

Key Points
Phenotypic TM isolation from unmanipulated donor apheresis via CD45RA depletion followed by CD8+ enrichment is feasible.
TM infusion for patients with relapse after allogeneic HCT was safe and resulted in minimal GVHD.
Abstract
Murine models showed that CD8+CD44hi memory T (TM) cells could eradicate malignant cells without inducing graft-versus-host disease (GVHD). We evaluated the feasibility and safety of infusing freshly isolated and purified donor-derived phenotypic CD8+ TM cells into adults with disease relapse after allogeneic hematopoietic cell transplantation (HCT). Phenotypic CD8 TM cells were isolated after unmobilized donor apheresis using a tandem immunomagnetic selection strategy of CD45RA depletion followed by CD8+ enrichment. Fifteen patients received CD8+ TM cells at escalating doses (1 × 106, 5 × 106, or 10 × 106 cells per kg). Thirteen received cytoreduction before CD8+ TM cell infusion, and 9 had active disease at the time of infusion. Mean yield and purity of the CD8+ TM infusion were 38.1% and 92.8%, respectively; >90% had CD8+ T effector memory phenotype, cytokine expression, and secretion profile. No adverse infusional events or dose-limiting toxicities occurred; GVHD developed in 1 patient (grade 2 liver). Ten patients (67%) maintained or achieved response (7 complete response, 1 partial response, 2 stable disease) for at least 3 months after infusion; 4 of the responders had active disease at the time of infusion. With a median follow-up from infusion of 328 days (range, 118-1328 days), median event-free survival and overall survival were 4.9 months (95% confidence interval [CI], 1-19.3 months) and 19.6 months (95% CI, 5.6 months to not reached), respectively. Collection and enrichment of phenotypic CD8+ TM cells is feasible, well tolerated, and associated with a low incidence of GVHD when administered as a manipulated infusion of donor lymphocytes in patients who have relapsed after HCT. This trial was registered at www.clinicaltrials.gov as #NCT01523223.
Visual Abstract
Introduction
Disease relapse remains the primary cause of failure after allogeneic hematopoietic cell transplantation (allo-HCT) for malignant diseases.1,2 Management options for post-HCT relapse include cessation of immunosuppressive medications, salvage therapy, second HCT, or donor lymphocyte infusion (DLI). Despite these conventional interventions, few patients achieve durable complete remission (CR), and survival after disease relapse remains poor, with less than 25% of patients alive at 2 years.3-8
The success of DLI to treat disease relapse after allo-HCT requires that the infused donor lymphocytes induce a clinically significant immune-mediated graft-versus-tumor (GVT) response without eliciting severe graft-versus-host disease (GVHD). Aside from chronic myeloid leukemia, the disease in which DLI proved most effective at inducing durable remissions,9,10 treatment of posttransplant relapse with DLI in other hematologic malignancies has been less effective.8,11,12
Dose-finding studies that used unmanipulated DLI showed that doses ≤1 × 107 CD3+ cells per kg resulted in reduced GVHD incidence but with minimal tumor response, and higher doses led to improved disease control but with the risk of severe GVHD.13 Manipulation of T-cell composition before DLI infusion (eg, total CD8+ T-cell depletion or enrichment of total CD4+ T cells) did not significantly influence GVHD risk or relapse.14,15
Studies from several groups that used murine models of bone marrow transplantation (BMT) demonstrated that phenotypic memory T (TM) cells, including CD4+ and CD8+ TM cells, induced significantly less GVHD than naive T (TN) cells (CD62LhiCD44lo) or combinations of TN and TM cells.16-21 Our group reported the CD8+CD44hi T-cell subset containing both central memory (TCM) and effector memory (TEM) cells mediated potent graft-versus-leukemia activity because total T cells had not yet induced severe GVHD.22 In these models, which included major histocompatibility–matched and –mismatched strain combinations, we showed that a highly enriched population of CD8+CD44hi TM cells can be used as therapeutic DLI in mice that have progressive lymphoma after BMT. In contrast, total TN cells, sorted CD4+ and CD8+ TN cells, CD4+ TM cells, and total TM cells either induced lethal GVHD or lacked potent antitumor activity.
We sought to translate the murine model to human transplantation and evaluated the feasibility and safety of infusing a freshly isolated and purified population of phenotypic CD8+ TM cells instead of an unmanipulated DLI into allo-HCT recipients who had relapsed after transplant. First, and as a prelude to the clinical trial, we used peripheral blood mononuclear cells from unstimulated apheresis collections and developed a tandem immunomagnetic selection strategy using iron-dextran beads conjugated to CD45RA to deplete naive cells followed by CD8 enrichment. In additional studies, we more fully characterized the phenotypic CD8+ TM cells and tested for immune reactivity in vitro by stimulation with irradiated allogeneic peripheral blood mononuclear cells from normal donors. The responder CD8+ TM cells showed little increase in 3H-thymidine incorporation in cultures with allogeneic stimulator cells, and the supernatants showed a marked increase in the concentration of interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) and a minimal increase in the concentration of interleukin-2 (IL-2). The results of the human mixed leukocyte response (MLR) experiments were consistent with the responses observed with CD8+ TM cells from mice in which the murine responder cells also showed little increase in 3H-thymidine incorporation after stimulation with allogeneic cells; IFN-γ production was considerably greater than IL-2 production.21 We then completed a phase 1 feasibility and safety study in which escalating doses of phenotypic CD8+ TM cells derived from the recipient’s original HLA-matched sibling donor were administered to 15 patients who had relapse of their hematologic malignancy after allo-HCT. The infusion of a purified population of phenotypic CD8+ TM cells was safe and did not induce GVHD. Efficacy was difficult to assess given the nature of the phase 1 safety and feasibility study, but durable CRs were observed in some patients.
Methods
Graft engineering: CD8+ TM selection
Phenotypic CD8+ TM cells (CD8+CD45RA–) were isolated from the original HLA-matched transplant donor after two 12-L unstimulated apheresis collections. The CD45RA– cells were obtained by labeling peripheral blood collections with research-grade CliniMACS CD45RA Microbeads (catalog No. 130-020-003) and Reagent (Miltenyi Biotec) followed by selection on the CliniMACS Plus Instrument using the Depletion 3.2 program with CliniMACS Depletion tubing sets. The flow-through CD45RA– cells were labeled with research-grade CliniMACS CD8 Microbeads (catalog No. 130-030-810) and Reagent and were selected by using the Enrichment 3.1 program with standard CliniMACS tubing sets. Postselection products were washed and resuspended in 100 mL of Normosol-R with 1% human serum albumin for 24 to 48 hours of storage at 4°C before infusion. No products were cryopreserved. Cell recovery and purity were determined by flow cytometric analysis. Release criteria included CD8+ TM cell products with >90% cell viability with no evidence of infection, and ≥80% of cells expressed CD8 memory phenotype (CD8+CD45RA–CD45RO+) with ≤5% of cells expressing the CD3+CD45RA+CD45RO– phenotype (LSR and FACS Vantage cytometers, Becton Dickinson). All products were infused within 48 hours after completing the last apheresis collection.
Antibodies and flow cytometric analysis
Flow cytometric analysis of cell subsets was performed at each selection step. Apheresis blood collections were sampled before selection and after CD45RA depletion and CD8 enrichment steps and were evaluated for expression of CD45, CD4, CD8, CD45RA, and CD45RO. Flow cytometric analysis was performed for CD8+CD45RA+selected cells, which were then evaluated for expression of CD45, CD4, CD8, CD44, CCR7, and CD62L. Reagents were obtained from BD Biosciences. Four single apheresis products from normal human donors were obtained before proceeding with the phase 1 clinical trial. Data were acquired on Influx cytometers and analyzed by using FlowJo software.
Cytokine expression, mixed lymphocyte reaction, and cytokine secretion
Cytokine expression of the T-cell subsets was evaluated according to BD Bioscience’s protocol, and as previously described.23 In brief, sorted T-cell populations were stimulated with ionomycin and phorbol myristate acetate for 6 hours and treated with Brefeldin A (Sigma-Aldrich) added after 2 hours. The cells were fixed, and membranes were permeabilized with a saponin-based reagent (Cytofix-Cytoperm Kit, BD Biosciences) and stained for intracellular cytokines with fluorescein isothiocyanate (FITC) anti-IL-2 monoclonal antibody (mAb), FITC anti-IFN-γ mAb, and FITC anti-TNF-α mAb according to the manufacturer’s instructions. The gated cells were analyzed for the percentage that were positive on staining for each cytokine.
Total CD4+ cells and the enriched CD8+ TM cell subset from healthy donors were used as responder cells and mixed with irradiated (5000 cGy) stimulator cells made from a pool of mononuclear cells obtained from 3 normal participants with MLR. The incorporation of 3H-thymidine during the last 24 hours of culture was measured after 7 days of incubation, and cytokine secretion in the supernatants was analyzed in a multiplex assay system with microsphere beads after the 7-day culture.24 Cytokine secretion for IL-2, TNF-α, and IFN-γ was assessed by Cytometric Bead Array (Becton Dickinson) according to the manufacturer’s guidelines.
Clinical trial eligibility
Eligible patients were age 18 to 75 years and had a hematologic malignancy that had relapsed after allo-HCT from an HLA-matched sibling donor. Treatment for disease relapse was allowed before CD8+ TM cell infusion but was not mandatory. Patients were required to have no active GVHD and to be receiving a stable dose of immunosuppressants or taking no immunosuppressants for 4 weeks before CD8+ TM cell infusion. Patients with active infection or inadequate organ function (liver function tests ≥4 times the upper limit of normal or a serum creatinine >2.5 mg/dL) were excluded. All patients provided written informed consent to participate in this study, which was approved by the Stanford University Institutional Review.
Study design
This single-institution, open-label, phase 1 clinical trial evaluated the feasibility, safety, and maximum-tolerated dose of allogeneic CD8+ TM cell infusion derived from HLA-matched sibling donors. The study design followed a standard 3+3 dose escalation, with patients enrolled at escalating dose levels of 1 × 106, 5 × 106, and 10 × 106 cells per kg. Dosing was chosen on the basis of results from the CD8+CD45RA– cell recovery obtained during the development of the CD8+ TM cell selection strategy. Dose-limiting toxicities (DLTs) were defined as grade 3 to 4 toxicity according to Common Toxicity Criteria v.4. Dose escalation required no DLTs for all 3 patients in a dose level cohort. An expanded 6-patient cohort was enrolled at the maximum-tolerated dose. Cells were infused fresh; premedication was not used. Patients were monitored every 30 minutes for a minimum of 2 hours after the infusion.
Study assessments
Toxicity evaluations were performed every 14 days for the first month and then every 28 days for a minimum of 6 months after cell infusion. Repeat disease evaluations were performed at 3 and 6 months after CD8+ TM cell infusion.
Results
CD8+ TM cell selection strategy
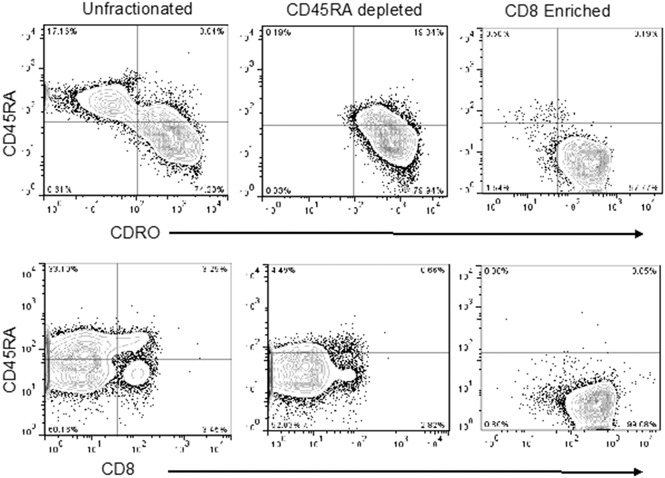
Experiments using 4 single apheresis products from normal human donors revealed that CD8+CD45RA– TM cells comprised approximately 3% (range, 1%-4%) of total nucleated cells or 6% of total CD3+ cells per apheresis collection (data not shown). Accordingly, we used the anti-CD45RA antibody-conjugated beads for negative selection to deplete naive cells. Thereafter, the flow-through CD45RA– cells were incubated with Miltenyi anti-CD8 antibody-conjugated beads to positively select the CD8+ cells. The postenrichment CD45RA–CD8+ T cells had high purity (>95%; Figure 1). After tandem selection, mean cell yield was 2.6 × 108 (range, 1.3-4.9 × 108) CD45RA–CD8+ T cells from a single apheresis product. We calculated that after 2 consecutive 12-L aphereses per donor, the highest feasible dose of CD8+ TM cells available for infusion would be 10 × 108 cells, assuming most recipients weighed <100 kg, and attainable numbers of CD8+ TM cells for the phase 1 dose-escalation trial were 1 × 106, 5 × 106, and 10 × 106 cells per kg.
Figure 1.

Representative flow analysis of peripheral blood apheresis collections from the preselection, post-CD45RA depletion, and CD8+enrichment steps. Cells were stained for expression of CD4, CD8, CD45RA, and CD45RO. Plots show CD45 gated events.
Flow cytometric analysis, cytokine expression, MLR, and cytokine secretion
In additional studies that used unstimulated products from 6 healthy human donors, we evaluated the composition and compartmentalization of TM CD4+ and CD8+ naive and TM cell subsets before selection, after CD45RA depletion, and after CD8+ enrichment (Figure 2A). In preselection apheresis samples, naïve and TCM cells were the dominant cell types and were in similar proportions among CD4+ gated T cells (Figure 2A), whereas TEM cells represented a minority of the cell composition. In contrast to the composition among CD4+ gated cells, TCM cells existed at very low frequencies (range, <1.0%-3.0%) in donor preselection samples among CD8+ gated T cells. Instead, TEM cells were dominant and ranged from 79.5% to 92.5% of CD45RO+ cells in CD8+ gated preselection samples. Thus, among CD8+ gated CD45RO+ total memory cells, the TEM:TCM ratio in preselection samples ranged from 3.9:1 to as high as 28.5:1. These results are in keeping with reports from others that showed TEM cells greatly outnumber TCM cell in CD8+ gated healthy donor blood samples.24,25 For a given individual and among CD8+ gated cells, the TEM:TCM ratio did not change with each step of cell processing: the ratio of CD8+ gated TEM:TCM cells in the preselection sample was maintained after CD45RA depletion and after CD8+ cell selection (Figure 2A). Flow cytometric analyses of the final CD45RA–CD8+-enriched cells were most consistent with phenotypic CD8+ TEM (CD45RO+CD44+CD62L–) cells (Figure 2A).
Figure 2.

Flow cytometric analysis, cytokine expression, mixed lymphocyte reaction, and cytokine secretion. (A) Representative flow analysis of 1 experiment repeated 5 times that shows cell composition and subsets from start to finish of preselection, post–CD45RA depletion, and post–CD8+ enrichment. Cells were stained for expression of CD45RA, CD45RO, and CD62L, and plots are shown for CD4+ and CD8+ gated cells at each step of the processing procedure. The surface phenotype of the CD45RA–CD8+-enriched cells is consistent with the phenotypic CD8+ TEM subset being predominantly CD45RA–CD45RO+CD62L–. The TEM cells ranged from 81.7% to 98.1% of the final cell composition. (B) Flow cytometric analysis of cytokine expression by enriched cell subsets. Cells were activated by ionomycin and phorbol myristate acetate, treated with monensin, and stained for expression of CD45, CD4, CD8, and CD45RA. Cells were fixed, permeabilized, and stained for INF-γ, and IL-2. Plots show comparison of IL-2 and IFN-γ expression in CD8+CD45RA–, CD8+CD45RA+, and CD4+CD45RA– cells as indicated. (C) Proliferation of CD8+CD45R– and CD4+ cells activated by co-culture with or without irradiated allogeneic stimulators and assessed by 3H-thymidine uptake during the last 24 hours of culture. Mean and standard deviations are shown (n = 4). (D) Cytokine secretion assessment in CD8+CD45RA– and CD4+ cells activated by co-culture with or without irradiated allogeneic stimulators. Supernatants from 7-day cultures were analyzed by flow cytometry using cytokine bead arrays for INF-γ, IL-2, and TNF-α (n = 4). Means with standard deviations are shown. Teff, T effector cells.
But a rigid subclassification of CD8+ TM cells based on expression of CD62L and CCR7 alone is unlikely to be all inclusive, because phenotypic heterogeneity within the CD8+ TM cell pool has been observed.25 The subclassification of CD8+ TM cell subsets is supported by characteristics of an immune response. In MLR, CD8+ TEM cells showed low-level proliferation in response to third-party stimulators and produced high levels of IFN-γ with little IL-2 compared with CD8+ TCM cells that proliferate and secrete IL-2 and IFN-γ.26 Consequently, we evaluated the cytokine expression in sorted phenotypic CD45RA–CD8+ TM cells upon activation by ionomycin and phorbol myristate acetate and observed high levels of IFN-γ and little detectable IL-2, whereas sorted CD45RA+CD8+ and CD45RA–CD4+ T cells expressed both IL-2 and IFN-γ (Figure 2B). In other studies, the sorted CD45RA–CD8+ T cells showed minimal proliferation in response to irradiated third-party allogeneic stimulators obtained from a pool of normal donors in MLR compared with the proliferation of CD4+ cells (Figure 2C). The supernatants from the CD45RA–CD8+ T-cell populations expressed high levels of IFN-γ and TNF-α but little detectable IL-2, consistent with the effector memory classification (Figure 2D). The results of these experiments were consistent with human CD8+ TEM responses and the responses observed with CD8+ TM cells from mice that had little increase in 3H-thymidine incorporation after stimulation with allogeneic cells, and the production of IFN-γ was considerably greater than production of IL-2.22,26
Patient characteristics
Fifteen patients with disease relapse after allo-HCT received phenotypic CD8+ TM cell infusion at 3 escalating dose levels (Table 1). The median time from allo-HCT to disease relapse was 508 days (range, 59-3214 days), and the median time from disease relapse to CD8+ TM cell infusion was 236 days (range, 7-3005 days). Thirteen patients (87%) received cytoreductive therapy before phenotypic CD8+ TM cell infusion. At the time of the CD8+ TM cell infusion, 6 patients (40%) were in CR, and the remaining 9 patients (60%) had active disease. No patients were receiving ongoing immunosuppressive therapy at the time of cell infusion.
Table 1.
Patient characteristics
| Patient | Dose level | Diagnosis | HCT regimen intensity | Time from HCT to relapse, d | Treatment before CD8+ TM cell infusion | Time from relapse to CD8+ TM cell infusion, d | Disease status at CD8+ TM cell infusion |
|---|---|---|---|---|---|---|---|
| 1 | 1 | AML | NMA | 865 | Chemotherapy | 236 | CR2 |
| 2 | 1 | AML | NMA | 118 | None | 7 | Active disease |
| 3 | 1 | AML | MA | 2325 | Chemotherapy | 183 | CR3 |
| 4 | 2 | AML | MA | 167 | Chemotherapy | 134 | CR2 |
| 5 | 2 | CML | MA | 1531 | Chemotherapy | 799 | CR3 |
| 6 | 2 | CLL | NMA | 437 | Chemotherapy | 498 | Active disease |
| 7 | 3 | AML | NMA | 59 | Chemotherapy | 98 | Active disease |
| 8 | 3 | AML | RIC | 214 | Chemotherapy | 135 | Active disease |
| 9 | 3 | NHL | NMA | 93 | Chemotherapy | 1106 | Active disease |
| 10 | 3 | NHL | NMA | 704 | Chemotherapy | 337 | Active disease |
| 11 | 3 | AML | MA | 3214 | Chemotherapy | 91 | CR2 |
| 12 | 3 | AML | MA | 1361 | Chemotherapy | 418 | CR3 |
| 13 | 3 | NHL | NMA | 508 | Chemotherapy | 855 | Active disease |
| 14 | 3 | MM | NMA | 878 | Chemotherapy | 3005 | Active disease |
| 15 | 3 | ALL | RIC | 303 | None | 125 | Active disease |
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; CR2, second complete remission; CR3, third complete remission; MA, myeloablative; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; NMA, non-myeloablative; RIC, reduced-intensity conditioning.
Clinical responses
Responses after CD8+ TM cell infusion are detailed in Table 2. Median follow-up for all patients from the time of CD8+ TM cell infusion was 328 days (range, 118-1328 days). Ten patients (67%) maintained or achieved response (7 CR, 1 partial response, 2 stable disease) for at least 3 months after CD8+ TM cell infusion; 4 of the responders had active disease at the time of infusion. Five patients (33%) had no response to CD8+ TM cell infusion; all 5 of these patients had active disease at the time of infusion. Among the 10 responders, 7 subsequently relapsed with a median time to relapse of 165 days (range, 90-973 days). Eight patients are currently alive; 3 are alive in CR and 5 are alive with disease. For the entire study cohort, the median event-free survival and overall survival after CD8+ TM cell infusion were 4.9 months (95% confidence interval, 1-19.3 months) and 19.6 months (95% confidence interval, 5.6 months to not reached), respectively (Figures 3 and 4).
Table 2.
Patient outcomes
| Patient | Diagnosis | Disease status at CD8+ TM cell infusion | CD8+ TM cell dose, × 106 cells per kg | 3-month response after CD8+ TM cell infusion | 6-month response after CD8+ TM cell infusion | Time from CD8+ TM cell infusion to death or last follow-up, d | Status at last follow-up |
|---|---|---|---|---|---|---|---|
| 1 | AML | CR2 | 1.0 | CR2 | CR | 1057 | Dead |
| 2 | AML | Active disease | 1.0 | CR2 | CR | 591 | Dead |
| 3 | AML | CR3 | 1.0 | CR3 | CR | 1328 | Alive in CR |
| 4 | AML | CR2 | 5.0 | CR2 | PD | 430 | Dead |
| 5 | CML | CR3 | 5.0 | CR3 | CR | 1069 | Alive in CR |
| 6 | CLL | Active disease | 5.0 | PD | PD | 168 | Dead |
| 7 | AML | Active disease | 7.8 | PD | PD | 128 | Dead |
| 8 | AML | Active disease | 8.0 | PD | PD | 151 | Dead |
| 9 | NHL | Active disease | 10.0 | PD | PD | 495 | Alive with disease |
| 10 | NHL | Active disease | 5.2 | SD | PD | 412 | Alive with disease |
| 11 | AML | CR2 | 10.0 | CR2 | CR | 328 | Alive in CR |
| 12 | AML | CR3 | 7.7 | CR3 | PD | 286 | Alive with disease |
| 13 | NHL | Active disease | 10.0 | SD | PD | 194 | Dead |
| 14 | MM | Active disease | 10.0 | PD | PD | 221 | Alive with disease |
| 15 | ALL | Active disease | 10.0 | PR | PD | 118 | Alive with disease |
CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Figure 3.

Kaplan-Meier estimates of event-free survival (EFS) for the 15 patients receiving CD8+TMcell infusion.
Figure 4.

Kaplan-Meier estimates of overall survival (OS) for the 15 patients receiving CD8+TMcell infusion.
CD8+ TM cell recovery and infusion
The recovery of phenotypic CD8+ TM cells after each step of the tandem selection process for each cohort is detailed in Table 3. Most of the loss of CD8+ TM cells occurred at the CD8+ enrichment step in all patients. The mean CD8+ TM cell purity of the infused product was 85.7%, 97.5%, and 93.1%, for dose levels 1, 2, and 3, respectively. All patients in dose levels 1 and 2 received the intended CD8+ TM cell doses. Five of the patients treated at dose level 3 received the planned 10 × 106 cells per kg CD8+ TM cell dose, whereas the dose could not be obtained for 4 patients who received 7.8 × 106, 8.0 × 106, 5.2 × 106, and 7.7 × 106 cells per kg.
Table 3.
CD8+ TM cell yield, purity, and infusion dose after tandem selection
| Dose level | Cell count, × 109 | Recovery of CD8+CD45RA– cells after tandem selection, % | CD8+CD45RA– cells in infused product, % | Infused product cell count, × 106 per kg | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Before CD8+ CD45RA– selection | After CD45RA reduction | After CD8 enrichment | ||||||||||
| Mean | Range | Mean | Range | Mean | Range | Mean | Range | Mean | Range | Mean | Range | |
| 1 | 1.3 | 0.9-1.6 | 0.7 | 0.6-0.9 | 0.4 | 0.2-0.9 | 30.7 | 12.5-54.1 | 85.7 | 80.1-96.8 | 1.0 | 1.0-1.0 |
| 2 | 3.3 | 2.4-4.3 | 3.0 | 1.2-4.6 | 1.18 | 0.5-1.9 | 38.8 | 34.6-44.3 | 97.5 | 96.4-99.7 | 5.0 | 5.0-5.0 |
| 3 | 3.3 | 0.9-7.8 | 2.8 | 0.9-6.9 | 1.2 | 0.4-3.1 | 39.6 | 27.7-61.5 | 93.1 | 80.5-98.6 | 8.7 | 5.2-10.0 |
Safety, GVHD, and adverse events
CD8+ TM cell infusions were well tolerated. One patient developed asymptomatic grade 2 GVHD of the liver after a CD8+ TM cell dose of 10 × 106 cells per kg. There were no infusion-related toxicities or DLTs attributed to the CD8+ TM cells in any patient.
Discussion
The primary objective of this study was to demonstrate the feasibility and safety of using donor-derived phenotypic CD8+ TM cells to treat disease relapse after allo-HCT. We developed a novel 2-step clinically compliant procedure that used unstimulated donor apheresis collections and depleted CD45RA+ cells followed by CD8+ cell selection to provide an enriched population of phenotypic CD8+ TEM cells and conducted a first-in-human 15-patient clinical trial.
The rationale for depleting CD45RA+ TN cells stemmed from murine models of BMT that consistently showed that TM cells (including CD4+ and/or CD8+ TM cells) induced significantly less GVHD than TN cells (CD62LhiCD44lo) from unprimed donors.16-19,27,28 Additional studies by our group showed that CD8+ TM cells, but not CD4+ TM cells, mediated potent antitumor reactions.22 In these models, the infusion of CD44hiCD8+ TM cells after major histocompatibility–matched and –mismatched transplantation eradicated BCL1 lymphoma, even after progressive tumor growth, just as well as an unmanipulated DLI but did not induce GVHD. Thus, TN depletion followed by CD8+ selection would best recapitulate the murine model and perhaps might also translate into an adoptive immunotherapy cell product that will maintain GVT reactions but not induce significant graft-versus-host (GVH) reactions.
The low incidence of GVHD in this trial paralleled the low incidence of GVHD seen in preclinical models. Only 1 patient developed postinfusion acute GVHD that was treated to resolution with corticosteroids. It is worth noting that a majority of patients included in this phase 1 trial had relapsed >1 year after HCT; late relapses have also shown an association with reduced risk of GVHD after DLI.29 The CD8+ TM DLI was otherwise well tolerated and safe, and no patient developed chronic GVHD. The impact of CD8+ TM cell infusions on donor chimerism was not assessable because nearly all patients had complete donor chimerism at the time of the infusion. In comparison, an unmanipulated DLI can invoke significant toxicity and GVHD-related mortality.11
It is not possible to draw conclusions regarding efficacy, given the intrinsic nature of a phase 1, 3+3 dose-escalation safety and feasibility trial design and heterogeneous study population. The CD8+ TM DLI had little or no impact on the 9 patients who had refractory disease, and these patients subsequently died of progressive disease. For the 6 patients whose disease returned to CR before the CD8+ TM cell infusion, 2 remain alive and in CR at last follow-up, and 4 had subsequent disease relapse. These observations are not unlike those from an experience using an unmanipulated DLI to treat relapse for diseases other than chronic myeloid leukemia, but the difference seems to be the lack of toxicity with CD8+ TM DLI.7,8,11,12
The processing method for CD8+ TM cell selection required 1 day in a routine hematopoietic stem cell laboratory and used clinical-grade immunomagnetic kits. The cell selection method was easier to use than alternative procedures currently being investigated that selectively deplete alloreactive T cells using immunotoxins or in vitro photodynamic purging.30,31 The purity of the final infusate in this trial was high, with a median value of >94.6% of cells expressing a CD8+ TM phenotype, of which the overwhelming majority were CD8+ TEM cells.32 A small percentage were CD8+ TCM cells, and even fewer were CD8+ natural killer cells and CD14+CD8+ monocytes. The main limitation of the selection procedure was the maximum dose of CD8+ TM cells that could be obtained. Even with batching 2 consecutive 12-L apheresis collections from each donor, we were unable to achieve the desired target dose (10 × 106 cells per kg) for all patients in cohort 3. Instead, we determined that a reliable dose for trial design using the current cell processing methods is 5 × 106 CD8+ TM cells per kg. The limitation in cell numbers arose because the CD8+ TM cell yield averaged about 40% of the starting number of cells. The majority of the cell loss occurred with CD8+ selection and not during CD45RA depletion. We posit that the CliniMACS CD8 Microbeads and Reagent Kit may be better suited for CD8+ depletion than for enrichment. Nonetheless, to achieve a dose of 5 × 106 CD8+ TM cells per kg, an unmanipulated DLI of at least 100 × 106 CD3+ cells per kg would be required because CD8+ TEM cells represent, on average, <5% of the nucleated cells in an unstimulated apheresis product. An unmanipulated DLI at this high dose has been associated with a substantial risk of precipitating severe acute GVHD.9,10,12,13,33 Therefore, in this trial, patients were infused with a dose of CD8+ TEM cells that would otherwise be untenable if an unmanipulated DLI were used.
Other groups previously performed clinical trials using TN-depleted products, but the cell harvesting method involved mobilization with granulocyte colony-stimulating factor, and the processing methods did not include CD8+ selection; consequently, the final cell product shared few similarities with the cell product in this trial. Bleakley et al34 reported the outcomes of acute leukemia patients transplanted with TN-depleted stem cell grafts from HLA-matched donors. Donors received granulocyte colony-stimulating factor for stem cell mobilization, and apheresis products were processed by using a 2-step immunomagnetic selection procedure that involved positive selection of CD34+ progenitor cells followed by depletion of CD45RA+ cells from the CD34– fraction. The final infusate consisted of the enriched CD34+ cells combined with the CD45RA-depleted product adjusted to infuse 10 × 106 CD3+CD45RA– T cells per kg. Analysis showed that among the CD3+CD45RA– T cells, the vast majority (80%) were CD4+ TM phenotype (CD3+CD4+CD45RA–CD45RO+) of which almost 70% were CCR7+CD27+CD28+, consistent with phenotypic TCM cells. The processing strategy relied on CD45RA depletion without positive selection; therefore, there was significant contamination of the final product with CD45RA– non–T cells. Acute grade 2 to 3 GVHD was observed in 23 of 35 recipients. It is possible that the cellular composition that included a high number of CD14+ monocytes and CD4+ TCM cells provided sufficient helper function to induce GVH reactions. In another study, 5 pediatric severe combined immunodeficiency patients received unstimulated BM grafts from 1-allele mismatched related or unrelated donors.35 The harvested BM was processed in a 2-step procedure that also involved CD34+ selection followed by CD45RA+ depletion of the CD34– flow-through fraction. The median dose of CD3+CD45RA– T cells infused was low (2.5 × 106 cells per kg), and >80% of the CD3+CD45RA– T cells were phenotypic CD4+ TCM cells. One of 5 patients developed clinically significant acute GVHD. In contrast to those 2 studies, in this trial, the final infused product was almost entirely composed of phenotypic CD8+ TEM cells. In murine models, TEM cells did not cause GVHD, whereas TCM cells did cause GVHD, albeit somewhat less severe than that caused by TN cells.20
It is unclear why phenotypic CD8+ TM cells from unprimed donors would possess antitumor activity. The ability of CD8+ TM cells to eradicate tumor may in part be dependent on their alloreactivity to host histocompatibility tissue antigens, because C57BL/6 CD8+ T cells tolerized to BALB/c alloantigens lose their graft antitumor activity against BCL1 lymphoma.36 Antitumor alloreactivity of CD8+ TM cells may also be explained by molecular mimicry and cross-reactivity with viral antigens in the environment that were shown to enhance immune responses to alloantigens on organ transplants.37-39 Alternatively, naive CD8+ T cells can masquerade as memory phenotype cells after homeostatic expansion and can maintain the naive T-cell tissue cross-reactivity repertoire.40 Consistent with retained antitumor activity, the CD8+ TM cells produced high levels of IFN-γ, a cytokine frequently required in models of antitumor activity.41
The main limitations to successful transplant outcomes are disease relapse, GVHD, and infections, and composite indexes such as GVHD–relapse free survival suggest that ∼25% of all recipients have uncomplicated posttransplant courses.42 Thus, there are compelling reasons to seek alternates to giving unmanipulated donor allografts. Within the limitations of this single-arm safety and feasibility trial, we developed a rapid and relatively easy cell processing method that provided a highly enriched population of CD8+ TEM cells. The cells were safe, did not induce significant GVHD, and may have a clinical signal of efficacy because durable remissions were observed in one-third of the recipients who returned to CR prior to the cell infusion. It is possible that lymphodepletion before the CD8+ TM cell infusion may potentiate efficacy without aggravating GVHD, and this approach is being evaluated in current and future planned studies of CD8+ TM cells. Alternatively, combining CD8+ TM cells with CD34+ selected grafts at the time of transplantation may be a reasonable consideration, and efficacy could potentially be further enhanced with additional strategies including checkpoint blockade. The generation of virus-specific and tumor-specific T-cell clones by immunizing donors with peptides before CD8+ TM cell collection may enhance protection from infection and relapse. We previously showed in a murine model of BMT that donor immunization with WT-1 peptide and adoptive transfer of CD8+ T cells protected recipients from posttransplant tumor challenge compared with CD8+ T cells from unimmunized donors.43 The results presented herein warrant further exploration of CD8+ TM cells with extension to other transplantation settings.
Acknowledgment
This study was funded by the National Institutes of Health, National Cancer Institute (P01 CA049605).
Footnotes
Presented as an abstract at the 58th annual meeting of the American Society of Hematology, San Diego, CA, 3-6 December 2016.
Authorship
Contribution: L.M. and R.L. drafted the manuscript; and all authors participated in revising the manuscript, in data collection and analysis, and approved the submitted manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Robert Lowsky, Division of Blood and Marrow Transplantation, Stanford University, 300 Pasteur Dr, H0101, Stanford, CA 94305; e-mail: rlowsky@stanford.edu.
References
- 1.Wingard JR, Majhail NS, Brazauskas R, et al. Long-term survival and late deaths after allogeneic hematopoietic cell transplantation. J Clin Oncol. 2011;29(16):2230-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Souza A, Zhu X Current Uses and Outcomes of Hematopoietic Cell Transplantation (HCT): CIBMTR Summary Slides, 2016. http://www.cibmtr.org/. Accessed 1 June 2017.
- 3.Mielcarek M, Storer BE, Flowers ME, Storb R, Sandmaier BM, Martin PJ. Outcomes among patients with recurrent high-risk hematologic malignancies after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2007;13(10):1160-1168. [DOI] [PubMed] [Google Scholar]
- 4.Oran B, Giralt S, Couriel D, et al. Treatment of AML and MDS relapsing after reduced-intensity conditioning and allogeneic hematopoietic stem cell transplantation. Leukemia. 2007;21(12):2540-2544. [DOI] [PubMed] [Google Scholar]
- 5.Schmid C, Labopin M, Nagler A, et al. Treatment, risk factors, and outcome of adults with relapsed AML after reduced intensity conditioning for allogeneic stem cell transplantation. Blood. 2012;119(6):1599-1606. [DOI] [PubMed] [Google Scholar]
- 6.Poon LM, Hamdi A, Saliba R, et al. Outcomes of adults with acute lymphoblastic leukemia relapsing after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2013;19(7):1059-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bejanyan N, Weisdorf DJ, Logan BR, et al. Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: a Center for International Blood and Marrow Transplant research study. Biol Blood Marrow Transplant. 2015;21(3):454-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmid C, Labopin M, Nagler A, et al. Donor lymphocyte infusion in the treatment of first hematological relapse after allogeneic stem-cell transplantation in adults with acute myeloid leukemia: a retrospective risk factors analysis and comparison with other strategies by the EBMT Acute Leukemia Working Party. J Clin Oncol. 2007;25(31):4938-4945. [DOI] [PubMed] [Google Scholar]
- 9.Kolb HJ, Mittermüller J, Clemm C, et al. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. 1990;76(12):2462-2465. [PubMed] [Google Scholar]
- 10.Bär BM, Schattenberg A, Mensink EJ, et al. Donor leukocyte infusions for chronic myeloid leukemia relapsed after allogeneic bone marrow transplantation. J Clin Oncol. 1993;11(3):513-519. [DOI] [PubMed] [Google Scholar]
- 11.Kolb HJ, Schattenberg A, Goldman JM, et al. ; European Group for Blood and Marrow Transplantation Working Party Chronic Leukemia. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood. 1995;86(5):2041-2050. [PubMed] [Google Scholar]
- 12.Collins RH Jr, Shpilberg O, Drobyski WR, et al. Donor leukocyte infusions in 140 patients with relapsed malignancy after allogeneic bone marrow transplantation. J Clin Oncol. 1997;15(2):433-444. [DOI] [PubMed] [Google Scholar]
- 13.Mackinnon S, Papadopoulos EB, Carabasi MH, et al. Adoptive immunotherapy evaluating escalating doses of donor leukocytes for relapse of chronic myeloid leukemia after bone marrow transplantation: separation of graft-versus-leukemia responses from graft-versus-host disease. Blood. 1995;86(4):1261-1268. [PubMed] [Google Scholar]
- 14.Alyea EP, Soiffer RJ, Canning C, et al. Toxicity and efficacy of defined doses of CD4(+) donor lymphocytes for treatment of relapse after allogeneic bone marrow transplant. Blood. 1998;91(10):3671-3680. [PubMed] [Google Scholar]
- 15.Giralt S, Hester J, Huh Y, et al. CD8-depleted donor lymphocyte infusion as treatment for relapsed chronic myelogenous leukemia after allogeneic bone marrow transplantation. Blood. 1995;86(11):4337-4343. [PubMed] [Google Scholar]
- 16.Anderson BE, McNiff J, Yan J, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest. 2003;112(1):101-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng H, Matte-Martone C, Li H, et al. Effector memory CD4+ T cells mediate graft-versus-leukemia without inducing graft-versus-host disease. Blood. 2008;111(4):2476-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Joe G, Zhu J, et al. Dendritic cell-activated CD44hiCD8+ T cells are defective in mediating acute graft-versus-host disease but retain graft-versus-leukemia activity. Blood. 2004;103(10):3970-3978. [DOI] [PubMed] [Google Scholar]
- 19.Chen BJ, Cui X, Sempowski GD, Liu C, Chao NJ. Transfer of allogeneic CD62L- memory T cells without graft-versus-host disease. Blood. 2004;103(4):1534-1541. [DOI] [PubMed] [Google Scholar]
- 20.Zheng H, Matte-Martone C, Jain D, McNiff J, Shlomchik WD. Central memory CD8+ T cells induce graft-versus-host disease and mediate graft-versus-leukemia. J Immunol. 2009;182(10):5938-5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dutt S, Tseng D, Ermann J, et al. Naive and memory T cells induce different types of graft-versus-host disease. J Immunol. 2007;179(10):6547-6554. [DOI] [PubMed] [Google Scholar]
- 22.Dutt S, Baker J, Kohrt HE, et al. CD8+CD44(hi) but not CD4+CD44(hi) memory T cells mediate potent graft antilymphoma activity without GVHD. Blood. 2011;117(11):3230-3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edinger M, Hoffmann P, Ermann J, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9(9):1144-1150. [DOI] [PubMed] [Google Scholar]
- 24.Strober S, Benike C, Krishnaswamy S, Engleman EG, Grumet FC. Clinical transplantation tolerance twelve years after prospective withdrawal of immunosuppressive drugs: studies of chimerism and anti-donor reactivity. Transplantation. 2000;69(8):1549-1554. [DOI] [PubMed] [Google Scholar]
- 25.Hikono H, Kohlmeier JE, Takamura S, Wittmer ST, Roberts AD, Woodland DL. Activation phenotype, rather than central- or effector-memory phenotype, predicts the recall efficacy of memory CD8+ T cells. J Exp Med. 2007;204(7):1625-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lefrançois L, Obar JJ. Once a killer, always a killer: from cytotoxic T cell to memory cell. Immunol Rev. 2010;235(1):206-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen BJ, Deoliveira D, Cui X, et al. Inability of memory T cells to induce graft-versus-host disease is a result of an abortive alloresponse. Blood. 2007;109(7):3115-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xystrakis E, Bernard I, Dejean AS, Alsaati T, Druet P, Saoudi A. Alloreactive CD4 T lymphocytes responsible for acute and chronic graft-versus-host disease are contained within the CD45RChigh but not the CD45RClow subset. Eur J Immunol. 2004;34(2):408-417. [DOI] [PubMed] [Google Scholar]
- 29.Bar M, Sandmaier BM, Inamoto Y, et al. Donor lymphocyte infusion for relapsed hematological malignancies after allogeneic hematopoietic cell transplantation: prognostic relevance of the initial CD3+ T cell dose. Biol Blood Marrow Transplant. 2013;19(6):949-957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boumédine RS, Roy DC. Elimination of alloreactive T cells using photodynamic therapy. Cytotherapy. 2005;7(2):134-143. [DOI] [PubMed] [Google Scholar]
- 31.Mielke S, Solomon SR, Barrett AJ. Selective depletion strategies in allogeneic stem cell transplantation. Cytotherapy. 2005;7(2):109-115. [DOI] [PubMed] [Google Scholar]
- 32.Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol. 2005;175(9):5895-5903. [DOI] [PubMed] [Google Scholar]
- 33.Kolb HJ, Schattenberg A, Goldman JM, et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood. 1995;86(5):2041-2050. [PubMed] [Google Scholar]
- 34.Bleakley M, Heimfeld S, Loeb KR, et al. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest. 2015;125(7):2677-2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Touzot F, Neven B, Dal-Cortivo L, et al. CD45RA depletion in HLA-mismatched allogeneic hematopoietic stem cell transplantation for primary combined immunodeficiency: A preliminary study. J Allergy Clin Immunol. 2015;135(5):1303-1309.e1-e3. [DOI] [PubMed] [Google Scholar]
- 36.Pillai A, Teo P, George T, Mukhopadhyay A, Dejbakhsh-Jones S, Strober S. Alloantigen recognition is critical for CD8 T cell-mediated graft anti-tumor activity against murine BCL1 lymphoma after myeloablative bone marrow transplantation. Bone Marrow Transplant. 2007;40(5):487-497. [DOI] [PubMed] [Google Scholar]
- 37.Ford ML, Kirk AD, Larsen CP. Donor-reactive T-cell stimulation history and precursor frequency: barriers to tolerance induction. Transplantation. 2009;87(9 Suppl):S69-S74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams AB, Pearson TC, Larsen CP. Heterologous immunity: an overlooked barrier to tolerance. Immunol Rev. 2003;196:147-160. [DOI] [PubMed] [Google Scholar]
- 39.Brehm MA, Markees TG, Daniels KA, Greiner DL, Rossini AA, Welsh RM. Direct visualization of cross-reactive effector and memory allo-specific CD8 T cells generated in response to viral infections. J Immunol. 2003;170(8):4077-4086. [DOI] [PubMed] [Google Scholar]
- 40.Murali-Krishna K, Ahmed R.. Cutting edge: naive T cells masquerading as memory cells. J Immunol. 2000;165(4):1733-1737. [DOI] [PubMed] [Google Scholar]
- 41.Bekisz J, Sato Y, Johnson C, Husain SR, Puri RK, Zoon KC. Immunomodulatory effects of interferons in malignancies. J Interferon Cytokine Res. 2013;33(4):154-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holtan SG, DeFor TE, Lazaryan A, et al. Composite end point of graft-versus-host disease-free, relapse-free survival after allogeneic hematopoietic cell transplantation. Blood. 2015;125(8):1333-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kohrt HE, Müller A, Baker J, et al. Donor immunization with WT1 peptide augments antileukemic activity after MHC-matched bone marrow transplantation. Blood. 2011;118(19):5319-5329. [DOI] [PMC free article] [PubMed] [Google Scholar]