

Abstract
The Human Proteome Organization (HUPO) Human Proteome Project (HPP) continues to make progress on its two overall goals: (1) completing the protein parts list, with an annual update of the HUPO draft human proteome, and (2) making proteomics an integrated complement to genomics and transcriptomics throughout biomedical and life sciences research. neXtProt version 2017-01-23 has 17,008 confident protein identifications (Protein Existence [PE] level 1) that are compliant with the HPP Guidelines v2.1 (https://hupo.org/Guidelines), up from 13,664 in 2012-12 and 16,518 in 2016-04. Remaining to be found by mass spectrometry and other methods are 2579 “missing proteins” (PE2+3+4), down from 2949 in 2016. PeptideAtlas 2017-01 has 15,173 canonical proteins, accounting for nearly all of the 15,290 PE1 proteins based on MS data. These resources have extensive data on PTMs, single amino acid variants, and splice isoforms. The Human Protein Atlas v16 has 10,492 highly-curated protein entries with tissue and subcellular spatial localization of proteins and transcript expression. Organ-specific popular protein lists have been generated for broad use in quantitative targeted proteomics using SRM-MS or DIA-SWATH-MS studies of biology and disease.
Keywords: Metrics, missing proteins, guidelines, neXtProt, PeptideAtlas, Human Proteome Project (HPP), long non-coding RNAs (lncRNAs)
For TOC Only
INTRODUCTION
The Human Proteome Organization (HUPO) (www.hupo.org) Human Proteome Project (HPP) is progressing toward two overall goals1: (1) completing stepwise the protein parts list, the draft human proteome, which is updated annually with an HPP Metrics publication2–5; and (2) integrating proteomics with genomics and transcriptomics for use throughout the biomedical/life sciences community. The protein parts list comprises at least one confidently identified protein product from each predicted protein-coding gene, along with post-translational modifications (PTMs), single amino acid variants (SAAVs), and splice isoforms of those proteins. The integration of proteomics with other ‘omics platforms has been enhanced through advances in assays, instrumentation, and knowledge bases for quantitative functional assessment of proteins and proteoforms in various biological systems. There are 50 HPP research teams worldwide organized by chromosome, mitochondria, biological processes, and disease categories, plus resource pillars for affinity-based protein capture, mass spectrometry, and knowledge bases. This Perspective is part of the 5th annual special issue of the Journal of Proteome Research2–5led by the Chromosome-centric HPP team (C-HPP).
Here we assess progress on identifying missing proteins (MP)—predicted proteins not previously reliably detected, having only neXtProt protein existence (PE) level 2, 3, or 4 evidence. We discuss applications of the HPP Guidelines v2.1 for Interpretation of MS data6 and emerging DIA-SWATH-MS data. Also, we highlight uncertainties in assessing claims of protein translation products from long non-coding RNAs.
The neXtProt and PeptideAtlas Metrics – Progress on the Human Proteome Parts List
PeptideAtlas7, 8 (www.peptideatlas.org) version 2017-01 and neXtProt version 2017-01-23 (www.neXtProt.org), using HPP Guidelines for Interpretation of MS Data v2.16 (https://hupo.org/Guidelines), provided the baseline for HPP investigators and others globally to identify MPs and to prepare HPP papers for this 2017 special issue. ProteomeXchange provides the platform for all proteomics investigators to contribute full raw datasets and metadata to the community through PRIDE and SRMAtlas, as well as GPMDB, MassIVE, jPOST, and ProteomicsDB, leading to standardized reanalyses by PeptideAtlas and GPMDB. The scheme for the HPP Data Workflow was published last year5 (see Supplementary Figure 1). ProteomeXchange9, 10 had 3496 publicly released datasets, of which 1478 are from human samples, up from 900 a year ago (www.proteomecentral.proteomexchange.org) as of 2017-04-27. The 2016 HPP Metrics paper5 presented substantial detail about the features of PeptideAtlas, GPMDB, and neXtProt, including PTMs, proteoforms defined by N and C termini, sequence variants, and splice isoforms.
neXtProt published an update on its many features in January 2017, with emphasis on phenotypic annotations (especially hereditary cancers and channelopathies), phosphorylation and acetylation, sequence variants, and access tools11. Notable among these tools, and of particular usefulness for HPP projects in meeting the Guidelines v2.1 for protein identification at the PE1 level, is the new neXtProt “peptide uniqueness checker”12. This tool matches submitted peptides against all human protein sequences and their variants and isoforms; it was designed to determine which peptides map to human protein sequences uniquely versus mapping as well or better to previously validated proteins. For example, the peptide “TKMGLYYSYFK”, which was proposed to be unique to the PE2 protein DPY19L2P1 (Q6NXN4), maps to the PE1 proteins DPY19L2 (Q6NUT2) and DPY19L1 (Q2PZI1) when SNPs are considered12.
Mass spectrometry datasets available via ProteomeXchange by October 2016 were included in PeptideAtlas 2017-01; a few details addressing nested peptides were added to the same datasets in version 2017-04-20. neXtProt uses the peptides evaluated by PeptideAtlas as the basis for mass spectrometry-based protein findings in its periodic updates. Table 1 shows the progression of highly confident protein identifications in neXtProt and in PeptideAtlas during the course of the HPP from 2012 to 2017.
Table 1.
Metrics of the Progress in the HPP Draft Human Proteome from 2012 to 2017. Numbers shown are of highly confident protein identifications in neXtProt and Peptide Atlas in each of the annual metrics reports in the special issues of the Journal of Proteome Research2–5.
| Chr. | neXtProt Protein Entries |
neXtProt PE1 Proteins |
Human PeptideAtlas Canonical |
|
|---|---|---|---|---|
| Dec 2012 | all | 20,059 | 13,664 | 12,509 |
| Sep 2013 | all | 20,123 | 15,646 | 13,377 |
| Oct 2014 | all | 20,055 | 16,491 | 14,928 |
| Apr 2016 | all | 20,055 | 16,518a | 14,629 |
| Jan 2017 | all | 20,159a | 17,008 | 15,173 |
PE 1–4 = 19,587
During 2016 UniProtKB/SwissProt added 82 human protein entries, while 22 were deprecated and a few others were merged or de-merged. The changes in these numbers contributed to the net increase in total neXtProt entries from 20,055 to 20,159. Most of the remaining increment is due to inclusion of a set of 107 entries for immunoglobulin-coding genes, which were previously excluded by neXtProt but have since been extensively revised and accepted by UniProtKB/SwissProt curators. The number of confidently identified PE1 proteins in neXtProt has grown from 13,664 in 2012 to 17,008 as of version 2017-01-23, and the canonical proteins in the Human PeptideAtlas have grown from 12,509 to 15,173, even after accommodating the more stringent guidelines between 2014 and 20165. From 2016 to 2017, PE1 proteins and canonical proteins increased by 490 and 544, respectively (Table 1).
The criteria for the HPP MS Guidelines v2.1 were extensively discussed in HUPO Congress meetings, Bioinformatics Hub sessions, and Proteomics Standards Initiative workshops, and proposed publicly for comments from the whole community. When combining the results from analyzing hundreds of millions of PSMs, false positives become difficult to control with PSM confidence metrics alone. The false discovery rate among proteins with only a single peptide identification is well known to be much higher than those for which there are multiple peptide identifications6. Therefore, in the pursuit of reliable evidence for difficult-to-observe missing proteins, the community consensus now requires two uniquely mapping non-nested peptides with a minimum length of 9 amino acids13. The guidelines offer provisions for exceptions for proteins that could truly never be identified using these requirements; as an extreme example, ataxin-8 (NX_Q156A1) has the sequence methionine + 79 glutamines (MQ79)! In many cases, researchers may meet the Guidelines v2.1 by using additional proteases14 or permitting missed cleavages or mis-cleavages.
Use of the more stringent v2.1 guidelines6 removed >400 proteins from PE1 status, as documented previously5; nearly all were attributed to requiring two uniquely-mapping non-nested peptides, instead of one, which is necessary to reduce the false discovery rate (FDR) of protein identification. The difference between minimal length of 9 aa versus 8 aa peptides accounted for the exclusion of only 17 proteinsFN1. Expert manual examination of these 17 (by EWD) indicated that about one-third are quite likely authentic detections, while two-thirds were interpreted as equivocal in several different ways. As described previously6, shorter peptides suffer from fewer fragmentation peaks, which causes a higher rate of false positives, and are more likely to map to multiple genomic locations, particularly when sequence variants are considered. There could also be mapping to immunoglobulin variable regions. For these 17 exclusions, more experimental data are needed to achieve sufficient confidence that the protein truly has been detected.
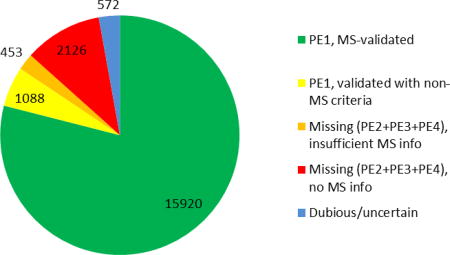
Table 2 presents the numbers of predicted proteins in neXtProt in each of the PE levels. Clear experimental evidence for the existence of a protein is based on mass spectrometry in accordance with the HPP Guidelines v2.1 for 15,920 proteins and on the following non-MS methods for a further 1088 (see Figure 1): Edman sequencing (107), biochemical studies (131), PTMs (127), protein-protein interactions (372), antibody-based techniques (37), 3D structures (63), and disease mutations (251). Thereby, 87% of the human proteome has been identified to level PE1.
Table 2.
neXtProt Status of the HUPO Draft Human Proteome from 2013 to 2017.
| PE Level | Sept 2013 |
Oct 2014 |
April 2016 |
Jan 2017 |
||
|---|---|---|---|---|---|---|
| 1: Evidence at protein levela | 15,646 | 16,491 | 16,518 | 17,008a | ||
| 2: Evidence at transcript level | 3570 | 2647 | 2290 | 1939 |
|
2579 |
| 3: Inferred from homology | 187 | 214 | 565 | 563 | Missing | |
| 4: Predicted | 87 | 87 | 94 | 77 | Proteins | |
| 5: Uncertain or dubious | 638 | 616 | 588 | 572 |
Percent of predicted proteins classified as PE1 by neXtProt = PE1/PE1+2+3+4 = 17,008/19,587 = 87%.
Figure 1.

Distribution of neXtProt predicted proteins by protein existence (PE) level and the nature of the evidence data as of version 2017-01. Supplementary Table 1 presents the status of neXtProt results by PE level for each chromosome as of version 2017-01. Supplementary Table 2 the classification of proteins in PeptideAtlas by chromosome (version 2017-04).
Figure 1 shows that the 2579 missing proteins (PE2+3+4) occur in two subsets: 453 with MS data that were excluded from PE1 due to the more stringent v2.1 Guidelines (down from 485 in 2016) and 2126 others lacking sufficient evidence for PE1. The list of the accession numbers of the 453 protein entries not promoted to PE1 under PE1Rules.PA_NP_2015_12 for both PeptideAtlas and neXtProt, but which would have been promoted to PE1 according to the older rules for neXtProt (PE1Rules.NP_2015_04) is available on the neXtProt ftp server at: ftp://ftp.nextprot.org/pub/current_release/custom/hpp/HPP_entries_with_unconfirmed_MS_data.txt. The difference between the 15,173 canonical proteins in PeptideAtlas 2017-01 and the 15,920 from MS in Figure 1 is accounted for by proteins curated by neXtProt from MS studies of PTMs.
The total of 2579 is down by 370 from 2949 a year ago, mostly from extensive findings in sperm15 and testis16, studies that were guided by the tissue-based proteome from Human Protein Atlas17, the antibody profiling resource pillar of the HPP. For the 1939 PE2 proteins in Table 2, neXtProt, Human Protein Atlas, GeneCards (www.genecards.org), and GTEx (www.gtexportal.org) provide annotations about tissues with their highest transcript expression; such information represents a guide to choose specimens in which to search for evidence of expression of the protein. The same is true for the 563 PE3 protein entries identified by homology in other species. However, proper analysis of the median correlation between mRNA and protein levels from individual genes has shown quite low values (r = 0.21), even ignoring the complexity of splice isoforms.18
Back in 2013, the HPP decided to exclude the PE5 category (572 entries) from the denominator, as those genome sequences are considered “dubious” or “uncertain” as candidates for protein-coding3 and about half are pseudogenes. If and when curated evidence supports moving PE5 entries higher, we will, of course, include such findings.
In 2016-17 the Human Protein Atlas released the Human Transcriptome19 and the Human Cell Atlas20, 21. The transcriptome map complements work from the Genome-based Tissue Expression Project GTEx (www.gtexportal.org) and Fantom consortium (fantom.gsc.riken.jp), plus several other repositories and maps. About 45% of the 18,864 transcripts are expressed in all tissues and 13% in many tissues, while 13% (2359) were highly enriched in just one tissue, predominantly in testis/male reproductive tract and next in brain. Fresh frozen tissue and postmortem tissue gave very similar results. Tissue-specificity may be influenced by complex gene-regulatory patterns, including alternative splicing; isoform analysis of tissue-specificity and protein networks might be revealing22, 23. The Human Protein Cell Atlas was built in a crowd-sourcing spatial proteomics project with 160,000 citizen-scientists examining immunohistochemical patterns for 12,000 proteins across 56 human cell lines, classified into 32 organelles and cellular structures (www.proteinatlas.org/cell; https://www.eveonline.com/discovery-proteins). A major quality control effort on antibodies has been launched by the International Working Group on Antibody Validation.24
The Search for Missing Proteins
To accelerate the completion of a high-quality draft of the human proteome, the C-HPP has mounted an initiative known as “The neXt-50 Challenge.” The aim is for each Chromosome team to plan how to identify up to 50 or more missing proteins from its respective chromosome (Supplementary Table 1) in time for the HUPO-2018 Congress. Noting that three chromosomes (13, 18, Y) and mitochondria had less than 50 total missing proteins and that many proteomics analyses are inherently chromosome-agnostic, teams were encouraged to share datasets for annotation and thereby contribute to the identification of additional missing proteins from other chromosomes. Three chromosomes (1, 11, 19) have >200 missing proteins. Progress is to be updated on the C-HPP Wiki (http://c-hpp.webhosting.rug.nl/tiki-index.php?page=Group%20composition).
Duek et al25 last year published a model analysis of the 134 and 93 missing proteins on chromosomes 2 and 14, respectively, a total of 227, and their strategy for identifying the likely most detectable missing proteins in spermatozoa. The authors put aside 29 predicted olfactory receptor proteins, 2 pseudogenes, 6 proteins refractory to tryptic digestion for MS, plus 27 newly validated proteins, 42 known to them to be in the neXtProt/SwissProt curation pipeline, and 22 with single proteotypic peptides (a total of 128). Then they identified 25 chromosome 2 and 15 chromosome 14 predicted proteins with highest priority for successful analyses in spermatozoa. This year Carapito et al (this issue) report a total of 12 new PE1 candidates from sperm for chromosomes 2 and 14. These results show how difficult it is to detect 50 missing proteins per chromosome, let alone all of the PE2-3-4 missing proteins in the entire proteome.
MissingProteinPedia26 (www.missingproteins.org) is a new on-line database from Australia with entries for missing proteins as of 2016. The database is populated with available information about 1482 of these proteins (as of 2017-07-24), including low-stringency MS hits, literature citations from a PubMed mining algorithm, and other information of variable reliability, which may provide clues that guide researchers to specific cells, tissues, or developmental stages for targeted investigation to find such missing proteins. Based on the MissingProteinPedia and neXtProt 2016-02, the 12 most numerous PE2–4 missing protein families are: G-protein coupled receptor 1 (including olfactory receptors), Krueppel C2H2-type zinc-finger, beta defensin, PRAME, G-protein coupled receptor T2R, HPIP, humanin, LCE, MS4A, NBPF, peptidase C19/USP17 subfamily, and peptidase type B retroviral polymerase/HERV class II K(HML-2)26. A focus on analyses of protein families could be a productive strategy; as reflected in the PeptideAtlas categories of ambiguous protein matches, however, there are many challenges in distinguishing highly homologous proteins with shared or subsumed peptides8. Another strategy, tied to chromosome-specific analyses, is the detection and characterization of all members of amplicons, such as the 21 genes of the ERBB2 amplicon on chromosome 17q12-2127.
Status of Evidence for Translation Products from lncRNAs and smORFs
The Encyclopedia of DNA Elements (ENCODE) Project28, The Cancer Genome Atlas (TCGA), and a growing literature have documented gene regulatory functions and RNA/protein interactions for a class of molecules known as long non-coding RNAs (lncRNAs)29 (https://genome.ucsc.edu/ENCODE/). These lncRNAs are transcribed primarily from intergenic regions and from intronic regions within protein-coding genes. Iyer et al30 created a landscape of lncRNAs in the human transcriptome from 25 independent studies with 91,000 expressed genome sequences, of which 59,000 coded for lncRNAs and 597 harbored ultraconserved elements. lncRNAs pose an important question for the HPP: Are any of these transcripts translated into polypeptides? If so, are the polypeptides present in biologically relevant amounts with distinct functions, or are they bystander products reflecting a dynamic system responding to cellular demands for a rapid protein translation capacity? Similar questions apply to small open-reading frames (smORFs), potentially yielding smORF-encoded polypeptides (SEPs) or micropeptides31. How should the HPP evaluate and potentially include these novel protein candidates as bona fide components of the human proteome and the HPP metrics? Also, the HPP lacks policies for classifying peptides versus proteins and for recognizing bioactive cleavage products from known proteins; examples include endostatin, derived from collagen XVIII, and asprosin, a C-terminal 140 aa polypeptide hormone secreted from white adipose tissue, annotated under its parent protein, fibrillin 1 (FBN1, NX_P35555)32.
Five prominent lncRNA researchers published a joint paper in Cell in 2013 concluding that ribosome profiling provides evidence that large noncoding RNAs do not encode proteins, even when they are shown to bind to 80S ribosomes33. Caviello34 similarly concluded that few lncRNAs generate protein products. In contrast, Slavoff et al35 presented evidence for 90 SEPs in human K562 cell line samples, extending the 2007 report from Oyama et al36; synthetic peptides were used, but many of the spectral matches appear suspect. The developers of the LNCipedia resource37 performed extensive re-analysis of data sets deposited in the PRIDE repository38 for 21,488 human lncRNA transcripts; they searched for peptide matches, but reported more decoy hits than forward matches.39, 40 Meanwhile, Wang et al41 reported assays of the “translatome”, mRNAs captured in the initiation of translation on polyribosomes; they have presented evidence for lncRNAs identified in the translatome at two 2017 C-HPP workshops. The scarcity of credible protein products may be because mass spectrometry techniques are biased against them, they have low abundance due to low efficiency of synthesis or rapid degradation, or they are really not present, with the deduced peptides mapping better to known proteins. Verheggen et al42 showed that potential MS biases due to protein length, amino acid composition, abundance, instability, or mRNA expression level are minimal.
Analyses of large numbers of speculative sequences are fraught with perils of false positives overwhelming true detections of variants and novel coding elements43. To address this problem, the HPP MS Dataset Interpretation Guidelines v2.1 were designed as an embodiment of the mantra “extraordinary claims require extraordinary evidence” (https://en.wikipedia.org/wiki/Sagan_standard); here claims of detecting proteins from lncRNAs are the “extraordinary claims.” Thus, for such “found proteins”, the Guidelines require two non-nested, uniquely-mapping peptides of at least 9 residues in length with excellent high-resolution spectral matches confirmed with synthetic peptide spectra, and with a process for ruling out alternative explanations for the confirmed peptide sequences, taking into account SAAVs and isobaric PTMs6. As yet, detection claims of SEPs and translated lncRNAs have not met the HPP guidelines. For some small products it may not be possible to obtain two non-nested uniquely-mapping peptides of at least 9 residues from protease digestion even when using alternative enzymes to trypsin14. Other corroborating evidence will be necessary in such cases, using specific antibodies, demonstrating biological function, or showing impacts on cellular processes from knockdowns. When evidence can be presented that meets the HPP Guidelines and neXtProt curation criteria, these sequences will enter the HPP knowledge base through PeptideAtlas and neXtProt. It will be interesting to see how this sub-field of proteogenomics progresses and whether lncRNAs contribute bona fide polypeptides with biological activity to the human proteome.
DIA-SWATH-MS Data
As discussed above, the HPP MS Dataset Interpretation Guidelines v2.1 provide specifics for validating claims of missing protein identifications or novel protein-coding elements via data-dependent analysis (DDA) and via SRM, both of which require using synthetic peptides44,45. Data-independent analysis (DIA) such as SWATH-MS (sequential window acquisition of all theoretical spectra) has emerged as an alternative workflow for protein identification and quantification46,47. Currently, the HPP Guidelines are silent on how to apply them to DIA data sets. This was intentional as it seemed too early in the rapid advancement of DIA techniques to apply the guidelines. Now, it is time to begin a process to clarify the application and adaptation of the guidelines to DIA data.
We briefly describe here our suggested interpretation of the guidelines as they should apply to DIA. Guidelines 1–9 (see Supplementary Table 3) are directly applicable: the checklist, data deposition in a ProteomeXchange repository with the required proteome reference, and issues related to false discovery rates, which, due to the high degree of multiplexing with SWATH-MS, may be even more important and challenging than for other workflows. Guidelines 10–15 apply only to claims of detection of proteins or other translation products that are not PE1. If a DIA data set is analyzed similar to a DDA data set, using tools such as DIA-Umpire48 or DISCO, then guidelines 10–12 apply. If a DIA dataset is analyzed similar to an SRM dataset, using tools such as Skyline49, OpenSWATH46, Spectronaut, or PeakView, then guideline 13 applies instead. In both of these cases the peptide signatures must show concordance with the corresponding synthetic peptide signatures. Guidelines 14–15 are independent of data type and apply to any analysis workflow, including DIA. As with the original Guidelines, we will promote a community discussion and refinement of these ideas, with the aim of releasing an updated version at year-end.
CONCLUDING REMARKS
The HUPO Human Proteome Project draft human proteome in neXtProt 2017-01-23 reached 17,008 high confidence neXtProt PE1 protein identifications, representing 87% of the neXtProt predicted PE1–4 proteins, with 15,920 from MS/MS and 1088 from other kinds of protein studies. Continuing progress will be reported regularly in the C-HPP Wiki, HUPO website, and in this and future HPP Special Issues of the Journal of Proteome Research. [The neXtProt 2017-04-20 update at www.nextprot.org has 17,045 PE1 proteins with 2563 PE2,3,4 missing proteins; clearly, the metrics are a moving target during each annual cycle.]
Meanwhile, the scheme from the Biology and Disease-driven HPP to utilize organ-specific lists of popular proteins in combination with quantitative multiplexed targeted proteomics assays50 is a stimulus for broader use of proteomics in many areas of life sciences research. Attention to the biological diversity enhanced by alternative proteoforms will advance understanding of strategies that have evolved to maintain homeostasis in the face of challenges from infection, disease, injury, aging, nutritional and environmental stress, and hormonal variations.
Given the importance of targeted proteomics with SRM or PRM-MS methods or DIA-SWATH-MS, the HPP is addressing the application of the HPP Guidelines for interpretation of DIA-SWATH MS data. Finally, we are considering the potential expansion of the proteome to include any credible translation products from long non-coding RNAs, small open reading frame sequences, and bioactive cleavage of known proteins.
Supplementary Material
Supplementary Table 1. The status of neXtProt results by PE level for each chromosome (from nextprot.org, version 2017-01)
Supplementary Table 2. Classification of Proteins in PeptideAtlas by Chromosome (from peptideatlas.org version 2017-04)
Supplementary Table 3. HPP Guidelines v2.1 for Interpretation of MS Data (ref. 6)
Supplementary Figure 1. Overview of the Human Proteome Project data workflow
Acknowledgments
We appreciate the guidance from the HPP Executive Committee and Dr. Amos Bairoch of neXtProt. We thank the UniProt groups at SIB, EBI, and PIR for providing high-quality annotations for the human proteins in UniProtKB/Swiss-Prot. The neXtProt server is hosted by VitalIT in Switzerland. G.S.O. acknowledges grant support from National Institutes of Health grants P30ES017885-01A1 and NIH U24CA210967; E.W.D. from NIH grants R01GM087221 and U54EB020406; L.L. and neXtProt from the SIB Swiss Institute of Bioinformatics; E.K.L., from the Knut and Alice Wallenberg Foundation and EU 7th Framework; and C.M.O. by a Canadian Institutes of Health Research 7-year Foundation Grant and a Canada Research Chair in Protease Proteomics and Systems Biology.
Footnotes
neXtProt accession numbers for the 17 excluded proteins: NX_L0R8F8, NX_O15375, NX_P18825, NX_Q6NSI1, NX_Q7Z769, NX_Q8IUB2, NX_Q8IWZ4, NX_Q8N878, NX_Q8N957, NX_Q8WTQ1, NX_Q96A84, NX_Q96BR6, NX_Q96DU9, NX_Q9BQI4, NX_Q9BRU2, NX_Q9P109, NX_Q9UNT1.
The following files are available free of charge at ACS website http://pubs.acs.org:
The authors declare no competing financial interest.
References
- 1.Legrain P, Aebersold R, Archakov A, Bairoch A, Bala K, Beretta L, Bergeron J, Borchers CH, Corthals GL, Costello CE, Deutsch EW, Domon B, Hancock W, He F, Hochstrasser D, Marko-Varga G, Salekdeh GH, Sechi S, Snyder M, Srivastava S, Uhlen M, Wu CH, Yamamoto T, Paik YK, Omenn GS. The Human Proteome Project: current state and future direction. Mol Cell Proteomics. 2011;10(7) doi: 10.1074/mcp.M111.009993. M111 009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marko-Varga G, Omenn GS, Paik YK, Hancock WS. A first step toward completion of a genome-wide characterization of the human proteome. J. Proteome Res. 2013;12(1):1–5. doi: 10.1021/pr301183a. [DOI] [PubMed] [Google Scholar]
- 3.Lane L, Bairoch A, Beavis RC, Deutsch EW, Gaudet P, Lundberg E, Omenn GS. Metrics for the human proteome project 2013–2014 and strategies for finding missing proteins. J. Proteome Res. 2014;13(1):15–20. doi: 10.1021/pr401144x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Omenn GS, Lane L, Lundberg EK, Beavis RC, Nesvizhskii AI, Deutsch EW. Metrics for the Human Proteome Project 2015: progress on the human proteome and guidelines for high-confidence protein identification. J. Proteome Res. 2015;14(9):3452–60. doi: 10.1021/acs.jproteome.5b00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Omenn GS, Lane L, Lundberg EK, Beavis RC, Overall CM, Deutsch EW. Metrics for the Human Proteome Project 2016: Progress on Identifying and Characterizing the Human Proteome, Including Post-Translational Modifications. J Proteome Res. 2016;15(11):3951–60. doi: 10.1021/acs.jproteome.6b00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deutsch EW, Overall CM, Van Eyk JE, Baker MS, Paik YK, Weintraub ST, Lane L, Martens L, Vandenbrouck Y, Kusebauch U, Hancock WS, Hermjakob H, Aebersold R, Moritz RL, Omenn GS. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 2.1. J Proteome Res. 2016;15(11):3961–70. doi: 10.1021/acs.jproteome.6b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desiere F, Deutsch EW, Nesvizhskii AI, Mallick P, King NL, Eng JK, Aderem A, Boyle R, Brunner E, Donohoe S, Fausto N, Hafen E, Hood L, Katze MG, Kennedy KA, Kregenow F, Lee H, Lin B, Martin D, Ranish JA, Rawlings DJ, Samelson LE, Shiio Y, Watts JD, Wollscheid B, Wright ME, Yan W, Yang L, Yi EC, Zhang H, Aebersold R. Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol. 2005;6(1):R9. doi: 10.1186/gb-2004-6-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deutsch EW, Sun Z, Campbell D, Kusebauch U, Chu CS, Mendoza L, Shteynberg D, Omenn GS, Moritz RL. State of the Human Proteome in 2014/2015 as viewed through PeptideAtlas: enhancing accuracy and coverage through the AtlasProphet. J Proteome Res. 2015;14(9):3461–73. doi: 10.1021/acs.jproteome.5b00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martens L, Vizcaíno JA. A golden age for working with public proteomics data. Trends in Biochemical Sciences. 2017;42(5):333–41. doi: 10.1016/j.tibs.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, Moritz RL, Carver JJ, Wang M, Ishihama Y, Bandeira N, Hermjakob H, Vizcaino JA. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45(D1):D1100–d1106. doi: 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaudet P, Michel PA, Zahn-Zabal M, Britan A, Cusin I, Domagalski M, Duek PD, Gateau A, Gleizes A, Hinard V, Rech de Laval V, Lin J, Nikitin F, Schaeffer M, Teixeira D, Lane L, Bairoch A. The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res. 2017;45(D1):D177–d182. doi: 10.1093/nar/gkw1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaeffer M, Gateau A, Teixeira D, Michel PA, Zahn-Zabal M, Lane L. The neXtProt peptide uniqueness checker: a tool for the proteomics community. Bioinformatics. 2017 May 17; doi: 10.1093/bioinformatics/btx318. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deutsch EW, Sun Z, Campbell DS, Binz PA, Farrah T, Shteynberg D, Mendoza L, Omenn GS, Moritz RL. Tiered human integrated sequence search databases for shotgun proteomics. J Proteome Res. 2016;15(11):4091–100. doi: 10.1021/acs.jproteome.6b00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giansanti P, Tsiatsiani L, Low TY, Heck AJ. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat Protoc. 2016;11(5):993–1006. doi: 10.1038/nprot.2016.057. [DOI] [PubMed] [Google Scholar]
- 15.Vandenbrouck Y, Lane L, Carapito C, Duek P, Rondel K, Bruley C, Macron C, Gonzalez de Peredo A, Coute Y, Chaoui K, Com E, Gateau A, Hesse AM, Marcellin M, Mear L, Mouton-Barbosa E, Robin T, Burlet-Schiltz O, Cianferani S, Ferro M, Freour T, Lindskog C, Garin J, Pineau C. Looking for missing proteins in the proteome of human spermatozoa: An Update. J Proteome Res. 2016;15(11):3998–4019. doi: 10.1021/acs.jproteome.6b00400. [DOI] [PubMed] [Google Scholar]
- 16.Wei W, Luo W, Wu F, Peng X, Zhang Y, Zhang M, Zhao Y, Su N, Qi Y, Chen L, Zhang Y, Wen B, He F, Xu P. Deep coverage proteomics identifies more low-abundance missing proteins in human testis tissue with Q-exactive HF mass spectrometer. J Proteome Res. 2016;15(11):3988–97. doi: 10.1021/acs.jproteome.6b00390. [DOI] [PubMed] [Google Scholar]
- 17.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 18.Fortelny N, Overall CM, Pavlidis P, Freue GVC. Can we predict protein from mRNA levels? Nature. 2017;547(7664):E19–e20. doi: 10.1038/nature22293. [DOI] [PubMed] [Google Scholar]
- 19.Uhlen M, Hallstrom BM, Lindskog C, Mardinoglu A, Ponten F, Nielsen J. Transcriptomics resources of human tissues and organs. Mol. Syst. Biol. 2016;12(4):862. doi: 10.15252/msb.20155865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM, Backstrom A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson A, Sjostedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Ponten F, von Feilitzen K, Lilley KS, Uhlen M, Lundberg E. A subcellular map of the human proteome. Science. 2017;356(6340) doi: 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 21.Lundberg E, Uhlen M. Creation of an antibody-based subcellular protein atlas. Proteomics. 2010;10(22):3984–96. doi: 10.1002/pmic.201000125. [DOI] [PubMed] [Google Scholar]
- 22.Li HD, Menon R, Eksi R, Guerler A, Zhang Y, Omenn GS, Guan Y. A network of splice isoforms for the mouse. Scientific Reports. 2016;6:24507. doi: 10.1038/srep24507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li HD, Omenn GS, Guan Y. A proteogenomic approach to understand splice isoform functions through sequence and expression-based computational modeling. Brief Bioinform. 2016;17(6):1024–31. doi: 10.1093/bib/bbv109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uhlen M, Bandrowski A, Carr S, Edwards A, Ellenberg J, Lundberg E, Rimm DL, Rodriguez H, Hiltke T, Snyder M, Yamamoto T. A proposal for validation of antibodies. Nat Methods. 2016;13(10):823–7. doi: 10.1038/nmeth.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duek P, Bairoch A, Gateau A, Vandenbrouck Y, Lane L. Missing protein landscape of human chromosomes 2 and 14: progress and current status. J Proteome Res. 2016;15(11):3971–78. doi: 10.1021/acs.jproteome.6b00443. [DOI] [PubMed] [Google Scholar]
- 26.Baker MS, Ahn SB, Mohamedali A, Islam MT, Cantor D, Verhaert PD, Fanayan S, Sharma S, Nice EC, Connor M, Ranganathan S. Accelerating the search for the missing proteins in the human proteome. Nat Commun. 2017;8:14271. doi: 10.1038/ncomms14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu S, Im H, Bairoch A, Cristofanilli M, Chen R, Deutsch EW, Dalton S, Fenyo D, Fanayan S, Gates C, Gaudet P, Hincapie M, Hanash S, Kim H, Jeong SK, Lundberg E, Mias G, Menon R, Mu Z, Nice E, Paik YK, Uhlen M, Wells L, Wu SL, Yan F, Zhang F, Zhang Y, Snyder M, Omenn GS, Beavis RC, Hancock WS. A chromosome-centric human proteome project (C-HPP) to characterize the sets of proteins encoded in chromosome 17. J Proteome Res. 2013;12(1):45–57. doi: 10.1021/pr300985j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan Y, Zhang Y, Xu S, Kong N, Zhou Y, Ren Z, Deng Y, Lin L, Ren Y, Wang Q, Zi J, Wen B, Liu S. Insights from ENCODE on missing proteins: why beta-defensin expression is scarcely detected. J Proteome Res. 2015;14(9):3635–44. doi: 10.1021/acs.jproteome.5b00565. [DOI] [PubMed] [Google Scholar]
- 29.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482(7385):339–46. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, Poliakov A, Cao X, Dhanasekaran SM, Wu YM, Robinson DR, Beer DG, Feng FY, Iyer HK, Chinnaiyan AM. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47(3):199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D'Lima NG, Ma J, Winkler L, Chu Q, Loh KH, Corpuz EO, Budnik BA, Lykke-Andersen J, Saghatelian A, Slavoff SA. A human microprotein that interacts with the mRNA decapping complex. Nat Chem Biol. 2017;13(2):174–80. doi: 10.1038/nchembio.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romere C, Duerrschmid C, Bournat J, Constable P, Jain M, Xia F, Saha PK, Del Solar M, Zhu B, York B, Sarkar P, Rendon DA, Gaber MW, LeMaire SA, Coselli JS, Milewicz DM, Sutton VR, Butte NF, Moore DD, Chopra AR. Asprosin, a fasting-induced glucogenic protein hormone. Cell. 2016;165(3):566–79. doi: 10.1016/j.cell.2016.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154(1):240–51. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calviello L, Mukherjee N, Wyler E, Zauber H, Hirsekorn A, Selbach M, Landthaler M, Obermayer B, Ohler U. Detecting actively translated open reading frames in ribosome profiling data. Nat Methods. 2016;13(2):165–70. doi: 10.1038/nmeth.3688. [DOI] [PubMed] [Google Scholar]
- 35.Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, Saghatelian A. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;9(1):59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oyama M, Kozuka-Hata H, Suzuki Y, Semba K, Yamamoto T, Sugano S. Diversity of translation start sites may define increased complexity of the human short ORFeome. Mol Cell Proteomics. 2007;6(6):1000–6. doi: 10.1074/mcp.M600297-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Volders PJ, Helsens K, Wang X, Menten B, Martens L, Gevaert K, Vandesompele J, Mestdagh P. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013;41(Database issue):D246–51. doi: 10.1093/nar/gks915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44(22):11033. doi: 10.1093/nar/gkw880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volders PJ, Verheggen K, Menschaert G, Vandepoele K, Martens L, Vandesompele J, Mestdagh P. An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic Acids Res. 2015;43(Database issue):D174–80. doi: 10.1093/nar/gku1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volders PJ, Verheggen K, Menschaert G, Vandepoele K, Martens L, Vandesompele J, Mestdagh P. An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic Acids Res. 2015;43(8):4363–4. doi: 10.1093/nar/gkv295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang T, Cui Y, Jin J, Guo J, Wang G, Yin X, He QY, Zhang G. Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic Acids Res. 2013;41(9):4743–54. doi: 10.1093/nar/gkt178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verheggen K, Volders PJ, Mestdagh P, Menschaert G, Van Damme P, Gevaert K, Martens L, Vandesompele J. Noncoding after all: biases in proteomics data do not explain observed absence of lncRNA translation products. J Proteome Res. 2017;16(7):2508–15. doi: 10.1021/acs.jproteome.7b00085. [DOI] [PubMed] [Google Scholar]
- 43.Nesvizhskii AI. Proteogenomics: concepts, applications and computational strategies. Nat Methods. 2014;11(11):1114–25. doi: 10.1038/nmeth.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kusebauch U, Campbell DS, Deutsch EW, Chu CS, Spicer DA, Brusniak MY, Slagel J, Sun Z, Stevens J, Grimes B, Shteynberg D, Hoopmann MR, Blattmann P, Ratushny AV, Rinner O, Picotti P, Carapito C, Huang CY, Kapousouz M, Lam H, Tran T, Demir E, Aitchison JD, Sander C, Hood L, Aebersold R, Moritz RL. Human SRMAtlas: a resource of targeted assays to quantify the complete human proteome. Cell. 2016;166(3):766–78. doi: 10.1016/j.cell.2016.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zolg DP, Wilhelm M, Schnatbaum K, Zerweck J, Knaute T, Delanghe B, Bailey DJ, Gessulat S, Ehrlich HC, Weininger M, Yu P, Schlegl J, Kramer K, Schmidt T, Kusebauch U, Deutsch EW, Aebersold R, Moritz RL, Wenschuh H, Moehring T, Aiche S, Huhmer A, Reimer U, Kuster B. Building ProteomeTools based on a complete synthetic human proteome. Nat Methods. 2017;14(3):259–62. doi: 10.1038/nmeth.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rost HL, Rosenberger G, Navarro P, Gillet L, Miladinovic SM, Schubert OT, Wolski W, Collins BC, Malmstrom J, Malmstrom L, Aebersold R. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32(3):219–23. doi: 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- 47.Schubert OT, Gillet LC, Collins BC, Navarro P, Rosenberger G, Wolski WE, Lam H, Amodei D, Mallick P, MacLean B, Aebersold R. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc. 2015;10(3):426–41. doi: 10.1038/nprot.2015.015. [DOI] [PubMed] [Google Scholar]
- 48.Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, Nesvizhskii AI. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods. 2015;12(3):258–64. doi: 10.1038/nmeth.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–8. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lam MP, Venkatraman V, Xing Y, Lau E, Cao Q, Ng DC, Su AI, Ge J, Van Eyk JE, Ping P. Data-driven approach to determine popular proteins for targeted proteomics translation of six organ systems. J Proteome Res. 2016;15(11):4126–34. doi: 10.1021/acs.jproteome.6b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. The status of neXtProt results by PE level for each chromosome (from nextprot.org, version 2017-01)
Supplementary Table 2. Classification of Proteins in PeptideAtlas by Chromosome (from peptideatlas.org version 2017-04)
Supplementary Table 3. HPP Guidelines v2.1 for Interpretation of MS Data (ref. 6)
Supplementary Figure 1. Overview of the Human Proteome Project data workflow