

Key Points
VLD rivaroxaban significantly reduces platelet-dependent thrombin generation and thrombus formation on top of DAPT in patients with ACS.
Adjunctive treatment with VLD rivaroxaban additionally reduced TG and thrombus formation in both clopidogrel responders and nonresponders.
Abstract
Very low–dose (VLD) factor Xa (FXa) inhibition, in combination with acetylsalicylic acid (ASA) and clopidogrel, is associated with improved outcomes in patients with acute coronary syndrome (ACS) with a tolerable bleeding risk profile. To date, there are no data documenting platelet inhibition and the anticoagulatory effects of VLD FXa inhibition on top of guideline-adherent dual-antiplatelet therapy (DAPT) in patients with ACS. Patients with non–ST-elevation myocardial infarction (NSTEMI) receiving oral DAPT (ASA + clopidogrel, n = 20; or ASA + ticagrelor, n = 20) were prospectively enrolled in a nonrandomized study. Coagulation- and platelet-dependent thrombin generation (TG), measured by means of the calibrated automated thrombogram, were significantly decreased after in vitro and in vivo addition of rivaroxaban. As shown by a total thrombus-formation analysis approach, rivaroxaban treatment led to a significantly decreased coagulation-dependent (AR-chip) thrombus formation in patients treated with ASA plus P2Y12 inhibitor (clopidogrel/ticagrelor), whereas the pure platelet-dependent (PL-chip) thrombus formation was not affected at all. Adjunctive rivaroxaban therapy was not associated with significant differences in platelet aggregation assessed by light-transmission aggregometry (LTA). Nevertheless, according to fluorescence-activated cell sorter analysis, VLD rivaroxaban treatment resulted in a significantly reduced expression of platelet HMGB-1, whereas P-selectin exposure was not affected. Furthermore, an enhanced effect of rivaroxaban on total thrombus formation and TG was observed in particular in clopidogrel nonresponder patients defined as adenosine 5′-diphosphate-induced LTA ≥40%. VLD rivaroxaban reduces thrombus formation and platelet-dependent TG in patients with ACS receiving DAPT, which can be of potential ischemic benefit. This trial was registered at www.clinicaltrials.gov as #NCT01417884.
Visual Abstract
Introduction
Platelets are critically involved in the pathophysiology of coronary thrombosis and atheroprogression.1,2 Factor Xa (FXa) inhibition on top of dual-antiplatelet therapy (DAPT) has been shown to reduce ischemic events in patients with acute coronary syndrome (ACS). The trade-off is the enhanced bleeding risk, and achieving the sweet spot of enhanced efficacy with tolerable safety profile represents a major clinical challenge.3 Of note, the APPRAISE-2 trial demonstrated that full-dose FXa inhibition with apixaban in combination with antiplatelet therapy (approximately 80% on acetylic salicylic acid [ASA] and a P2Y12 inhibitor, mostly clopidogrel) was associated with an unacceptable bleeding risk, including increased risk for intracranial and fatal bleeding.4 Recently, 2 randomized trials demonstrated a tolerable bleeding risk profile with very low–dose (VLD; ie, 2.5 mg) twice-daily dosing of rivaroxaban with DAPT.5,6 In the ATLAS-ACS 2 TIMI 51 trial, rivaroxaban 2.5 mg twice daily in combination with DAPT (ASA + clopidogrel) was associated with a significant reduction of ischemic events, including stent thrombosis, and with higher bleeding events, including intracranial hemorrhages, but without an increase in fatal bleeding. In the PIONEER-AF PCI trial, a VLD rivaroxaban regimen plus DAPT (ASA + clopidogrel) was associated with lower bleeding rates compared with a vitamin K antagonist plus DAPT regimen and with similar bleeding rates compared with low-dose (15 mg once daily) rivaroxaban plus P2Y12 inhibitor in patients with atrial fibrillation undergoing percutaneous coronary intervention. It is noteworthy that the index event was ACS in more than half (51.5%) of the patients.
In 2 very recent trials, VLD rivaroxaban was tested in combination with single-antiplatelet therapy against standard antiplatelet therapy. In the phase 2 GEMINI-ACS-1 trial, VLD rivaroxaban was compared with ASA on the background of P2Y12 inhibition (clopidogrel or ticagrelor, based on investigator preference).7 This trial showed comparable bleeding rates with VLD rivaroxaban compared with ASA without excess in ischemic events. The COMPASS trial demonstrated superiority of the combination of VLD (2.5 mg twice daily) rivaroxaban plus ASA compared with ASA or rivaroxaban monotherapy in reducing the primary composite endpoint of cardiovascular (CV) death, stroke, or myocardial infarction with a favorable net clinical benefit in patients with stable coronary artery disease.8
The mechanistic effects of VLD FXa inhibition in addition to standard DAPT, including contemporary P2Y12 antagonism, on the inhibition of platelet function and coagulation have not been investigated in patients with ACS. Importantly, there are no mechanistic data on the added benefit of rivaroxaban on ASA plus ticagrelor, and very limited evidence of added benefit on ASA plus clopidogrel in patients with ACS. Therefore, the REVEAL (Rivaroxaban—Evaluation of Variables Enhancing Antithrombotic Efficacy and Longterm-Outcome after Non ST-Elevation Myocardial Infarction) study sought to assess the added effects of in vitro and in vivo VLD rivaroxaban in patients with ACS while receiving DAPT.
Material and methods
Study subjects
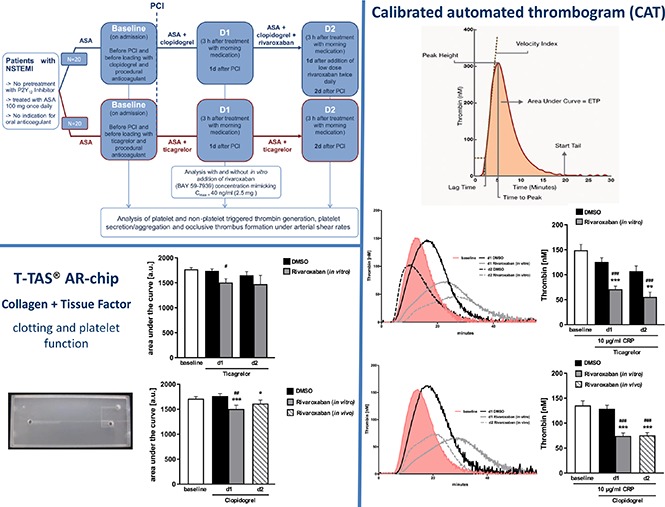
In total, 40 consecutive patients presenting with non–ST-elevation myocardial infarction (NSTEMI) in the chest pain unit of the university hospital in Tübingen, Germany, were prospectively enrolled in this prospective, nonrandomized study. All patients underwent coronary angiography confirming the presence of relevant coronary artery disease (eg, stenosis >50% in 1 or more coronary arteries). Twenty patients with NSTEMI were treated with ticagrelor, and 20 patients with NSTEMI with contraindications for ticagrelor and prasugrel were treated with clopidogrel (600-mg loading dose and 75-mg daily maintenance dose), as indicated in the flowchart (Figure 1). Reasons for avoiding administration of ticagrelor or prasugrel were older age, history of sick sinus syndrome, second- or third-degree atrioventricular block or bradycardia-related syncope not protected by a pacemaker, low body weight, chronic pulmonary disease/severe episodes of dyspnea in patient history, and potential interactions with preexisting comedication. Patients were selected for VLD rivaroxaban in combination with DAPT according to the drug label and current guidelines.9
Figure 1.

Flowchart of REVEAL study.
Preparation of platelet-rich and platelet-poor plasma
Citrated anticoagulated human blood, drawn from patients by venous puncture, was centrifuged for 3 minutes at 290g at room temperature. Supernatant was collected, and platelet-rich plasma (PRP) was obtained by a further centrifugation at 655g for 30 seconds without deceleration at room temperature. PRP was collected and platelet count was determined by means of a KX-21N hematology analyzer (Sysmex, Norderstedt, Germany). Platelet-poor plasma (PPP) was obtained afterward by centrifugation of the blood samples at 2560g for 5 minutes at room temperature.
Spiking of rivaroxaban
In vitro treatment of samples with rivaroxaban (Bayer AG, Wuppertal, Germany) or the corresponding vehicle control (dimethyl sulfoxide [DMSO]) was performed by spiking immediately before starting the measurements. On the basis of previous pharmacokinetic profile studies, 40 ng/mL rivaroxaban was chosen as final concentration to simulate VLD rivaroxaban.10
Platelet aggregation
Platelet count in citrate anticoagulated PRP was adjusted to a final concentration of 2.5 × 105/µL with corresponding PPP, and aggregation was measured with a lumi-aggregometer (ChronoLog Model 700; Chrono-Log Corp., Havertown, PA). Samples were treated with DMSO control or rivaroxaban at the indicated times and concentrations. After calibration, agonists were added and aggregation was measured for 10 minutes with a stir speed of 1000 rpm at 37°C. Maximum platelet aggregation (PA), as well as the area under the curve (AUC), was quantified using the aggrolink8 software (ChronoLog).
In the group of clopidogrel-treated patients, samples showing at least 40% of the aggregation as compared with baseline after adenosine 5′-diphosphate (ADP; 5 µM) stimulation were defined as clopidogrel nonresponders at day 2 (d2).11,12
Fluorescence-activated cell sorter analysis of platelet surface abundance of P-selectin and HMGB-1
Two-color flow cytometry analysis was conducted using monoclonal phycoerythrin-labeled antibodies for expression of glycoprotein Ib (CD42b; Beckman Coulter) as a platelet marker and a fluorescein isothiocyanate-conjugated monoclonal antibody detecting surface expression of platelet P-selectin (CD62P; R&D Systems), or an Alexa Fluor 488-conjugated monoclonal antibody detecting platelet high-mobility group box-1 protein (HMGB-1; R&D Systems), as described in detail previously.13
Calibrated automated thrombogram
Thrombin generation in human PPP as well as human PRP was measured by using the calibrated automated thrombogram method by Hemker.14 For that purpose, citrated anticoagulated PRP was adjusted to a final platelet concentration of 1.5 × 105/µL with corresponding PPP, and measurements were performed with a Thrombinoscope system and reagents from Stago (Stago, Düsseldorf, Germany) according to the manufacturer’s protocol. Where indicated, samples were spiked with DMSO or rivaroxaban 40 ng/mL, and collagen-related peptide (CRP; 10 µg/mL) was added. The whole procedure was performed at 37°C, and calibration was performed using a thrombin calibrator (Stago, Düsseldorf, Germany). Finally, the thrombin peak height (maximal rate of thrombin formation), time to peak (TTP), and velocity were analyzed by means of the Thrombinoscope software (Stago, Düsseldorf, Germany).
Total thrombus-formation analysis
Thrombus formation was determined by using the total thrombus formations analysis system (Fujimori Kogyo Co Ltd, Shinjuku, Japan). Measurements were performed on a collagen- and tissue factor-coated slide (AR-chip, Fujimori Kogyo Co Ltd), using 450 µL recalcified citrated human blood, or on a collagen-coated slide (PL-chip, Fujimori Kogyo Co Ltd), using 320 µL hirudin anticoagulated blood, according to the manufacturer’s specifications. Where indicated, DMSO or rivaroxaban was added to the blood samples at the specified concentration. Analysis was performed with total thrombus-formation analysis (T-TAS) software (Fujimori Kogyo Co Ltd, Shinjuku; Japan), calculating time to start occlusion (T10), occlusion time (OT), and the AUC.
Statistical analysis
Analysis was performed with GraphPad prism 7.03, using multiple comparison test (analysis of variance), with a significance level of P < .05. Nonparametric data were compared using the Tukey’s multiple comparison test. The primary hypothesis was to test whether rivaroxaban added in vitro has an effect on platelet-dependent TG in patients under treatment with DAPT after NSTEMI (ASA + clopidogrel/ticagrelor). The primary outcome variable consisted of change of TG in PRP triggered by CRP. Secondary outcome variables consisted of change of PA in response to various agonists (collagen-related peptide, ADP, and thrombin). For sample size calculation, we assumed at least a relative reduction of CRP-induced TG by 15%15,16 by spiking with rivaroxaban (change post- to preincubation) as relevant. An estimated minimum sample size of 15 patients in each treatment group (ticagrelor or clopidogrel) would be sufficient to detect this difference with 90% power at a 2-sided α value of 5% (IBM SPSS Sample Power Version 3; IBM Corp., Armonk, NY).
Ethical issues
All patients gave informed consent. The study was approved by the institutional ethics committee at the University Hospital Tübingen (270/2011BO1; www.clinicaltrials.gov #NCT01417884) and complies with the Declaration of Helsinki and good clinical practice guidelines.
Results
Baseline characteristics of patients enrolled in the study are presented in Table 1. Eighty percent (16/20) of the patients in the ticagrelor group vs 55% (11/20) in the clopidogrel group were male. Because of this difference in male:female ratio between the 2 groups, we determined whether there were any differences in test outcomes potentially related to sex, but we did not find any such effects (data not shown). Patients in the clopidogrel group were significantly older compared with patients treated with ticagrelor (73.2 ± 11.2 years vs 63.1 ± 11.7 years; P = .008). There were no significant differences with regard to CV risk factors, comedication, procedural characteristics, renal function, and hemostatic parameters. There were no major or clinically relevant bleeding events in both groups during hospital stay.
Table 1.
Baseline characteristics of the patient cohort
| Clopidogrel group (n = 20) | Ticagrelor group (n = 20) | P | |
|---|---|---|---|
| Age, mean ± standard deviation, y | 73.2 ± 11.2 | 63.1 ± 11.7 | .008 |
| Female sex, no (%) | 9/20 (45) | 4/20 (20) | .09 |
| History, CV risk factors, n (%) | |||
| Previous MI | 4/20 (25) | 3/20 (15) | .68 |
| History of CAD | 4/20 (20) | 5/20 (25) | .71 |
| History of PAD | 2/20 (10) | 1/20 (5) | .55 |
| Diabetes mellitus | 3/20 (15) | 6/20 (30) | .26 |
| Current smoker | 2/20 (10) | 3/20 (15) | .63 |
| Arterial hypertension | 18/20 (90) | 17/20 (85) | .63 |
| Hyperlipidemia | 6/20 (30) | 8/20 (40) | .51 |
| Medication, n (%) | |||
| ACE-I/ARB | 17/20 (85) | 20/20 (100) | .07 |
| Lipid lowering agents | 16/20 (80) | 19/20 (95) | .15 |
| β blocker | 15/20 (75) | 19/20 (95) | .08 |
| MRA | 3/20 (15) | 3/20 (15) | 1.0 |
| PPI | 10/20 (50) | 6/20 (30) | .20 |
| Procedural characteristics | |||
| Stent implantation, n (%) | 14/20 (70) | 18/20 (90) | .114 |
| DES only, n (%) | 14/14 (100) | 18/18 (100) | |
| Normal LV function (EF > 55%), n (%) | 11/20 (55) | 9/20 (45) | .27 |
| Moderately reduced LV function (EF 45-55%), n (%) | 8/20 (40) | 10/20 (50) | |
| Severely reduced LV function (EF<35%), n (%) | 1/20 (5) | 1/20 (5) | |
| Bleeding complication, n (%) | 0/20 (0) | 0/20 (0) | 1.0 |
| Renal function Baseline CrCl, mean ± standard deviation, mL/min | 78.4 ± 23.3 | 82.8 ± 28.1 | .60 |
| Baseline C-reactive protein, mg/dL | 0.74 ± 0.9 | 1.86 ± 4.25 | .26 |
| INR, baseline | 1.1 ± 0.08 | 1.1 ± 0.05 | .36 |
| Platelet count, mean ± standard deviation, ×109/L | 227 ± 54.5 | 239 ± 74.7 | .59 |
ACE-I, angiotensin-converting enzyme inhibitors; ARB, angiotensin receptor blockers; CAD, coronary artery disease; CrCl, creatinine clearance according to Modification of Diet in Renal Disease formula; DES, drug-eluting stents; EF, ejection fraction; INR, international normalized ratio; MI, myocardial infarction; LV-function, left-ventricular function; MRA, mineralocorticoid receptor antagonists; PAD, peripheral artery disease; PPI, proton pump inhibitors.
Thrombin generation
To investigate the influence on coagulation-dependent TG, calibrated automated thrombogram measurements were executed using PPP. For that purpose, baseline patient samples were measured on admission (+ASA) before starting ticagrelor (baseline) and at days 1 and 2 (d1, d2) after initiation of ticagrelor, respectively. Samples at d1 and d2 were spiked in vitro with control (DMSO) or 40 ng/mL rivaroxaban. At d1, the lag time (3.08 ± 0.18 vs 4.87 ± 0.36 minutes; P = .0005) and TTP (6.00 ± 0.36 vs 10.90 ± 1.00 minutes; P = .0149) were significantly increased, whereas peak (254.25 ± 14.27 vs 114.29 ± 12.81 nM; n = 20; P = .0001) and velocity (109.97 ± 14.32 vs 31.71 ± 6.64 nM/min; n = 20; P = .0001) were significantly decreased after rivaroxaban spiking compared with the vehicle control. Furthermore, in vitro rivaroxaban spiking at d1 significantly changed the lag time (3.20 ± 0.21 vs 4.87 ± 0.36; P = .0002), TTP (6.88 ± 0.59 vs 10.90 ± 1.00; P = .0011), peak (205.866 ± 19.97 vs 114.29 ± 12.81; P = .0004), and velocity (87.21 ± 13.66 vs 31.71 ± 6.64; P = .0010) compared with baseline. The same effects were observed at d2 of ticagrelor medication (Figure 2). PPP samples from patients with ACS treated with clopidogrel + ASA at d1 showed significantly reduced TG, as lag time (3.22 ± 0.25 vs 5.01 ± 0.45; P < .0001) and TTP (6.31 ± 0.42 vs 11.31 ± 0.98; P < .0001) were significantly increased, whereas peak (219.58 ± 13.86 vs 96.75 ± 10.92; P < .0001) and velocity (92.50 ± 13.32 vs 25.41 ± 5.04; P < .0001) were significantly decreased after rivaroxaban spiking compared with the vehicle control. Medication with clopidogrel + ASA and 2.5 mg rivaroxaban twice daily at d2 also led to significantly changed lag time (3.28 ± 0.28 vs 5.15 ± 0.39; P < .0001), TTP (6.02 ± 0.49 vs 11.92 ± 0.89; P < .0001), peak (182.43 ± 14.10 vs 89.42 ± 11.69; P < .0001), and velocity (74.67 ± 7.70 vs 21.42 ± 5.54; P < .0001) compared with baseline (Figure 3).
Figure 2.

TG in PPP of patients with NSTEMI treated with ticagrelor in the presence or absence of VLD rivaroxaban. (A) Representative tracings and (B) arithmetic means ± SEM (n = 20) of lag time, TTP, peak (thrombin), and velocity of TG in PPP of patients with NSTEMI before (baseline) and after medication with ticagrelor + ASA for 1 and 2 days (d1 and d2) in the absence (DMSO) or presence of 40 ng/mL rivaroxaban (in vitro). **P < .01 and ***P < .001 indicate statistically significant differences from solvent control at the indicated time point. #P < .05, ##P < .01, and ###P < .001 indicate significant differences from baseline.
Figure 3.

TG in PPP of patients with NSTEMI treated with clopidogrel in the presence or absence of VLD rivaroxaban. (A) Representative tracings and (B) arithmetic means ± SEM (n = 20) of lag time, TTP, peak (thrombin), and velocity of TG in PPP of patients with NSTEMI before (baseline) and after medication with clopidogrel + ASA at d1 in the absence (DMSO) or presence of 40 ng/mL rivaroxaban (in vitro) as well as following medication with clopidogrel + ASA and 2.5 mg rivaroxaban twice daily at d2 (in vivo). ***P < .001 indicates statistically significant differences from solvent control at the indicated time point. ##P < .01 and ###P < .001 indicate significant differences from baseline.
TG is known to be platelet-dependent.17 Thus, platelet-dependent TG was measured in PRP of patients on DAPT with ASA + ticagrelor/clopidogrel. In ticagrelor-treated and resting PRP at d1, lag time (9.29 ± 0.47 vs 16.61 ± 2.10; P = .0013) and TTP (23.45 ± 1.15 vs 36.32 ± 1.86; P < .0001) were significantly increased, whereas peak (84.50 ± 4.81 vs 52.49 ± 5.09; P < .0001) and velocity (6.14 ± 0.66 vs 2.63 ± 0.31; P = .0003) were significantly reduced after rivaroxaban spiking compared with the vehicle control. Rivaroxaban treatment on top at d1 led to significant changes in lag time (10.72 ± 1.20 vs 16.61 ± 2.10; P = .02), TTP (21.9994 ± 1.36 vs 36.32 ± 1.86; P < .001), peak (79.69 ± 5.72 vs 52.49 ± 5.09; P = .001), and velocity (7.10 ± 0.70 vs 2.63 ± 0.31; P = .0001) compared with the baseline. The same effects were observed at d2 of DAPT (Figure 4A-B). Stimulation of PRP with 10 µg/mL CRP resulted in decreased TG at d1 (lag time, 7.44 ± 0.41 vs 9.82 ± 0.53 [P < .001]; TTP, 16.35 ± 0.78 vs 26.00 ± 1.51 [P < .0001]; peak, 125.80 ± 8.21 vs 71.13 ± 6.02 [P < .0001]; and velocity, 14.73 ± 1.43 vs 5.13 ± 0.88 [P < .0001]) and d2 after rivaroxaban spiking (Figure 4C-D).
Figure 4.
TG in PRP of patients with NSTEMI treated with ticagrelor in the presence or absence of VLD rivaroxaban. (A). Representative tracings of TG in PRP of patients with NSTEMI before (baseline) and after medication with ticagrelor + ASA for 1 and 2 days (d1 and d2) in the absence (DMSO) or presence of 40 ng/mL rivaroxaban (in vitro) with resting platelets and (C) with 10 µg/ml CRP-stimulated platelets. (B) Arithmetic means ± SEM (n = 20) of lag time, TTP, peak (thrombin), and velocity of TG in PRP of patients with NSTEMI before (baseline) and after treatment with ticagrelor for 1 and 2 days (d1 and d2) in the absence (DMSO) and presence of 40 ng/mL rivaroxaban (in vitro) with resting platelets and (D) with 10 µg/mL CRP stimulated platelets. *P < .05, **P < .01, and ***P < .001 indicate statistically significant differences from solvent control at the indicated time point. #P < .05 and ###P < .001 indicate significant differences from baseline.


Treatment with clopidogrel + ASA also showed decreased TG in resting PRP at d1 compared with the solvent control as lag time (9.84 ± 0.83 vs 15.82 ± 2.01; P = .0008), TTP (22.86 ± 1.83 vs 34.17 ± 1.80; n = 20; P < .0001), peak (80.24 ± 6.07 vs 45.58 ± 5.16; P < .0001), and velocity (7.70 ± 1.16 vs 2.70 ± 0.36; P = .0009) were reduced after rivaroxaban treatment. Medication with clopidogrel + ASA and 2.5 mg rivaroxaban twice daily at d2 also led to significantly changed lag time (10.09 ± 0.84 vs 14.76 ± 0.57; P = .0001), TTP (21.47 ± 1.39 vs 34.58 ± 1.26; P < .0001), peak (78.76 ± 4.91 vs 57.81 ± 5.42; P = .0042), and velocity (7.64 ± 0.68 vs 3.30 ± 0.48; P = .0010) compared with baseline (Figure 5A-B). Stimulation of PRP with 10 µg/mL CRP resulted in decreased TG at d1 (lag time, 6.90 ± 0.50 vs 9.94 ± 0.73 [P = .0001]; TTP, 13.58 ± 0.83 vs 23.95 ± 1.57 [P < .0001]; peak, 135.60 ± 9.09 vs 75.56 ± 5.53 [P < .0001]; and velocity, 22.90 ± 2.63 vs 6.14 ± 0.70 [P < .0001]), and after spiking with rivaroxaban ex vivo and after oral administration at d2 compared with baseline (Figure 5C-D). Finally, rivaroxaban (in vivo and ex vivo) led to a marked reduction of platelet-dependent TG, in particular in clopidogrel nonresponders at d1 and especially at d2 of clopidogrel treatment (supplemental Figure 2A-B). Especially after CRP stimulation, clopidogrel nonresponders showed a more pronounced reduction of TG at d2 when rivaroxaban was administered in vivo (lag time, 7.01 ± 0.38 vs 12.76 ± 1.96 [n = 5; P = .002]; TTP, 13.20 ± 0.34 vs 29.14 ± 4.19 [n = 5; P < .002]; peak, 140.65 ± 16.33 vs 63.83 ± 15.02 [n = 5; P < .001]; and velocity, 24.50 ± 2.48 vs 5.11 ± 1.05 [n = 5; P < .0006]) compared with the baseline (supplemental Figure 2B).
Figure 5.
TG in PRP of patients with NSTEMI treated with clopidogrel in the presence or absence of VLD rivaroxaban. (A) Representative tracings of TG in PRP of patients with NSTEMI before (baseline) and after medication with clopidogrel + ASA at d1 in the absence (DMSO) or presence of 40 ng/mL rivaroxaban (in vitro), as well as after medication with clopidogrel + ASA and 2.5 mg rivaroxaban twice daily at d2 (in vivo) with resting platelets and (C) with 10 µg/mL CRP-stimulated platelets. (B) Arithmetic means ± SEM (n = 20) of lag time, TTP, peak (thrombin), and velocity of TG in PRP of patients with NSTEMI before (baseline) and after medication with clopidogrel + ASA at d1 in the absence (DMSO) or presence of 40 ng/mL rivaroxaban (in vitro) as well as after medication with clopidogrel + ASA and 2.5 mg rivaroxaban twice daily at d2 (in vivo) with resting platelets and (D) with 10 µg/mL CRP-stimulated platelets. *P < .05, **P < .01, and ***P < .001 indicate statistically significant differences from solvent control at the indicated time point. #P < .05 and ###P < .001 indicate significant differences from baseline.


In vitro thrombus formation
The coagulation- and platelet-dependent thrombus formation under high shear rates in vitro was investigated by means of the T-TAS by perfusing recalcified citrated blood from patients with ACS on DAPT over a collagen- and tissue factor-coated surface (AR-chip). Additional spiking of rivaroxaban on top of ticagrelor treatment resulted in decreased thrombus formation as occlusion start time (T10; 443.12 ± 26.83 vs 603.75 ± 45.89; n = 12; P = .0058) was increased at d1 as well as d2, whereas the OT only showed a tendency without reaching significance, and the AUC (1739.98 ± 37.53 vs 1505.07 ± 75.78; n = 12; P = .0108) was diminished compared with the baseline, whereas there was no significant effect of rivaroxaban treatment compared with the solvent control on d1 and d2 (Figure 6).
Figure 6.

Total thrombus formation of patients with NSTEMI treated with ticagrelor in the presence or absence of VLD rivaroxaban. (A) Representative tracings and (B) arithmetic means ± SEM (n = 12) of occlusion start time (T10), AUC and OT of total thrombus formation on a collagen and tissue factor coated (AR) chip in recalcified citrated whole blood from patients with NSTEMI before (baseline) and after medication with ticagrelor + ASA for 1 and 2 days (d1 and d2) in the absence (DMSO) or presence of 40 ng/mL rivaroxaban (in vitro). #P < .05 indicates significant differences from baseline.
Additional in vitro rivaroxaban treatment of patients with ACS with clopidogrel + ASA resulted in significantly decreased thrombus formation compared with DMSO as T10 (417.43 ± 33.18 vs 601.14 ± 53.98; n = 14; P = .0021) and OT (545.33 ± 38.89 vs 752.36 ± 61.86; n = 14; P = .0009) were increased at d1 and the AUC (1764.56 ± 47.70 vs 1505.45 ± 75.72; n = 14; P = .008) was decreased. Compared with the baseline, rivaroxaban treatment at d1 also unraveled a significant effect, as T10 (457.78 ± 29.95 vs 601.14 ± 53.98; n = 14; P = .0283) and OT (584.21 ± 35.10 vs 752.36 ± 61.86; n = 14; P = .0259) were significantly increased at d1 and the AUC (1713.23 ± 42.84 vs 1505.45 ± 75.72; n = 14; P = .0245) was significantly decreased. Moreover, medication with clopidogrel + ASA and rivaroxaban at d2 also resulted in significantly reduced thrombus formation compared with the baseline or DMSO control (Figure 7). Of note, after in vitro rivaroxaban treatment, a significant reduction of thrombus formation was observed, especially in clopidogrel nonresponders at d1 of clopidogrel treatment, as documented by changes in AUC (1588.22 ± 66.86 vs 1226.90 ± 79.93; n = 5; P = .02) and significant reduction of OT (691.23 ± 53.49 vs 976.20 ± 67.04; n = 5; P = .03) compared with the baseline (supplemental Figure 1C).
Figure 7.

Total thrombus formation of patients with NSTEMI treated with clopidogrel in the presence or absence of VLD rivaroxaban. (A) Representative tracings and (B) arithmetic means ± SEM (n = 14) of occlusion start time (T10), AUC, and OT of total thrombus formation on a collagen- and tissue factor-coated (AR) chip in recalcified citrated whole blood from patients with NSTEMI before (baseline) and after medication with clopidogrel + ASA at d1 in the absence (DMSO) or presence of 40 ng/mL rivaroxaban (in vitro), as well as after medication with clopidogrel + ASA and 2.5 mg rivaroxaban twice daily at d2 (in vivo). *P < .05, **P < .01, and ***P < .001 indicate statistically significant differences from solvent control at the indicated time point. #P < .05 and ##P < .01 indicate significant differences from baseline.
Interestingly, the thrombus formation on a collagen-coated surface (PL-chip) reflecting the coagulation-independent and platelet-dependent thrombus formation alone was not affected by subsequent rivaroxaban spiking in vitro in patients with ACS receiving DAPT with either ticagrelor or clopidogrel (supplemental Figure 2).
Platelet aggregation
To investigate effects on PA after rivaroxaban treatment, light-transmission aggregometry measurements were performed. As illustrated in supplemental Figure 2, there were no additional effects of rivaroxaban on top of DAPT observable in patients with ACS. Neither ticagrelor treatment with subsequent rivaroxaban spiking at d1 and d2 nor clopidogrel medication at d1 and clopidogrel + rivaroxaban medication at d2 resulted in significantly changed PA (supplemental Figure 3).
Platelet surface abundance of thrombo-inflammatory markers
Because platelets are the major regulators of vascular inflammation and atherothrombosis in patients with coronary artery disease, we studied the effect of VLD rivaroxaban on platelet P-selectin surface expression, as well as the expression of platelet-derived HMGB-1. According to fluorescence-activated cell sorter analysis, platelet surface abundance of P-selectin was not significantly affected, whereas platelet HMGB-1 expression was significantly reduced after spiking with VLD rivaroxaban in clopidogrel- as well as ticagrelor-treated patients (supplemental Figure 4). P2Y12 inhibition with ticagrelor (mean fluorescence intensity [MFI], 9.24 ± 0.24 vs 10.42 ± 0.54; n = 14; P = .16) or clopidogrel (MFI 10.02 ± 0.35 vs 10.46 ± 0.44; n = 10; P = .83), in addition to ASA, showed no significant reduction of platelet HMGB-1 expression. In vitro spiking of blood samples from patients treated with ASA + ticagrelor with 40 ng/mL rivaroxaban resulted in a significant reduction of platelet HMGB-1 expression both at d1 (MFI, 8.10 ± 0.40 vs 10.42 ± 0.54; n = 14; P < .0003) and at d2 (MFI, 8.22 ± 0.30 vs 10.42 ± 0.54; n = 14; P = .0006) compared with baseline. In patients treated with DAPT (ASA + clopidogrel), the addition of VLD rivaroxaban significantly decreased platelet HMGB-1 abundance in vitro (MFI, 8.92 ± 0.25 vs 10.46 ± 0.44; n = 10; P = .0325) and in vivo (MFI, 7.98 ± 0.38 vs 10.46 ± 0.44; n = 10; P = .0004) compared with baseline. In vivo VLD rivaroxaban on d2 also resulted in significantly decreased platelet HMGB-1 abundance (MFI, 7.98 ± 0.38 vs 10.02 ± 0.35; n = 10; P = .0035) compared with control (DMSO) spiking on d1.
Discussion
The major findings of the present study are that rivaroxaban, at an equivalent dose of 2.5 mg, effectively reduced various parameters of TG on the background of both clopidogrel and ticagrelor + ASA (the results were consistent when rivaroxaban was added to clopidogrel in vivo [ATLAS-like regimen] or to ticagrelor in vitro); that another major finding of this study is that on top of DAPT (neither with clopidogrel nor with ticagrelor), treatment with VLD rivaroxaban (in vitro and in vivo) did not result in any significantly changed PA ex vivo, induced by ADP, thrombin receptor-activating peptide (TRAP), or CRP; that we found a significant effect of rivaroxaban on thrombus formation measured by the T-TAS system using the AR-chip; and that VLD rivaroxaban treatment significantly reduced platelet HMGB-1 surface expression, and thus might result in a reduction of platelet-triggered arterial thrombo-inflammation.
The extent of TG correlates with atheroprogression and is associated with short- and long-term CV events in a broad spectrum of vascular disease.18 In particular, after an ACS, markers of TG have been found persistently increased in patients receiving antiplatelet therapy19,20 and were associated with prognostic outcome, including the occurrence of stent thrombosis.21,22 Previously, dose-dependent synergistic effects of in vitro rivaroxaban and P2Y12 inhibitors, including ticagrelor on TG, were documented in blood of healthy volunteers.16 To date, there are no data on the effects of rivaroxaban on TG on the background of oral DAPT (clopidogrel/ticagrelor + ASA) in patients with ACS. Increased antithrombotic efficacy of VLD rivaroxaban on top of clopidogrel and ASA might explain the observed clinical effects of this regimen compared with DAPT with clopidogrel and ASA in the ATLAS-TIMI-51 trial, including a pronounced reduction of stent thrombosis.5 Remarkably, in that trial, rivaroxaban was particularly effective in preventing spontaneous type 1 MIs, thought to be mainly triggered by PA and subsequent platelet-driven activation of the coagulation cascade.23 In the present study, we could further confirm the additional effects of VLD rivaroxaban on inhibition of TG in patients treated with DAPT including ASA + ticagrelor. Conversely, initiation of DAPT led to a marked prolongation of T10 and decrease of AUC with the PL-chip, but not with the AR-chip in accordance with previous reports.24
In a previous study by Perzborn et al, PA induced by tissue factor was attenuated by rivaroxaban in spiking concentrations of 15 and 30 ng/mL, in addition to in vitro ticagrelor at concentrations of 1 and 3 µg/mL.16 In contrast to this study, different agonists to induce PA (ADP, TRAP, CRP) were used in the present study. Furthermore, ticagrelor was administered orally, and rivaroxaban-spiking effects on PA were measured on the background of clopidogrel/ticagrelor + ASA. Although activated platelets substantially trigger TG25 and thrombin represents a potent platelet activator mainly via protease-activated receptor 1 and protease-activated receptor 4 on human platelets, we did not observe a parallel effect of suppression of TG and inhibition of PA. Recently, the platelet collagen receptor GPVI has been identified as receptor for polymerized fibrin by amplifying TG.26 Nevertheless, a specific activation of platelets via GPVI by stimulation with CRP was not affected by addition of rivaroxaban. Probably, inhibition of PA induced by major platelet agonists (TRAP, ADP) was already effectively suppressed by DAPT without rivaroxaban, as shown by low levels of PA by clopidogrel/ticagrelor + ASA, respectively, in most of the patients. A recent study failed to show a clear effect of direct thrombin inhibitors on ADP-induced platelet function in patients treated with DAPT.27 Of note, there are no compelling results documenting a correlation of markers of TG and PA in patients treated with DAPT.28 In a subset of patients identified as poor responders to clopidogrel, a significant reduction of ADP-induced PA was observed pinpointing to favorable antiplatelet effects by VLD rivaroxaban in patients with impaired response to P2Y12 receptor antagonists, although numbers seem low and larger studies are needed.
As expected, there was no effect on pure platelet thrombus formation with the PL-chip, neither following rivaroxaban spiking nor under oral treatment compared with DAPT. This observation is in line with previous investigations showing no effect of spiking with FXa inhibitors on platelet (collagen only)-dependent thrombus formation with the PL-chip, but on tissue-factor dependent thrombus formation using the T-TAS AR-chip.29,30
Because platelets are the major regulators of vascular inflammation in atherothrombosis, we studied the effect of VLDR on the platelet P-selectin and HMGB-1 surface expression. P-selectin CD62P is the ligand for the leukocyte receptor PSGL-1 initiating tethering and adhesion of leukocyte/platelet complexes to the endothelium.31 HMGB-1 is a damage-associated molecular pattern protein that is upregulated by activated platelets in multiple inflammatory diseases and is a critical regulator of thrombosis.32,33 HMGB-1 is highly expressed in platelet-rich human coronary artery thrombi34 and a key mediator of reciprocal communication between platelet and neutrophils, as well as monocytes facilitating formation of prothrombotic neutrophil extracellular traps and production of monocyte-derived tissue factor.33 We found that P-selectin exposure was not affected by VLD rivaroxaban treatment, findings that are in line with previous studies35,36 that both found no significant effect of rivaroxaban on platelet P-selectin expression and release. However, in the present study, we could show that platelet surface abundance of HMGB-1 was significantly reduced after treatment with VLD rivaroxaban pointing to a potential effect on thrombo-inflammatory processes triggered by platelets in patients with coronary artery disease. Further studies will be necessary to address this hypothesis.
The current study has several limitations. First, given the nonrandomized design, we cannot rule out a significant bias by patient selection. Decisions for treatment of patients with ASA + clopidogrel or ticagrelor first, and then ASA, clopidogrel, and rivaroxaban 2.5 mg twice daily, were made on the basis of clinical characteristics and contraindications or cautions for treatment with more potent P2Y12 antagonist (eg, patients with dyspnea, rhythm disturbances, drug intolerance). Despite older age in patients receiving DAPT (clopidogrel + ASA) in combination with rivaroxaban, we did not observe significant differences between patients in both groups concerning demographic factors, clinical risk factors (eg, diabetes, renal failure), and comedication. Second, in vitro spiking with rivaroxaban at a concentration of 40 ng/mL cannot completely simulate the in vivo situation under treatment with 2.5 mg twice-daily rivaroxaban, in particular where interindividual variation in activity levels likely occurs. The rationale for choosing 40 ng/mL for spiking experiments was based on previous analyses of the pharmacokinetic and pharmacodynamic profile of VLD rivaroxaban.10 Of note, we did observe very consistent results in particular with the TG assays between in vivo treatment and in vitro spiking of rivaroxaban in patients treated with ASA + clopidogrel. Third, we mainly focused on a rather platelet-dependent activation pathway, using CRP as stimulus, and did not investigate the effect of tissue factor-induced TG. Fourth, we did not investigate the effects of rivaroxaban in combination with ASA alone or a P2Y12 receptor antagonist alone. It is tempting to speculate that these combinations might represent a sweet spot with enhanced efficacy while maintaining a tolerable safety profile in high-risk patients with CV, as investigated in the previous GEMINI-ACS-1 trial and in the recently published COMPASS trial.7,8 Finally, the study was not powered for clinical events, in particular, bleeding events, and one cannot extrapolate these data to the clinical setting. Interestingly, the extent of inhibition of thrombus formation parameters with VLD rivaroxaban in addition to DAPT in this study was in the range of predicting low bleeding risk using T-TAS, as reported recently.37
In conclusion, the present study demonstrates consistent effects by addition of rivaroxaban at an ex vivo dose equivalent to the VLD (2.5 mg) oral regimen to standard DAPT with both clopidogrel and ticagrelor on markers of TG and shear-dependent thrombus formation in real-world non–ST-elevation patients with ACS. There were no additional effects of VLD rivaroxaban in combination with DAPT on platelet aggregation induced by major platelet agonists (via protease-activated receptor 1, P2Y12, and GPVI) and on collagen-dependent thrombus formation under high arterial shear rates, using the PL-chip. These data indicate that VLD rivaroxaban provides additional, thrombin-generation-related antithrombotic effects compared with DAPT, even when more potent P2Y12 inhibition (ticagrelor) is used. Therefore, adding VLD rivaroxaban might be considered worth testing in high-risk patients with CV after careful consideration of the bleeding risk. Current trials will show whether combination therapy using VLD rivaroxaban in combination with 1 single antiplatelet regimen (ie, a P2Y12 receptor antagonist, as in the GEMINI-ACS trial, or aspirin, as in the COMPASS trial) will provide enhanced antithrombotic efficacy with an acceptable bleeding risk as compared with standard antiplatelet therapy.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Fujimori Kogyo Co Ltd, Shinjuku, Japan, and Erich Gorges (Probe & go Labordiagnostica GmbH, Lemgo, Germany) for providing the Automated Microchip Flow Chamber System (T-TAS) and, to a great extent, Daniela Eißler for excellent technical assistance and support.
The authors acknowledge support by Deutsche Forschungsgemeinschaft and Open Access Publishing Fund of the University of Tübingen. This project was supported in part by the Deutsche Forschungsgemeinschaft (Klinische Forschungsgruppe-KFO-274: “Platelets-Molecular Mechanisms and Translational Implications”). The project was additionally funded through a restricted grant by Bayer Vital GmbH.
Authorship
Contribution: O.B. contributed to the study concept, drafting the study protocol, collection of data, data analysis, and drafting the manuscript; P.M. performed laboratory measurements, contributed to collection of data, performed data analysis, and drafted the manuscript; N.A. performed laboratory measurements, contributed to collection of data, performed data analysis, and drafted the manuscript; S.G. performed laboratory measurements; R.T., D.R., and M.D. contributed to the collection of data; P.S. contributed to the collection of data and assisted in drafting the manuscript; S.H. contributed to the study concept; J.W.M.H., L.K.J., R.F.S., D.J.A., B.R., H.S., and H.T.C. assisted in drafting the manuscript; M.G. contributed to the study concept and assisted in drafting the manuscript; and T.G. contributed to the study concept, drafting the study protocol, collection of data, data analysis, and drafting the manuscript.
Conflict-of-interest disclosure: O.B. received research grants and personal fees from Bayer Healthcare. S.H. is an employee of Bayer AG. R.F.S. reports receiving institutional research grants/support from AstraZeneca and PlaqueTec; consultancy fees from Actelion, AstraZeneca, Avacta, Bayer, Bristol-Myers Squibb/Pfizer, Novartis, PlaqueTec, and The Medicines Company; and honoraria from AstraZeneca. D.J.A. reports receiving payments as an individual for a consulting fee or honorarium from Amgen, Aralez, AstraZeneca, Bayer, Biosensors, Bristol-Myers Squibb, Chiesi, Daiich-Sankyo, Eli Lilly, Janssen, Merck, PLx Pharma, Pfizer, Sanofi, and The Medicines Company; for participation in review activities from CeloNova and St. Jude Medical; and receiving institutional payments for grants from Amgen, AstraZeneca, Bayer, Biosensors, CeloNova, CSL Behring, Daiichi-Sankyo, Eisai, Eli Lilly, Gilead, Janssen, Matsutani Chemical Industry Co., Merck, Novartis, Osprey Medical, and Renal Guard Solutions. B.R. received institutional research grants from the Italian Medicines Agency, consultancy fees from Bayer AG, and speaker fees from Amgen, Celgene, Daiichi Sankyo Italia, Novartis Farma, and Sanofi. H.T.C. is a consultant to Stago; received research support from Bayer, Boehringer, and Pfizer/BMS; serves on the advisory boards for Bayer and Daiichi Sankyo; and is the chair of the board for the Dutch Federation of Anticoagulation clinics (unpaid). T.G. received personal fees from AstraZeneca, Boehringer Ingelheim, Pfizer, and MSD; grants and personal fees from Bayer Healthcare, Bristol-Myers Squibb, Daiichi Sankyo, Eli Lilly, and The Medicines Company; and grants from Siemens Healthcare and Spartan Bioscience, outside of the submitted work. The remaining authors declare no competing financial interests.
Correspondence: Tobias Geisler, Department of Cardiology and Cardiovascular Medicine, University of Tübingen, Otfried Müller-Str 10, 72076 Tübingen, Germany; e-mail: tobias.geisler@med.uni-tuebingen.de.
References
- 1.Murphy AJ, Tall AR. Disordered haematopoiesis and athero-thrombosis. Eur Heart J. 2016;37(14):1113-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chatterjee M, Rath D, Schlotterbeck J, et al. Regulation of oxidized platelet lipidome: implications for coronary artery disease. Eur Heart J. 2017;38(25):1993-2005. [DOI] [PubMed] [Google Scholar]
- 3.Oldgren J, Wallentin L, Alexander JH, et al. New oral anticoagulants in addition to single or dual antiplatelet therapy after an acute coronary syndrome: a systematic review and meta-analysis. Eur Heart J. 2013;34(22):1670-1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexander JH, Lopes RD, James S, et al. ; APPRAISE-2 Investigators. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med. 2011;365(8):699-708. [DOI] [PubMed] [Google Scholar]
- 5.Mega JL, Braunwald E, Wiviott SD, et al. ; ATLAS ACS 2–TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366(1):9-19. [DOI] [PubMed] [Google Scholar]
- 6.Gibson CM, Mehran R, Bode C, et al. Prevention of bleeding in patients with atrial fibrillation undergoing PCI. N Engl J Med. 2016;375(25):2423-2434. [DOI] [PubMed] [Google Scholar]
- 7.Ohman EM, Roe MT, Steg PG, et al. Clinically significant bleeding with low-dose rivaroxaban versus aspirin, in addition to P2Y12 inhibition, in acute coronary syndromes (GEMINI-ACS-1): a double-blind, multicentre, randomised trial. Lancet. 2017;389(10081):1799-1808. [DOI] [PubMed] [Google Scholar]
- 8.Eikelboom JW, Connolly SJ, Bosch J, et al. ; COMPASS Investigators. Rivaroxaban with or without aspirin in stable cardiovascular disease. N Engl J Med. 2017;377(14):1319-1330. [DOI] [PubMed] [Google Scholar]
- 9.Roffi M, Patrono C, Collet JP, et al. ; Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). Eur Heart J. 2016;37(3):267-315. [DOI] [PubMed] [Google Scholar]
- 10.Mueck W, Stampfuss J, Kubitza D, Becka M. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban. Clin Pharmacokinet. 2014;53(1):1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tantry US, Bonello L, Aradi D, et al. ; Working Group on On-Treatment Platelet Reactivity. Consensus and update on the definition of on-treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J Am Coll Cardiol. 2013;62(24):2261-2273. [DOI] [PubMed] [Google Scholar]
- 12.Cuisset T, Cayla G, Frere C, et al. Predictive value of post-treatment platelet reactivity for occurrence of post-discharge bleeding after non-ST elevation acute coronary syndrome. Shifting from antiplatelet resistance to bleeding risk assessment? EuroIntervention. 2009;5(3):325-329. [DOI] [PubMed] [Google Scholar]
- 13.Vogel S, Rath D, Borst O, et al. Platelet-derived high-mobility group box 1 promotes recruitment and suppresses apoptosis of monocytes. Biochem Biophys Res Commun. 2016;478(1):143-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hemker HC, Giesen P, AlDieri R, et al. The calibrated automated thrombogram (CAT): a universal routine test for hyper- and hypocoagulability. Pathophysiol Haemost Thromb. 2002;32(5-6):249-253. [DOI] [PubMed] [Google Scholar]
- 15.Hosokawa K, Ohnishi T, Sameshima H, et al. Comparative evaluation of direct thrombin and factor Xa inhibitors with antiplatelet agents under flow and static conditions: an in vitro flow chamber model. PLoS One. 2014;9(1):e86491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perzborn E, Heitmeier S, Laux V. Effects of rivaroxaban on platelet activation and platelet-coagulation pathway interaction: in vitro and in vivo studies. J Cardiovasc Pharmacol Ther. 2015;20(6):554-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vanschoonbeek K, Feijge MA, Van Kampen RJ, et al. Initiating and potentiating role of platelets in tissue factor-induced thrombin generation in the presence of plasma: subject-dependent variation in thrombogram characteristics. J Thromb Haemost. 2004;2(3):476-484. [DOI] [PubMed] [Google Scholar]
- 18.Ten Cate H, Hemker HC. Thrombin Generation and Atherothrombosis: What Does the Evidence Indicate? J Am Heart Assoc. 2016;5(8):e003553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szczeklik A, Dropinski J, Radwan J, Krzanowski M. Persistent generation of thrombin after acute myocardial infarction. Arterioscler Thromb. 1992;12(5):548-553. [DOI] [PubMed] [Google Scholar]
- 20.Ardissino D, Merlini PA, Bauer KA, et al. Coagulation activation and long-term outcome in acute coronary syndromes. Blood. 2003;102(8):2731-2735. [DOI] [PubMed] [Google Scholar]
- 21.Granger CB, Becker R, Tracy RP, et al. Thrombin generation, inhibition and clinical outcomes in patients with acute myocardial infarction treated with thrombolytic therapy and heparin: results from the GUSTO-I Trial. GUSTO-I Hemostasis Substudy Group. Global Utilization of Streptokinase and TPA for Occluded Coronary Arteries. J Am Coll Cardiol. 1998;31(3):497-505. [DOI] [PubMed] [Google Scholar]
- 22.Loeffen R, Godschalk TC, van Oerle R, et al. The hypercoagulable profile of patients with stent thrombosis. Heart. 2015;101(14):1126-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cavender MA, Gibson CM, Braunwald E, et al. The effect of rivaroxaban on myocardial infarction in the ATLAS ACS 2 - TIMI 51 trial. Eur Heart J Acute Cardiovasc Care. 2015;4(5):468-474. [DOI] [PubMed] [Google Scholar]
- 24.Arima Y, Kaikita K, Ishii M, et al. Assessment of platelet-derived thrombogenicity with the total thrombus-formation analysis system in coronary artery disease patients receiving antiplatelet therapy. J Thromb Haemost. 2016;14(4):850-859. [DOI] [PubMed] [Google Scholar]
- 25.Agbani EO, van den Bosch MT, Brown E, et al. Coordinated membrane ballooning and procoagulant spreading in human platelets. Circulation. 2015;132(15):1414-1424. [DOI] [PubMed] [Google Scholar]
- 26.Mammadova-Bach E, Ollivier V, Loyau S, et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood. 2015;126(5):683-691. [DOI] [PubMed] [Google Scholar]
- 27.Olivier CB, Weik P, Meyer M, et al. Dabigatran and rivaroxaban do not affect AA- and ADP-induced platelet aggregation in patients receiving concomitant platelet inhibitors. J Thromb Thrombolysis. 2016;42(2):161-166. [DOI] [PubMed] [Google Scholar]
- 28.Gremmel T, Panzer S, Steiner S, et al. Response to antiplatelet therapy is independent of endogenous thrombin generation potential. Thromb Res. 2013;132(1):e24-e30. [DOI] [PubMed] [Google Scholar]
- 29.Sugihara H, Idemoto Y, Kuwano T, et al. Evaluation of the antithrombotic effects of rivaroxaban and apixaban using the total thrombus-formation analysis system®: in vitro and ex vivo studies. J Clin Med Res. 2016;8(12):899-907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishii M, Kaikita K, Ito M, et al. Direct oral anticoagulants form thrombus different from warfarin in a microchip flow chamber system. Sci Rep. 2017;7(1):7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McEver RP, Cummings RD. Perspectives series: cell adhesion in vascular biology. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100(3):485-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogel S, Bodenstein R, Chen Q, et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest. 2015;125(12):4638-4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stark K, Philippi V, Stockhausen S, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128(20):2435-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahrens I, Chen YC, Topcic D, et al. HMGB1 binds to activated platelets via the receptor for advanced glycation end products and is present in platelet rich human coronary artery thrombi. Thromb Haemost. 2015;114(5):994-1003. [DOI] [PubMed] [Google Scholar]
- 35.Steppich B, Dobler F, Brendel LC, et al. Effect of the FXa inhibitors Rivaroxaban and Apixaban on platelet activation in patients with atrial fibrillation. J Thromb Thrombolysis. 2017;43(4):490-497. [DOI] [PubMed] [Google Scholar]
- 36.Zemer-Wassercug N, Haim M, Leshem-Lev D, et al. The effect of dabigatran and rivaroxaban on platelet reactivity and inflammatory markers. J Thromb Thrombolysis. 2015;40(3):340-346. [DOI] [PubMed] [Google Scholar]
- 37.Oimatsu Y, Kaikita K, Ishii M, et al. Total thrombus-formation analysis system predicts periprocedural bleeding events in patients with coronary artery disease undergoing percutaneous coronary intervention. J Am Heart Assoc. 2017;6(4):e005263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.