

Key Points
VKORC1 is more sensitive than VKORC1L1 to OAC inhibition, whereby 4-hydroxycoumarin rodenticides are equally effective.
In silico and in vitro analysis revealed OAC binding sites that are different for VKORC1 and VKORC1L1.
Abstract
Vitamin K reduction is catalyzed by 2 enzymes in vitro: the vitamin K 2,3-epoxide reductase complex subunit 1 (VKORC1) and its isozyme VKORC1-like1 (VKORC1L1). In vivo, VKORC1 reduces vitamin K to sustain γ-carboxylation of vitamin K-dependent proteins, including coagulation factors. Inhibition of VKORC1 by oral anticoagulants (OACs) is clinically used in therapy and in prevention of thrombosis. However, OACs also inhibit VKORC1L1, which was previously shown to play a role in intracellular redox homeostasis in vitro. Here, we report data for the first time on specific inhibition of both VKOR enzymes for various OACs and rodenticides examined in a cell-based assay. Effects on endogenous VKORC1 and VKORC1L1 were independently investigated in genetically engineered HEK 293T cells that were knocked out for the respective genes by CRISPR/Cas9 technology. In general, dose-responses for 4-hydroxycoumarins and 1,3-indandiones were enzyme-dependent, with lower susceptibility for VKORC1L1 compared with VKORC1. In contrast, rodenticides exhibited nearly identical dose-responses for both enzymes. To explain the distinct inhibition pattern, we performed in silico modeling suggesting different warfarin binding sites for VKORC1 and VKORC1L1. We identified arginine residues at positions 38, 42, and 68 in the endoplasmatic reticulum luminal loop of VKORC1L1 responsible for charge-stabilized warfarin binding, resulting in a binding pocket that is diametrically opposite to that of VKORC1. In conclusion, our findings provide insight into structural and molecular drug binding on VKORC1, and especially on VKORC1L1.
Visual Abstract
Introduction
In the 1940s, dicoumarol, a naturally occurring 4-hydroxycoumarin in fodder contaminated with spoiled clover, was identified as the cause of spontaneous hemorrhage in livestock.1 Further homologs were synthesized and characterized by Link and colleagues for intended use as rodenticides and, later, as human oral anticoagulants (OACs).2 Since the 1950s, OACs have been frequently prescribed to treat and prevent thromboembolism. Warfarin is the oral anticoagulant drug of choice in the United States, acenocoumarol in southern Europe and Asia, and phenprocoumon in Central Europe.3 Fluindione, a substituted 1,3-indandione, is used frequently in French-speaking countries.4 Dosing and efficacy of OACs depend on the specific compound and patient’s pharmacogenetic and pharmacokinetic profile.5
4-Hydroxycoumarin-based OACs and rodenticides, as well as indandiones, directly inhibit vitamin K 2,3-epoxide reductase complex subunit 1 (VKORC1).6-8 VKORC1 catalyzes the reduction of vitamin K 2,3-epoxide (K>O) and vitamin K quinone (K) to vitamin K hydroquinone (KH2), which is essential to support γ-carboxylation, and thereby activation of vitamin K-dependent (VKD) proteins (eg, coagulation factor IX, FIX).9 Therefore, inhibition of VKORC1 limits the availability of KH2, leading to reduced posttranslational modifications of VKD clotting factors.10,11 The isozyme of VKORC1, VKORC1-like 1 (VKORC1L1), possesses the same enzymatic reactions as VKORC1 in vitro.12-14 Although VKORC1 is essential for blood clotting, it was shown that VKORC1L1 cannot substitute for this function, and likely has other biological roles.14-16 Recent studies suggest that the unique function of VKORC1 may be a result of its specific tissue distribution and high expression in hepatocytes where VKD clotting factors are synthesized.12,17,18 Both enzymes were found to exhibit different affinities with regard to warfarin.14,17 However, detailed knowledge of the molecular mechanism and structural site of OAC action for VKORC1L1 are lacking.
In this study, we established VKORC1 and VKORC1L1 knockout (KO) HEK 293T cell lines to investigate specific inhibition of endogenously expressed VKORC1L1 and VKORC1, respectively, by various OACs and rodenticides. We used in silico modeling and docking to investigate OAC drug binding and verified putative interacting residues for VKORC1L1. Our findings demonstrate enzyme-specific differences in warfarin binding for VKORC1 and VKORC1L1 that are consistent with lower OAC sensitivity for VKORC1L1.
Material and methods
Generation of VKORC1 and VKORC1L1 single- and double-KO HEK 293T cell lines
HEK 293T cell lines with single and double gene KOs for VKORC1 and VKORC1L1 were generated using CRISPR/Cas9-based gene editing.19 Guide RNA target sequence was GCTCTACGCGCTGCACGTGAAGG for VKORC1 and CTTCTCCCGCTCCACGTGGTAGG for VKORC1L1. HEK 293T cells were maintained in Dulbecco’s modified Eagle medium (Thermo Fisher Scientific, Dreieich, Germany) containing 1% vol/vol penicillin/streptomycin, 1× nonessential amino acids (Thermo Fisher Scientific) and 10% vol/vol fetal bovine serum (PAN, Aidenbach, Germany). Cells were seeded (25 000 cells per 96-well plate) and transfected with 200 ng plasmid containing mCherry Cas9 and a U6 promoter-driven guide RNA against VKORC1 or VKORC1L1 with Lipofectamine 2000 (Thermo Fisher Scientific). Three days after transfection, cells were seeded under limiting conditions to obtain single-cell clones. Single-cell clones were expanded for maintenance and analysis. For next-generation sequencing (Illumina, MiSeq, San Diego, CA), ∼200 different single-cell clones were lysed in 100 µL lysis buffer (0.2 mg/mL proteinase K, 1 mM CaCl2, 3 mM MgCl2, 1 mM EDTA, 1% Triton X-100, 10 mM Tris at pH 7.5) for genomic DNA preparation. Dual polymerase chain reaction barcoding and deep sequencing were performed as described by Schmidt et al.20 Deep sequencing data were evaluated using the OutKnocker.org web tool, and positive clones were verified by next-generation sequencing deep sequencing, as described earlier.21,22
Mammalian expression vectors
For solely human F9 expression, a pCMV6-XL4 vector was used (Origene, Rockville, MD). The pIRES vectors harboring cDNA of human VKORC1 (hVKORC1) or human VKORC1L1 (hVKORC1L1), together with human F9, were generated as previously described.23,24 The VKORC1 loop swap construct was created by replacing amino acids Lys30 to Ser79 of VKORC1 with Glu37 to Pro86 of VKORC1L1, and vice versa. Variants of VKORC1L1 were generated by site-directed mutagenesis. For expression analysis, VKOR variants were cloned into pcDNA3.1/myc-His (Thermo Fisher Scientific).
Determination of vitamin K reductase and VKOR activity and IC50 values
Wild-type (WT), VKORC1 KO and VKORC1L1 KO HEK 293T cells were transfected with F9 cDNA to assess endogenous vitamin K reductase (VKR) and vitamin K 2,3-epoxide reductase (VKOR) activity of VKORC1L1 and VKORC1. Double-KO (DKO) HEK 293T cells were transfected with pIRES vectors containing cDNAs of F9 together with VKOR variants (or cotransfected with F9 [pCMV6-XL4] and VKOR myc-tagged variants [pcDNA3.1/myc-His]). After 4 hours, transfection medium was replaced by expression medium (OptiMEM [Thermo Fisher Scientific], including 0.25% BSA [protease-free; Sigma-Aldrich, Munich, Germany] and 2.5 mM CaCl2 [Sigma-Aldrich]) supplemented with 12 µM vitamin K1 quinone (K1; in dimethyl sulfoxide, Sigma-Aldrich) or 12 µM vitamin K1 2,3-epoxide (K1>O; in ethanol, Sigma-Aldrich). For determination of half-maximum inhibitory concentrations (IC50), various concentrations of 4-hydroxycoumarins or 1,3-indandiones were added. Stock solutions of acenocoumarol (Cayman, Ann Arbor, MI), warfarin, coumatetralyl, coumachlor, phenindione, coumarin, brodifacoum, bromadiolone (Sigma-Aldrich), dicoumarol (Merck, Darmstadt, Germany), fluindione (BOC Sciences, New York, NY), phenprocoumon (Roche Diagnostics, Mannheim, Germany), 4-hydroxycoumarin (Dr. Ehrenstorfer GmbH, Wesel, Germany), and ferulenol (Santa Cruz Biotechnology, Heidelberg, Germany) were prepared in ethanol. Seventy-two hours after transfection, medium was collected and concentrated (VivaSpin 6; 10-kDa cutoff; Sartorius, Göttingen, Germany), and FIX activity was assessed on the basis of an aPTT test, using the coagulation analyzer BCS XP (Siemens Healthcare, Erlangen, Germany). The FIX activity was normalized to negative control (ie, cells secreting FIX in the absence of OACs), and activated FIX (FIXa) values were plotted against OAC concentration. Statistical analyses were performed using GraphPad Prism 5.01 (GraphPad Software Inc., La Jolla, CA) to impose nonlinear regression dose-response curves with variable slope.
Determination of expression level of overexpressed VKOR variants
For quantification of overexpressed VKOR variants, HEK 293T cells were transfected with myc-tagged VKOR variants, using Lipofectamine 2000. Forty-eight hours after transfection, cells were harvested using lysis buffer (150 mM NaCl, 1% NP-40, 50 mM Tris⋅HCl at pH 8.0) containing protease inhibitor (cOmplete mini, EDTA-free, Roche Diagnostics). Expression was detected by western blot, using an anti-c-myc- antibody (M4439, Sigma-Aldrich), including a loading control (ERGIC-53, sc-271517, Santa Cruz Biotechnology, Heidelberg, Germany). The detection antibody was a goat anti-mouse HPR-conjugated antibody in both reactions (sc-2005, Santa Cruz Biotechnology).
Molecular modeling of human VKORC1 and VKORC1L1 with comparative dockings of OACs
We constructed hVKORC1L1 homology models based on the 3.6-Å resolution X-ray crystallographic structure of the prokaryotic (Synechococcus sp.) homolog of VKORC1 (PDB ID: 3KP9), using the I-TASSER automated modeling server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/).25,26 Of 5 generated models, the best one was chosen on the basis of the calculated C-score of −0.46. Stereochemical quality of the model and template structures were assessed and compared, using the MOLPROBITY server (http://molprobity.biochem.duke.edu/). Comparison of backbone Cα alignment between the model and template structures was assessed using the Mustang function embedded in the YASARA molecular modeling software.27 Visualization, graphical rendering, and structural analysis were performed using YASARA and Chimera software.27,28
We used the automated AutoDock module (The Scripps Research Institute) with default settings for small-molecule docking embedded in YASARA (ver. 12.9.27; Elmar Krieger) to search for predicted warfarin binding poses on the hVKORC1L1 model.27,29 Structure data format files (.sdf) for the ligands: acenocoumarol (CID 54676537), bromadiolone (CID 547539), brodifacoum (CID 54680676), coumachlor (CID 54682651), coumatetralyl (CID 54678504), dicoumarol (CID 54676038), fluindione (CID 68942), phenindione (CID 4760), phenprocoumon (CID 54680692), and S-warfarin (CID 54688261) were downloaded from the Pubchem database (https://pubchem.ncbi.nlm.nih.gov/search/) and converted to PDB format (.pdb), using the AUTOSMILES function of YASARA. A course-screened rigid rolling ball global search was performed, allowing the energy-optimized, rigid warfarin DCO isomer configuration (deprotonated, open side-chain) to move relative to the fixed, rigid hVKORC1L1 model. In addition, 10 OAC structures were flexibly docked on the hVKORC1L1, as well as on a previously generated hVKORC1 model.24 Representative docked poses on hVKORC1L1 were selected on the basis of lowest pseudo-energy function scores and the relative orientation with respect to predicted physiological positively charged arginine residues on hVKORC1L1. Docking poses on hVKORC1 for all inhibitors except warfarin were chosen on the basis of their similarity to a published warfarin docking pose identified for hVKORC1.24 All selected docking poses were subjected to 100 ns membrane-embedded simulation with a membrane composition of 70% PC (phosphatidylcholine) and 30% (phosphatidylethanolamine). The docked complexes with the lowest energy in the simulation trajectory were chosen as the final docked structures and analyzed for identifying putative binding residues.
Measurement of cell viability
The influence of oxidative stress was assessed using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Mannheim, Germany). Thereby, a tetrazolium compound is converted exclusively by viable cells into formazan, which is measured at 490 nm. WT, VKORC1 KO, and VKORC1L1 KO and DKO HEK 293T cells were seeded on 96-well plate (5 × 105 cells/mL). Cells were incubated with different concentrations of H2O2 (0-250 µM) or 75 µM H2O2 combined without K1 or K1>O (12 µM). After 18 hours, 15 µL of reagent was added and incubated for 2 to 4 hours, and absorbance was measured at 490 nm (Synergy 2; BioTek Germany, Bad Friedrichshall, Germany).
Results
Characterization of VKORC1 and VKORC1L1 single-KO and DKO HEK cell lines
Figure 1A summarizes the genetic profile of VKORC1 and VKORC1L1 single-KO and DKO cell lines. Both single-KO cell lines showed similar FIX activities (Figure 1B) compared with WT HEK 293T cells, whereas the DKO cell line exhibited no FIX activity when transfected with F9 cDNA solely and supplemented with 12 µM K1. Thus, single-KO cells are suitable to determine specific inhibition of endogenous VKORC1 (in VKORC1L1 KO cells) and endogenous VKORC1L1 activity (in VKORC1 KO cells). Alternatively, the DKO cell line allows investigation of any VKOR variant without confounding endogenous VKR and VKOR activity at a substrate level below 12 µM.
Figure 1.

CRISPR/Cas9-mediated gene editing of VKORC1 and VKORC1L1 in HEK293T cells. (A) The wild-type sequence for both genes (c.70-116 of VKORC1 and c.88-131 of VKORC1L1) is shown on the top, deletions are indicated by dashes, and insertion and substitution is shown in bold type. (B) Measurement of FIX activity after transfection of human F9 cDNA and supplementation of 12 µM vitamin K1 in different genetically engineered cells. Columns represent measurements of endogenous VKORC1 and VKORC1L1 activity in HEK 293T WT, endogenous VKORC1 activity (VKORC1L1 KO), endogenous VKORC1L1 activity (VKORC1 KO), and in cells lacking both VKOR enzymes (VKORC1/VKORC1L1 DKO cells). Columns show mean of triplicates ± standard error of the mean.
Determination of IC50 values of OACs and rodenticides for endogenous VKORC1 and VKORC1L1
IC50 data for all compounds investigated in single VKOR KO cells and treated with K1 are reported in Table 1, including the ratio of IC50 values for endogenous VKORC1L1 with respect to endogenous VKORC1. Dose-response curves are shown in supplemental Figures 1 and 2. Four-parameter logistic curves could not fit to the coumarin and dicoumarol data. Dicoumarol dose-response for both VKOR enzymes were biphasic, and single-concentration data were used to estimate IC50 concentration (VKORC1, 49.1% FIX activity at 5 nM; VKORC1L1, 48.2% FIX activity at 100 nM).
Table 1.
IC50 values of vitamin K antagonists
| Compound class | Compound | Chemical structure | VKORC1 IC50 nM (95% CI) | VKORC1L1 IC50 nM (95% CI) | Ratio L1/C1 |
|---|---|---|---|---|---|
| Inactive backbone | Coumarin | 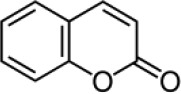 |
Not inhibitory* | Not inhibitory* | NA |
| Active 4HC backbone | 4-hydroxycoumarin | 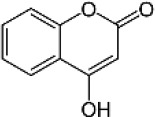 |
1978 (1837-2130) | 25110 (18530-34020) | 12.7 |
| Naturally occurring and therapeutic 4HC | Dicoumarol | 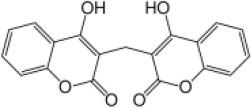 |
5.0† | 100.0† | 20.0 |
| Human therapeutic 4HC | Warfarin | 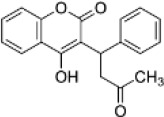 |
1.9 (1.8-2.1) | 25.2 (23.1-27.4) | 13.4 |
| Human therapeutic 4HC | Acenocoumarol | 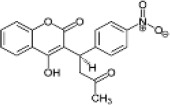 |
1.5 (1.4-1.7) | 5.8 (5.4-6.2) | 3.9 |
| Human therapeutic 4HC | Phenprocoumon | 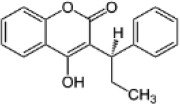 |
3.6 (3.3-3.9) | 10.5 (8.8-12.4) | 2.9 |
| Human therapeutic indandione | Phenindione | 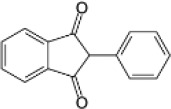 |
2.9 (2.6-3.3) | 258.0 (213.4-312.0) | 89.0 |
| Human therapeutic indandione | Fluindione | 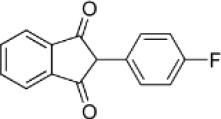 |
4.8 (4.3-5.2) | 268.5 (198.4-363.4) | 55.9 |
| First-generation rodenticide | Coumatetralyl | 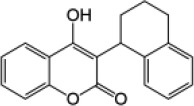 |
1.9 (1.8-2.1) | 2.8 (2.7-3.0) | 1.5 |
| First-generation rodenticide | Coumachlor | 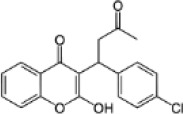 |
0.7 (0.6-0.8) | 1.0 (0.8-1.1) | 1.4 |
| Second-generation rodenticide | Bromadiolone | 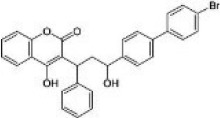 |
1.6 (1.5-1.7) | 0.8 (0.8-0.9) | 0.5 |
| Second-generation rodenticide | Brodifacoum |  |
0.5 (0.5-0.6) | 0.4 (0.4-0.5) | 0.8 |
| Naturally occurring 4HC | Ferulenol |  |
0.8 (0.8-0.9) | 3.0 (2.4-3.8) | 3.8 |
Overview of OACs investigated in the study, including their compound class as well as IC50 assessed in VKORC1L1 KO and VKORC1 KO HEK 293T cells, using K1 as substrate. Measurements were performed in triplicates.
4HC, 4-hydroxycoumarin; 95% CI, 95% confidence interval; C1, VKORC1; L1, VKORC1L1; NA, not applicable.
VKR enzyme activities assessed up to 1 mM coumarin.
Biphasic dose-response, no-curve fit with logistic function (VKORC1, 49.1%; VKORC1L1, 48.2% mean normalized activities with respect to 0 nM dicoumarol).
For all OACs VKORC1L1 uniformly required higher drug concentrations for inhibition (three- to ∼100-fold greater; Table 1). Of the 4-hydroxycoumarin-based drugs, warfarin showed the highest difference in IC50 concentrations between VKORC1 and VKORC1L1 (VKORC1L1 exhibits ∼13-fold greater IC50 than VKORC1). Acenocoumarol and phenprocoumon exhibited similar specificity (VKORC1L1 was three- to fourfold less sensitive than VKORC1). In contrast, the 1,3-indandiones showed markedly greater enzyme specificity. VKORC1 was inhibited at low nanomolar concentrations, whereas VKORC1L1 yielded up to 100-fold greater IC50 values.
Notably, all rodenticides showed similar IC50 values for both VKOR enzymes in low nanomolar range (Table 1; supplemental Figure 2).
To investigate the inhibitory potential on VKOR activity, warfarin, fluindione, and coumachlor were tested in the same assay setup, using 12 µM K1>O (instead of K1) as substrate (supplemental Table 1; supplemental Figure 3). For warfarin and coumachlor, similar IC50 values were obtained compared with K1-treated cells. Interestingly, VKORC1L1 KO cells showed markedly lower susceptibility to fluindione under K1>O supplementation.
In silico modeling of hVKORC1L1
The highest scored model from ITASSER for hVKORC1L1 showed a 4-transmembrane (4TM) structure, as previously described.13 Stereochemical parameters of this model agree favorably with the crystal structure represented by 3KP9. Structural alignment of the model with the 3KP9 structure showed a strong similarity of root-mean-square deviation <1Å (ie, 0.86 Å) over 110 aligned residues. Similarly, alignment of the hVKORC1 and hVKORC1L1 model also showed strong structural similarity (ie, 0.96 Å over 146 aligned residues).24 Apart from the TM domains, the loops of both models appear similar in structure but very different to the thioredoxin-like domain on 3KP9 (Figure 2A-B).
Figure 2.

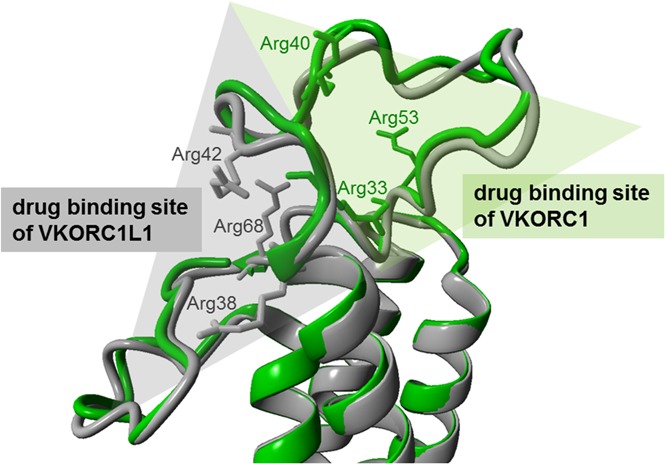
The binding poses for warfarin on VKORC1 and VKORC1L1. (A) Favored docking pose of warfarin on VKORC1L1 (gray-colored ribbon format). The electrostatic surface charge distribution is illustrated based on columbic charge distribution (blue, positive charge; red, negative charge). The clustered arginines on the endoplasmatic reticulum (ER) luminal loop are depicted in blue and warfarin in beige-colored stick forms. (B) Favored docking pose of warfarin on VKORC1 (green-colored ribbon format). Charge distribution, arginines, and warfarin are colored as in panel A. (C) Interactions of the bound warfarin (beige stick form) with arginine residues (blue-colored stick forms) of VKORC1L1 (gray-colored ribbon format). Hydrophobic interactions between warfarin and VKORC1L1 residues are depicted as green lines, and charge-based (cation-π) interactions are depicted with blue lines of different intensities. (D) Structural alignment of the VKORC1 and VKORC1L1 models. The backbone of the VKORC1 and VKORC1L1 models are colored as in panels A and B. The arginine residues clustered around the favored warfarin binding site of each model are depicted in stick format. Opposite drug binding sites are highlighted with shaded triangles (gray for VKORC1L1, green for VKORC1).
Docking predictions for warfarin and other OACs on VKORC1 and VKORC1L1
The dockings performed on the previously generated VKORC1 model showed poses with pseudo-energy scores consistent for all OACs surveyed, and that are located similar to the published warfarin pose.24,30 The predicted pose on hVKORC1L1 is located in the ER luminal loop, similar to the docking site on hVKORC1, but lies on the opposite site of the loop (Figure 2A-D). The ER luminal loops of hVKORC1L1 and hVKORC1 (Figure 2D) are similar in backbone structure, but differ greatly in their amino acid composition, especially with respect to the distribution of positively charged arginine residues. Because OACs bear a formal negative charge at physiological pH, the location of the predicted binding poses appears to be of great consequence with respect to possible charge interactions with the adjacent lying arginine residues that bear positive charges. Therefore, Arg38, Arg42, and Arg68 are likely to contribute to the OAC binding interactions with hVKORC1L1 (supplemental Figure 4). Moreover, the predicted hVKORC1L1 OAC binding pocket lacks any stacking interactions and hydrogen bonds similar to those observed between warfarin and hVKORC1.30
Mutation of specific loop residues of VKORC1L1 to identify warfarin binding site
In a first approach, 2 variants were generated in which the loop of VKORC1L1 was swapped into VKORC1 cDNA and vice versa (supplemental Figure 6). In the DKO cells, IC50 of VKORC1, VKORC1L1, and both loop swap variants were determined by overexpression of these variants together with F9 (in a bicistronic vector or by coexpression of VKOR myc-tagged variants with F9). Western blot analysis of myc-tagged VKOR variants revealed similar protein expression (Figure 3C). Warfarin dose-response of VKORC1 loop swap shifted to that for VKORC1L1 WT, and VKORC1L1 loop swap shifted to that of VKORC1 WT (Figure 3A; supplemental Table 2). We further hypothesized that introducing the previously published VKORC1 warfarin binding interface II (located in the loop) with Phe55 as major warfarin-binding residue in VKORC1L1 leads to higher OAC susceptibility.24,30,31 Neither single (Leu62Phe) nor multi-VKORC1L1 variants (Ala60Arg+Ala61Val+Leu62Phe) showed increased warfarin sensitivity. Indeed, these VKORC1L1 variants required three- to fourfold higher warfarin IC50 concentrations compared with VKORC1L1 WT (Figure 3C; supplemental Table 2).
Figure 3.

Inhibition curves for warfarin assessed in DKO cells. Exogenous VKR activity determined in DKO cells and assessed by overexpression of VKOR variants and F9. (A) Warfarin dose-response curves for VKORC1, VKORC1L1, and loop swap variants. (B) Warfarin dose-response curves for arginine variants of VKORC1L1. Measurements were performed in triplicates, values are shown as mean and error bars are represented as standard error of the mean. The coefficient of determination (R2) calculated for each inhibition curve: (A) VKORC1, R2 = 0.9935; VKORC1L1, R2 = 0.9628; VKORC1 loop swap L1, R2 = 0.9826. (B) VKORC1L1, R2 = 0.9628; Arg38Glu, R2 = 0.9715; Arg42Glu, R2 = 0.9875; Arg38Glu+ Arg42Glu, R2 = 0.9140. (C-D) Expression of all VKOR variants detected by α-myc antibody (vertical lines have been inserted to indicate a repositioned gel lane). As loading control, α-ERGIC-53 was used.
For verification of the in silico-predicted binding residues, positively charged arginine residues (Arg38, Arg42, Arg68) were mutated to negatively charged glutamate to maximize potential shifts in the respective dose-response curves. Warfarin dose-response data for single mutations Arg38Glu and Arg42Glu were shifted to greater values with respect to VKORC1L1 WT, whereas the values of Arg68Glu do not fall below 50% over the entire range of warfarin concentrations (Figure 3B). Arg38Glu+Arg42Glu revealed an eightfold higher IC50, whereas the triple mutant Arg38Glu+Arg42Glu+Arg68Glu was completely resistant (Figure 3B). Of note, the triple mutant showed reduced activities, yielding about 12% absolute FIX activity compared with ∼25% of double arginine mutant and ∼44% of VKORC1L1 WT. However, western blot analysis showed no difference in expression level for the VKORC1L1 variants (Figure 3D).
VKORC1L1 KO cells are most sensitive to oxidative stress
Assessment of cell viability revealed that all cell lines were sensitive toward H2O2 treatment, whereby VKORC1L1 KO cells were most sensitive. VKORC1 KO and DKO cells exhibited similar cell viability as WT HEK cells (Figure 4A). K1 supplementation seems to compensate cell stress for VKORC1 KO cells, whereby K1>O had no effect on cell viability. WT, DKO, and VKORC1L1 KO cells showed no difference when supplemented with K1 or K1>O (Figure 4B).
Figure 4.

Effect of cell stress on VKOR KO cells. (A) Cell viability with regard to H2O2 treatment was assessed in WT (purple), VKORC1 KO (red), VKORC1L1 KO (blue), and DKO (green) HEK 293T. Curves showing normalized values for 3 replicates including error bars representing standard error of the mean. (B) Effect of K1 and K1>O treatment on cell viability. Negative control without H2O2; positive control with 75 µM H2O2. Cells were incubated with 75 µM H2O2 and K1 or K1>O (each 12 µM).
Discussion
Here, we present a cell-based assay to independently study endogenous VKORC1 and VKORC1L1 inhibition by several compounds that affect vitamin K recycling. Previous studies have used different approaches to examine the capability of VKORC1 and VKORC1L1 to reduce K or K>O by 2 different types of assays (Table 2). In the dithiothreitol (DTT) assay, direct conversion rates of K>O to K are measured using HPLC.14,17,32,33 Alternatively, in the cell-based assays, a reporter protein that requires γ-carboxylation for biological activity is used as marker for VKR and VKOR activity.34-37 In several studies, WT FIX and chimeric FIX Gla domain-containing proteins were used as reporter for VKOR/VKR activity and were shown to reflect the in vivo situation properly.34,36-38 Until now, the majority of the studies could not discriminate between VKOR/VKR activities from endogenously expressed VKOR enzymes. Our assay analyzes endogenous inhibition of VKORC1 and VKORC1L1 independently by using genetically engineered VKORC1L1 and VKORC1 KO HEK 293T cell lines, respectively. Drug inhibition was not influenced by metabolic activity, as cytochrome P450 monooxygenases known to be involved in metabolism of vitamin K and warfarin are not expressed in HEK 293 cells.37
Table 2.
Comparison of published VKOR inhibition data
| Gebauer32 | Tie et al34 | Westhofen et al14 | Tie et al35 | Fregin et al36 | Hammed et al17 | Bevans et al33 | Haque et al37 | Present study | Present study | Present study | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Expression system/cell line | Rat liver microsomes | HEK 293 | HEK 293T | HEK 293 WT and C1 KO | HEK 293T | P. pastoris | P. pastoris | HEK 293-C3 | C1 KO and L1 KO HEK 293T | C1 KO and L1 KO HEK 293T | DKO HEK 293T |
| VKOR expression | Endogenous | Endogenous | Overexpression of both VKOR enzymes | Endogenous | Overexpression of VKORC1 | Overexpression of both VKOR enzymes | Overexpression of VKORC1 | Endogenous | Endogenous | Endogenous | Overexpression of both VKOR enzymes |
| Type of measurement | Direct | Indirect | Direct | Indirect | Indirect | Direct | Direct | Indirect | Indirect | Indirect | Indirect |
| Activity measurement | K>O conversion | FIXgla-PC | K>O conversion | FIXgla-PC | FIX | K>O conversion | K>O conversion | F9CH | FIX | FIX | FIX |
| Reporter expression | NA | Stable | NA | Stable | Transient | NA | NA | Inducible, stable | Transient | Transient | Transient |
| Substrate | 25 mM K1>O | 5 µM K1>O | 50 µM K1>O | 5 µM K1>O | 10 µM K1 | Up to 0.2 mM K1>O | Up to 1 µM K1>O | 50 nM K1 | 12 µM K1 | 12 µM K1>O | 12 µM K1 |
| Type of assay | DTT assay | ELISA | DTT assay | ELISA | Clotting assay | DTT assay | DTT assay | FACS | Clotting assay | Clotting assay | Clotting assay |
| Discrimination between C1 and L1 | No | No | Yes (but remaining endogenous activity) | Yes (in KO cell line) | Yes (but remaining endogenous activity) | Yes | Yes | No | Yes | Yes | Yes |
| Warfarin | IC50 2.2 µM | IC50 100 nM | At 5 µM: C1: 53% inhibition L1: 29% inhibition | IC50 < 50 nM | IC50 25 nM | Ki ratios: hL1/C1: 29× rL1/C1: 54× | IC50 3-24 µM | IC50 125 nM | IC50: C1: 1.9 nM L1: 25.2 nM | IC50: C1: 2.4 nM L1: 27 nM | IC50: C1: 17.3 nM L1: 474 nM |
Published data regarding inhibition of VKORC1 or VKORC1L1 by warfarin are summarized.
Ki, inhibitory constant.
VKORC1 and VKORC1L1 exhibit different inhibition characteristics for various OACs
A panel of 13 compounds with 4-hydroxycoumarin and 1,3-indandione backbones were analyzed for their inhibitory effect on VKORC1 and VKORC1L1 (Table 1). Consistent with in vivo data, naturally occurring coumarin showed no anticoagulatory effect in our in vitro assay.1,39 In contrast, VKORC1 and VKORC1L1 were both inhibited by 4-hydroxycoumarin at micromolar concentrations. The addition of 1 or more aromatic hydrocarbons of the variable 3-position side chains of 4-hydroxycoumarins correlates with decreased IC50 values, especially for VKORC1 (eg, 4-hydroxycoumarin [VKORC1 IC50 2000 nM] and acenocoumarol [VKORC1 IC50 1.6 nM]). It was recently shown that VKORC1 Phe55 participates in hydrophobic and stacking interactions to the 3-aromatic hydrocarbon of warfarin, thus explaining the increased affinities toward OACs with additional aromatic hydrocarbons.30
In general, VKORC1 was more sensitive to therapeutic 4-hydroxycoumarin derivatives compared with VKORC1L1. The same inhibitory trend of coumarin derivatives regarding VKORC1 was shown by several in vitro and in vivo studies (Table 2).14,17,32-37 However, most studies relied on overexpressed VKOR enzymes. Tie et al were the first measuring endogenous VKORC1L1 inhibition in VKORC1 KO HEK 293 cells.35 However, they detected no difference in inhibition between WT and VKORC1 KO HEK 293 cells, and VKORC1L1 KO cells were not examined. Hammed et al studied inhibition of hVKORC1 and hVKORC1L1 by overexpression in Pichia pastoris, using the DTT assay that revealed ∼30-fold higher susceptibility to warfarin for VKORC1 compared with VKORC1L1.17 In accordance with these results, we found a 30-fold difference in VKORC1 inhibition compared with VKORC1L1 when enzymes were overexpressed in DKO cells (Figure 3A; supplemental Table 1). Interestingly, measurement of endogenous VKOR inhibition by our assay revealed only 13-fold difference in warfarin sensitivity with lower IC50 values (Tables 1; supplemental Table 1). This might explain why studies using overexpressed VKOR enzymes, in general, reported higher IC50 values relative to the current study results.14,17,33,36 Various warfarin IC50 values, primarily for VKORC1, have been published and range between 25 nM and 24 µM.13,32,33,35-37 Differences may arise from the type of assay used (DTT vs cell-based), the type of substrate (K1 vs K1>O) and its concentration, and the relative protein expression levels (endogenously vs overexpressed), and source of cells (kidney- vs liver-derived) (Table 2). We observed that the type of substrate (K1 or K1>O) did not affect warfarin dose responses when using the same molarity (Tables 1; supplemental Table 1). Moreover, higher IC50 values detected for VKORC1 in other studies are probably a result of simultaneously measured endogenous VKORC1L1 activity, especially in cell-based assays. The low dose-response values and ability to discriminate between both VKOR enzymes indicate that our method using VKORC1 and VKORC1L1 KO cells is highly sensitive. Notably, polyploid endogenous VKORC1 expression may be similar in HEK 293T cells and native hepatocytes, the cells responsible for producing substantially all VKD coagulation factors.40
Among the therapeutically used 4-hydroxycoumarins, acenocoumarol was most effective on VKORC1L1 inhibition, whereas warfarin required the greatest IC50 concentration. This suggests that at effective therapeutic dosages that lower VKORC1-mediated VKR activity and, hence, lower VKD coagulation factor activities, it is likely that VKORC1L1-mediated VKR activity is not appreciably affected in liver, and especially in nontarget tissues. However, the effect of VKORC1L1 inhibition in vivo at therapeutic OAC drug levels is not known yet. Although the cell-based assays are sensitive and reflect the in vivo situation, it is still unclear whether VKORC1L1 provides KH2 for γ-carboxylation of VKD proteins in vivo, too. Results from mice studies indicate that VKORC1L1 is not able to compensate lack of VKORC1 activity, as bleeding phenotype in constitutive VKORC1−/− was not rescued.15 In liver, where coagulation factors are synthesized, VKORC1L1 expression is very low and is not upregulated in VKORC1−/− mice.17 Assessment of VKOR activity in VKORC1−/− liver microsomes showed markedly reduced activity (by 97%) compared with WT.17 However, VKORC1L1−/− in osteoblasts revealed no difference in vitamin K-dependent osteocalcin levels compared with control mice.41 In an in vitro study, Westhofen et al postulated that VKORC1L1 plays a role in intracellular antioxidation.14 Therefore, we investigated whether our VKOR KO cell lines are sensitive toward oxidative stress, as well. Our results point into the same direction of Westhofen et al, with VKORC1L1 KO cells being most sensitive toward H2O2 treatment (Figure 4A). Moreover, supplementation of K1 slightly reversed H2O2 treatment in VKORC1 KO cells, but not K1>O. In contrast, DKO and VKORC1L1 KO cells were not rescued by K1 treatment, indicating that VKORC1L1 is the only enzyme responsible for this pathway. However, in WT HEK cells, endogenous VKORC1L1 did not improve cell viability under K1 supplementation (Figure 4B). Both VKOR enzymes are present in those cells catalyzing K1, which probably leads to lower substrate availability for VKORC1L1. Another possibility could be that VKORC1L1 is upregulated in VKORC1 KO cells. More studies are needed to investigate the effects of VKOR enzymes on oxidative stress. However, our results indicate that inhibition of VKORC1L1 may result in reduced antioxidative properties.14
For phenindione and fluindione, IC50 values were almost similar to that of 4-hydroxycoumarin drugs regarding VKORC1 inhibition when determined in presence of K1. In contrast, investigated indandiones required up to 50-fold greater concentrations compared with coumarin-based compounds for VKORC1L1 inhibition. Interestingly, VKORC1 exhibited greatly increased IC50 values for fluindione in the presence of K1>O (4.8 nM for K1 vs 204 nM for K1>O; supplemental Figure 6; Table 1; supplemental Table 1). Therefore, fluindione seems to have different binding characteristics in presence of K1>O compared with K1, as well as compared with 4-hydroxycoumarin-based drugs. We further conclude that K1>O accumulation may affect the inhibitory potential of fluindione.
Inhibition of endogenous VKORC1 and VKORC1L1 by rodenticides and ferulenol
Rodenticides are long-lasting coumarin derivatives, whereas second-generation rodenticides emerged because of resistance against compounds of the first generation.42 The 3-position substitutions by long, phenyl side-chains yielded tremendous increase in potency and duration of action.43 Our data show that rodenticides have increased affinities for both VKORC1 and VKORC1L1 that may explain their potency. The difference between first- and second-generation rodenticides was not as great as shown by in vivo studies in rodents (Table 1; supplemental Figure 2).43 Hence, studies with VKORC1 mutations should be performed to determine which rodenticide is most potent for inhibition of resistance phenotypes.
Moreover, ferulenol, a plant-derived 4-hydroxycoumarin, was reported to cause hemorrhagic symptoms in mammals.44 Inhibitory potency of ferulenol was confirmed by our results, but values were not as high as described in literature.32,44 It is the most potent naturally occurring coumarin and is similarly effective as rodenticides and more effective than OACs on VKORC1.
VKORC1L1 is less OAC sensitive than VKORC1 because of different drug binding sites
To elucidate the basis for the different OAC susceptibilities of VKORC1 and VKORC1L1, the ER luminal loop of VKORC1L1 was swapped in VKORC1 cDNA and vice versa (Figure 3A; supplemental Figure 5). The loop swap variants are consistent with VKOR models comprising either 3 or 4 TM domains.45,46 Determination of IC50 values for warfarin and fluindione for VKORC1 loop swap yielded shifted doses responses toward VKORC1L1 (Figure 3A; supplemental Figure 6; supplemental Table 2). In accordance, VKORC1L1 loop swap turned out to be as sensitive as VKORC1 WT when using warfarin. These results strongly suggest that the loops of both VKOR enzymes play an important structural role in coumarin-based OAC binding. Interestingly, when using fluindione, dose responses of VKORC1L1 loop swap did not completely shift to that observed for VKORC1 (supplemental Figure 6). We also detected differences in fluindione IC50 values for VKORC1 WT when supplemented with K1>O compared with K1, suggesting that fluindione might have other drug binding characteristics on VKORC1 than 4-hydroxycoumarins (supplemental Figure 3; supplemental Table 1).
In a previously published warfarin binding pocket on VKORC1, 1 warfarin binding surface comprises residues Ser52 through Phe55 of the ER luminal loop, with Phe55 being the major amino acid binding warfarin.24,30,31 Therefore, we decided to swap this interface into VKORC1L1. The idea was to probe whether these warfarin interacting residues in VKORC1 contribute to higher binding affinity in VKORC1L1. Neither introducing the phenylalanine into the corresponding sequence of VKORC1L1 (VKORC1L1: Leu62Phe) nor complete swap of the binding surface (VKORC1L1: Ser59-Phe62) showed different susceptibility to warfarin, indicating that VKORC1 warfarin binding surface is different from VKORC1L1 (Figure 2D). Comparative visualization of the calculated charge distribution for the ER luminal loops of the VKORC1 and VKORC1L1 models suggests that the primary difference in binding sites might be attributed to the different clustering of positively charged arginines contributing to overall binding affinity of the anionic OAC drugs. In VKORC1, 3 arginine residues are clustered around the reported warfarin binding pocket and were found to result in resistance when mutated to glutamate residues (Arg33, Arg40, and Arg53), whereas other arginine residues in the loop of VKORC1 had no effect on warfarin binding (Arg61, Arg35).30 In contrast, in hVKORC1L1, 3 arginine residues at amino acid positions 38, 42, and 68 cluster around a binding pocket that is diametrically opposite to that one in VKORC1 (ie, on the other side of the short helix; Figure 2D). Substitution of these arginine residues by glutamate residues resulted in greater IC50 values up to complete resistance. Interestingly, the corresponding arginine residues found in VKORC1L1 to affect warfarin binding have been shown to not affect OAC binding in VKORC1 (VKORC1L1:Arg68 = VKORC1:Arg61, VKORC1L1:Arg42 = VKORC1:Arg35; supplemental Figure 5).30 These results, along with the loop-swapping data, are consistent with our prediction that this region is the binding site for warfarin in VKORC1L1 (Figures 2 and 3; supplemental Table 2). But why is OAC binding to VKORC1L1 weaker than to VKORC1? In VKORC1, where warfarin is stabilized by a combination of hydrogen bonds, aromatic stacking interactions, hydrophobic interactions, and charge-based interactions, unlike warfarin binding in VKORC1L1, appear to be stabilized only by charge-based and hydrophobic interactions. This might explain the poorer sensitivity of VKORC1L1 toward warfarin when compared with VKORC1. The distinct binding sites of VKORC1 and VKORC1L1 with comparably more amino acids being involved in OAC binding in VKORC1 may explain their different affinities to OACs. Furthermore, in silico docking predictions for other 4-hydroxycoumarins showed favorable docking poses in close apposition to all 3 experimentally determined arginines (supplemental Figure 4).
In conclusion, we have established KO HEK 293T cell lines that express either endogenously VKORC1 or VKORC1L1, and found that VKORC1 is more sensitive to OACs than VKORC1L1, whereas rodenticides apparently inhibit both VKOR enzymes equally. In silico analysis of VKORC1L1 and corresponding in vitro data strongly suggest that the warfarin binding sites for VKORC1 and VKORC1L1 are located in the loop but are different with respect to position and interacting residues.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported, in part, by funding from BONFOR 2015-1-3 (K.J.C.) and Baxter Germany GmbH (J.O.), and travel support from Bayer (K.J.C., K.L., and J.O.) and from the Deutsche Forschungsgemeinschaft grant Ol100 5 (M.W. and J.O.).
Authorship
Contribution: K.J.C. and K.L. provided experimental design; K.L. performed data collection, data analysis, and figure production; A.B. provided protein modeling; K.J.C., K.H., and V.H. provided CRISPR/Cas9 gene editing of HEK 293T cells; and K.J.C., K.L., K.H., V.H., M.W., and J.O. provided manuscript drafting and editing.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Johannes Oldenburg, Institute of Experimental Haematology and Transfusion Medicine, University Clinic Bonn, Sigmund-Freud Str 25, 53127 Bonn, Germany; e-mail: johannes.oldenburg@ukbonn.de.
References
- 1.Link KP. The discovery of dicumarol and its sequels. Circulation. 1959;19(1):97-107. [DOI] [PubMed] [Google Scholar]
- 2.Overman RS, Stahmann MA, Huebner CF, et al. . Studies on the hemorrhagic sweet clover disease: XIII. Anticoagulant activity and structure in the 4-hydroxycoumarin group. J Biol Chem. 1944;153:5-24. [Google Scholar]
- 3.Ufer M. Comparative pharmacokinetics of vitamin K antagonists: warfarin, phenprocoumon and acenocoumarol. Clin Pharmacokinet. 2005;44(12):1227-1246. [DOI] [PubMed] [Google Scholar]
- 4.Ansell J. Recommendations on bleeding and nonbleeding complications of vitamin K antagonists. Clin Adv Hematol Oncol. 2007;5(7):517-518. [PubMed] [Google Scholar]
- 5.Oldenburg J, Bevans CG, Fregin A, Geisen C, Müller-Reible C, Watzka M. Current pharmacogenetic developments in oral anticoagulation therapy: the influence of variant VKORC1 and CYP2C9 alleles. Thromb Haemost. 2007;98(3):570-578. [PubMed] [Google Scholar]
- 6.Rost S, Fregin A, Ivaskevicius V, et al. . Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427(6974):537-541. [DOI] [PubMed] [Google Scholar]
- 7.Chu P-H, Huang T-Y, Williams J, Stafford DW. Purified vitamin K epoxide reductase alone is sufficient for conversion of vitamin K epoxide to vitamin K and vitamin K to vitamin KH2. Proc Natl Acad Sci USA. 2006;103(51):19308-19313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li T, Chang C-Y, Jin D-Y, Lin P-J, Khvorova A, Stafford DW. Identification of the gene for vitamin K epoxide reductase. Nature. 2004;427(6974):541-544. [DOI] [PubMed] [Google Scholar]
- 9.Shearer MJ, Newman P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J Lipid Res. 2014;55(3):345-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furie B, Furie BC. Molecular basis of vitamin K-dependent gamma-carboxylation. Blood. 1990;75(9):1753-1762. [PubMed] [Google Scholar]
- 11.Berkner KL, Runge KW. The physiology of vitamin K nutriture and vitamin K-dependent protein function in atherosclerosis. J Thromb Haemost. 2004;2(12):2118-2132. [DOI] [PubMed] [Google Scholar]
- 12.Oldenburg J, Watzka M, Bevans CG. VKORC1 and VKORC1L1: why do vertebrates have two vitamin K 2,3-epoxide reductases? Nutrients. 2015;7(8):6250-6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tie J-K, Jin D-Y, Stafford DW. Conserved loop cysteines of vitamin K epoxide reductase complex subunit 1-like 1 (VKORC1L1) are involved in its active site regeneration. J Biol Chem. 2014;289(13):9396-9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westhofen P, Watzka M, Marinova M, et al. . Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function. J Biol Chem. 2011;286(17):15085-15094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spohn G, Kleinridders A, Wunderlich FT, et al. . VKORC1 deficiency in mice causes early postnatal lethality due to severe bleeding. Thromb Haemost. 2009;101(6):1044-1050. [PubMed] [Google Scholar]
- 16.Van Horn WD. Structural and functional insights into human vitamin K epoxide reductase and vitamin K epoxide reductase-like1. Crit Rev Biochem Mol Biol. 2013;48(4):357-372. [DOI] [PubMed] [Google Scholar]
- 17.Hammed A, Matagrin B, Spohn G, Prouillac C, Benoit E, Lattard V. VKORC1L1, an enzyme rescuing the vitamin K 2,3-epoxide reductase activity in some extrahepatic tissues during anticoagulation therapy. J Biol Chem. 2013;288(40):28733-28742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caspers M, Czogalla KJ, Liphardt K, et al. . Two enzymes catalyze vitamin K 2,3-epoxide reductase activity in mouse: VKORC1 is highly expressed in exocrine tissues while VKORC1L1 is highly expressed in brain. Thromb Res. 2015;135(5):977-983. [DOI] [PubMed] [Google Scholar]
- 19.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt T, Schmid-Burgk JL, Hornung V. Synthesis of an arrayed sgRNA library targeting the human genome. Sci Rep. 2015;5(1):14987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmid-Burgk JL, Schmidt T, Gaidt MM, et al. . OutKnocker: a web tool for rapid and simple genotyping of designer nuclease edited cell lines. Genome Res. 2014;24(10):1719-1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrangou R, Birmingham A, Wiemann S, Beijersbergen RL, Hornung V, Smith A. Advances in CRISPR-Cas9 genome engineering: lessons learned from RNA interference. Nucleic Acids Res. 2015;43(7):3407-3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Unger T, Jacobovitch Y, Dantes A, Bernheim R, Peleg Y. Applications of the Restriction Free (RF) cloning procedure for molecular manipulations and protein expression. J Struct Biol. 2010;172(1):34-44. [DOI] [PubMed] [Google Scholar]
- 24.Czogalla KJ, Biswas A, Wendeln A-C, et al. . Human VKORC1 mutations cause variable degrees of 4-hydroxycoumarin resistance and affect putative warfarin binding interfaces. Blood. 2013;122(15):2743-2750. [DOI] [PubMed] [Google Scholar]
- 25.Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature. 2010;463(7280):507-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krieger E, Vriend G. YASARA View: molecular graphics for all devices: from smartphones to workstations. Bioinformatics. 2014;30(20):2981-2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pettersen EF, Goddard TD, Huang CC, et al. . UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605-1612. [DOI] [PubMed] [Google Scholar]
- 29.Morris GM, Goodsell DS, Halliday RS, et al. . Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639-1662. [Google Scholar]
- 30.Czogalla KJ, Biswas A, Höning K, et al. . Warfarin and vitamin K compete for binding to Phe55 in human VKOR. Nat Struct Mol Biol. 2017;24(1):77-85. [DOI] [PubMed] [Google Scholar]
- 31.Shen G, Cui W, Zhang H, et al. . Warfarin traps human vitamin K epoxide reductase in an intermediate state during electron transfer. Nat Struct Mol Biol. 2017;24(1):69-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gebauer M. Synthesis and structure-activity relationships of novel warfarin derivatives. Bioorg Med Chem. 2007;15(6):2414-2420. [DOI] [PubMed] [Google Scholar]
- 33.Bevans CG, Krettler C, Reinhart C, et al. . Determination of the warfarin inhibition constant Ki for vitamin K 2,3-epoxide reductase complex subunit-1 (VKORC1) using an in vitro DTT-driven assay. Biochim Biophys Acta. 2013;1830(8):4202-4210. [DOI] [PubMed] [Google Scholar]
- 34.Tie J-K, Jin D-Y, Straight DL, Stafford DW. Functional study of the vitamin K cycle in mammalian cells. Blood. 2011;117(10):2967-2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tie J-K, Jin D-Y, Tie K, Stafford DW. Evaluation of warfarin resistance using transcription activator-like effector nucleases-mediated vitamin K epoxide reductase knockout HEK293 cells. J Thromb Haemost. 2013;11(8):1556-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fregin A, Czogalla KJ, Gansler J, et al. . A new cell culture-based assay quantifies vitamin K 2,3-epoxide reductase complex subunit 1 function and reveals warfarin resistance phenotypes not shown by the dithiothreitol-driven VKOR assay. J Thromb Haemost. 2013;11(5):872-880. [DOI] [PubMed] [Google Scholar]
- 37.Haque JA, McDonald MG, Kulman JD, Rettie AE. A cellular system for quantitation of vitamin K cycle activity: structure-activity effects on vitamin K antagonism by warfarin metabolites. Blood. 2014;123(4):582-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hallgren KW, Qian W, Yakubenko AV, Runge KW, Berkner KL. r-VKORC1 expression in factor IX BHK cells increases the extent of factor IX carboxylation but is limited by saturation of another carboxylation component or by a shift in the rate-limiting step. Biochemistry. 2006;45(17):5587-5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wardrop D, Keeling D. The story of the discovery of heparin and warfarin. Br J Haematol. 2008;141(6):757-763. [DOI] [PubMed] [Google Scholar]
- 40.Milne LS. The histology of liver tissue regeneration. J Pathol. 1909;13(1):127-160. [Google Scholar]
- 41.Ferron M, Lacombe J, Germain A, Oury F, Karsenty G. GGCX and VKORC1 inhibit osteocalcin endocrine functions. J Cell Biol. 2015;208(6):761-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.King N, Tran M-H. Long-acting anticoagulant rodenticide (superwarfarin) poisoning: a review of its historical development, epidemiology, and clinical management. Transfus Med Rev. 2015;29(4):250-258. [DOI] [PubMed] [Google Scholar]
- 43.Hadler MR, Buckle AP Forty-five years of anticoagulant rodenticides: past, present and future trends. In: Borrecco JE, Marsh RE, eds. Proceedings of the Fifteenth Vertebrate Pest Conference. Davis, CA, University of California, Davis; 1992:149-155.
- 44.Louvet M-S, Gault G, Lefebvre S, et al. . Comparative inhibitory effect of prenylated coumarins, ferulenol and ferprenin, contained in the ‘poisonous chemotype’ of Ferula communis on mammal liver microsomal VKORC1 activity. Phytochemistry. 2015;118:124-130. [DOI] [PubMed] [Google Scholar]
- 45.Tie J-K, Nicchitta C, von Heijne G, Stafford DW. Membrane topology mapping of vitamin K epoxide reductase by in vitro translation/cotranslocation. J Biol Chem. 2005;280(16):16410-16416. [DOI] [PubMed] [Google Scholar]
- 46.Czogalla KJ, Watzka M, Oldenburg J. Structural modeling insights into human VKORC1 phenotypes. Nutrients. 2015;7(8):6837-6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.