm)2 would form intermediate 430, then the peroxide 431 and finally the α-hydroxy product. Similarly, this system was competent in the reduction of a carbonyl group towards the alcohol product; thus, reaction in the presence of oxygen gave 98% yield whereas only 57% was obtained under argon.445
In summary, detailed studies on the peroxidation of alkenes suggested the involvement of free radical intermediates subsequent to hydrometallation. Nojima also supported the formation of metal-alkyl and metal-peroxy complexes by independent preparation of these precursors. In contrast, Magnus observed the generation of a metal enolate when using α,β-unsaturated substrates, which could be protonated with i-PrOH. These studies also suggested the formation of distinct reducing agents in the presence or the absence of oxygen.
7.8 Studies on Catalytic Alkene Hydrogenation
During the studies on the peroxidation/hydration of α,β-unsaturated substrates shown in Figure 185, Magnus noticed the reduction of some of the alkenes when the reaction was performed in the absence of oxygen.107,221 As mentioned, the authors suggested the formation of HMn(dpm)2 as the active species of this transformation, which would attack the double bond and lead to the final product 433 after protonation. The use of isopropanol was crucial during this transformation: methanol led to rapid gas evolution (presumably H2) forming PhSi(OMe)3 along with no reduction of the substrate and t-BuOH was really slow. Interestingly, deuterium-labeling experiments using PhSiD3 revealed incorporation of one deuterium in the β-position (d-436), which demonstrated that the hydridic addition was irreversible according to the authors, and none in the α-position suggesting that the origin was i-PrOH (Figure 186). This process was non-stereospecific (d-437).
Figure 186.

Deuterium incorporation in the reduction of α,β-unsaturated ketones.
As mentioned throughout this manuscript, hydrogenation of alkenes has been observed as a byproduct in many of the aforementioned hydrofunctionalization protocols. Iwasaki et al. envisioned that stepwise hydrogen atom addition361 might serve as an alternative to dissolving metal reactions, which allow obtaining the thermodynamically favored configuration446 from an alkene when steric constraints favored the kinetic alkane product (Figure 141, see Section 6.1).24 The original hydration conditions of Mukaiyama under argon were not competent to reduce unactivated alkenes but excellent yields were obtained when TBHP was used as activator.38,99 Moreover, treatment of 1,6-dienes like 438 led to the reductive cyclization products (439 and 440), confirming the involvement of radical intermediates.
Reaction with radicals had been described as a diastereoselective process; a non-planar ground state was proposed, which evolved to pyramidalization in the transition state.447 Thus, Giese and Zipse had investigated the reactivity of substituted cyclohexyl radicals and the effect of the methyl-groups in the ratio of the axial/equatorial attack (Figure 188).379 The results suggested that the diastereoselectivity was highly controlled by steric effects, generally leading to equatorial attack with bulkier groups. However, substituents on the ring could tune this reactivity: whereas α-substitution increased the amount of axial isomer because of 1,3-interactions, α-substitution led to a pronounced reduction and almost no effects were observed with γ-substituents.
Figure 188.

Diastereoselective radical addition to α,β-unsaturated substrates.
In 2016, Obradors et al. observed that PhSiH3 was not the kinetically preferred reductant in the presence of a metal-complex and an alcohol as Ph(i-PrO)SiH2 reacted much faster than any of the silyl derivatives observed by GC (Figure 189): Ph(i-PrO)2SiH, PhSi(i-PrO)3, (Ph(i-PrO)2Si)2O, Ph(i-PrO)(t-BuO)SiH and Ph(i-PrO)(dpm)SiH, among others. The team had speculated that the alcohol was necessary to increase the hydritic character of the silane via a pentavalent intermediate but these results suggested that they functioned as important silane ligands,438,439 presumably as a result of increased Si elecrophilicity and more rapid ligand exchange with the catalyst. This novel reductant (Ph(i-PrO)SiH2) improved the yields and rates of different metal-catalyzed radical hydrofunctionalizations38,116,264,20 and allowed catalyst loadings and temperatures to be lowered. Functional group tolerance increased and diverse aprotic solvents could be used with manganese and iron catalysts. In the absence of TBHP, some substrates underwent a competitive hydrosilylation reaction, presumably via radical chain pathway.448 Thus, the carbon-centered radical from 292 could react with PhSiH3 to generate a silyl radical capable to further adding into a double bond, to give 441. This new reactivity showed multiple roles for TBHP including reoxidation of the catalyst, suppression of side-reactions and probably acceleration of ligand exchange on the catalyst.449 However, a full understanding of the complete mechanism under these conditions has not become available.
Figure 189.

Mechanistic tests for the catalytic hydrogenation of alkenes.
Since Norton had demonstrated that some cobalt hydrides existed as equilibrium of Co-H and O-H tautomers when bearing non-innocent dimethylglyoxime (dmg) ligands shown in Figure 176,302 the potential non-innocence of β-diketonate ligands was questioned. The Ph(i-PrO)SiH2 reductant allowed reactions to be carried out in hexanes, which prevented H-exchange between ligand and protic solvent. Using Mn(d-dpm)3 no deuterium incorporation was observed in 442 (Figure 190). This result suggested that β-diketonate was not a redox-active ligand. As expected, hydrogenation of 442 with PhSiD3 led to complete incorporation in the terminal position of the alkene but only 63% of deuterium was observed in the internal one (i.e. d-443). Exhaustive studies excluded i-PrOH as well as TBHP as competing hydrogen sources and showed that the remaining hydrogen derived from scrambling of the substrate. This process was highly dependent on the structure of the alkene, specifically the steric congestion and the BDE of the C-H bonds adjacent to the newly formed radical carbon.450 Thus, substrate 444 revealed 100% of incorporation in the terminal position (d-445) but 83% in the internal one along with 11% of scrambling.
Figure 176.

Tautomerization of metal complexes with redox active ligands.
Figure 190.

Deuterium incorporation in the reduction of unactivated alkenes.
Few reports had explored the origin of the second hydrogen atom transfer under Mukaiyama’s conditions. Besides Boger’s studies (Figure 182),26 Herzon performed deuterium-labeling experiments in the hydrogenation of alkenyl halides with a related protocol using Co(acac)2/Et3SiH/TBHP/1,4-CHD/n-PrOH (Figure 191).27 In this case, use of Et3SiD in the reduction of 446 led to partial incorporation in the terminal position (d-447), but no deuterium was observed at the internal position, probably due to the multiple hydrogen sources present in the reaction conditions. Use of deuterated solvent led to the same result.
Figure 191.

Deuterium incorporation in the reduction of haloalkenes.
Finally, a competition experiment between PhSiH3 and PhSiD3 (1:1) with 442 revealed major deuterium incorporation in the terminal position of d-443 whereas no preference was observed in the internal one (Figure 190).116,451 Similarly, the kinetic profiles showed an overall rate slightly faster for PhSiD3. These results suggested an inverse KIE present in the catalytic cycle, probably during the hydrogen atom transfer or the formation of the metal hydride. However, the relatively small size of the observed KIE might indicate a tempering effect of normal KIE at other points in the catalytic cycle.
In summary, metal-catalyzed hydrogenation of alkenes via radical intermediates allowed thermodynamic diastereoselectivity under very mild reaction conditions. Shenvi suggested a hydrogen atom transfer to generate this species and later reported a full study on the transformation along with the development of a new exceptional reductant, Ph(i-PrO)SiH2. Deuterium labelling experiments revealed scrambling of the substrate, which was highly dependent on the structure of the alkene. Competition experiments suggested an inverse KIE present in the catalytic cycle.
7.9 Studies of the Reverse Hydrogen Atom Transfer
Isotope scrambling during the hydrogenation of alkenes and anthracene suggested that the hydrogen atom transfer could be a reversible process (Figures 168, 169 and 172).360,361,426,298 Thus, abstraction of the hydrogen adjacent to organic radical could be effected by some metal complexes, which would result in reformation of a metal hydride and olefin. This concept was extensively exploited in the cobalt-catalyzed radical polymerization of activated alkenes.297,452 As an example, radical polymerization of vinyl acetate (452) was described using a porphyrinate-cobalt(II) complex (LCoII) in the presence of AIBN as initiator (Figure 193).453 These processes showed an induction period with paramagnetic species (CoII) prior to the polymerization step, when the solution turned diamagnetic (CoIII).454 Computational studies on the initiation step performed by Bruin and Wayland suggested the thermal generation of radical 448 from AIBN, which would react with LCoII to form cobalt hydride 450 (BDE = 50.6 kcal/mol) or complex 449 by radical collapse (ΔG° = −4.5 kcal/mol).455 The reverse hydrogen atom transfer to form 450 and 451 proceeded via a single transition state with structural features resembling the products and a very low overall barrier (ΔG≠ = 3.8 kcal/mol). The insertion to methacrylonitrile (451) or vinyl acetate (452) also showed low activation barriers (ΔG≠ = 8.9 kcal/mol and ΔG≠ = 11.4 kcal/mol, respectively). However, radical 454 was ca. 11 kcal/mol less stabilized than 448, resulting in preferential formation of complex 453: dissociation of 449 occurred at 2·102 s−1 (Co–C = 2.020 Å) whereas 453 dissociated at 1·10−5 s−1 (Co–C = 1.982 Å). Complex 453 would then initiate the polymerization step by reacting with another equivalent of vinyl acetate (452), presumably through an associative radical exchange mechanism. Importantly, the study indicated a non-concerted multistep and very efficient hydrogen atom transfer from the metal hydride to the unsaturated substrate and the authors suggested that this pathway should be always considered even with complexes with a vacant cis-coordination site because of its low activation energy.
Figure 193.

Initiation steps for the radical polymerization of alkenes.
Crossley et al. reasoned that this principle could be used in the isomerization of alkenes (Figure 194).28,397,416 The reaction could be initiated both with catalytic amounts of silane or AIBN to generate the metal hydride 450, which would then undergo hydrogen atom transfer to alkene 458 to form a radical pair (456). Careful selection of the ligand and the solvent allowed the isomerization to occur via a reverse hydrogen atom transfer to generate 455. The persistence of the carbon-centered radical depended on the stability of the metal radical counterpart (LCoII in 456) and decreased the competing hydrogenation pathway. Interestingly, mono-substituted alkenes required elevated temperatures to obtain synthetically useful yields, presumably in order to provide the activation energy to promote Co-C bond homolysis of parasitic organometallics species like 457. This complex would be formed via radical collapse with less sterically hindered substrates. However, cycloisomerization of dienes (460 to 461)456 and retrocycloisomerization of strained rings (362 to 363) was also efficient under those conditions, indicating the presence of the radical intermediate. Moreover, electron-rich ligands increased the cyclization product according to the expected persistence of 456. These results suggested a kinetically-controlled irreversible process, which discriminated against steric bulk and appeared to be limited to terminal and 2,2-disubstituted alkenes.
Figure 194.

Isomerization of alkenes via reverse HAT.
In possibly related work, cobalt complex 459 reacted with PhSiH3 to isomerize internal alkenes to the terminal position instead.457 The resulting alkene then underwent an anti-Markovnikov hydrosilylation (Figure 189). The multistep pathway for this transformation was not clarified, although similar olefin isomerization to linear products was precedented with cobalt, iron and other metals via insertion/β-hydride elimination.400,458 In the former case, the process could not be distinguished from a radical pathway.
In summary, based on the reverse hydrogen atom transfer used in the initiation of free radical polymerization of alkenes, Shenvi developed an isomerization protocol along with the cycloisomerization of dienes and retrocycloisomerization of strained rings. The study showed the generation of organometallic species with monosubstituted alkenes, which underwent homolytic cleavage at 60 °C.
7.10 Studies of the Radical Attack to a Carbon Atom
Addition of carbon-centered radicals to electron-deficient alkenes was described decades ago and very attractive methodologies arose for the formation of the sterically demanding quaternary C-C bonds.459,460 Detailed studies, reviewed by Giese in 1983, showed that the radical attack was an exothermic process with a very early transition state and the rates were controlled by both steric and polar effects.461 However, α- and β-substitution caused different behavior, which suggested an asymmetric approach of the radical towards the double bond. Moreover, addition of tertiary radicals was much faster that primary and secondary indicating the importance of electron density and SOMO-LUMO interactions.462,463 In the case of prochiral substrates, the reaction could also be highly diastereoselective.
The Baran research group developed a new method for reductive olefin cross coupling by presumably generating a carbon-centered radical from a metal hydride and an electron-rich alkene, which could later attack an electron-deficient alkene and be reduced (Figure 195).20,21 The difference in electron densities between the two alkenes involved was crucial in the success of the reaction; thus, the suggested iron hydride 463 reacted preferentially with alkene 385, which would preferentially react with alkene 385. Interestingly, the authors suggested the reduction of radical 464 with FeII and therefore no oxidant was required in this transformation.
Figure 195.

Proposed catalytic cycle for the reductive olefin coupling.
Reaction between substrate 465 and methyl acrylate (466) confirmed the presence of radical intermediates by the formation of 467, albeit in 6% yield (Figure 196). More efficiently, β-ionone (468) formed the cyclopropane-fused bicycle 207. Finally, analogous to Magnus experiments,221 deuterium labeling identified the source of the first hydrogen as PhSiH3 whereas the second one came from the -OH of ethanol, which indicated the formation of a carbanion at some point of the catalytic cycle. Therefore, this transformation could be mechanistically considered as a radical-polar crossover reaction.
Figure 196.

Radical trap experiments in the conjugate addition.
In contrast, nitroolefins can be attacked with opposite regioselectivity: radical addition to substrate 470 could lead to intermediate 471 and the product 472 with a sequential elimination of a nitro radical (Figure 197). 266 In this case, the authors suggest the formation of complex 469 prior to the homolytic cleavage, in analogy to Mukaiyama’s suggestion and without invocation of HAT. They also proposed that FeII could be reoxidized to FeIII species along with the formation of the nitrite anion. The presence of radical intermediates was confirmed by addition of TEMPO, which completely inhibited the transformation, but further evidence for this proposal was not provided.
Figure 197.

Proposed catalytic cycle for the radical addition to β-nitroalkenes.
Similarly, the carbon-centered radical could attack hydrazone 473, generated from formaldehyde and a hydrazine, in order to form the C-C bond in intermediate 474 (Figure 198).265 This hydrazide was not isolated but could be detected by LC-MS and underwent elimination of sulfinic acid and nitrogen after addition of methanol at 60 °C.317 Overall, this transformation allows hydromethylation of alkenes. Isotope-labeling experiments confirmed the origin of the new carbon as deriving from formaldehyde (CD2O gave 100% D incorporation). However, only partial incorporation was observed when using CD3OD, presumably due to incomplete deuteration of intermediate 474.
Figure 198.

Proposed catalytic cycle for the hydromethylation of alkenes.
Prior to this work, Carreira had reported the reaction between an alkene and tosyl cyanide in the presence of catalytic amounts of a cobalt complex and a silane in ethanol (Figure 102, Section 4.3).22 Although the mechanism was not discussed in detail, the authors related this protocol to previous methods for radical addition to electrophiles. It had been reported by Barton and Theodorakis that formation of C-C bonds could occur by means of radical addition to tosyl cyanide, for example, from light-induced photolysis of Barton esters.464 In Carreira’s hydrocyanation, a metal hydride would be presumably generated and react with the alkene to form a radical intermediate capable of attacking the cyanide, although HAT was not invoked. However, the direct analogy between these alkene hydrofunctionalizations and decades of literature on radical C-C bond formation was clear.
In summary, radical addition to sp2 and sp carbon atoms led to useful methodologies that allowed the conjugate addition, methylation, styrenylation and hydrocyanation of alkenes, among others. The involvement of radical intermediates was supported by radical clock experiments, use of radical traps and deuterium labelling experiments.
7.11 Studies of the Radical Attack to a Nitrogen Atom
Following Mukaiyama’s precedent in the hydration of alkenes via reaction of a radical intermediate with molecular oxygen, Carreira’s research group reasoned that the same intermediates could be trapped with a N=N bond to generate a new set of synthetically useful transformations. The first example involved hydrohydrazination of a wide variety of alkenes with PhSiH3 and an azocarboxylate in ethanol using cobalt catalysis as shown in Sections 3.2 and 3.3.252 The authors initially suggested the formation of a metal hydride 450 followed by hydrometallation of an alkene to afford organocobalt species 426 (Figure 199). Nucleophilic addition to an azocarboxylate (479) would form intermediate 477 leading to 475 after σ-bond metathesis with the silane. This last step would also regenerate the active species 450 to turn over the catalytic cycle.
Figure 199.

Proposed catalytic cycle for the hydrohydrazination and hydroazidation of alkenes.
Ligand design allowed the selective construction of the C-N bond in 476 when di-tert-butyl azodicarboxylate was used as the trapping agent: the steric congestion was proposed to avoid the premature reduction of 479 to 480. Manganese was also competent for this transformation and led to faster albeit less selective reactions:255 an extremely rare example of competitive formation of the anti-Markovnikonv product for a metal-catalyzed radical hydrofunctionalization of an alkene.
Analogously, a hydroazidation of olefins was developed by using tosyl azide as the radical trap forming 481 (Section 3.3);267 this protocol required 30 mol % of TBHP since a much faster catalyst deactivation occurred due to precipitation of apparently oligomeric Co-sulfinate salts. However, the formation of the metal hydride 450 appeared indiscriminate to the cobalt source and its oxidation state. Thereafter, the authors undertook mechanistic study to accompany this work.115 1H NMR-monitoring of the stoichiometric hydrohydrazination towards 476 revealed the first insight: complex 169 in CD3OD showed no changes after the addition of an alkene or the axodicarboxylate whereas new species were immediately formed with the addition of PhSiH3 (Figure 200). Interestingly, broadening of the signals simultaneously occurred, which suggested partial reduction of CoIII to CoII, but the structure of the new complex could not be identified. Moreover, no changes were observed when the olefin was added to the new cobalt species but fast conversion to the hydrohydrazination product 476 along with formation of hydrazine occurred with the addition of the azodicarboxylate.
Figure 200.

Mechanistic tests for the hydrohydrazination reaction.
Reactions with PhSiD3 led to complete deuterium incorporation in the terminal position of the alkene for both the hydrohydrazination and the hydroazidation, confirming that this was the hydrogen source for the hydrofunctionalization and discarding an initial addition of a Co-N complex to the alkene. However, isotope labeling of indene 481 showed a mixture 1:1 between the syn and anti product 482. This result suggested the involvement of radical species before the attack of the azocarboxylate and after the addition of the metal hydride to the alkene.
Moreover, a normal KIE (2.2) was observed for the hydrohydrazination reaction, which the authors attributed to the formation of the C-H bond in the product as the rate-determining step. However, a significantly smaller normal KIE (1.65) was observed during the hydroazidation. The authors did not discuss the common inverse kinetic isotope effect observed in metal-mediated hydrogen atom transfers for their interpretations.375
The presence of radical species was also confirmed by hydrohydrazination of vinylcyclopropane (483), which afforded the rearranged product 484, and with a diene (485), which afforded cis-product 424 as expected for a radical 5-exo-trig cyclization.465 Thus, the mechanism was more complex than previously predicted (Figure 199): intermediate 426 was proposed to undergo Co-C homolytic cleavage to generate a radical and later react with 479 to form intermediate 478 and/or complex 417. Reaction with the silane would then regenerate cobalt hydride 450 and release 475 to form the final product 476 by reaction with ethanol. (PhSiHx(OEt)y species were detected as by-products).
Moreover, the orders of the reagents were measured using the van’t Hoff plots of the initial rates and confirmed a complicated scenario (Figure 201). In the case of hydrohydrazination, 0.98 was obtained for the substrate, suggesting that it was involved in the rate-determining step, and the rate was completely independent from the azocarboxylate (479). Otherwise, 0.22 was obtained for the silane and 0.54 for the cobalt complex showing that the situation was not so clear. In the case of hydroazidation, 0.43 was observed for the substrate, suggesting that several steps contributed equally to the reaction rate, and zero for the tosyl azide. Then, 0.33 was obtained for the silane and 0.82 for the catalyst so the influence of the complex was stronger than for the hydrohydrazination reaction. In the case of the peroxide (TBHP), an order of 0.64 was measured indicating that it was not only important as initiator but during the course of the reaction. The fact that the hydrohydrazination required no additives suggested that TBHP was involved after the olefin hydrocobaltation step, which could be due to activation of Co-alkyl species or regeneration of the Co-H complex. The authors suggested that a (partial) rate-determining catalyst regeneration step would be consistent with the lower kinetic isotope effect and the mixed order in alkene. Thus, a ‘hydrocobaltation’ step was assigned as rate-determining and the following amination step was faster, a proposal explaining why the Co-alkyl complex was never observed. However, addition of the alkene to cobalt species 169 led to no reaction in the NMR experiments so two scenarios were envisaged: either an insertion event was reversible (no isomerization observed) or the nitrogen source was required to generate the active catalyst. Notably, stoichiometric experiments should be treated carefully for the interpretation of catalytic reactions since the relative concentrations of the reagents change, which may have a significant effect on relative rates in a multistep transformation.
Figure 201.

Further mechanistic details for the hydrohydrazination and hydroazidation of alkenes.
Finally, Arrhenius plots for the reactions determined the corresponding activation energies: 76 KJ/mol for hydrohydrazination and 60 KJ/mol for hydroazidation. These values are significantly smaller than the results for the direct hydroamination reaction via olefin activation using Ni-catalysis (108 kJ/mol),466 which was the lowest of all the metals examined. Although the mechanistic understanding of these transformations remained partially speculative, the volume and impact of this study was and still is extremely significant for the overall development of the metal-catalyzed radical hydrofunctionalizations of alkenes.
The cobalt-catalyzed nitrosation of alkenes had also been developed using either a silane or sodium borohydride as the reducing agents (Section 3.1).235,236 Thus, Okamoto and Tanimoto reported the regioselective synthesis of keto-oximes from styrenes and ethyl nitrite (Figure 202). The authors observed that the steric size of the alkyl nitrite 487 influenced the reactivity of the nitrosation, which suggested that generation of free NO under the reaction conditions was unlikely.467 Interestingly, reaction with cyclopropylphenylethylene led to complete consumption of the substrate but no oxime product, suggesting a competitive alternative pathway. These results indicated that the presence of radical intermediates was plausible, but the authors decided that isotope-labeling experiments connoted the possible formation of an alkylcobalt complex such as 488. That is, reaction with NaBD4 led to partial deuterium incorporation (mixture of one, two and three deuterium atoms) as a result of equilibrium reaction between cobalt hydride 450 and organometallic species 488. Therefore, the authors suggested a non-chain free radical mechanism along with the intermediate formation of alkylcobalt complexes. Independent preparation of complex 489 and treatment with ethyl nitrite formed the oxime product along with styrene. Although the retarding effect of polar solvents suggested a nonpolar rate-determining step such as Co-C homolysis, the heterolytic mechanism was not completely eliminated.
Figure 202.

Proposed catalytic cycle for the nitrosation of styrenes.
Baran described the hydroamination of alkenes presumably via reaction of a carbon-centered radical with a nitroarene (Figure 203).264,278 Generation of metal hydride 463 with Fe(acac)3/PhSiH3 in ethanol was proposed to initiate the reaction via reduction of both the alkene and also the nitro group to form an aniline. Whereas the aniline was not reactive towards the hydroamination product 494, the nitroso intermediate 490 was a competent precursor. Thus, radical attack to the nitroso group was suggested, which would lead towards the formation of 492 or 493.272,468 Product 493 was indeed isolated as a by-product of the reaction and converted to 494 by treatment of Zn/HCl at 60 °C. Intermediate 492 was then presumably reduced with iron(II) affording 494 as well as reoxidizing the catalyst. Although further evidence for this hypothesis was not provided, heat flow calorimetry showed an internal temperature rise (2 °C) upon addition of the silane to the reaction mixture with no induction period.
Figure 203.

Proposed catalytic cycle for the hydroamination of alkenes.
In summary, radical addition to N=N bonds was studied in detail by Carreira et al. during the hydrohydrazination and hydroaziridation of alkenes. These transformations were monitored by NMR spectroscopy although neither a metal hydride nor a metal-alkyl complex could be observed. Radical clock and deuterium labelling experiments supported their proposal, which were accompanied with Van’t Hoff and Arrhenius plots. Analogous studies were also reported for the synthesis of keto-oximes and the hydroamination of alkenes.
7.12 Studies of the Radical Attack to Other Atoms
Similar to the hydrocyanation and hydroazidation protocols, Carreira developed the hydrochlorination reaction using tosyl chloride as the radical trap (Figure 204). 340 The authors propose that phenylsilane in the presence of a cobalt complex reacts to form a metal hydride (450) that could insert into an alkene to form complex 426 regioselectively (Figure 183). The subsequent steps to the chlorinated product remained unclear; the authors suggested by analogy that free-radical intermediates could be involved via homolytic cleavage of the Co-C bond followed by attack to the tosyl chloride. Thus, sulfonyl radicals (495) would form as co-products, which could dimerize to unstable sulfinylsulfonates 496 and react with ethanol. Accordingly, formation of 4-methylbenzenesulfinate (497) was observed during the course of the reaction (detected during the hydrocyanation as well). Support for this hypothesis was based on reaction with PhSiD3, which led to complete deuterium incorporation in the terminal position of the alkene. However, the steps to reoxidize CoII and regenerate the cobalt hydride were not known. Interestingly, two protocols for this transformation were reported: when complex 21 was used as precursor, no initiator was required, but for ligand 175, TBHP was needed. In contrast, previous methods showed the formation of metal hydrides only from CoIII species; therefore, elucidation of the hydrogen/chloride-transfer process remained unresolved.
Following the same approach, Herzon reported the bromination, iodination and selenation of alkenes and suggested hydrogen atom transfer between a metal hydride and the double bond as the key step of the process.354 Girijavallabhan also developed a thioetherification using Carreira’s conditions.359 Interestingly, the combination of complex 21 and PhSiH3 required the use of tosyl sulfides instead of diphenyl disulfide or other sulfur precursors because only hydrogenation was observed in these cases. In contrast, Kano had described the thioetherification of styrenes using a porphirine-iron (III) complex with NaBH4 and diphenyl disulfide (Figure 205).358 The authors suggested that the reaction proceeded via organometallic species with carbanion character such as 498. The transformation led to an FeII species but the multistep pathway was not clear by UV spectroscopy. However, dimerization was observed as a side reaction (499) and the rate of the reaction was substantially decreased in the presence of TEMPO leading to 500. Thus, the authors claimed that a radical mechanism could not be completely ruled out (nor could a possible radical-polar reaction).
Figure 205.

Mechanistic tests for the hydrosulfurination of alkenes.
Kojo and Sano reported in 1981 a very interesting study on the hydrothioetherification of an iron porphyrin complex 501, in which the catalyst was also the substrate (Figure 206).469 Thus, treatment of 501 with L-cysteine, sodium borohydride and oxygen in a pH=8.1 solution of cetyltrimethylammonium bromide (CTAB) led to complex 502 in moderate yield with Markovnikov regioselectivity. The reaction did not occur in the absence of iron or oxygen or when the borohydride was replaced for another reductant. CTAB improved the efficiency of the transformation by preventing demetallation of complex 501 through generation of micelles. Interestingly, addition of cyanide anion or carbon monoxide completely inhibited the formation of 502, indicating again that the iron was essential for the reaction. Otherwise, in the absence of L-cysteine the consumption of 501 was very slow and led to a mixture of the reduction and the hydration of the alkene. Therefore, the authors suggested the formation of a common radical intermediate for these processes via a “free hydrogen atom,” generated by reduction of iron(III) with sodium borohydride, which adds to the double bond. Combination of the radical intermediate with a persistent cysteinyl radical would lead to the formation of the sulphide bond in 502. Oxygen was presumably involved in the oxidation of iron(II) to iron(III) but also in the formation of the cysteinyl radical. UV-vis spectroscopy confirmed this proposal and suggested coordination of the L-cysteine to iron in order to facilitate the reduction of the metal. Finally, involvement of a radical intermediate was further supported by use of deuterated sodium borohydride in the absence of L-cysteine: complete incorporation was observed at the β-position whereas only 50% was observed at the α-carbon since the borohydride was competing for the radical intermediate with other hydrogen atom donors in the reaction mixture.
Figure 206.

Hydrothiolation of iron complex 501.
Shigehisa reported a cobalt-catalyzed hydrofluorination of unactivated olefins via radical fluorine transfer in trifluorotoluene (Figure 207).271 The catalytic cycle was presumably initiated by generation of Co-F complex 503 using an electrophilic fluorine source (23), which reacted with (Me2SiH)2O to form a cobalt hydride 450. The driving force of this process was suggested as the strong Si-F bonding energy and was previously observed by Holland et al.470 Initially, complex LCoII was bright red and the solution became olive green upon the addition of 23. The authors proposed insertion of the cobalt hydride 450 into the olefin to form complex 504, but efforts to detect this intermediate were unsuccessful. Complex 504 would then undergo homolytic Co-C bond cleavage to release a free radical intermediate. Reaction with 23 would form the final product 505 along with the amino cation radical 506 (2,4,6-trimethylpyridinium tetrafluorobrate (507) was isolated as by-product). Further evidence for the presence of radical intermediates was provided by the presence of isomerized and hydrated byproducts. Moreover, treatment of dienes led to mixtures of the cis- and trans-cyclized products via radical olefin cyclization.
Figure 207.

Proposed catalytic cycle for the hydrofluorination of alkenes.
Prior to this work, Boger had reported the alkene hydrofluorination reaction using Fe2(ox)3 with NaBH4 and Selectfluor (Figure 208).349 The presence of free radical intermediates was characterized by the 5-exo-trig cyclization of diene 508 to pyrrolidine 509 and the non-diastereoselective hydrofluorination of 510 to 511when NaBD4 was used. Moreover, under the same conditions, other radical traps could be used leading to the corresponding hydrofunctionalized olefins, for example, an azide, chlorine, a cyano and a nitroso group, TEMPO, molecular oxygen and cyanate or thiocyanate groups.81 Boger characterized the reactions as proceeding via Fe-mediated hydrogen atom radical addition to alkenes as the initial step in the reaction, consistant with their previously established mechanistic paradigm.26
Figure 208.

Mechanistic tests for the iron-mediated hydrofluorination of alkenes.
In summary, radical attack has been suggested for the hydrochlorination, bromination, iodination, selenation, sulfurination and fluorination of alkenes. However, due to the distinctive features of each catalytic system, slightly different proposals arose regarding the generation of the metal hydride and the turn over of the catalytic cycle.
7.13 Radical-Polar Crossover Mechanisms
As mentioned previously, radical addition to α,β-unsaturated substrates led to deuterium incorporation at the α-position when using an isotope-labeled alcoholic solvent, for example, in the hydrogenation of alkenes reported by Magnus or the conjugate addition described by Baran (Figures 186 and 195).20,221 These transformations could be considered radical-anion cross-over reactions since a metallo-enolate appears to be involved in a protonation step. Anionic character was also suggested for some metal-alkyl complexes bearing electron-rich ligands on the metal, for example, in the sulfurination of styrenes with PhSSPh (Figure 205).358
A few examples in the literature propose trapping of aldehydes building a new C-C bond following the same approach.326 Krische et al. reported a cobalt-catalyzed intramolecular reductive cyclization between a α,β-unsaturated ketone and an aldehyde (Figure 209).337 Thus, substrate 514 led to 512 with high levels of syn-diastereoselectivity when treated with PhSiH3/Co(dpm)2 in DCE.
Figure 209.

Proposed catalytic cycle for the reductive cycloaddition to an aldehyde.
The catalytic cycle proposed to initiate via the formation of a metal hydride intermediate which hydrometalated an enone.471 The interaction between Co(dpm)2 and the silane was a key feature of the mechanism; tetrahedral cobalt(II) complexes could potentially undergo single electron oxidative addition or disproportonation. The latter pathway was supported by HRMS analysis since an intense signal consistent with Co(dpm)3 was detected. Generation of complex LCoI was proposed, which could potentially undergo oxidative addition with PhSiH3 to form hydride 513. Thus, a CoI-CoIII cycle was envisaged as a working model. Complex 513 would undergo hydrometalation of the enone leading to complex 515, which would add to the aldehyde providing cobalt-alkoxide 516 after the formation of the new C-C bond. Oxygen-silicon reductive elimination was suggested to form product 512 and regenerate complex LCoI.
Gas evolution (presumably H2) was observed throughout the reaction indicating competitive dehydrogenative coupling of the silane.472 The authors also noted that the proposed mechanism bore similarity to the Chalk-Harrod process,473 which is commonly accepted for alkene hydrosilylation. Deuterium labeling experiments using PhSiD3 led to complete incorporation in the β-position of the enone, which was consistent with the previous proposal (Figure 210). An equimolar mixture of stereoisomers was obtained for this deuterium-incorporation reaction (single-crystal neutron diffraction analysis). This result suggested π-facial interconversion of the kinetically formed metallo-enolate (518/519) was faster than the aldehyde addition.
Figure 210.

Mechanistic tests for the reductive cycloaddition to an aldehyde.
Also consistent with this mechanism, the stereochemical outcome of the transformation was independent of the alkene geometry. Both trans- and cis-enone 517 led to the formation of the syn-product 521. The observed syn-diastereoselectivity was accounted for on the basis of a Zimmerman-Traxler type transition state. Coordination of the reacting partners in the form of their higher haptomers resulted in chelates of normal ring size; Z-enolate formation was preferred because of allylic 1,2-strain. The collective experiments also revealed competitive enone reduction pathways. Similarly, reaction between two enones led to the cycloaddition product.338 Interestingly, Pronin reported the analogous addition of an iron-enolate to an aldehyde subsequent to the radical conjugate addition reported by Baran in a tandem fashion to form the polycyclized core of an indole diterpene.342
Oxidation of a radical intermediate could potentially lead to radical-cationic transformations. Shigehisa reported a very different outcome for his hydrofluorination protocol when it was performed in an alcoholic solvent (Figure 211).156 Under these conditions, treatment of an unactivated olefin with cobalt complex 21, PhSiH3 and 23 (c.f. Figure 207) led to formation of the Markovnikov alkoxylation product (523). The authors proposed conversion of radical intermediate to carbocation 522. Deuterium-labeling experiments showed incorporation of deuterium at the terminal position using PhSiD3 (d1-525a). The authors suggest that rearrangement of 526 to 527 under the conditions of their hydroalkoxylation provide evidence of radical intermediates. Substrate 528 delivered cyclized product 529, which indicated the presence of cationic species, and exclusive incorporation of deuterium on the methyl in d3-525a indicated that solvent did not contribute a hydrogen to the alkene. Later, the proposed carbocation was also trapped with secondary amines in an intramolecular fashion.270 Oxidation of the radical intermediate to 522 was suggested in order to explain these observations with a hypothetical cationic complex [LCoIIIBF4]. Although this process had been previously reported by Kochi with other cationic cobalt species, the specific pathway for this step remain speculative since the transformation required very high temperatures (80–100 °C).474 Nevertheless, the idea that easily-generated radicals can be converted to high energy carbocations is extremely promising.
Figure 211.

Mechanistic tests for the hydroalkoxylation of alkenes.
In summary, the formation of a metal enolate was also proposed for the addition of α,β unsaturated substrates to aldehydes and Krische suggested a Co(I)-Co(III) cycle based on deuterium labelling experiments. In contrast, Shigehisa reported the hydroalkoxylation of alkenes via oxidation of a radical intermediate. The proposal was based on isotope exchange and radical clock experiments.
8. Conclusion
Metal-hydride hydrogen atom transfer (MH HAT) has emerged from a curiosity of metal carbonyls to become a useful tool for preparative synthetic chemistry – the radical equivalent of Brønsted acid proton transfer. Many of the synthetic methods to arise from early exploration of cofactor mimics appear to have been mischaracterized as hydrometallation reactions, as noted by Boger.26 The retrospective overlay of HAT pathways onto several decades-worth of observations brings some clarity to this area. From our analysis of the literature, Norton first explicitly linked Halpern’s transition metal-hydride hydrogen atom transfer (TM HAT) mechanism to the cofactor-mimetic reactions typified by Mukaiyama hydration in a passing reference during a rate analysis in 2007.298 However, this mechanistic link was not adapted by the community until the publication in 2014 of two hydrogenation reactions24,27 that directly addressed the likelihood of reactivity overlap. It can only be hoped that this newfound coherence can be brought to bear on challenging problems in synthetic chemistry and to the better understanding of the reactions of metal hydrides.
As pointed out by a referee of this review, although HAT is likely to underlie this array of reactions, such a mechanism leads to an energy conundrum. The radicals generated from α-olefins would possess adjacent C-H bonds with bond strengths less than 40 kcal/mol. This BDE is substantially weaker than the lower limit for valence-saturated hydrides calculated by Landis475 and weaker than the weakest characterized metal hydrides,395 potentially increasing the endothermicity of an already endothermic process. Already, cage pairs of carbon and metal radicals can revert to stable metal hydrides and alkenes with rates approaching the vibrational limit.417 So how unstable must a metal hydride be to produce an unstablized carbon radical at a reasonable rate and what structural factors in the ligand govern such instability? What other species lie on the catalytic cycle that might drive the reactions to product? Are the concentrations of metal hydride low enough to prevent hydrogen evolution or does ligand structure also prevent this off-path reaction?
Despite the rich precedence and recent, burgeoning interest in this area of radical hydrofunctionalization chemistry, there remain many promising avenues for future research. For example, clear cases of applying this chemistry to the formation of Markovnikov C-B, C-Si and C-P bonds remain unknown. Similarly, the development of radical polycyclization cascades, enantioselective bond formation, and coupling of catalytic TM HAT cycles with other cross-coupling catalytic cycles would be highly enabling. Fundamentally, the field would benefit immensely from the expertise of inorganic and organometallic chemists, who could further elucidate structural and electronic parameters that govern the fascinating capacity for certain manganese, iron, cobalt and other transition metals to generate carbon-centered radicals from alkenes. We hope that this review brings further coherence to a field that has already required a diverse range of chemical expertise, and we anticipate the community will realize the aforementioned challenges and many others yet unknown.
Figure 2.

Relative rates of radical addition to different alkenes.
Figure 7.

Structures of the cobalt(II) tetraphenylporphyrine (CoTPP) and cobalt(II) bis[3-(salicylideneimino)propyl]methylamine (CoSalMDPT) complexes.
Figure 68.

Zhu and Yu’s synthesis of linckosides A (139) and B (140).
Figure 104.

Radical addition into a polarized unsaturated electrophile.
Figure 109.

Hydromethylation via radical addition / functional-group ablation.
Figure 111.

Radical conjugate addition-elimination to give β-substituted styrenes.
Figure 149.

Some structural motifs accessible through TM HAT isomerization.
Figure 162.

Mukai et al. observed sulfonyl reduction under Co(acac)2 reduction conditions.
Figure 187.

Catalytic hydrogenation of unactivated alkenes via HAT.
Figure 192.

Competition experiment between PhSiH3 and PhSiD3.
Acknowledgments
Funding Sources
Financial support for this work was provided by the NIH (GM105766, GM104180 and F31 GM111050 to R.M.), the NSF (CHE – 1352587) and NSERC (PGS-D3 fellowship to S.C.). Additional support was provided by Eli Lilly, Novartis, Bristol-Myers Squibb, Amgen, Boehringer-Ingelheim, the Sloan Foundation and the Baxter Foundation.
Biographies
Steven W. M. Crossley received his B.Sc. in chemistry with honors (first class) from the University of British Columbia in Vancouver in 2012. As an undergraduate student, he explored dinitrogen activation, diiminepyridine, and β-diimininate chemistry of group 9 metals, and indium allyl chemistry in the laboratories of Michael Fryzuk, Peter Budzelaar, and Parisa Mehrkodavandi, respectively. He also investigated C H functionalization chemistry in the laboratory of Marco Ciufolini. He is currently a NSERC PGS fellow with Ryan Shenvi at The Scripps Research Institute in La Jolla, California. At Scripps, he gets to pursue his fascination with both organometallic chemistry and organic synthesis through his research on the development of organotransition metal radical chemistry for use in organic synthesis.
Dr. Carla Obradors was born in 1987 in Manresa (Spain). She obtained her B.S. degree in chemistry from Universitat Autònoma de Barcelona in 2010 and she was also a research student in Institut de Química Avançada de Catalunya (CSIC). In 2011, she obtained her M.S. degree in synthesis and catalysis from Universitat Rovira i Virgili in Tarragona. She earned her Ph.D. in 2014 as a FPU predoctoral fellow with Antonio M. Echavarren at the Institute of Chemical Research of Catalonia (ICIQ). Meanwhile, she performed an international internship with Ryan Shenvi at The Scripps Research Institute and since 2015 she has been working in his laboratory as a postdoctoral fellow. Carla is currently involved in the development of new methods based on HAT for the synthesis of biologically active compounds.
Ruben M. Martinez earned his B.S. degree in applied chemistry (2010) at the University of California, Davis, where he did undergraduate research in the group of Jared T. Shaw. From there he moved to the San Francisco bay area and worked for three years as a Research Associate in the Medicinal Chemistry department at Gilead Sciences, where he worked on drug design teams in the areas of oncology, cardiovascular disease, and hepatitis C virus (HCV). He is currently an NIH NRSA F31 fellow pursuing his PhD in the lab of Ryan A. Shenvi, where his research is focused on reaction development and natural product synthesis.
Ryan Shenvi is an Associate Professor in the Department of Chemistry at The Scripps Research Institute. He earned his B.S. degree from Penn State University and obtained his Ph.D. with Phil Baran at The Scripps Research Institute. After postdoctoral studies with E. J. Corey at Harvard University, Ryan returned to Scripps to start his own lab. The Shenvi lab devises chemical syntheses of secondary metabolites that are relevant to human health. These efforts frequently require the invention of new chemical reactions and so lead to discoveries in chemistry and biology.
References
- 1.Hegedus LS. Transition Metals in the Synthesis of Complex Organic Molecules. University Science Books; Sausalito, CA: 1999. [Google Scholar]
- 2.Mathey F. Transition Metal Organometallic Chemistry. Springer; New York, NY: 2013. [Google Scholar]
- 3.Collman JP, Hegedus LS, Norton JR, Finke RG. Principles and Applications of Organotransition Metal Chemistry. University Science Books; Mill Valley, CA: 1987. [Google Scholar]
- 4.Hartung J, Norton J. In: Catalysis Without Precious Metals. Bullock RM, editor. Wiley; Weinheim: 2010. [Google Scholar]
- 5.Hoffmann RW. Markovnikov Free Radical Addition Reactions, a Sleeping Beauty Kissed to Life. Chem Soc Rev. 2016;45:577–583. doi: 10.1039/c5cs00423c. [DOI] [PubMed] [Google Scholar]
- 6.Simonneau A, Oestreich M. Fascinating Hydrogen Atom Transfer Chemistry of Alkenes Inspired by Problems in Total Synthesis. Angew Chem Int Ed. 2015;54:3556–3558. doi: 10.1002/anie.201500557. [DOI] [PubMed] [Google Scholar]
- 7.Streuff J, Gansäuer A. Metal-Catalyzed β-Functionalization of Michael Acceptors through Reductive Radical Addition Reactions. Angew Chem Int Ed. 2015;54:14232–14242. doi: 10.1002/anie.201505231. [DOI] [PubMed] [Google Scholar]
- 8.Trost BM. Selectivity: A Key to Synthetic Efficiency. Science. 1983;219:245–250. doi: 10.1126/science.219.4582.245. [DOI] [PubMed] [Google Scholar]
- 9.Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. Wiley; New York: 1995. [Google Scholar]
- 10.Faraday M. On New Compounds of Carbon and Hydrogen, and on Certain Other Products Obtained during Decomposition of Oil by Heat. Phil Trans R Soc. 1825;115:440–466. [Google Scholar]
- 11.Hennell H, Sevullas H. On the Mutual Action of Sulphuric Acid and Alcohol, and on the Nature of the Process by which Ether is Formed. Phil Trans R Soc. 1828;118:365–371. [Google Scholar]
- 12.Toteva MM, Richard JP. Mechanism of Nucleophilic Substitution and Elimination Reactions at Tertiary Carbon in Largely Aqueous Solutions: Lifetime of a Simple Tertiary Carbocation. J Am Chem Soc. 1996;118:11434–11445. [Google Scholar]
- 13.Reed CA. Carborane Acids. New “Strong Yet Gentle” Acids for Organic and Inorganic Chemistry. Chem Commun. 2005:1669–1677. doi: 10.1039/b415425h. [DOI] [PubMed] [Google Scholar]
- 14.Giese B. Formation of CC Bonds by Addition of Free Radicals to Alkenes. Angew Chem Int Ed Engl. 1983;22:753–764. [Google Scholar]
- 15.Lewis FM, Mayo FR, Hulse WF. Copolymerization. II. The Copolymerization of Acrylonitrile, Methyl Methacrylate, Styrene and Vinylidene Chloride. J Am Chem Soc. 1945;67:1701–1705. [Google Scholar]
- 16.Curran DP, Eichenberger E, Collis M, Roepel MG, Thoma G. Group Transfer Addition Reactions of Methyl(phenylseleno)malononitrile to Alkenes. J Am Chem Soc. 1994;116:4279–4288. [Google Scholar]
- 17.Munger K, Fischer H. Separation of Polar and Steric Effects on Absolute Rate Constants and Arrhenius Parameters for the Reaction of tert-Butyl Radicals with Alkenes. Int J Chem Kinet. 1985;17:809–829. [Google Scholar]
- 18.Roberts BP. Polarity-Reversal Catalysis of Hydrogen-Atom Abstraction Reactions: Concepts and Applications in Organic Chemistry. Chem Soc Rev. 1999;28:25–35. [Google Scholar]
- 19.Evans DA. Chemistry 206: Advanced Organic Chemistry, Handout 27A. Harvard University; 2001. An Organizational Format for the Classification of Functional Groups. Application to the Construction of Difunctional Relationships. [Google Scholar]
- 20.Lo JC, Yabe Y, Baran PS. A Practical and Catalytic Reductive Olefin Coupling. J Am Chem Soc. 2014;136:1304–1307. doi: 10.1021/ja4117632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lo JC, Gui J, Yabe Y, Pan CM, Baran PS. Functionalized Olefin Cross-Coupling to Construct Carbon–Carbon Bonds. Nature. 2014;516:343–348. doi: 10.1038/nature14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaspar B, Carreira EM. Mild Cobalt-Catalyzed Hydrocyanation of Olefins with Tosyl Cyanide. Angew Chem Int Ed. 2007;46:4519–4522. doi: 10.1002/anie.200700575. [DOI] [PubMed] [Google Scholar]
- 23.Gaspar B, Carreira EM. Cobalt Catalyzed Functionalization of Unactivated Alkenes: Regioselective Reductive C–C Bond Forming Reactions. J Am Chem Soc. 2009;131:13214–13215. doi: 10.1021/ja904856k. [DOI] [PubMed] [Google Scholar]
- 24.Iwasaki K, Wan KK, Oppedisano A, Crossley SWM, Shenvi RA. Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol. J Am Chem Soc. 2014;136:1300–1303. doi: 10.1021/ja412342g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sears JE, Boger DL. Total Synthesis of Vinblastine, Related Natural Products, and Key Analogues and Development of Inspired Methodology Suitable for the Systematic Study of Their Structure–Function Properties. Acc Chem Res. 2015;48:653–662. doi: 10.1021/ar500400w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. Total Synthesis of Vinblastine, Vincristine, Related Natural Products, and Key Structural Analogues. J Am Chem Soc. 2009;131:4904–4916. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.King SM, Ma X, Herzon SB. A Method for the Selective Hydrogenation of Alkenyl Halides to Alkyl Halides. J Am Chem Soc. 2014;136:6884–6887. doi: 10.1021/ja502885c. [DOI] [PubMed] [Google Scholar]
- 28.Crossley SWM, Barabé F, Shenvi RA. Simple, Chemoselective, Catalytic Olefin Isomerization. J Am Chem Soc. 2014;136:16788–16791. doi: 10.1021/ja5105602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gansauer A, Shi L, Otte M, Huth I, Rosales A, Sancho-Sanz I, Padial NM, Oltra JE. Hydrogen Atom Donors: Recent Developments. Top Curr Chem. 2012;320:93–120. doi: 10.1007/128_2011_124. [DOI] [PubMed] [Google Scholar]
- 30.Gridnev AA, Ittel SD. Catalytic Chain Transfer in Free-Radical Polymerizations. Chem Rev. 2001;101:3611–3660. doi: 10.1021/cr9901236. [DOI] [PubMed] [Google Scholar]
- 31.Valyaev DA, Lavigne G, Lugan N. Manganese Organometallic Compounds in Homogeneous Catalysis: Past, Present, and Prospects. Coord Chem Rev. 2016;308:191–235. [Google Scholar]
- 32.Bauer I, Knölker HJ. Iron Catalysis in Organic Synthesis. Chem Rev. 2015;115:3170–3387. doi: 10.1021/cr500425u. [DOI] [PubMed] [Google Scholar]
- 33.Greenhalgh MD, Jones AS, Thomas SP. Iron-Catalysed Hydrofunctionalisation of Alkenes and Alkynes. ChemCatChem. 2015;7:190–222. [Google Scholar]
- 34.Gilbert BC, Parsons AF. The Use of Free Radical Initiators Bearing Metal–Metal, Metal–Hydrogen and Non-Metal–Hydrogen Bonds in Synthesis. J Chem Soc, Perkin Trans. 2002;2:367–387. [Google Scholar]
- 35.Maity A, Teets TS. Main Group Lewis Acid-Mediated Transformations of Transition-Metal Hydride Complexes. Chem Rev. 2016 doi: 10.1021/acs.chemrev.6b00034. Online early access. [DOI] [PubMed] [Google Scholar]
- 36.Halpern J. Determination and Significance of Transition Metal-Alkyl Bond Dissociation Energies. Acc Chem Res. 1982;15:238–244. [Google Scholar]
- 37.Halpern J. Free Radical Mechanisms in Organometallic and Bioorganometallic Chemistry. Pure Appl Chem. 1986;58:575–584. [Google Scholar]
- 38.Mukaiyama T, Yamada T. Recent Advances in Aerobic Oxygenation. Bull Chem Soc Jpn. 1995;68:17–35. [Google Scholar]
- 39.Ullrich V. Enzymatic Hydroxylations with Molecular Oxygen. Angew Chem Int Ed Engl. 1972;11:701–712. doi: 10.1002/anie.197207011. [DOI] [PubMed] [Google Scholar]
- 40.Matsuura T. Bio-Mimetic Oxygenation. Tetrahedron. 1977;33:2869–2905. [Google Scholar]
- 41.Mimoun H. The Role of Peroxymetallation in Selective Oxidative Processes. J Mol Catal. 1980;7:1–29. [Google Scholar]
- 42.Mansuy D. Cytochrome P-450 and synthetic models. Pure & Appl Chem. 1987;59:759–770. [Google Scholar]
- 43.Punniyamurthy T, Velusamy S, Iqbal J. Recent Advances in Transition Metal Catalyzed Oxidation of Organic Substrates with Molecular Oxygen. Chem Rev. 2005;105:2329–2364. doi: 10.1021/cr050523v. [DOI] [PubMed] [Google Scholar]
- 44.Fenton HJH. Oxidation of Tartaric Acid in Presence of Iron. J Chem Soc Trans. 1894;65:899–910. [Google Scholar]
- 45.Kornweitz H, Burg A, Meyerstein D. Plausible Mechanisms of The Fenton-Like Reactions, M = Fe(II) and Co(II), in the Presence of RCO2− Substrates: are •OH Radicals Formed in the Process? J Phys Chem A. 2015;119:4200–4206. doi: 10.1021/jp512826f. [DOI] [PubMed] [Google Scholar]
- 46.Simic MG. Free Radical Mechanisms in Autooxidation Processes. J Chem Educ. 1981;58:125–131. [Google Scholar]
- 47.Lyons JE. Oxidation of Olefins in the Presence of Transition Metal Complexes. ACS Adv Chem Ser. 1974;132:64–89. [Google Scholar]
- 48.Walsh C. Enzymatic Reaction Mechanisms. Chapters 12–15 W.H. Freeman and Co; 1979. [Google Scholar]
- 49.Karlin KD, Cruse RW, Gultneh Y, Farooq A, Hayes JC, Zubieta J. Dioxygen-Copper Reactivity. Reversible Binding of O2 and CO to a Phenoxo-Bridged Dicopper(I) Complex. J Am Chem Soc. 1987;109:2668–2679. [Google Scholar]
- 50.Kitajima N, Koda T, Hashimoto S, Kitagawa T, Moro-oka Y. An Accurate Synthetic Model of Oxyhaemocyanin. J Chem Soc, Chem Commun. 1988:151–152. [Google Scholar]
- 51.Omura T, Sato R. The Carbon Monoxide-Binding Pigment of Liver Microsomes: II. Solubilization, Purification, and Properties. J Biol Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- 52.Tabushi I, Koga N. P-450 Type Oxygen Activation by Porphyrin-Manganese Complex. J Am Chem Soc. 1979;101:6456–6458. [Google Scholar]
- 53.Klingenberg M. Pigments of Rat Liver Microsomes. Arch Biochem Biophys. 1958;75:376–386. doi: 10.1016/0003-9861(58)90436-3. [DOI] [PubMed] [Google Scholar]
- 54.Uchida K, Soma M, Naito S, Onishi T, Tamaru K. Manganese Phthalocyanine as a Model of Tryptophan-2,3-Dioxygenase. Chem Lett. 1978;7:471–474. [Google Scholar]
- 55.Costantini M, Dromard A, Jouffret M, Brossard B, Varagnat J. Selective Oxidation of 3,5,5-trimethylcyclohexene-3 one ({β}-isophorone) to 3,5,5-trimethylcyclohexene-1,4: Diones by Oxygen Catalyzed by MnII OR CoII Chelates. J Mol Catal. 1980;7:89–97. [Google Scholar]
- 56.Dufour MN, Crumbliss AL, Johnston G, Gaudemer A. Reaction of Indoles with Molecular Oxygen Catalyzed by Metalloporphyrins. J Mol Catal. 1980;7:277–287. [Google Scholar]
- 57.Perree-Fauvet M, Gaudemer A. Manganese Porphyrin-Catalysed Oxidation of Olefins to Ketones by Molecular Oxygen. J Chem Soc, Chem Commun. 1981:874–875. [Google Scholar]
- 58.Shimizu M, Orita H, Hayakawa T, Takehira K. The Oxidation of Olefins with O2 and NaBH4 Catalyzed by Manganese Meso-tetrakis(p-sulfonatophenyl)porphin. J Mol Catal. 1988;45:85–90. [Google Scholar]
- 59.Shimizu M, Orita H, Hayakawa T, Takehira K. Aromatic Olefin Oxygenation with Tetrahydroborate and Dioxygen Catalyzed by a Manganese Porphyrin. J Mol Catal. 1989;53:165–172. [Google Scholar]
- 60.Nishiki M, Satoh T, Sakurai H. Biomimetic Models of Cytochrome P-450 Monooxygenases: Studies on Cyclohexene Oxygenation in Metalloporphyrin-Sodium Borohydride-Microcrystalline Cellulose-Oxygen Systems. J Mol Catal. 1990;62:79–91. [Google Scholar]
- 61.Takeuchi M, Kano K. Mechanisms for (Porphinato)manganese(III)-Catalyzed Oxygenation and Reduction of Styrenes in Benzene–Ethanol Containing Sodium Borohydride. Bull Chem Soc Jpn. 1994;67:1726–1733. [Google Scholar]
- 62.Santa T, Mori T, Hirobe M. Oxygen Activation and Olefin Oxygenation by Iron(III) porphyrin as a Model of Cytochrome P-450. Chem Pharm Bull (Tokyo) 1985;33:2175–2178. doi: 10.1248/cpb.33.2175. [DOI] [PubMed] [Google Scholar]
- 63.Kano K, Takagi H, Takeuchi M, Hashimoto S, Yoshida Z-i. Porphinatoiron-Catalyzed Oxygenation of Styrene in Aqueous Solution. Chem Lett. 1991;20:519–522. [Google Scholar]
- 64.Takeuchi M, Kano K. (σ-Alkyl)iron Complexes as Intermediates in (Porphinato)Iron-Mediated Reduction of Alkenes and Alkynes with Sodium Borohydride. Organometallics. 1993;12:2059–2064. [Google Scholar]
- 65.Setsune J-I, Ishimaru Y, Sera A. Synthesis of -(Vinyl)iron(III) Porphyrins and (Dialkylcarbene)iron(II) Porphyrins through the Hydrometallation of Alkynes with Iron(III) Porphyrins and NaBH4. J Chem Soc, Chem Commun. 1992:328–329. [Google Scholar]
- 66.Setsune JI, Ishimaru Y, Sera A. 1H NMR Study of the Reaction of Iron(III) Porphyrins with NaBH4 in the Presence of Alkenes. Formation of Organoiron(III) Porphyrins. Chem Lett. 1992;21:377–380. [Google Scholar]
- 67.Takeuchi M, Kodera M, Kano K, Yoshida Z-i. Mechanisms for (Porphyrinato)Iron(III)-Catalyzed Oxygenation of Styrenes by O2 in Presence of BH4−. J Mol Catal A: Chem. 1996;113:51–59. [Google Scholar]
- 68.Howard JAK. Dorothy Hodgkin and her Contributions to Biochemistry. Nature Rev Molec Cell Biol. 2003;4:891–896. doi: 10.1038/nrm1243. [DOI] [PubMed] [Google Scholar]
- 69.Schrauzer GN, Kohnle J. Coenzyme B12 Models. Chem Ber. 1964;97:3056–3064. [Google Scholar]
- 70.Schrauzer GN, Windgassen RJ. On Cobaloxime (II) and its Relationship to Vitamin B12r. Chem Ber. 1966;99:602–610. doi: 10.1002/cber.19660990234. [DOI] [PubMed] [Google Scholar]
- 71.Schrauzer GN, Windgassen RJ. Alkylcobaloximes and Their Relation to Alkylcobalamins. J Am Chem Soc. 1966;88:3738–3743. [Google Scholar]
- 72.Schrauzer GN, Windgassen RJ. Cobalamin Model Compounds. Preparation and Reactions of Substituted Alkyl- and Alkenylcobaloximes and Biochemical Implications. J Am Chem Soc. 1967;89:1999–2007. [Google Scholar]
- 73.Crabtree RH. The Organometallic Chemistry of the Transition Metals. Chapter 16 Wiley and Sons Inc; 2014. [Google Scholar]
- 74.Okamoto T, Oka S. Cobalt-Catalyzed Regiospecific Conversion of Aryl-Substituted Olefins into Alcohols using Molecular Oxygen and Tetrahydroborate. the Reaction of Oxygen with Activated Substrates. Tetrahedron Lett. 1981;22:2191–2194. [Google Scholar]
- 75.Okamoto T, Oka S. Oxygenation of Olefins under Reductive Conditions. Cobalt-Catalyzed Selective Conversion of Aromatic Olefins to Benzylic Alcohols by Molecular Oxygen and Tetrahydroborate. J Org Chem. 1984;49:1589–1594. [Google Scholar]
- 76.Ohkatsu Y, Ohno M, Ooi T, Inoue S. Hydroxylation Reactions of Olefins with Cobalt(II) Complex-NaBH4-O2 Systems. Nippon Kagaku Kaishi. 1985:387–393. [Google Scholar]
- 77.Setsune J-I, Ishimaru Y, Moriyama T, Kitao T. Hydrometallation of Alkenes and Alkynes by the Combination of Cobalt(II) Porphyrins, NaBH4 and Oxidizing Agents. J Chem Soc, Chem Commun. 1991:555–556. [Google Scholar]
- 78.Sugimori T, Horike S-i, Tsumura S, Handa M, Kasuga K. Catalytic Oxygenation of Olefin with Dioxygen and Tetra-t-Butylphthalocyanine Complexes in the Presence of Sodium Borohydride. Inorg Chim Acta. 1998;283:275–278. [Google Scholar]
- 79.Sorokin AB. Phthalocyanine Metal Complexes in Catalysis. Chem Rev. 2013;113:8152–8191. doi: 10.1021/cr4000072. [DOI] [PubMed] [Google Scholar]
- 80.Ishikawa H, Colby DA, Boger DL. Direct Coupling of Catharanthine and Vindoline to Provide Vinblastine: Total Synthesis of (+)- and ent-(−)-Vinblastine. J Am Chem Soc. 2008;130:420–421. doi: 10.1021/ja078192m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leggans EK, Barker TJ, Duncan KK, Boger DL. Iron(III)/NaBH4-Mediated Additions to Unactivated Alkenes: Synthesis of Novel 20′-Vinblastine Analogues. Org Lett. 2012;14:1428–1431. doi: 10.1021/ol300173v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ahouari H, Rousse G, Rodríguez-Carvajal J, Sougrati MT, Saubanère M, Courty M, Recham N, Tarascon JM. Unraveling the Structure of Iron(III) Oxalate Tetrahydrate and Its Reversible Li Insertion Capability. Chem Mater. 2015;27:1631–1639. [Google Scholar]
- 83.Sakamoto N, Tan H, Hata E, Kihara N. Process for the Preparation of Binary Indole Alkaloids. 5,432,279. US Patent. 1995
- 84.Tan H, Sakamoto N, Hata E, Ishitoku T, Kihara N. Method for Production of Dimeric Alkaloids. 5,037,977. US Patent. 1991
- 85.Vukovic J, Goodbody A. Production of Alkaloid Dimers using Ferric Ion. 4,778,885. US Patent. 1988
- 86.Vukovic J, Goodbody AE, Kutney JP, Misawa M. Production of 3′, 4′ Anhydrovinblastine: a Unique Chemical Synthesis. Tetrahedron. 1988;44:325–331. [Google Scholar]
- 87.Taniguchi T, Goto N, Nishibata A, Ishibashi H. Iron-Catalyzed Redox Radical Cyclizations of 1,6-Dienes and Enynes. Org Lett. 2010;12:112–115. doi: 10.1021/ol902562j. [DOI] [PubMed] [Google Scholar]
- 88.Hashimoto T, Hirose D, Taniguchi T. Direct Synthesis of 1,4-Diols from Alkenes by Iron-Catalyzed Aerobic Hydration and C-H Hydroxylation. Angew Chem Int Ed Engl. 2014;53:2730–2734. doi: 10.1002/anie.201308675. [DOI] [PubMed] [Google Scholar]
- 89.Nam W, Lim MH, Oh SY, Lee JH, Lee HJ, Woo SK, Kim C, Shin W. Remarkable Anionic Axial Ligand Effects of Iron(III) Porphyrin Complexes on the Catalytic Oxygenations of Hydrocarbons by H2O2 and the Formation of Oxoiron(IV) Porphyrin Intermediates by m-Chloroperoxybenzoic Acid. Angew Chem Int Ed. 2000;39:3646–3649. doi: 10.1002/1521-3773(20001016)39:20<3646::aid-anie3646>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 90.Nam W, Lim MH, Oh S-Y. Effect of Anionic Axial Ligands on the Formation of Oxoiron(IV) Porphyrin Intermediates. Inorg Chem. 2000;39:5572–5575. doi: 10.1021/ic000502d. [DOI] [PubMed] [Google Scholar]
- 91.Nam W, Jin SW, Lim MH, Ryu JY, Kim C. Anionic Ligand Effect on the Nature of Epoxidizing Intermediates in Iron Porphyrin Complex-Catalyzed Epoxidation Reactions. Inorg Chem. 2002;41:3647–3652. doi: 10.1021/ic011145p. [DOI] [PubMed] [Google Scholar]
- 92.Zombeck A, Hamilton DE, Drago RS. Novel Catalytic Oxidations of Terminal Olefins by Cobalt(II)-Schiff Base Complexes. J Am Chem Soc. 1982;104:6782–6784. [Google Scholar]
- 93.Corden BB, Drago RS, Perito RP. Steric and Electronic Effects of Ligand Variation on Cobalt Dioxygen Catalysis. J Am Chem Soc. 1985;107:2903–2907. [Google Scholar]
- 94.Hamilton DE, Drago RS, Zombeck A. Mechanistic Studies on the Cobalt(II) Schiff Base Catalyzed Oxidation of Olefins by O2. J Am Chem Soc. 1987;109:374–379. [Google Scholar]
- 95.Talsi EP, Zimin YS, Nekipelov VM. Coordination of Molecular Oxygen by Cobalt(II) β-Diketonates. React Kinet Catal Lett. 1985;27:361–364. [Google Scholar]
- 96.Tamagaki S, Kanamaru Y, Ueno M, Tagaki W. Physical and Chemical Properties of Mononuclear Cobalt Dioxygen Complexes with Tetraimidazolyl-Substituted Pyridine Chelates. Bull Chem Soc Jpn. 1991;64:165–174. [Google Scholar]
- 97.Mukaiyama T, Isayama S, Inoki S, Kato K. Oxidation-Reduction Hydration of Olefins with Molecular Oxygen and 2-Propanol Catalyzed by Bis(acetylacetonato)cobalt(II) Chem Lett. 1989;18:449–452. [Google Scholar]
- 98.Inoki S, Kato K, Takai T, Isayama S, Yamada T, Mukaiyama T. Bis(trifluoroacetylacetonato)cobalt(II) Catalyzed Oxidation-Reduction Hydration of Olefins Selective Formation of Alcohols from Olefins. Chem Lett. 1989;18:515–518. [Google Scholar]
- 99.Kato K, Yamada T, Takai T, Inoki S, Isayama S. Catalytic Oxidation-Reduction Hydration of Olefin with Molecular Oxygen in the Presence of Bis(1,3-diketonato)cobalt(II) Complexes. Bull Chem Soc Jpn. 1990;63:179–186. [Google Scholar]
- 100.Isayama S, Mukaiyama T. Hydration of Olefins with Molecular Oxygen and Triethylsilane Catalyzed by Bis(trifluoroacetylacetonato)cobalt(II) Chem Lett. 1989;18:569–572. [Google Scholar]
- 101.Citron JD. Reductions with Organosilicon Hydrides. III. Reduction of Acyl Fluorides to Esters. J Org Chem. 1971;36:2547–2548. [Google Scholar]
- 102.Kato J-i, Iwasawa N, Mukaiyama T. A Novel Method for the Preparation of Symmetrical and Unsymmetrical Ethers. Trityl Perchlorate Promoted Reduction of Carbonyl Compounds with Triethylsilane. Chem Lett. 1985;14:743–746. [Google Scholar]
- 103.Isayama S, Mukaiyama T. Novel Method for the Preparation of Triethylsilyl Peroxides from Olefins by the Reaction with Molecular Oxygen and Triethylsilane Catalyzed by Bis(1,3-diketonato)cobalt(II) Chem Lett. 1989;18:573–576. [Google Scholar]
- 104.Isayama S. An Efficient Method for the Direct Peroxygenation of Various Olefinic Compounds with Molecular Oxygen and Triethylsilane Catalyzed by a Cobalt(II) Complex. Bull Chem Soc Jpn. 1990;63:1305–1310. [Google Scholar]
- 105.Isayama S, Mukaiyama T. A New Method for Preparation of Alcohols from Olefins with Molecular Oxygen and Phenylsilane by the use of Bis(acetylacetonato)cobalt(II) Chem Lett. 1989;18:1071–1074. [Google Scholar]
- 106.Inoki S, Kato K, Isayama S, Mukaiyama T. A New and Facile Method for the Direct Preparation of α-Hydroxycarboxylic Acid Esters from α,β-Unsaturated Carboxylic Acid Esters with Molecular Oxygen and Phenylsilane Catalyzed by Bis(dipivaloylmethanato)manganese(II) Complex. Chem Lett. 1990;19:1869–1872. [Google Scholar]
- 107.Magnus P, Payne AH, Waring MJ, Scott DA, Lynch V. Conversion of α,β-Unsaturated Ketones into α-Hydroxy Ketones using an MnIII Catalyst, Phenylsilane and Dioxygen: Acceleration of Conjugate Hydride Reduction by Dioxygen. Tetrahedron Lett. 2000;41:9725–9730. [Google Scholar]
- 108.Magnus P, Scott DA, Fielding MR. Direct Conversion of α,β-Unsaturated Nitriles into Cyanohydrins Using Mn(dpm)3 Catalyst, Dioxygen and Phenylsilane. Tetrahedron Lett. 2001;42:4127–4129. [Google Scholar]
- 109.Sato M, Gunji Y, Ikeno T, Yamada T. Stereoselective Preparation of α–Hydroxycarboxamide by Manganese Complex Catalyzed Hydration of α,β-Unsaturated Carboxamide with Molecular Oxygen and Phenylsilane. Chem Lett. 2004;33:1304–1305. [Google Scholar]
- 110.Lee HN, Baik JS, Han; S-b. (Salen)Mn-Catalyzed Oxygenation of Vinyl Arenes to the Corresponding Alcohols in the Presence of Sodium Borohydride. Bull Korean Chem Soc. 1999;20:867–868. [Google Scholar]
- 111.Lee NHBJC, Baik JS, Han C-H, Han S-b. Development of Mn(III)(Schiff Base) Complexes for the Catalyst of Olefin Oxygenation to Alcohols in the Presence of NaBH4. Bull Korean Chem Soc. 2002;23:1365–1366. [Google Scholar]
- 112.Lee NH, Baik JS, Han S-h. Development of Manganese(III) Acetate along with Schiff-Base Ligands as the Catalyst for the Oxygenation of Olefins in the O2/NaBH4 System. Bull Korean Chem Soc. 2004;25:1455–1456. [Google Scholar]
- 113.Baik JS, Lee NH. Mechanistic Studies on the O2-mediated Oxidation of Olefins in the Presence of (Schiff-base)Mn(III) Catalyst and NaBH4. Bull Korean Chem Soc. 2006;27:765–766. [Google Scholar]
- 114.Baik JS, Han S-b, Lee NH. (Schiff-Base)Mn(III)-Catalyzed Hydroxylation of α, β-Unsaturated Esters Using Molecular Oxygen in the Presence of Metal Hydrides. Bull Korean Chem Soc. 2006;27:333–334. [Google Scholar]
- 115.Waser J, Gaspar B, Nambu H, Carreira EM. Hydrazines and Azides via the Metal-Catalyzed Hydrohydrazination and Hydroazidation of Olefins. J Am Chem Soc. 2006;128:11693–11712. doi: 10.1021/ja062355+. [DOI] [PubMed] [Google Scholar]
- 116.Obradors C, Martinez RM, Shenvi RA. Ph(i-PrO)SiH2: An Exceptional Reductant for Metal-Catalyzed Hydrogen Atom Transfers. J Am Chem Soc. 2016;138:4962–4971. doi: 10.1021/jacs.6b02032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.O’Neill PM, Hindley S, Pugh MD, Davies J, Bray PG, Park BK, Kapu DS, Ward SA, Stocks PA. Co(thd)2: a Superior Catalyst for Aerobic Epoxidation and Hydroperoxysilylation of Unactivated Alkenes: Application to the Synthesis of Spiro-1,2,4-Trioxanes. Tetrahedron Lett. 2003;44:8135–8138. [Google Scholar]
- 118.Tokuyasu T, Kunikawa S, Masuyama A, Nojima M. Co(III)-Alkyl Complex- and Co(III)-Alkylperoxo Complex-Catalyzed Triethylsilylperoxidation of Alkenes with Molecular Oxygen and Triethylsilane. Org Lett. 2002;4:3595–3598. doi: 10.1021/ol0201299. [DOI] [PubMed] [Google Scholar]
- 119.Wu JM, Kunikawa S, Tokuyasu T, Masuyama A, Nojima M, Kim HS, Wataya Y. Co-catalyzed Autoxidation of Alkene in the Presence of Silane. the Effect of the Structure of Silanes on the Efficiency of the Reaction and on the Product Distribution. Tetrahedron. 2005;61:9961–9961. [Google Scholar]
- 120.Tokuyasu T, Kunikawa S, McCullough KJ, Masuyama A, Nojima M. Synthesis of Cyclic Peroxides by Chemo- and Regioselective Peroxidation of Dienes with Co(II)/O2/Et3SiH. J Org Chem. 2005;70:251–260. doi: 10.1021/jo048359j. [DOI] [PubMed] [Google Scholar]
- 121.Tokuyasu T, Kunikawa S, Abe M, Masuyama A, Nojima M, Kim HS, Begum K, Wataya Y. Synthesis of Antimalarial Yingzhaosu A Analogues by the Peroxidation of Dienes with Co(II)/O2/Et3SiH. J Org Chem. 2003;68:7361–7367. doi: 10.1021/jo030107f. [DOI] [PubMed] [Google Scholar]
- 122.Tokuyasu T, Masuyama A, Nojima M, McCullough KJ, Kim HS, Wataya Y. Yingzhaosu A Analogues: Synthesis by the Ozonolysis of Unsaturated Hydroperoxides, Structural Analysis and Determination of Anti-Malarial Activity. Tetrahedron. 2001;57:5979–5989. [Google Scholar]
- 123.Hamada Y, Tokuhara H, Masuyama A, Nojima M, Kim HS, Ono K, Ogura N, Wataya Y. Synthesis and Notable Antimalarial Activity of Acyclic Peroxides, 1-(Alkyldioxy)-1-(methyldioxy)cyclododecanes. J Med Chem. 2002;45:1374–1378. doi: 10.1021/jm010473w. [DOI] [PubMed] [Google Scholar]
- 124.Kim HS, Begum K, Ogura N, Wataya Y, Tokuyasu T, Masuyama A, Nojima M, McCullough KJ. Antimalarial Activity of Yingzhaosu A Analogues. J Med Chem. 2002;45:4732–4736. doi: 10.1021/jm020208q. [DOI] [PubMed] [Google Scholar]
- 125.Ito T, Tokuyasu T, Masuyama A, Nojima M, McCullough KJ. Synthesis of Novel Macrocyclic Peroxides by Bis(sym-collidine)iodine (I) Hexafluorophosphate-Mediated Cyclization of Unsaturated Hydroperoxides and Unsaturated Alcohols. Tetrahedron. 2003;59:525–536. [Google Scholar]
- 126.Masuyama A, Nojima M. Development of New Antimalarial Reagents based on Cyclic Peroxides. Kagaku to Kogyo (Osaka, Jpn) 2005;79:250–258. [Google Scholar]
- 127.Gemma S, Martí F, Gabellieri E, Campiani G, Novellino E, Butini S. Synthetic Studies Toward 1,2-Dioxanes as Precursors of Potential Endoperoxide-Containing Antimalarials. Tetrahedron Lett. 2009;50:5719–5722. [Google Scholar]
- 128.Gemma S, Gabellieri E, Coccone SS, Martí F, Taglialatela-Scafati O, Novellino E, Campiani G, Butini S. Synthesis of Dihydropakortin, 6-epi-Dihydroplakortin, and their C10-Desethyl Analogues. J Org Chem. 2010;75:2333–2340. doi: 10.1021/jo1001559. [DOI] [PubMed] [Google Scholar]
- 129.Dai P, Dussault PH. Intramolecular Reactions of Hydroperoxides and Oxetanes: Stereoselective Synthesis of 1,2-Dioxolanes and 1,2-Dioxanes. Org Lett. 2005;7:4333–4335. doi: 10.1021/ol051407h. [DOI] [PubMed] [Google Scholar]
- 130.Barnych B, Vatèle JM. Exploratory Studies toward the Synthesis of Peroxylactone Unit of Plakortolides. Tetrahedron. 2012;68:3717–3724. [Google Scholar]
- 131.Ghorai P, Dussault PH, Hu C. Synthesis of Spiro-bisperoxyketals. Org Lett. 2008;10:2401–2404. doi: 10.1021/ol800657m. [DOI] [PubMed] [Google Scholar]
- 132.Boto A, Hernández R, Velázquez SM, Suárez E, Prangé T. Stereospecific Synthesis of 1,2-Dioxolanes by Alkoxy Radical β-Fragmentation of Steroidal Cyclic Peroxyhemiacetals. J Org Chem. 1998;63:4697–4705. [Google Scholar]
- 133.Laurent SAL, Boissier J, Coslédan F, Gornitzka H, Robert A, Meunier B. Synthesis of “Trioxaquantel”® Derivatives as Potential New Antischistosomal Drugs. Eur J Org Chem. 2008;89:895–913. [Google Scholar]
- 134.Jung M, Lee K, Kim H, Park M. Recent Advances in Artemisinin and Its Derivatives as Antimalarial and Antitumor Agents. Curr Med Chem. 2004;11:1265–1284. doi: 10.2174/0929867043365233. [DOI] [PubMed] [Google Scholar]
- 135.Xiao S-h, Mei J-y, Jiao P-y. Schistosoma japonicum-Infected Hamsters (Mesocricetus auratus) used as a Model in Experimental Chemotherapy with Praziquantel, Artemether, and OZ Compounds. Parasitol Res. 2011;108:431–437. doi: 10.1007/s00436-010-2084-7. [DOI] [PubMed] [Google Scholar]
- 136.Boissier J, Coslédan F, Robert A, Meunier B. In Vitro Activities of Trioxaquines against Schistoma mansoni. Antimicrob Agents Chemother. 2009;53:4903–4906. doi: 10.1128/AAC.00640-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pradines V, Portela J, Boissier J, Cosledan F, Meunier B, Robert A. Trixaquine PA1259 Alkylates Heme in the Blood-Feeding Parasite Schistosoma mansoni. Antimicrob Agents Chemother. 2011;55:2403–2405. doi: 10.1128/AAC.00082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maurya R, Soni A, Anand D, Ravi M, Raju KSR, Taneja I, Naikade NK, Puri SK, Wahajuddin K, Anojiya S, et al. Synthesis and Antimalarial Activity of 3,3-Sprioanellated 5,6-Disubstituted 1,2,4-Trioxanes. Med Chem Lett. 2013;4:165–169. doi: 10.1021/ml300188t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.O’Neil PM, Pugh M, Davies J, Ward SA, Park BK. Regioselective Mukaiyama hydroperoxysilylation of 2-alkyl- or 2-aryl-prop-2-en-1-ols: application to the synthesis of 1,2,4-trioxanes. Tetrahedron Lett. 2001;42:4569–4571. [Google Scholar]
- 140.Oh CH, Kang JH. A Convenient Synthesis of Substituted 1,2,4-Trioxepanes via Co(II) Catalyzed Oxygenation of Cinnamyl Alcohol. Tetrahedron Lett. 1998;39:2771–2774. [Google Scholar]
- 141.Brindisi M, Gemma S, Kunjir S, Di Cerbo L, Brogi S, Parapini S, D’Alessandro S, Taramelli D, Habluetzel A, Tapanelli S, et al. Synthetic spirocyclic endoperoxides: new antimalarial scaffolds. Med Chem Commun. 2015;6:357–362. [Google Scholar]
- 142.Terent’ev AO, Borisov DA, Vil VA, Dembitsky VM. Synthesis of Five- and Six-Membered Cyclic Organic Peroxides: Key Transformations into Peroxide Ring-Retaining Products. Beilstein J Org Chem. 2014;10:34–114. doi: 10.3762/bjoc.10.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.McCullough KJ, Nojima M. Recent Advances in the Chemistry of Cyclic Peroxides. Curr Org Chem. 2001;5:601–636. [Google Scholar]
- 144.Hurlocker B, Miner MR, Woerpel KA. Synthesis of Silyl Monoperoxyketals by Regioselective Cobalt-Catalyzed Peroxidation of Silyl Enol Ethers: Application to the Synthesis of 1,2-Dioxolanes. Org Lett. 2014;16:4280–4283. doi: 10.1021/ol5020015. [DOI] [PubMed] [Google Scholar]
- 145.Hilf JA, Witthoft LW, Woerpel KA. An SN1-type Reaction to Form the 1,2-Dioxepane Ring: Synthesis of 10,12-Peroxycalamenene. J Org Chem. 2015;80:8262–8267. doi: 10.1021/acs.joc.5b01326. [DOI] [PubMed] [Google Scholar]
- 146.Matsushita Y-i, Matsui T, Sugamoto K. Cobalt(II) Porphyrin-Catalyzed Oxidation of Olefins to Ketones with Molecular Oxygen and Triethylsilane in 2-Propanol. Chem Lett. 1992;21:1381–1384. [Google Scholar]
- 147.Matsushita Y-i, Sugamoto K, Matsui T. Novel Method for Preparation of 4-Oxo-2-alkenoic Acid Derivatives from 2,4-Alkadienoic Acid Derivatives by Cobalt(II) Porphyrin-catalyzed Oxygenation. Chem Lett. 1992;21:2165–2168. [Google Scholar]
- 148.Matsusita Y-i, Sugamoto K, Matsui T. One-Pot Preparation of Alcohols from Aromatic Olefins and Acrylic Acid Derivatives by Cobalt(II) Porphyrin-Catalyzed Reductive Oxygenation Followed by Reduction with Trimethyl Phosphite. Chem Lett. 1993;22:925–928. [Google Scholar]
- 149.Matsushita Y-i, Sugamoto K, Nakama T, Matsui T. Synthesis of γ-Hydroperoxy-α,β–Unsaturated Carbonyl Compounds from α,β,γ,δ-Unsaturated Carbonyl Compounds by Cobalt(II) Porphyrin-catalysed Hydroperoxygenation. J Chem Soc, Chem Commun. 1995:567–568. [Google Scholar]
- 150.Matsushita Y-i, Sugamoto K, Nakama T, Sakamoto T, Matsui T, Nakayama M. γ-Selective Hydroxylation of α,β,γ,δ-Unsaturated Carbonyl Compounds and its Application to Syntheses of (±)-6-Hydroxyshogaol and Related Furanoids. Tetrahedron Lett. 1995;36:1879–1882. [Google Scholar]
- 151.Sugamoto K, Matsushita Y-i, Matsui T. Direct Hydroperoxygenation of Conjugated Olefins Catalyzed by Cobalt(II) Porphyrin. J Chem Soc, Perkin Trans. 998:3989–3998. [Google Scholar]
- 152.Kato K, Mukaiyama T. A Convenient Method for the Direct Preparation of Ketones from Vinylsilanes with Molecular Oxygen Catalyzed by a Cobalt(II) Complex. Chem Lett. 1989;18:2233–2236. [Google Scholar]
- 153.Taylor NH, Thomas EJ. 1,5-Induction in Reactions of 4-Alkoxy-2-trimethylsilylalk-2-enyl(tributyl)stannanes with Aldehydes. Tetrahedron. 1999;55:8757–8768. [Google Scholar]
- 154.Nagatomo M, Koshimizu M, Masuda K, Tabuchi T, Urabe D, Inoue M. Total Synthesis of Ryanodol. J Am Chem Soc. 2014;136:5916–5919. doi: 10.1021/ja502770n. [DOI] [PubMed] [Google Scholar]
- 155.Nagatomo M, Hagiwara K, Masuda K, Koshimizu M, Kawamata T, Matsui Y, Urabe D, Inoue M. Symmetry-Driven Strategy for the Assembly of the Core Tetracycle of (+)-Ryanodine: Synthetic Utility of a Cobalt-Catalyzed Olefin Oxidation and α-Alkoxy Bridgehead Radical Reaction. Chem Eur J. 2016;22:222–229. doi: 10.1002/chem.201503640. [DOI] [PubMed] [Google Scholar]
- 156.Shigehisa H, Aoki T, Yamaguchi S, Shimizu N, Hiroya K. Hydroalkoxylation of Unactivated Olefins with Carbon Radicals and Carbocation Species as Key Intermediates. J Am Chem Soc. 2013;135:10306–10309. doi: 10.1021/ja405219f. [DOI] [PubMed] [Google Scholar]
- 157.Shigehisa H. Functional Group Tolerant Markovnikov-Selective Hydrofunctionalization of Unactivated Olefins Using a Cobalt Complex as Catalyst. Synlett. 2015;26:2479–2484. [Google Scholar]
- 158.Shigehisa H, Kikuchi H, Hiroya K. Markovnikov-Selective Addition of Fluorous Solvents to Unactivated Olefins Using a Co Catalyst. Chem Pharm Bull. 2016;64:371–374. doi: 10.1248/cpb.c15-01024. [DOI] [PubMed] [Google Scholar]
- 159.Merlic CA, Hietbrink BN, Houk KN. Donor and Acceptor Properties of the Chromium Tricarbonyl Substitutent in Benzylic and Homobenzylic Anions, Cations and Radicals. J Org Chem. 2001;66:6738–6744. doi: 10.1021/jo010620y. [DOI] [PubMed] [Google Scholar]
- 160.Matsushita Y-i, Furusawa H, Matsui T, Nakayama M. Total Synthesis of (−)-Pyrenophorin via Cobalt(II) Porphyrin-Catalyzed Oxygenation of Ethyl (2E,4E,7R)-7-Acetoxy-2,4-octadienoate. Chem Lett. 1994;23:1083–1084. [Google Scholar]
- 161.Matsushita Y-i, Sugamoto K, Nakama T, Matsui T, Hayashi Y, Uenakai K. Enantioselective Syntheses of 10-Oxo-11(E)-octadecen-13-olide and Related Fatty Acid. Tetrahedron Lett. 1997;38:6055–6058. [Google Scholar]
- 162.Tanaka M, Mitsuhashi H, Maruno M, Wakamatsu T. First Total Synthesis of Optically Pure Metabolites of Gomisin A. Synlett. 1994:604–606. [Google Scholar]
- 163.Tanaka M, Ikeya Y, Mitsuhashi H, Maruno M, Wakamatsu T. Total Syntheses of the Metabolites of Schizandrin. Tetrahedron. 1995;51:11703–11724. [Google Scholar]
- 164.Tanaka M, Mukaiyama C, Mitsuhashi H, Maruno M, Wakamatsu T. Synthesis of Optically Pure Gomisi Lignans: The Total Synthesis of (+)-Schizandrin, (+)-Gomisin A, and (+)-Isoschizandrin in Naturally Occurring Forms. J Org Chem. 1995;60:4339–4352. [Google Scholar]
- 165.Wakamatsu T, Tanaka M, Mitsuhashi H, Maruno M. Total Synthesis of the Major Metabolites of Gomisin A. Synthesis of Homochiral Met A-II, Met A-III, and Met F. Heterocycles. 1996;42:359–374. [Google Scholar]
- 166.Tietze LF, Raschke T. Enantioselective Total Synthesis of a Natural Norsesquiterpene of the Calamene Group by a Silane-Terminated Intramolecular Heck Reaction. Synlett. 1995:597–598. [Google Scholar]
- 167.Xu XX, Dong HQ. Enantioselective Total Syntheses and Stereochemical Studies of All Four Stereoisomers of Yingzhaosu C. J Org Chem. 1995;60:3039–3044. [Google Scholar]
- 168.Enders D, Ridder A. First Asymmetric Synthesis of Stigmolone: The Fruiting Body Inducing Pheromone of the Myxobacterium Stigmatella Aurantiaca. Synthesis. 2000:1848–1851. [Google Scholar]
- 169.Enders D. In: Asymmetric Synthesis. Morrision JD, editor. 3B. Academic; Orlando: 1984. p. 275. [Google Scholar]
- 170.Magnus P, Westlund N. Synthesis of (±)-Lahadinine B and (±)-11-Methoxykopsilongine. Tetrahedron Lett. 2000;41:9369–9372. [Google Scholar]
- 171.Magnus P, Hobson LA, Westlund N, Lynch V. Synthesis of (±)-Demethoxypauciflorine B and (±)-Pauciflorine B from (±)-11,12-Demethoxylahadinine B and (±)-Lahadinine B, Respectively via a Peroxycarbanolamine Fragmentation Reaction. Tetrahedron Lett. 2001;42:993–997. [Google Scholar]
- 172.Magnus P, Gazzard L, Hobson L, Payne AH, Rainey TJ, Westlund N, Lynch V. Synthesis of the Kopsia Alkaloids (±)-Pauciflorine B, (±)-Lahadinine B, (±)-Kopsidasine, (±)-Kopsidasine-N-oxide, (±)-Kopsijasminilam and (±)-11-Methoxykopsilongine. Tetrahedron. 2002;58:3423–3443. [Google Scholar]
- 173.Walker AJ, Bruce NC. Combined Biological and Chemical Catalysis in the Preparation of Oxycodone. Tetrahedron. 2004;60:561–568. [Google Scholar]
- 174.Bondar D, Liu J, Müller T, Paquette LA. Pectenotoxin-2 Synthetic Studies. 2. Construction and Conjoining of ABC and DE Eastern Hemisphere Subtargets. Org Lett. 2005;7:1813–1816. doi: 10.1021/ol0504291. [DOI] [PubMed] [Google Scholar]
- 175.Halim R, Brimble MA. Synthetic Studies Towards the Pectenotoxins: a Review. Org Biomol Chem. 2006;4:4048–4058. doi: 10.1039/b611531b. [DOI] [PubMed] [Google Scholar]
- 176.Kuramochi A, Usuda H, Yamatsugu K, Kanai M, Shibasaki Total Synthesis of (±)-Garsubellin A. M. J Am Chem Soc. 2005;127:14200–14201. doi: 10.1021/ja055301t. [DOI] [PubMed] [Google Scholar]
- 177.Njardarson JT. Synthetic Efforts Toward [3.3. 1] Bridged Bicyclic Phloroglucinol Natural Products. Tetrahedron. 2011;67:7631–7666. doi: 10.1016/j.tet.2011.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shenvi RA, Guerrero CA, Shi J, Li CC, Baran PS. Synthesis of (+)-Cortistatin A. J Am Chem Soc. 2008;130:7241–7243. doi: 10.1021/ja8023466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Shi J, Shigehisa H, Guerrero CA, Shenvi RA, Li CC, Baran PS. Stereodivergent Synthesis of 17-α and 17-β-Aryl Steroids: Application and Biological Evaluation of D-Ring Cortistatin Analogues. Angew Chem, Int Ed. 2009;48:4328–4331. doi: 10.1002/anie.200901116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Shi J, Manolikakes G, Yeh CH, Guerrero CA, Shenvi RA, Shigehisa H, Baran PS. Scalable Synthesis of Cortistatin A and Related Structures. J Am Chem Soc. 2011;133:8014–8027. doi: 10.1021/ja202103e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chen DYK, Tseng CC. Chemistry of the Cortistatins- a Novel Class of Anti-Angiogenic Agents. Org Biomol Chem. 2010;8:2900–2911. doi: 10.1039/c003935g. [DOI] [PubMed] [Google Scholar]
- 182.Schindler CS, Stephenson CRJ, Carreira EM. Enantioselective Synthesis of the Core of Banyaside, Suomilide, and Spumigin HKVV. Angew Chem Int Ed. 2008;47:8852–8855. doi: 10.1002/anie.200803655. [DOI] [PubMed] [Google Scholar]
- 183.Schindler CS, Bertschi l, Carreira EM. Total Synthesis of Nominal Banyaside B: Structural Revision of the Glycosylation Site. Angew Chem Int Ed. 2010;49:9229–9232. doi: 10.1002/anie.201004047. [DOI] [PubMed] [Google Scholar]
- 184.Va P, Campbell EL, Robertson WM, Boger DL. Total Synthesis and Evaluation of a Key Series of C5-Substituted Vinblastine Derivatives. J Am Chem Soc. 2010;132:8489–8495. doi: 10.1021/ja1027748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sasaki Y, Kato D, Boger DL. Asymmetric Total Synthesis of Vindorosine, Vindoline, and Key Vinblastine Analogues. J Am Chem Soc. 2010;132:13533–13544. doi: 10.1021/ja106284s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gotoh H, Duncan KK, Robertson WM, Boger DL. 10′-Fluorovinblastine and 10′-Fluorovincristine: Synthesis of a Key Series of Modified Vinca Alkaloids. ACS Med Chem Lett. 2011;2:948–952. doi: 10.1021/ml200236a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Barker TJ, Duncan KK, Otrubova K, Boger DL. Potent Vinblastine C20′ Ureas Displaying Additionally Improved Activity Against a Vinblastine-Resistant Cancer Cell Line. ACS Med Chem Lett. 2013;4:985–988. doi: 10.1021/ml400281w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Leggans EK, Duncan KK, Barker TJ, Schleicher KD, Boger DL. A Remarkable Series of Vinblastine Analogues Displaying Enhanced Activity and an Unprecedented Tubulin Binding Steric Tolerance: C20′ Urea Derivatives. J Med Chem. 2013;56:628–639. doi: 10.1021/jm3015684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Schleicher KD, Sasaki Y, Tam A, Kato D, Duncan KK, Boger DL. Total Synthesis and Evaluation of Vinblastine Analogues Containing Systematic Deep-Seated Modifications in the Vindoline Subunit Ring System: Core Redesign. J Med Chem. 2013;56:483–495. doi: 10.1021/jm3014376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Allemann O, Brutsch M, Lukesh JC, Brody DM, Boger DL. Synthesis of a Potent Vinblastine: Rationally Designed Added Benign Complexity. J Am Chem Soc. 2016 doi: 10.1021/jacs.6b04330. Online early access. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cassayre J, Winkler T, Pitterna T, Quaranta L. Application of Mn(III)-Catalysed Olefin Hydration Reaction to the Selective Functionalisation of Avermectin B1. Tetrahedron Lett. 2010;51:1706–1709. [Google Scholar]
- 192.Liu G, Romo D. Total Synthesis of (+)-Omphadiol. Angew Chem Int Ed. 2011;50:7537–7540. doi: 10.1002/anie.201102289. [DOI] [PubMed] [Google Scholar]
- 193.Peng F, Danishefsky SJ. Toward the Total Synthesis of Maoecrystal V: an Intramolecular Diels–Alder Route to the Maoecrystal V Pentacyclic Core with the Appropriate Relative Stereochemistry. Tetrahedron Lett. 2011;52:2104–2106. doi: 10.1016/j.tetlet.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Herzon SB, Calandra NA, King SM. Efficient Entry to the Hasubanan Alkaloids: First Enantioselective Total Syntheses of (−)-Hasubanonine, (−)-Runanine, (−)-Delavayine, and (+)-Periglaucine B. Angew Chem Int Ed. 2011;50:8863–8868. doi: 10.1002/anie.201102226. [DOI] [PubMed] [Google Scholar]
- 195.Calandra NA, King S, Herzon SB. Development of Enantioselective Synthetic Routes to the Hasubanan and Acutumine Alkaloids. J Org Chem. 2013;78:10031–10057. doi: 10.1021/jo401889b. [DOI] [PubMed] [Google Scholar]
- 196.King SM, Herzon SB. Substrate-Modified Functional Group Reactivity: Hasubanan and Acutumine Alkaloid Syntheses. J Org Chem. 2014;79:8937–8947. doi: 10.1021/jo501516x. [DOI] [PubMed] [Google Scholar]
- 197.Endoma-Arias MAA, Hudlicky T. A Short Synthesis of Nonracemic Iodocyclohexene Carboxylate Fragment for Kibdelone and Congeners. Tetrahedron Lett. 2011;52:6632–6634. [Google Scholar]
- 198.Farcet JB, Himmelbauer M, Mulzer J. A Non-Photochemical Approach to the Bicyclo[3.2.0]heptane Core of Bielschowskysin. Org Lett. 2012;14:2195–2197. doi: 10.1021/ol3006419. [DOI] [PubMed] [Google Scholar]
- 199.Jeker OF, Carreira EM. Total Synthesis and Stereochemical Reassignment of (±)-Indoxamycin B. Angew Chem Int Ed. 2012;51:3474–3477. doi: 10.1002/anie.201109175. [DOI] [PubMed] [Google Scholar]
- 200.Barnych B, Vatèle JM. Total Synthesis of seco-Plakortolide E and (−)-ent-Plakortolide I: Absolute Configurational Revision of Natural Plakortolide I. Org Lett. 2012;14:564–567. doi: 10.1021/ol203185f. [DOI] [PubMed] [Google Scholar]
- 201.Vatèle JM, Barnych B. Synthetic Studies toward the Bicyclic Peroxylactone Core of Plakortolides. Synlett. 2011;2011:1912–1916. [Google Scholar]
- 202.Barnych B, Fenet B, Vatèle JM. Asymmetric Synthesis of Andavadoic Acid via Base-Catalyzed 5-exo-tet Cyclization of a β-Hydroperoxy Epoxide. Tetrahedron. 2013;69:334–340. [Google Scholar]
- 203.Xu ZJ, Tan DX, Wu Y. Synthesis and Structural Reassignment of Plakinidone. Org Lett. 2015;17:5092–5095. doi: 10.1021/acs.orglett.5b02599. [DOI] [PubMed] [Google Scholar]
- 204.Chen HJ, Wu Y. Expeditious Entry to the Chamigrane Endoperoxide Family of Natural Products. Org Lett. 2015;17:592–595. doi: 10.1021/ol503603t. [DOI] [PubMed] [Google Scholar]
- 205.Gregg C, Gunawan C, Ng AWY, Wimala S, Wickremasinghe S, Rizzacasa MA. Formal Total Synthesis of Spirangien A. Org Lett. 2013;15:516–519. doi: 10.1021/ol3033253. [DOI] [PubMed] [Google Scholar]
- 206.Zahel M, Metz P. A Concise Enantioselective Synthesis of the Guaiane Sesquiterpene (−)-Oxyphyllol. Beilstein J Org Chem. 2013;9:2028–3032. doi: 10.3762/bjoc.9.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Renata H, Zhou Q, Baran PS. Strategic Redox Relay Enables A Scalable Synthesis of Ouabagenin, A Bioactive Cardenolide. Science. 2013;339:59–63. doi: 10.1126/science.1230631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Renata H, Zhou Q, Dünstl G, Felding J, Merchant RR, Yeh CH, Baran PS. Development of a Concise Synthesis of Ouabagenin and Hydroxylated Corticosteroid Analogues. J Am Chem Soc. 2015;137:1330–1340. doi: 10.1021/ja512022r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Shigehisa H, Suwa Y, Furiya N, Nakaya Y, Fukushima M, Ichihashi Y, Hiroya K. Stereocontrolled Synthesis of Trichodermatide A. Angew Chem Int Ed. 2013;52:3646–3649. doi: 10.1002/anie.201210099. [DOI] [PubMed] [Google Scholar]
- 210.Shigehisa H, Kikuchi H, Suzuki T, Hiroya K. The Revised Structure of Trichodermatide A. Eur J Org Chem. 2015;2015:7670–7673. [Google Scholar]
- 211.Tietze LF, Jackenkroll S, Hierold J, Ma L, Waldecker B. A Domino Approach to the Enantioselective Total Syntheses of Blennolide C and Gonytolide C. Chem Eur J. 2014;20:8628–8635. doi: 10.1002/chem.201402495. [DOI] [PubMed] [Google Scholar]
- 212.Urabe D, Nagatomo M, Hagiwara K, Masuda K, Inoue M. Symmetry-Driven Synthesis of 9-Demethyl-10,15-dideoxyryanodol. Chem Sci. 2013;4:1615–1619. [Google Scholar]
- 213.Cherney EC, Lopchuk JM, Green JC, Baran PS. A Unified Approach to ent-Atisane Diterpenes and Related Alkaloids: Synthesis of (−)-Methyl Atisenoate, (−)-Isoatisine, and the Hetidine Skeleton. J Am Chem Soc. 2014;136:12592–12595. doi: 10.1021/ja507321j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Michaudel Q, Journot G, Regueiro-Ren A, Goswami A, Guo Z, Tully TP, Zou L, Ramabhadran RO, Houk KN, Baran PS. Improving Physical Properties via C-H Oxidation: Chemical and Enzymatic Approaches. Angew Chem Int Ed. 2014;53:12091–12096. doi: 10.1002/anie.201407016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Concepción JI, Francisco CG, Hernμndez R, Salazar JA, Suárez E. Intramolecular Hydrogen Abstraction. Iodosobenzene Diacetate an Efficient and Convenient Reagent for Alkoxy Radical Generation. Tetrahedron Lett. 1984;25:1953–1956. [Google Scholar]
- 216.Hu X, Maimone TJ. Four-Step Synthesis of the Antimalarial Cardamom Peroxide via an Oxygen Stitching Strategy. J Am Chem Soc. 2014;136:5287–5290. doi: 10.1021/ja502208z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tran DN, Cramer N. Biomimetic Synthesis of (+)-Ledene, (+)-Viridiflorol, (−)-Palustrol, (+)-Spathulenol, and Psiguadial A, C, and D via the Platform Terpene (+)-Bicyclogermacrene. Chem Eur J. 2014;20:10654–10660. doi: 10.1002/chem.201403082. [DOI] [PubMed] [Google Scholar]
- 218.Cramer N, Seiser T. β-Carbon Elimination from Cyclobutanols: A Clean Access to Alkylrhodium Intermediates Bearing a Quaternary Stereogenic Center. Synlett. 2011;4:449–460. [Google Scholar]
- 219.Feng X, Jiang G, Xia Z, Hu J, Wan X, Gao JM, Lai Y, Xie W. Total Synthesis of (−)-Conolutinine. Org Lett. 2015;17:4428–4431. doi: 10.1021/acs.orglett.5b02046. [DOI] [PubMed] [Google Scholar]
- 220.Xu Z, Wang Q, Zhu J. Total Syntheses of (−)-Mersicarpine, (−)-Scholarisine G, (+)-Melodinine E, (−)-Leuconoxine, (−)-Leuconolam, (−)-Leuconodine A, (+)-Leuconodine F, and (−)-Leuconodine C: Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon for Scholarisine G and Leuconodines A and C. J Am Chem Soc. 2015;137:6712–6724. doi: 10.1021/jacs.5b03619. [DOI] [PubMed] [Google Scholar]
- 221.Magnus P, Waring MJ, Scott DA. Conjugate Reduction of α,β-Unsaturated Ketones using an MnIII Catalyst, Phenylsilane and Isopropyl Alcohol. Tetrahedron Lett. 2000;41:9731–9733. [Google Scholar]
- 222.Feng Y, Holte D, Zoller J, Umemiya S, Simke LR, Baran PS. Total Synthesis of Verruculogen and Fumitremorgin A Enabled by Ligand-Controlled C-H Borylation. J Am Chem Soc. 2015;137:10160–10163. doi: 10.1021/jacs.5b07154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.See YY, Herrmann AT, Aihara Y, Baran PS. Scalable C-H Oxidation with Copper: Synthesis of Polyoxypregnanes. J Am Chem Soc. 2015;137:13776–13779. doi: 10.1021/jacs.5b09463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhu D, Yu B. Total Synthesis of Linckosides A and B, the Representative Starfish Polyhydroxysteroid Glycosides with Neuritogenic Activities. J Am Chem Soc. 2015;137:15098–15101. doi: 10.1021/jacs.5b11276. [DOI] [PubMed] [Google Scholar]
- 225.Song L, Zhu G, Liu Y, Liu B, Qin S. Total Synthesis of Atisane-Type Diterpenoids: Application of Diels-Alder Cycloadditions of Podocarpane-Type Unmasked ortho-Benzoquinones. J Am Chem Soc. 2015;137:13706–13714. doi: 10.1021/jacs.5b08958. [DOI] [PubMed] [Google Scholar]
- 226.Liu XY, Qin Y. Ongoing Pursuit of Diterpenoid Alkaloids: A Synthetic View. Asian J Org Chem. 2015;4:1010–1019. [Google Scholar]
- 227.Keβberg A, Metz P. Utilizing an o-Quinone Methide in Asymmetric Transfer Hydrogenation: Enantioselective Synthesis of Brosimine A, Brosimine B, and Brosimacutin L. Angew Chem Int Ed. 2016;55:1160–1163. doi: 10.1002/anie.201507269. [DOI] [PubMed] [Google Scholar]
- 228.Kawamura S, Chu H, Felding J, Baran PS. Nineteen-Step Total Synthesis of (+)-Phorbol. Nature. 2016;532:90–93. doi: 10.1038/nature17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Yang P, Yao M, Li J, Li Y, Li A. Total Synthesis of Rubriflordilactone B. Angew Chem Int Ed. 2016;55:6964–6968. doi: 10.1002/anie.201601915. [DOI] [PubMed] [Google Scholar]
- 230.Chung W-j, Carlson JS, Vanderwal CD. General Approach to the Synthesis of the Chlorosulfolipids Danicalipin A, Mytilipin A, and Malhamensilipin A in Enantioenriched Form. J Org Chem. 2014;79:2226–2241. doi: 10.1021/jo5000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Zhang Y, Wu JP, Li G, Fandrick KR, Gao J, Tan Z, Johnson J, Li W, Sanyal S, Wang J, Sun X, Lorenz JC, Rodriguez S, Reeves JT, Grinberg N, Lee H, Yee N, Lu BZ, Senanayake CH. An Enantioselective Synthesis of an 11-β-HSD-1 Inhibitor via an Asymmetric Methallylation Catalyzed by (S)-3,3′-F2-BINOL. J Org Chem. 2016;81:2665–2669. doi: 10.1021/acs.joc.6b00189. [DOI] [PubMed] [Google Scholar]
- 232.Liu W, Li H, Cai PJ, Wang Z, Yu ZX, Lei X. Scalable Total Synthesis of rac-Jungermannenones B and C. Angew Chem Int Ed. 2016;55:3112–3116. doi: 10.1002/anie.201511659. [DOI] [PubMed] [Google Scholar]
- 233.Hartung J. Organic Radical Reactions Associated with Nitrogen Monoxide. Chem Rev. 2009;109:4500–4517. doi: 10.1021/cr900085j. [DOI] [PubMed] [Google Scholar]
- 234.Okamoto T, Kobayashi K, Oka S, Tanimoto S. Cobalt-Catalyzed Reaction of Nitric Oxide with Aryl-Substituted Olefins in the Presence of Tetrahydroborate Ion. J Org Chem. 1987;52:5089–5092. [Google Scholar]
- 235.Okamoto T, Kobayashi K, Oka S, Tanimoto S. A High-Yield Regiospecific Synthesis of Keto Oximes from Aryl-Conjugated Ethylenes and Ethyl Nitrite in the Presence of Cobalt Complex and BH4− Ion. J Org Chem. 1988;53:4897–4901. [Google Scholar]
- 236.Kato K, Mukaiyama T. Nitrosation of α,β-Unsaturated Carboxamide with Nitric Oxide and Triethylsilane Catalyzed by Cobalt(II) Complex. Chem Lett. 1990;19:1395–1398. [Google Scholar]
- 237.Kato K, Mukaiyama T. A Novel Method for the Preparation of 2-Hydroxyiminocarboxylic Acid Esters. Cobalt(II) Catalyzed α-Oximation of α,β-Unsaturated Esters with Butyl Nitrite and Phenylsilane. Chem Lett. 1990;19:1917–1920. [Google Scholar]
- 238.Kato K, Mukaiyama T. A Novel and Efficient Method for the Preparation of α-Hydroxyimino Carbonyl Compounds from α,β-Unsaturated Carbonyl Compounds with Butyl Nitrite and Phenylsilane Catalyzed by a Cobalt(II) Complex. Bull Chem Soc Jpn. 1991;64:2948–2953. [Google Scholar]
- 239.Kato K, Mukaiyama T. Iron(III) Complex Catalyzed Nitrosation of Terminal and 1,2-Disubstituted Olefins with Butyl Nitrite and Phenylsilane. Chem Lett. 1992;21:1137–1140. [Google Scholar]
- 240.Emmons WD. The Synthesis of Nitrosoalkane Dimers. J Am Chem Soc. 1957;79:6522–6524. [Google Scholar]
- 241.Gowenlock BG, Richter-Addo GB. Preparations of C-Nitroso Compounds. Chem Rev. 2004;104:3315–3340. doi: 10.1021/cr030450k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Hata E, Kato K, Yamada T, Mukaiyama T. Nitrosation and Nitration of Olefins Using Nitrogen Monoxide. J Synth Org Chem Jpn. 1996;54:728–739. [Google Scholar]
- 243.Sugamoto K, Hamasuna Y, Matsushita Y-i, Matsui T. A Synthesis of Oximes from Olefins by Cobalt(II) Porphyrin-Catalyzed Reduction-Nitrosation. Synlett. 1998;1998:1270–1272. [Google Scholar]
- 244.Prateeptongkum S, Jovel I, Jackstell R, Vogl N, Weckbecker C, Beller M. First Iron-Catalyzed Synthesis of Oximes from Styrenes. Chem Commun. 2009:1990–1992. doi: 10.1039/b900326f. [DOI] [PubMed] [Google Scholar]
- 245.Ray R, Chowdhury AD, Maiti D, Lahiri GK. Iron Catalysed Nitrosation of Olefins to Oximes. Dalton Trans. 2014;43:38–41. doi: 10.1039/c3dt51764k. [DOI] [PubMed] [Google Scholar]
- 246.Desimoni G, Faita G, Righetti PP, Sfulcini A, Tsyganov D. Solvent Effect in Pericyclic Reactions. IX. The Ene Reaction. Tetrahedron. 1994;50:1821–1832. [Google Scholar]
- 247.Jacobsen BM, Arvanitis GM, Eliasen CA, Mitelman R. Ene Reactions of Conjugated Dienes. 2. Dependence of Rate on Degree of Hydrogen Removed and s-Cis or s-Trans Diene Character. J Org Chem. 1985;50:194–201. [Google Scholar]
- 248.Ohno M, Ishazaki K, Eguchi S. Synthesis Of Adamantane Derivatives by Bridgehead Radical Addition to Electron-Deficient Unsaturated Bonds. J Org Chem. 1988;53:1285–1288. [Google Scholar]
- 249.Amaoka Y, Kamijo S, Hoshikawa T, Inoue M. Radical Amination of C(sp3)–H Bonds Using N-Hydroxyphthalimide and Dialkyl Azodicarboxylate. J Org Chem. 2012;77:9959–9969. doi: 10.1021/jo301840e. [DOI] [PubMed] [Google Scholar]
- 250.An DK, Kirakawa H, Okamoto S, Sato F. Electrophilic Amination of Racemic and Non-Racemic Allenyltitaniums. One-Pot Synthesis of α-Hydrazinoalkynes from Propargylic Alcohol Derivatives. Tetrahedron Lett. 1999;40:3737–3740. [Google Scholar]
- 251.Velarde-Ortiz R, Guijarro A, Rieke RD. Electrophilic Amination of Organozinc Halides. Tetrahedron Lett. 1998;39:9157–9160. [Google Scholar]
- 252.Waser J, Carreira EM. Convenient Synthesis of Alkylhydrazides by the Cobalt-Catalyzed Hydrohydrazination Reaction of Olefins and Azodicarboxylates. J Am Chem Soc. 2004;126:5676–5677. doi: 10.1021/ja048698u. [DOI] [PubMed] [Google Scholar]
- 253.Zhirov AM, Aksenov AV. Azodicarboxylates: Synthesis and Functionalization of Organic Compounds. Russ Chem Rev. 2014;83:502–522. [Google Scholar]
- 254.Punniyamurthy T, Bhatia B, Reddy MM, Maikap GC, Iqbal J. A Versatile Cobalt(II)-Schiff Base Catalyzed Oxidation of Organic Substrates with Dioxygen: Scope and Mechanism. Tetrahedron. 1997;53:7649–7670. [Google Scholar]
- 255.Waser J, Carreira EM. Catalytic Hydrohydrazination of a Wide Range of Alkenes with a Simple Mn Complex. Angew Chem Int Ed. 2004;43:4099–4102. doi: 10.1002/anie.200460811. [DOI] [PubMed] [Google Scholar]
- 256.Waser J, González-Gómez JC, Nambu H, Huber P, Carreira EM. Cobalt-Catalyzed Hydrohydrazination of Dienes and Enynes: Access to Allylic and Propargylic Hydrazides. Org Lett. 2005;7:4249–4252. doi: 10.1021/ol0517473. [DOI] [PubMed] [Google Scholar]
- 257.Sato M, Gunji Y, Ikeno T, Yamada T. Stereoselective α-Hydrazination of α,β-Unsaturated Carboxylates Catalyzed by Manganese(III) Complex with Dialkylazodicarboxylate and Phenylsilane. Chem Lett. 2005;34:316–317. [Google Scholar]
- 258.Bunker KD, Sach NW, Huang Q, Richardson PF. Scalable Synthesis of 1-Bicyclo[1.1.1]pentylamine via a Hydrohydrazination Reaction. Org Lett. 2011;13:4746–4748. doi: 10.1021/ol201883z. [DOI] [PubMed] [Google Scholar]
- 259.Gianatassio R, Lopchuk JM, Wang J, Pan CM, Malins LR, Prieto L, Brandt TA, Collins MR, Gallego GM, Sach NW, et al. Strain-release amination. Science. 2016;351:241–246. doi: 10.1126/science.aad6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Zheng J, Qi J, Cui S. Fe-Catalyzed Olefin Hydroamination with Diazo Compounds for Hydrazone Synthesis. Org Lett. 2016;18:128–131. doi: 10.1021/acs.orglett.5b03317. [DOI] [PubMed] [Google Scholar]
- 261.Li W, Liu X, Hao X, Hu X, Chu Y, Cao W, Qin S, Hu C, Lin L, Feng X. New Electrophilic Addition of α-Diazoesters with Ketones for Enantioselective C–N Bond Formation. J Am Chem Soc. 2011;133:15268. doi: 10.1021/ja2056159. [DOI] [PubMed] [Google Scholar]
- 262.Li L, Chen JJ, Li YJ, Bu XB, Liu Q, Zhao YL. Activation of α-Diazocarbonyls by Organic Catalysts: Diazo Group Acting as a Strong N-Terminal Electrophile. Angew Chem Int Ed. 2015;54:12107–12111. doi: 10.1002/anie.201505064. [DOI] [PubMed] [Google Scholar]
- 263.Ford A, Miel H, Ring A, Slattery CN, Maguire AR, McKervey MA. Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem Rev. 2015;115:9981–10080. doi: 10.1021/acs.chemrev.5b00121. [DOI] [PubMed] [Google Scholar]
- 264.Gui J, Pan CM, Jin Y, Qin T, Lo JC, Lee BJ, Spergel SH, Mertzman ME, Pitts WJ, La Cruz TE, et al. Practical Olefin Hydroamination with Nitroarenes. Science. 2015;348:886–891. doi: 10.1126/science.aab0245. [DOI] [PubMed] [Google Scholar]
- 265.Dao HT, Li C, Michaudel Q, Maxwell BD, Baran PS. Hydromethylation of Unactivated Olefins. J Am Chem Soc. 2015;137:8046–8049. doi: 10.1021/jacs.5b05144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zheng J, Wang D, Cui S. Fe-Catalyzed Reductive Coupling of Unactivated Alkenes with β-Nitroalkenes. Org Lett. 2015;17:4572–4575. doi: 10.1021/acs.orglett.5b02294. [DOI] [PubMed] [Google Scholar]
- 267.Waser J, Nambu H, Carreira EM. Cobalt-Catalyzed Hydroazidation of Olefins: Convenient Access to Alkyl Azides. J Am Chem Soc. 2005;127:8294–8295. doi: 10.1021/ja052164r. [DOI] [PubMed] [Google Scholar]
- 268.Carreira E, Gaspar B, Waser J. Cobalt-Catalyzed Synthesis of Tertiary Azides from α,α-Disubstituted Olefins under Mild Conditions Using Commercially Available Reagents. Synthesis. 2007;2007:3839. [Google Scholar]
- 269.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 270.Shigehisa H, Koseki N, Shimizu N, Fujisawa M, Niitsu M, Hiroya K. Catalytic Hydroamination of Unactivated Olefins Using a Co Catalyst for Complex Molecule Synthesis. J Am Chem Soc. 2014;136:13534–13537. doi: 10.1021/ja507295u. [DOI] [PubMed] [Google Scholar]
- 271.Shigehisa H, Nishi E, Fujisawa M, Hiroya K. Cobalt-Catalyzed Hydrofluorination of Unactivated Olefins: A Radical Approach of Fluorine Transfer. Org Lett. 2013;15:5158–5161. doi: 10.1021/ol402696h. [DOI] [PubMed] [Google Scholar]
- 272.Hosogai T, Inamoto N, Okazaki R. Abnormal Addition of Free Radicals to Sterically Hindered Nitrosobenzenes. J Chem Soc C. 1971:3399–3401. [Google Scholar]
- 273.Corey EJ, Gross AW. Methods for the Synthesis of Chiral Hindered Amines. J Org Chem. 1985;50:5391–5393. [Google Scholar]
- 274.Janzen EG. Spin Trapping. Acc Chem Res. 1971;4:31–40. [Google Scholar]
- 275.Inamoto N, Simamura O. Reactions of Nitro Compounds with 1-Cyano-1-methylethyl Radicals Produced by the Decomposition of α,α′-Azobisisobutyronitrile. J Org Chem. 1958;23:408–410. [Google Scholar]
- 276.Gingras BA, Waters WA. Properties and Reactions of Free Alkyl Radicals in Solution. Part VII. Reactions with Quinone Imides, Nitric Oxide, and Nitroso-Compounds. J Chem Soc. 1954:1920–1924. [Google Scholar]
- 277.Zhu K, Shaver MP, Thomas SP. Amine-bis(phenolate) Iron(III)-Catalyzed Formal Hydroamination of Olefins. Chem Asian J. 2016;11:977–980. doi: 10.1002/asia.201501098. [DOI] [PubMed] [Google Scholar]
- 278.Zhu K, Shaver MP, Thomas SP. Chemoselective Nitro Reduction and Hydroamination using a Single Iron Catalyst. Chem Sci. 2016;7:3031–3035. doi: 10.1039/c5sc04471e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Ali A, Gill GB, Pattenden G, Roan GA, Kam T-S. Palladium- and Cobalt-Mediated Cyclisations of Halo-Polyenes: a Comparative Study. J Chem Soc, Perkin Trans. 1996;1:1081–1093. [Google Scholar]
- 280.Okabe M, Tada M. The Reaction of Bis(dimethylglyoximato)(pyridine)cobalt(I), Cobaloxime(I), with 2-(Allyloxy)ethyl Halides and the Photolysis of the Resulting Organo-cobaloximes. Bull Chem Soc Jpn. 1982;55:1498–1503. [Google Scholar]
- 281.Baldwin JE, Li C-S. Cobalt-Mediated Cyclization of Amino Acid Derivatives. Application to the Kainoids. J Chem Soc, Chem Commun. 1987:166–168. [Google Scholar]
- 282.Branchaud BP, Detlefsen WD. Cobaloxime-Catalyzed Radical Alkyl-Styryl Cross Couplings. Tetrahedron Lett. 1991;32:6273–6276. [Google Scholar]
- 283.Busato S, Tinembart O, Zhang Z-d, Scheffold R. Vitamin B129 a Catalyst in the Synthesis Of Prostaglandins. Tetrahedron. 1990;46:3155–3166. [Google Scholar]
- 284.Hu C, Qiu Y. Cobaloxime-Catalyzed Hydroperfluoroalkylation of Electron-Deficient Alkenes with Perfluoroalkyl Halides: Reaction and Mechanism. J Org Chem. 1992;57:3339–3342. [Google Scholar]
- 285.Inokuchi T, Kawafuchi H, Aoki K, Yoshida A, Torii S. Indirect Electrochemical Radical Cyclization of Bromo Acetals by the Combined Use of Cobaloxime and Sacrificial Electrode. Bull Chem Soc Jpn. 1994;67:595–598. [Google Scholar]
- 286.Wayland BB, Poszmik G, Mukerjee SL, Fryd M. Living Radical Polymerization of Acrylates by Organocobalt Porphyrin Complexes. J Am Chem Soc. 1994;116:7943–7944. [Google Scholar]
- 287.Giedyk M, Goliszewska K, Gryko D. Vitamin B12 catalyzed reactions. Chem Soc Rev. 2015;44:3391–3404. doi: 10.1039/c5cs00165j. [DOI] [PubMed] [Google Scholar]
- 288.Shey J, McGinley CM, McCauley KM, Dearth AS, Young BT, van der Donk WA. Mechanistic Investigation of a Novel Vitamin B12-Catalyzed Carbon Carbon Bond Forming Reaction, the Reductive Dimerization of Arylalkenes. J Org Chem. 2002;67:837–846. doi: 10.1021/jo0160470. [DOI] [PubMed] [Google Scholar]
- 289.McGinley CM, Relyea HA, van der Donk WA. Vitamin B12 Catalyzed Radical Cyclizations of Arylalkenes. Synlett. 2006:211–214. [Google Scholar]
- 290.Hartung J, Pulling ME, Smith DM, Yang DX, Norton JR. Initiating radical cyclizations by H• transfer from transition metals. Tetrahedron. 2008;64:11822–11830. [Google Scholar]
- 291.Kuo JL, Hartung J, Han A, Norton JR. Direct generation of oxygen-stabilized radicals by H* transfer from transition metal hydrides. J Am Chem Soc. 2015;137:1036–1039. doi: 10.1021/ja511883b. [DOI] [PubMed] [Google Scholar]
- 292.Han A, Spataru T, Hartung J, Jr, Li G, Norton JR. Effect of Double-Bond Substituents on the Rate of Cyclization of α-Carbomethoxyhex-5-enyl Radicals. J Org Chem. 2014;79:1938–1946. doi: 10.1021/jo402499w. [DOI] [PubMed] [Google Scholar]
- 293.Abramo GP, Norton JR. Catalysis by C5Ph5Cr(CO)3· of Chain Transfer during the Free Radical Polymerization of Methyl Methacrylate. Macromolecules. 2000;33:2790–2792. [Google Scholar]
- 294.Tang L, Norton JR, Edwards JC. Inverse Temperature Dependence of Chain Transfer Rate Constant for a Chromium Metalloradical in Polymerization of MMA. Macromolecules. 2003;36:9716–9720. [Google Scholar]
- 295.Tang L, Papish ET, Abramo GP, Norton JR, Baik MH, Friesner RA, Rappe A. Kinetics and Thermodynamics of H• Transfer from (η5-C5R5)Cr(CO)3H (R = Ph, Me, H) to Methyl Methacrylate and Styrene. J Am Chem Soc. 2003;125:10093–10102. doi: 10.1021/ja034927l. [DOI] [PubMed] [Google Scholar]
- 296.Tang L, Norton JR. Effect of Steric Congestion on the Activity of Chromium and Molybdenum Metalloradicals as Chain Transfer Catalysts during MMA Polymerization. Macromolecules. 2004;37:241–243. [Google Scholar]
- 297.Tang L, Norton JR. Factors Affecting the Apparent Chain Transfer Rate Constants of Chromium Metalloradicals: Mechanistic Implications. Macromolecules. 2006;39:8229–8235. [Google Scholar]
- 298.Choi J, Tang L, Norton JR. Kinetics of Hydrogen Atom Transfer from (η5-C5H5)Cr(CO)3H to Various Olefins: Influence of Olefin Structure. J Am Chem Soc. 2007;129:234–240. doi: 10.1021/ja066325i. [DOI] [PubMed] [Google Scholar]
- 299.Smith DM, Pulling ME, Norton JR. Tin-Free and Catalytic Radical Cyclizations. J Am Chem Soc. 2007;129:770–771. doi: 10.1021/ja0673276. [DOI] [PubMed] [Google Scholar]
- 300.Estes DP, Norton JR, Jockusch S, Sattler W. Mechanisms by which Alkynes React with CpCr(CO)3H. Application to Radical Cyclization. J Am Chem Soc. 2012;134:15512–15518. doi: 10.1021/ja306120n. [DOI] [PubMed] [Google Scholar]
- 301.Norton JR, Spataru T, Camaioni DM, Lee SJ, Li G, Choi J, Franz JA. Kinetics and Mechanism of the Hydrogenation of the CpCr(CO)3·/[CpCr(CO)3]2 Equilibrium to CpCr(CO)3H. Organometallics. 2014;33:2496–250. [Google Scholar]
- 302.Estes DP, Grills DC, Norton JR. The Reaction of Cobaloximes with Hydrogen: Products and Thermodynamics. J Am Chem Soc. 2014;136:17362–17365. doi: 10.1021/ja508200g. [DOI] [PubMed] [Google Scholar]
- 303.Beckwith ALJ, Schiesser CH. Regio- and Stereo-Selectivity of Alkenyl Radical Ring Closure: A Theoretical Study. Tetrahedron. 1985;41:3925–3941. [Google Scholar]
- 304.Spellmeyer DC, Houk KN. A Force-Field Model for Intramolecular Radical Additions. J Org Chem. 1987;52:959–974. [Google Scholar]
- 305.Fang JM, Chen MY. Free Radical Type Addition of Toluenesulfonyl Cyanide to Unsaturated Hydrocarbons. Tetrahedron Lett. 1987;28:2853–2856. [Google Scholar]
- 306.Barton DHR, Jaszberenyi JCs, Theodorakis EA. Radical Nitrile Transfer with Methanesulfonyl Cyanide or p-Toluenesulfonyl Cyanide to Carbon Radicals Generated from the Acyl Derivatives of N-Hydroxy-2-Thiopyridone. Tetrahedron Lett. 1991;32:3321–3324. [Google Scholar]
- 307.Kim S, Song HJ. Tin-free Radical Cyanation of Alkyl Iodides and Alkyl Phenyl Tellurides. Synlett. 2002;12:2110–2112. [Google Scholar]
- 308.Schaffner AP, Darmency V, Renaud P. Radical-Mediated Alkenylation, Alkynylation, Methanimination, and Cyanation of B-Alkylcatecholboranes. Angew Chem Int Ed. 2006;45:5847–5849. doi: 10.1002/anie.200601206. [DOI] [PubMed] [Google Scholar]
- 309.Giese B, Meister J. Die Addition von Kohlenwasserstoffen an Olefine Eine neue synthetische Methode. Chem Ber. 1977;110:2588–2600. [Google Scholar]
- 310.Ramaiah M. Radical Reactions in Organic Synthesis. Tetrahedron. 1987;43:3541–3676. [Google Scholar]
- 311.Srikanth GSC, Castle SL. Advances in Radical Conjugate Additions. Tetrahedron. 2005;61:10377–10441. [Google Scholar]
- 312.Giese B, Kretzschmar G. Steric Effects in the Addition of Alkyl Radicals to Alkenes. Angew Chem Int Ed Engl. 1981;20:967–967. [Google Scholar]
- 313.Shen Y, Qi J, Mao Z, Cui S. Fe-Catalyzed Hydroalkylation of Olefins with para-Quinone Methides. Org Lett. 2016;18:2722–2725. doi: 10.1021/acs.orglett.6b01173. [DOI] [PubMed] [Google Scholar]
- 314.Parnes ZN, Bolestova GI, Akhrem IS, Vol’pin ME, Kursanov DN. Alkyl Groups Migration from Tetra-Alkyl-Silanes, -Germanes, -Stannanes to Carbenium Ions, Effected by Lewuis Acids: A Novel Method for Synthesising Hydrocarbons with a Quaternary Carbon Atom. J Chem Soc, Chem Commun. 1980:748–748. [Google Scholar]
- 315.Terao J, Watanabe T, Saito K, Kambe N, Sonoda N. Zirconocene-Catalyzed Alkylation of Aryl Alkenes with Alkyl Tosylates, Sulfates And Bromides. Tetrahedron Lett. 1998;39:9201–9204. [Google Scholar]
- 316.Fontaine FG, Tilley TD. Control of Selectivity in the Hydromethylation of Olefins via Ligand Modification in Scandocene Catalysts. Organometallics. 2005;24:4340–2342. [Google Scholar]
- 317.Kim S, Cho JR. Radical Cyclization of Mesitylsulfonylhydrazones. Synlett. 1992:629–630. [Google Scholar]
- 318.Baldwin JE, Adlington RM, Newington IM. Azo Anions in Synthesis: α-Amino Carbanion Equivalents from t-Butyldiphenyl-Methylhydrazones. J Chem Soc, Chem Commun. 1986:176–178. [Google Scholar]
- 319.Ying H, Yao-Zeng H, Cheng-Ming Z. An Unexpected Reaction Between 2-Aryl-1-Nitro-1-Alkenes and Trialkylgallium Compounds. Tetrahedron Lett. 1996;37:3347–3350. [Google Scholar]
- 320.Yao CF, Chu CM, Liu JT. Free-Radical Reactions of Trialkylboranes with β-Nitrostyrenes to Generate Alkenes. J Org Chem. 1998;63:719–722. doi: 10.1021/jo9716901. [DOI] [PubMed] [Google Scholar]
- 321.Liu JY, Liu JT, Yao CF. Novel Synthesis of Alkenes via Triethylaluminum-Induced Free Radical Reactions of Alkyl Iodides and β-nitrostyrenes. Tetrahedron Lett. 2001;42:3613–3615. [Google Scholar]
- 322.Jang YJ, Wu J, Lin YF, Yao CF. A Highly Diastereoselective Tandem Radical Reaction. Facile Three-Component Routes to Protected (E)-Polysubstituted Homoallylic Alcohols. Tetrahedron. 2004;60:6565–6574. [Google Scholar]
- 323.Russell GA, Tashtoush H, Ngoviwatchai P. Alkylation of β-Substituted Styrenes by a Free Radical Addition-Elimination Sequence. J Am Chem Soc. 1984;106:4622–4623. [Google Scholar]
- 324.Gui Q, Hu L, Chen X, Liu J, Tan Z. Synthesis of Oxindoles via Iron-Mediated Hydrometallation-Cyclization of N-Arylacrylamides. Asian J Org Chem. 2015;4:870–874. [Google Scholar]
- 325.Crossley SWM, Martinez RM, Guevara-Zuluaga S, Shenvi RA. Synthesis of the Privileged 8-Arylmenthol Class by Radical Arylation of Isopulegol. Org Lett. 2016;18:2620–2623. doi: 10.1021/acs.orglett.6b01047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Isayama S, Mukaiyama T. Cobalt(II) Catalyzed Coupling Reaction of α,β–Unsaturated Compounds with Aldehydes by the Use of Phenylsilane. New Method for Preparation of β-Hydroxy Nitriles, Amides, and Esters. Chem Lett. 1989;18:2005–2008. [Google Scholar]
- 327.Denmark SE, Stavenger RA. Aymmetric Catalysis of Aldol Reactions with Chiral Lewis Bases. Acc Chem Res. 2000;33:432–440. doi: 10.1021/ar960027g. [DOI] [PubMed] [Google Scholar]
- 328.Huddleston RR, Krische MJ. Enones as Latent Enolates in Catalytic Processes: Catalytic Cycloreductions, Cycloaddition and Cycloisomerization. Synlett. 2003:12–21. [Google Scholar]
- 329.Ooi T, Doda K, Sakai D, Maruoka K. Unique Property of Copper(I) Chloride as a Radical Initiator as well as a Lewis Acid: Application to CuCl-Catalyzed Aldol Reaction of α,β–Unsaturated Ketones with Bu3SnH. Tetrahedron Lett. 1999;40:2133–2136. [Google Scholar]
- 330.Nokami J, Mandai T, Watanabe H, Ohyama H, Tsuji J. The Palladium-Catalyzed Directed Aldol Reaction of Aldehydes with Ketone Enolates Generated by the Decarboxylation of Allyl β-Keto Carboxylates under Neutral Conditions. J Am Chem Soc. 1989;111:4126–4127. [Google Scholar]
- 331.Kiyooka S, Shimizu A, Torii S. A Mild Aldol Reaction of Aryl Aldehydes through Palladium-Catalyzed Hydrosilation of α,β-Unsaturated Carbonyl Compounds with Trichlorosilane. Tetrahedron Lett. 1998;39:5237–5238. [Google Scholar]
- 332.Zhao CX, Duffey MO, Taylor SJ, Morken JP. Enantio- and Diastereoselective Reductive Aldol Reactions with Iridium Pybox Catalysts. Org Lett. 2001;3:1829–1831. doi: 10.1021/ol015859f. [DOI] [PubMed] [Google Scholar]
- 333.Revis A, Hilty TK. Novel Synthesis of α,β-Siloxy Esters by Condensation of Carbonyls and Trimethylsilane with α,β-Unsaturated Esters Catalyzed by RhCl3. Tetrahedron Lett. 1987;28:4809–4812. [Google Scholar]
- 334.Slough GA, Bergman RG, Heathcock H. Synthesis of η1-Oxygen-Bound Rhodium Enolates. Applications to Catalytic Aldol Chemistry. J Am Chem Soc. 1989;111:938–949. [Google Scholar]
- 335.Taylor SJ, Duffey MO, Morken JP. Rhodium-Catalyzed Enantioselective Reductive Aldol Reaction. J Am Chem Soc. 2000;122:4528–4529. [Google Scholar]
- 336.Hayashi C, Hayashi T, Kikuchi S, Yamada T. Cobalt-Catalyzed Reductive Carboxylation of αβ–Unsaturated Nitriles with Carbon Dioxide. Chem Lett. 2014;43:565–567. [Google Scholar]
- 337.Baik TG, Luis AL, Wang L-C, Krische MJ. Diastereoselective Cobalt-Catalyzed Aldol and Michael Cycloreductions. J Am Chem Soc. 2001;123:5112–5113. doi: 10.1021/ja0040971. [DOI] [PubMed] [Google Scholar]
- 338.Baik TG, Luis AL, Wang L-C, Krische MJ. Diastereoselective Metal-Catalyzed [2+2] Cycloaddition of Bis-enones. J Am Chem Soc. 2001;123:6716–6717. doi: 10.1021/ja010800p. [DOI] [PubMed] [Google Scholar]
- 339.Kim S, Kim S. Tin-Free Radical Carbon–Carbon Bond-Forming Reactions Based on α-Scission of Alkylsulfonyl Radicals. Bull Chem Soc Jpn. 2007;80:809–822. [Google Scholar]
- 340.Gaspar B, Carreira EM. Catalytic Hydrochlorination of Unactivated Olefins with Para-Toluenesulfonyl Chloride. Angew Chem Int Ed. 2008;47:5758–5760. doi: 10.1002/anie.200801760. [DOI] [PubMed] [Google Scholar]
- 341.Ruider SA, Sandmeier T, Carreira EM. Total Synthesis of (+/−)-Hippolachnin A. Angew Chem Int Ed Engl. 2015;54:2378–2382. doi: 10.1002/anie.201410419. [DOI] [PubMed] [Google Scholar]
- 342.George DT, Kuenstner EJ, Pronin SV. A Concise Approach to Paxilline Indole Diterpenes. J Am Chem Soc. 2015;137:15410–15413. doi: 10.1021/jacs.5b11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. Applications of Fluorine in Medicinal Chemistry. J Med Chem. 2015;58:8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- 344.Olah GA, Nojima M, Kerekes I. Synthetic Methods and Reactions II. Hydrofluorination of Alkenes, Cyclopropane and Alkynes with Poly-Hydrogen Fluoride/Pyridine (Trialkamine) Reagents. Synthesis. 1973;1973:779–780. [Google Scholar]
- 345.Leung JCT, Chatalova-Sazepin C, West JG, Rueda-Becerril M, Paquin JF, Sammis GM. Photo-fluorodecarboxylation of 2-Aryloxy and 2-Aryl Carboxylic Acids. Angew Chem Int Ed. 2012;51:10804–10807. doi: 10.1002/anie.201206352. [DOI] [PubMed] [Google Scholar]
- 346.Rueda-Becerril M, Chatalova Sazepin C, Leung JCT, Okbinoglu T, Kennepohl P, Paquin JF, Sammis GM. Fluorine Transfer to Alkyl Radicals. J Am Chem Soc. 2012;134:4026–4029. doi: 10.1021/ja211679v. [DOI] [PubMed] [Google Scholar]
- 347.Yin F, Wang Z, Li Z, Li C. Silver-Catalyzed Decarboxylative Fluorination of Aliphatic Carboxylic Acids in Aqueous Solution. J Am Chem Soc. 2012;134:10401–10404. doi: 10.1021/ja3048255. [DOI] [PubMed] [Google Scholar]
- 348.Baudoux J, Cahard D. Organic Reactions. Vol. 69. John Wiley & Sons, Inc; 2004. Electrophilic Fluorination with N–F Reagents; pp. 347–672. [Google Scholar]
- 349.Barker TJ, Boger DL. Fe(III)/NaBH4-Mediated Free Radical Hydrofluorination of Unactivated Alkenes. J Am Chem Soc. 2012;134:13588–13591. doi: 10.1021/ja3063716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Stavber S, Jereb M, Zupan M. Efficient Synthesis of 4-Fluorocyclohexa-2,5-dienone Derivatives Using N-Fluoro-1,4-diazoniabicyclo[2.2. 2]octane Salt Analogues. Synlett. 1999;1999:1375–1378. [Google Scholar]
- 351.Chan LY, Kim S, Park Y, Lee PH. Iron(III)-Catalyzed Conia–Ene Cyclization of 2-Alkynic 1,3-Dicarbonyl Compounds. J Org Chem. 2012;77:5239–5244. doi: 10.1021/jo300957q. [DOI] [PubMed] [Google Scholar]
- 352.Yeh MCP, Fang CW, Lin HH. Facile Synthesis of Azaspirocycles via Iron Trichloride-Promoted Cyclization/Chlorination of Cyclic 8-Aryl-5-aza-5-tosyl-2-en-7-yn-1-ols. Org Lett. 2012;14:1830–1833. doi: 10.1021/ol300434m. [DOI] [PubMed] [Google Scholar]
- 353.Ekomié A, Lefèvre G, Fensterbank L, Lacôte E, Malacria M, Ollivier C, Jutand A. Iron-Catalyzed Reductive Radical Cyclization of Organic Halides in the Presence of NaBH4: Evidence of an Active Hydrido Iron(I) Catalyst. Angew Chem Int Ed. 2012;51:6942–6946. doi: 10.1002/anie.201200589. [DOI] [PubMed] [Google Scholar]
- 354.Ma XS, Herzon SB. Non-Classical Selectivities in the Reduction of Alkenes by Cobalt-Mediated Hydrogen Atom Transfer. Chem Sci. 2015;6:6250–6255. doi: 10.1039/c5sc02476e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Ogawa A. Hydrofunctionalization. In: Ananikov VP, Tanaka M, editors. Topics in Organometallics Chemistry. Vol. 43. Springer; Berlin: 2012. pp. 325–360. [Google Scholar]
- 356.Ikeda T, Tamai T, Daitou M, Minamida Y, Mitamura T. Highly Regioselective Palladium-Catalyzed Double Hydroselenation of Terminal Alkynes with Benzeselenolin the Presence of Acetic Acid. Chem Lett. 2013;42:1383–1385. [Google Scholar]
- 357.Kano K, Takeuchi M, Hashimoto S, Yoshida Z. Hemin-Catalyzed Addition Reactions of Thiophenols to Styrene. Chem Lett. 1990;19:1381–1384. [Google Scholar]
- 358.Takeuchi M, Shimakoshi H, Kano K. (Porphinato)iron-Catalyzed Addition Reactions of Thiols to Alkenes via (σ-Alkyl)iron(II) Complexes. Organometallics. 1994;13:1208–1213. [Google Scholar]
- 359.Girijavallabhan V, Alvarez C, Njoroge FG. Regioselective Cobalt-Catalyzed Addition of Sulfides to Unactivated Alkenes. J Org Chem. 2011;76:6442–6446. doi: 10.1021/jo201016z. [DOI] [PubMed] [Google Scholar]
- 360.Feder HM, Halpern J. Mechanism of the Cobalt Carbonyl-Catalyzed Homogeneous Hydrogenation of Aromatic Hydrocarbons. J Am Chem Soc. 1975;97:7186–7188. [Google Scholar]
- 361.Sweany RL, Halpern J. Hydrogenation of α-Methylstyrene by Hydridopentacarbonylmanganese (I). Evidence for a Free-Radical Mechanism. J Am Chem Soc. 1977;99:8335–8337. [Google Scholar]
- 362.Cornils B, Herrmann WA, Rasch M. Otto Roelen, Pioneer in Industrial Homogeneous Catalysis. Angew Chem Int Ed Engl. 1994;33:2144–2163. [Google Scholar]
- 363.Adkins H, Krsek G. Hydroformylation of Unsaturated Compounds with a Cobalt Carbonyl Catalyst. J Am Chem Soc. 1949;71:3051–3055. [Google Scholar]
- 364.Wender I, Levine R, Orchin M. Chemistry of the Oxo and Related Reactions. II. Hydrogenation1. J Am Chem Soc. 1950;72:4375–4378. [Google Scholar]
- 365.Wender I, Greenfield H, Orchin M. Chemistry of the Oxo and Related Reactions. IV. Reductions in the Aromatic Series1. J Am Chem Soc. 1951;73:2656–2658. [Google Scholar]
- 366.Friedman S, Metlin S, Svedi A, Wender I. Selective Hydrogenation of Polynuclear Aromatic Hydrocarbons. J Org Chem. 1959;24:1287–1289. [Google Scholar]
- 367.Kwiatek J. Reactions Catalyzed by Pentacyanocobaltate(II) Catalysis Revs. 1968;1:37–72. [Google Scholar]
- 368.Iguchi M. Studies on the Catalytic Oxido-reduction by Some Metallic Complex Salts. XVII. Nippon Kagaku Kaishi. 1942;63:634–643. [Google Scholar]
- 369.Kwiatek J, Mador IL, Seyler JK. Catalytic Hydrogenation of Organic Compounds by Pentacyanocobaltate(II) J Am Chem Soc. 1962;84:304–305. [Google Scholar]
- 370.Kwiatek J, Mador IL, Seyler JK. Catalytic Hydrogenation by Pentacyanopentacobaltate (II) Adv Chem Ser. 1962;37:201–215. [Google Scholar]
- 371.Halpern J. Homogeneous Catalysis by Coordination Compounds. Homogeneous Catalysis: Industrial Applications and Implications. Adv Chem Ser. 1974;70:1–24. [Google Scholar]
- 372.Halpern J, Wong LY. Kinetics of the Addition of Hydridopentacyanocobaltate(III) to some α β-Unsaturated Compounds. J Am Chem Soc. 1968;90:6665–6669. [Google Scholar]
- 373.Simandi L, Nagy F. Homogeneous Catalytic Activators of the H2 Molecule, IV. Acta Chim Hung. 1965;46:138–149. [Google Scholar]
- 374.Halpern J. Free Radical Mechanisms in Organometallic and Bioorganometallic Chemistry. Pure Appl Chem. 1986;58:575–584. [Google Scholar]
- 375.Eisenberg DC, Norton JR. Hydrogen-Atom Transfer Reactions of Transition-Metal Hydrides. Isr J Chem. 1991;31:55–66. [Google Scholar]
- 376.Wan KK, Iwasaki K, Umotoy JC, Wolan DW, Shenvi RA. Nitrosopurines en route to Potently Cytotoxic Asmarines. Angew Chem Int Ed Engl. 2015;54:2410–2415. doi: 10.1002/anie.201411493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Shenvi R, Wan K. Conjuring a Supernatural Product – DelMarine. Synlett. 2016;27:1145–1146. [Google Scholar]
- 378.Ficini J, Francilette J, Touzin AM. Stereochemistry of Reduction of Methylenecyclohexanes by Dissolving Metals. J Chem Research (S) 1979:150–151. [Google Scholar]
- 379.Damm W, Giese B, Hartung J, Hasskerl T, Houk KN, Hueter O, Zipse H. Diastereofacial Selectivity in Reactions of Substituted Cyclohexyl Radicals. An Experimental and Theoretical Study. J Am Chem Soc. 1992;114:4067–4079. [Google Scholar]
- 380.Gonzaĺez-Sierra M, Laborde MA, Rúveda EA. Alternative and Stereoselective Synthesis of 8β(H)-Drimane, A Bicyclic Sesquiterpane of Widespread Occurrence in Petroleum. Synth Commun. 1987;17:431–441. [Google Scholar]
- 381.King Sandra M, Calandra Nicholas A, Seth B. Herzon Total Syntheses of (−)-Acutumine and (−)-Dechloroacutumine. Angew Chem Int Ed. 2013;52:3642–3645. doi: 10.1002/anie.201210076. [DOI] [PubMed] [Google Scholar]
- 382.Chung SK. Selective Reduction of Mono- and Disubstituted Olefins by Sodium Borohydride and Cobalt(II) J Org Chem. 1979;44:1014–1016. [Google Scholar]
- 383.Periasamy M, Thirumalaikumar M. Methods of Enhancement of Reactivity and Selectivity of Sodium Borohydride for Applications in Organic Synthesis. J Organomet Chem. 2000;609:137–151. [Google Scholar]
- 384.Osby JO, Heinzman SW, Ganem B. Studies on the Mechanism of Transition-Metal-Assisted Sodium Borohydride and Lithium Aluminum Hydride Reductions. J Am Chem Soc. 1986;108:67–72. [Google Scholar]
- 385.Caggiano TJ. Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2001. Cobalt Boride. [Google Scholar]
- 386.Cane DE, Tandon M. Total Synthesis of (±)-Epicubenol. Tetrahedron Lett. 1994;35:5351–5354. [Google Scholar]
- 387.Suh YG, Koo BA, Kim EN, Choi NS. Construction of Oxocane Skeleton via Unusually Regioselective C-C Bond Forming Cyclization: Synthesis of (+) and (−)-Lauthisan. Tetrahedron Lett. 1995;36:2089–2092. [Google Scholar]
- 388.Dankwardt JW, Dankwardt SM, Schlessinger RH. Synthesis of the C19 through C27 Segment of Okadaic Acid Using Vinylogous Urethane Aldol Chemistry: Part III. Tetrahedron Lett. 1998;39:4979. [Google Scholar]
- 389.Ghosh AK, Swanson LM, Cho H, Leshchenko S, Hussain KA, Kay S, Walters DE, Koh Y, Mitsuya H. Structure-Based Design: Synthesis and Biological Evaluation of a Series of Novel Cycloamide-Derived HIV-1 Protease Inhibitors. J Med Chem. 2005;48:3576–3585. doi: 10.1021/jm050019i. [DOI] [PubMed] [Google Scholar]
- 390.Chandrasekhar B, Prasada Rao J, Venkateswara Rao B, Naresh P. [2, 3]-Wittig Rearrangement Approach to Iminosugar C-glycosides: 5-epi-Ethylfagomine 2-epi-5-Deoxyadenophorine and Formal Synthesis of Indolizidine 167B and 209D. Tetrahedron Lett. 2011;52:5921–5925. [Google Scholar]
- 391.Hugelshofer CL, Magauer T. Total Synthesis of the Leucosceptroid Family of Natural Products. J Am Chem Soc. 2015;137:3807–3810. doi: 10.1021/jacs.5b02021. [DOI] [PubMed] [Google Scholar]
- 392.Kano K, Takeuchi M, Hashimoto S, Yoshida Z-i. Porphyrinatoiron-Catalysed Reduction of Styrene with Sodium Borohydride: Proposed [σ]-Alkyliron(III) Complex as an Intermediate. J Chem Soc, Chem Commun. 1991:1728–1729. [Google Scholar]
- 393.MacNair AJ, Tran MM, Nelson JE, Sloan GU, Ironmonger A, Thomas SP. Iron-Catalysed, General and Operationally Simple Formal Hydrogenation using Fe(OTf)3 and NaBH4. Org Biomol Chem. 2014;12:5082–5088. doi: 10.1039/c4ob00945b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Biswas S. Mechanistic Understanding of Transition-Metal Catalyzed Olefin Isomerization: Transition-Metal-Catalyzed Olefin Isomerization: Metal-Hydride Insertion-Elimination vs. π-Allyl Pathways. Comments on Inorganic Chemistry. 2015;35:301–331. [Google Scholar]
- 395.Choi J, Pulling ME, Smith DM, Norton JR. Unusually Weak Metal-Hydrogen Bonds in HV(CO)4(P-P) and their Effectiveness as H• Donors. J Am Chem Soc. 2008;130:4250–4252. doi: 10.1021/ja710455c. [DOI] [PubMed] [Google Scholar]
- 396.Lacy DC, Roberts GM, Peters JC. The Cobalt Hydride that Never Was: Revisiting Schrauzer’s ‘Hydridocobaloxime’. J Am Chem Soc. 2015;137:4860–4864. doi: 10.1021/jacs.5b01838. [DOI] [PubMed] [Google Scholar]
- 397.Li G, Han A, Pulling ME, Estes DP, Norton JR. Evidence for Formation of a Co-H Bond from (H2O)2Co(dmgBF2)2 under H2: Application to Radical Cyclizations. J Am Chem Soc. 2012;134:14662–14665. doi: 10.1021/ja306037w. [DOI] [PubMed] [Google Scholar]
- 398.Li G, Estes DP, Norton JR, Ruccolo S, Sattler A, Sattler W. Dihydrogen Activation by Cobaloximes with Various Axial Ligands. Inorg Chem. 2014;53:10743–10747. doi: 10.1021/ic501975r. [DOI] [PubMed] [Google Scholar]
- 399.Li G, Kuo JL, Han A, Abuyuan JM, Young LC, Norton JR, Palmer JH. Radical Isomerization and Cycloisomerization Initiated by H• Transfer. J Am Chem Soc. 2016;138:7698–7704. doi: 10.1021/jacs.6b03509. [DOI] [PubMed] [Google Scholar]
- 400.Obligacion JV, Chirik PJ. Bis(imino)pyridine Cobalt-Catalyzed Alkene Isomerization–Hydroboration: A Strategy for Remote Hydrofunctionalization with Terminal Selectivity. J Am Chem Soc. 2013;135:19107–19110. doi: 10.1021/ja4108148. [DOI] [PubMed] [Google Scholar]
- 401.Larsen CR, Grotjahn DB. Stereoselective Alkene Isomerization over One Position. J Am Chem Soc. 2012;134:10357–10360. doi: 10.1021/ja3036477. [DOI] [PubMed] [Google Scholar]
- 402.Chen C, Dugan TR, Brennessel WW, Weix DJ, Holland PL. Z-Selective Alkene Isomerization by High-Spin Cobalt(II) Complexes. J Am Chem Soc. 2014;136:945–955. doi: 10.1021/ja408238n. [DOI] [PubMed] [Google Scholar]
- 403.Bosch C, Fiser B, Gómez-Bengoa E, Bradshaw B, Bonjoch J. Approach to cis-Phlegmarine Alkaloids via Stereodivergent Reduction: Total Synthesis of (+)-Serratezomine E and Putative Structure of (−)-Huperzine N. Org Lett. 2015;17:5084. doi: 10.1021/acs.orglett.5b02581. [DOI] [PubMed] [Google Scholar]
- 404.Mukai C, Ueda M, Takahashi Y, Inagaki F. Concise Construction of Bicyclo[6.4.0] and -[7.4.0] Frameworks by [4+2] Cycloaddition of 3,4-Dimethylene-2,5-bis(phenylsulfonyl)cycloalk-1-enes. European J Org Chem. 2015;2015:4412–4422. [Google Scholar]
- 405.Lu HH, Pronin SV, Antonova-Koch Y, Meister S, Winzeler EA, Shenvi RA. Synthesis of (+)-7,20-Diisocyanoadociane and Liver Stage Antiplasmodial Activity of the ICT Class. J Am Chem Soc. 2016;138:7268–7271. doi: 10.1021/jacs.6b03899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Bohen HW. Hydroformylation of Alkenes: An Industrial View of the Status and Importance. Adv Catal. 2002;47:1–64. [Google Scholar]
- 407.Sanfilippo D, Rylander PN. Hydrogenation and Dehydrogenation. Ullmann’s Encyclopedia of Industrial Chemistry. 2009 [Google Scholar]
- 408.Pearson RG, Ford PC. The Brønsted acidity of Transition Metal Hydrides. Comments on Inorganic Chemistry. 1982;1:270–291. [Google Scholar]
- 409.Pearson RG. The Transition-Metal-Hydrogen Bond. Chem Rev. 1985;85:41–49. [Google Scholar]
- 410.Baird MC. Seventeen-Electron Metal-Centered Radicals. Chem Rev. 1988;88:1217–1227. [Google Scholar]
- 411.Ungvary F, Marko L. Reaction of HCo(CO)4 and CO with Styrene. Mechanism of (α-Phenylpropionyl)- and (β-Phenylpropionyl)cobalt Tetracarbonyl Formation. Organometallics. 1982;1:1120–1125. [Google Scholar]
- 412.Bockman TM, Garst JF, King RB, Markó L, Ungváry F. CIDNP Evidence for Radical Intermediates in the Hydroformylation and Reduction of Styrene by HCo(CO)4/CO. J Organomet Chem. 1985;279:165–169. [Google Scholar]
- 413.Galiano-Roth AS, Michaelides EM, Collum DB. 6Li, 13C and 15N NMR Spectroscopic Studies of Lithium Dialkylamides. Solution Structure of Lithium Isopropylcyclohexylamide (LICA) in Tetrahydrofuran. J Am Chem Soc. 1988;110:2658–2660. [Google Scholar]
- 414.Nalesnik TE, Orchin M. Free Radical Reactions of Tetracarbonylhydridocobalt. Organometallics. 1982;1:222–223. [Google Scholar]
- 415.Heck RF, Breslow DS. Acylcobalt Carbonyls and their Triphenylphosphine Complexes. J Am Chem Soc. 1962;84:2499–2502. [Google Scholar]
- 416.Heck RF, Breslow DS. The Reaction of Cobalt Hydrotetracarbonyl with Olefins. J Am Chem Soc. 1961;83:4023–4027. [Google Scholar]
- 417.Jacobsen EN, Bergman RG. Synthesis and Chemistry of a Bridging Vinylidenedicobalt Complex. Evidence for a Nonchain Radical Mechanism in Its Reaction with Metal Hydrides to Give Heteronuclear Clusters. J Am Chem Soc. 1985;107:2023–2032. [Google Scholar]
- 418.Garst JF, Bockman TM, Batlaw R. Tetramethylallene and 2,4-Dimethyl-1,3-Pentadiene as Hydrogen-Atom Acceptors in Reactions with HMn(CO)5 and HCo(CO)4. J Am Chem Soc. 1986;108:1689–1691. [Google Scholar]
- 419.Connolly JW. Reaction between Hydridotetracarbonyl(trichlorosilyl)iron, HFe(CO)4SiCl3, and Conjugated Dienes. Evidence for a Free Radical Mechanism. Organometallics. 1984;3:1333–1337. [Google Scholar]
- 420.Sweany R, Butler SC, Halpern J. The Hydrogenation of 9,10-Dimethylanthracene by Hydridopentacarbonylmanganese(I). Evidence for a Free-Radical Mechanism. J Organomet Chem. 1981;213:487–492. [Google Scholar]
- 421.Nalesnik TE, Freudenberger JH, Orchin M. Stoichiometric Radical Hydrogenations with HCo(CO)4, HMn(CO)5 and their Deuterio Analogs. J Mol Catal. 1982;16:43–49. [Google Scholar]
- 422.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; 2006. [Google Scholar]
- 423.Eisenberg DC, Lawrie CJ, Moody AE, Norton JR. Relative Rates of H• Transfer from Transition-Metal Hydrides to Trityl Radicals. J Am Chem Soc. 1991;113:4888–4895. [Google Scholar]
- 424.Mader EA, Davidson ER, Mayer JM. Large Ground-State Entropy Changes for Hydrogen Atom Transfer Reactions of Iron Complexes. J Am Chem Soc. 2007;129:5153–5166. doi: 10.1021/ja0686918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Bullock RM, Samsel EG. Hydrogen Atom Transfer Reactions of Transition-Metal Hydrides. Utilization of a Radical Rearrangement in the Determination of Hydrogen Atom Transfer Rates. J Am Chem Soc. 1987;109:6542–6544. [Google Scholar]
- 426.Bullock RM, Samsel EG. Hydrogen Atom Transfer Reactions of Transition-Metal Hydrides. Kinetics and Mechanism of the Hydrogenation of α-Cyclopropylstyrene by Metal Carbonyl Hydrides. J Am Chem Soc. 1990;112:6886–6898. [Google Scholar]
- 427.Fischer H, Radom L. Factors Controlling the Addition of Carbon-Centered Radicals to Alkenes – An Experimental and Theoretical Perspective. Angew Chem Int Ed. 2001;40:1340–1371. doi: 10.1002/1521-3773(20010417)40:8<1340::aid-anie1340>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 428.Halpern J. Mechanisms of Coenzyme B12-Dependent Rearrangements. Science. 1985;227:869–875. doi: 10.1126/science.2857503. [DOI] [PubMed] [Google Scholar]
- 429.Ittel SD, Gridnev AA. Initiation of Polymerization by Hydrogen Atom Donation. 7,022,792. US Patent. 2006 Apr 4;
- 430.Weiss ME, Kreis LM, Lauber A, Carreira EM. Cobalt-Catalyzed Coupling of Alkyl Iodides with Alkenes: Deprotonation of Hydridocobalt Enables Turnover. Angew Chem Int Ed. 2011;50:11125–11128. doi: 10.1002/anie.201105235. [DOI] [PubMed] [Google Scholar]
- 431.Szeverényi Z, Budó-Záhonyi É, Simándi LI. Autocatalytic Reduction of Pyridinecobaloxime(III) by Molecular Hydrogen. J Coord Chem. 1980;10:41–45. [Google Scholar]
- 432.Schrauzer GN, Holland RJ. Hydridocobaloximes. J Am Chem Soc. 1971;93:1505–1506. [Google Scholar]
- 433.Bhattacharjee A, Chavarot-Kerlidou M, Andreiadis ES, Fontecave M, Field MJ, Artero V. Combined Experimental-Theoretical Characterization of the Hydrido-Cobaloxime [HCo(dmgH)2(PnBu3)] Inorg Chem. 2012;51:7087–7093. doi: 10.1021/ic2024204. [DOI] [PubMed] [Google Scholar]
- 434.Dempsey JL, Winkler JR, Gray HB. Mechanism of H2 Evolution from a Photogenerated Hydridocobaloxime. J Am Chem Soc. 2010;132:16774–16776. doi: 10.1021/ja109351h. [DOI] [PubMed] [Google Scholar]
- 435.Nishida Y, Tanaka N, Okazaki M. Chemical Mechanism of Olefin Oxygenation Reaction Catalyzed by Bis(acetylacetonato)nikel(II) or Cobalt(II) Compounds in the Presence of Reducing Agents. Polyhedron. 1994;13:2245–2249. [Google Scholar]
- 436.Yamada T. Enantioselective Borohydride Reduction Catalyzed by Cobalt Complexes: Discovery, Analysis, and Design for a Halogen-Free System. Synthesis. 2008;2008:1628–1640. [Google Scholar]
- 437.Huffman JW. Metal-Ammonia Reductions of Cyclic Aliphatic Ketones. Acc Chem Res. 1983;16:399–405. [Google Scholar]
- 438.O’Donnell K, Bacon R, Chellappa KL, Schowen RL, Lee JK. Catalysis in Organosilicon Chemistry. III. Catalytic Mode, Solvent Isotope Effects, and Transition-State Structure in Hydride Expulsion from Silicon. J Am Chem Soc. 1972;94:2500–2505. [Google Scholar]
- 439.Revunova K, Nikonov GI. Base-Catalyzed Hydrosilylation of Ketones and Esters and Insight into the Mechanism. Chem– Eur J. 2014;20:839–845. doi: 10.1002/chem.201302728. [DOI] [PubMed] [Google Scholar]
- 440.Gotoh H, Sears JE, Eschenmoser A, Boger DL. New Insights into the Mechanism and an Expanded Scope of the Fe(III)-Mediated Vinblastine Coupling Reaction. J Am Chem Soc. 2012;134:13240–13243. doi: 10.1021/ja306229x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Saussine L, Brazi E, Robine A, Mimoun H, Fischer J, Weiss R. Cobalt(III) Alkylperoxy Complexes. Synthesis, X-Ray Structure, and Role in the Catalytic Decomposition of Alkyl Hydroperoxides and in the Hydroxylation of Hydrocarbons. J Am Chem Soc. 1985;107:3534–3540. [Google Scholar]
- 442.Jensen FR, Kiskis RC. Stereochemistry and Mechanism of the Photochemical and Thermal Insertion of Oxygen into the Carbon-Cobalt Bond of Alkyl(pyridine)cobaloximes. J Am Chem Soc. 1975;97:5825–5831. [Google Scholar]
- 443.Taft KL, Caneschi A, Pence LE, Delfs CD, Papaefthymiou GC, Lippard SJ. Iron and Manganese Alkoxide Cubes. J Am Chem Soc. 1993;115:11753–11766. [Google Scholar]
- 444.Kitajima N, Komatsuzaki H, Hikichi S, Osawa M, Moro-oka Y. A Monomeric Side-On Peroxo Manganese(III) Complex: Mn(O2)(3,5-i-Pr2pzH)(HB(3,5-i-Pr2pz)3) J Am Chem Soc. 1994;116:11596–11597. [Google Scholar]
- 445.Magnus P, Fielding MR. Acceleration of the Reduction of Aldehydes and Ketones Using Mn(dpm)3 Catalyst and Phenylsilane in the Presence of Dioxygen. Tetrahedron Lett. 2001;42:6633–6636. [Google Scholar]
- 446.Whitesides GM, Ehmann WJ. Reduction of Olefins Using Sodium-Hexamethylphosphoramide-Tert-Butyl Alcohol. J Org Chem. 1970;35:3565–3567. [Google Scholar]
- 447.Lloyd RV, Williams RV. Bridgehead Decalin Radicals: Evidence for Nonplanarity at the Radical Site. J Phys Chem. 1985;89:5379–5381. [Google Scholar]
- 448.Chatgilialoglu C. Organosilanes as Radical-Based Reducing Agents in Synthesis. Acc Chem Res. 1992;25:188–194. [Google Scholar]
- 449.Chatgilialoglu C. Structural and Chemical Properties of Silyl Radicals. Chem Rev. 1995;95:1229–1251. [Google Scholar]
- 450.Mayer JM. Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc Chem Res. 2011;44:36–46. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Simmons EM, Hartwig JF. On the Interpretation of Deuterium Kinetic Isotope Effects in C-H Bond Functionalizations by Transition-Metal Complexes. Angew Chem Int Ed. 2012;51:3066–3072. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- 452.Gridnev AA, Ittel SD. Catalytic Chain Transfer in Free-Radical Polymerizations. Chem Rev. 2001;101:3611–3660. doi: 10.1021/cr9901236. [DOI] [PubMed] [Google Scholar]
- 453.Wayland BB, Poszmik G, Mukerjee SL, Fryd M. Living Radical Polymerization of Acrylates by Organocobalt Porphyrin Complexes. J Am Chem Soc. 1994;116:7943–7944. [Google Scholar]
- 454.Li S, de Bruin B, Peng CH, Fryd M, Wayland BB. Exchange of Organic Radicals with Organo-Cobalt Complexes Formed in the Living Radical Polymerization of Vinyl Acetate. J Am Chem Soc. 2008;130:13373–13381. doi: 10.1021/ja804010h. [DOI] [PubMed] [Google Scholar]
- 455.De Bruin B, Dzik WI, Li S, Wayland BB. Hydrogen-Atom Transfer in Reactions of Organic Radicals with [Co(II)(por)]· (Por = Porphyrinato) and in Subsequent Addition of [Co(H)(por)] to Olefins. Eur J Chem. 2009;15:4312–4320. doi: 10.1002/chem.200802022. [DOI] [PubMed] [Google Scholar]
- 456.Clark AJ. Atom Transfer Radical Cyclisation Reactions Mediated by Copper Complexes. Chem Soc Rev. 2002;31:1–11. doi: 10.1039/b107811a. [DOI] [PubMed] [Google Scholar]
- 457.Chen C, Hecht MB, Kavara A, Brennessel WW, Mercado BQ, Weix DJ, Holland PL. Rapid, Regioconvergent, Solvent-Free Alkene Hydrosilylation with a Cobalt Catalyst. J Am Chem Soc. 2015;137:13244–13247. doi: 10.1021/jacs.5b08611. [DOI] [PubMed] [Google Scholar]
- 458.Tondreau AM, Atienza CCH, Weller KJ, Nye SA, Lewis KM, Delis JGP, Chirik PJ. Iron Catalysts for Selective Anti-Markovnikov Alkene Hydrosilylation Using Tertiary Silanes. Science. 2012;335:567–570. doi: 10.1126/science.1214451. [DOI] [PubMed] [Google Scholar]
- 459.Renaud P, Sibi MP. Radicals in Organic Synthesis. Vol. 2. Wiley-VCH; Weinheim: 2001. [Google Scholar]
- 460.Zard SZ. Radical Reactions in Organic Synthesis. Oxford; New York: 2003. [Google Scholar]
- 461.Giese B. Formation of C-C Bonds by Addition of Free Radicals to Alkenes. Angew Chem Int Ed. 1983;22:753–764. [Google Scholar]
- 462.Fischer H, Paul H. Rate Constants for Some Prototype Radical Reactions in Liquids by Kinetic Electron Spin Resonance. Acc Chem Res. 1987;20:200–206. [Google Scholar]
- 463.Beckwith ALJ, Poole JS. Factors Affecting the Rates of Addition of Free Radicals to Alkenes. Determination of Absolute Rate Coefficients Using the Persistent Aminoxyl Method. J Am Chem Soc. 2002;124:9489–9497. doi: 10.1021/ja025730g. [DOI] [PubMed] [Google Scholar]
- 464.Barton DHR, Jaszberenyi JC, Theodorakis EA. The Invention of Radical Reactions. Part XXIII New Reactions: Nitrile and Thiocyanate Transfer to Carbon Radicals from Sulfonyl Cyanides and Sulfonyl Isothiocyanates. Tetrahedron. 1992;48:2613–2626. [Google Scholar]
- 465.Spellmeyer DC, Houk KN. A Force-Field Model for Intramolecular Radical Additions. J Org Chem. 1987;52:959–974. [Google Scholar]
- 466.Senn HM, Blöchl PE, Togni A. Toward an Alkene Hydroamination Catalyst: Static and Dynamic Ab Initio DFT Studies. J Am Chem Soc. 2000;122:4098–4107. [Google Scholar]
- 467.Kharasch MS, Meltzer TH, Nudenberg W. Reactions of Atoms and Free Radicals in Solution. XXXIX. The Reaction of Diacetyl Peroxide with Sec-Butyl Nitrite and 3-Amyl Nitrite. J Org Chem. 1957;22:37–39. [Google Scholar]
- 468.Russell GA, Yao CF. Deoxygenation of Nitro and Nitrosoaromatics by Photolysis With t-BuHgl/Kl. Regiochemistry Oftert-Butyl Radical Addition to Nitrosoaromatics. Heteroat Chem. 1993;4:433–444. [Google Scholar]
- 469.Kojo S, Sano S. Mechanism of a Novel Synthesis of Haemin c from Protohaemin and L-Cysteine. A Markownikoff-Type Radical Addition Reaction. J Chem Soc Perkin I. 1981:2864–2870. [Google Scholar]
- 470.Ding K, Dugan TR, Brennessel WW, Bill E, Holland PL. Synthesis, Properties, and Reactivity of Diketiminate-Supported Cobalt Fluoride Complexes. Organometallics. 2009;28:6650–6656. [Google Scholar]
- 471.Wang LC, Jang HY, Roh Y, Lynch V, Schultz AJ, Wang X, Krische MJ. Diastereoselective Cycloreductions and Cycloadditions Catalyzed by Co(dpm)2 -Silane (Dpm = 2,2,6,6-Tetramethylheptane-3,5-Dionate): Mechanism and Partitioning of Hydrometallative versus Anion Radical Pathways. J Am Chem Soc. 2002;124:9448–9453. doi: 10.1021/ja020223k. [DOI] [PubMed] [Google Scholar]
- 472.Rosenberg L, Davis CW, Yao J. Catalytic Dehydrogenative Coupling of Secondary Silanes Using Wilkinson’s Catalyst. J Am Chem Soc. 2001;123:5120–5121. doi: 10.1021/ja015697i. [DOI] [PubMed] [Google Scholar]
- 473.Chalk AJ, Harrod JF. Homogeneous Catalysis. II. The Mechanism of the Hydrosilation of Olefins Catalyzed by Group VIII Metal Complexes. J Am Chem Soc. 1965;87:16–21. [Google Scholar]
- 474.Lande SS, Kochi JK. Formation and Oxidation of Alkyl Radicals by Cobalt(III) Complexes. J Am Chem Soc. 1968;90:5196–5207. [Google Scholar]
- 475.Uddin J, Morales CM, Maynard JH, Landis CR. Computational Studies of Metal Ligand Bond Enthalpies across the Transition Metal Series. Organometallics. 2006;25:5566–5581. [Google Scholar]