

Abstract
Background
Apremilast, an oral phosphodiesterase‐4 inhibitor, has demonstrated efficacy in patients with moderate to severe psoriasis.
Objective
To evaluate long‐term efficacy and safety of apremilast in biologic‐naive patients with moderate to severe plaque psoriasis and safety of switching from etanercept to apremilast in the phase 3b LIBERATE trial.
Methods
Two hundred fifty patients were randomized to placebo, apremilast 30 mg BID or etanercept 50 mg QW through Week 16; thereafter, all patients continued or switched to apremilast through Week 104 (extension phase). Skin, scalp and nail involvement at Weeks 16, 52 and 104 were assessed using the Psoriasis Area and Severity Index (PASI; 0–72), Scalp Physician Global Assessment (ScPGA; 0–5) and Nail Psoriasis Severity Index (NAPSI; 0–8); patient‐reported outcomes (PROs) were assessed using the Dermatology Life Quality Index (DLQI; 0–32) and pruritus visual analog scale (VAS; 0–100 mm).
Results
The apremilast‐extension phase (Weeks 16–104) included 226 patients in the placebo/apremilast (n = 73), apremilast/apremilast (n = 74) and etanercept/apremilast (n = 79) groups, and at Week 104, 50.7%, 45.9% and 51.9% of these patients, respectively, maintained ≥75% reduction from baseline in PASI score (based on last‐observation‐carried‐forward analysis). Across treatment groups, ScPGA 0 (clear) or 1 (minimal) was achieved by 50.0%–59.2% of patients; NAPSI mean change from baseline was −48.1% to −51.1%; DLQI score ≤5 was achieved by 66.0%–72.5% of patients; and pruritus VAS mean change from baseline was −24.4 to −32.3. AEs in ≥5% of patients (diarrhoea, nausea, nasopharyngitis, upper respiratory tract infection and headache) did not increase with prolonged apremilast exposure.
Conclusions
Apremilast demonstrated significant and sustained improvements in skin, scalp, nails and PROs (pruritus and quality of life) over 104 weeks in patients with moderate to severe plaque psoriasis. Safety was consistent with the known safety profile of apremilast.
Introduction
LIBERATE (NCT01690299), a global phase 3b study in biologic‐naive patients with moderate to severe plaque psoriasis, demonstrated that significantly more patients receiving apremilast for 16 weeks achieved PASI‐75 (≥75% reduction from baseline in Psoriasis Area and Severity Index [PASI]) vs. placebo.1 A post hoc analysis found no significant difference in response rates among patients given apremilast vs. etanercept, an anti‐tumour necrosis factor‐α biologic agent.1
This report describes efficacy and safety outcomes from the LIBERATE apremilast‐extension phase (Weeks 16–104) in patients who continued apremilast treatment through 104 weeks, including those who switched from etanercept or placebo.
Materials and methods
Patients and study design
Patient selection, study design and study methods have been described in detail.1 Briefly, patients in the LIBERATE trial were adults in the United States, Canada, Europe (Belgium, Czech Republic, Estonia, Germany, Great Britain, Hungary, Latvia, the Netherlands) or Australia with chronic, moderate to severe plaque psoriasis with inadequate response, inability to tolerate or contraindication to treatment with ≥1 conventional systemic agent, and who were biologic‐naive. Eligible patients were randomized (1 : 1 : 1) to double‐blind treatment with apremilast 30 mg twice daily, etanercept subcutaneous injection 50 mg once weekly, or placebo for 16 weeks. At Week 16, placebo and etanercept patients were switched to apremilast (placebo, without titration; etanercept, with 1‐week titration); apremilast patients continued apremilast treatment through Week 104.
Efficacy and safety assessments
The primary efficacy end point, the proportion of patients who achieved PASI‐75 at Week 16, was previously reported.1 Efficacy of continued apremilast treatment was evaluated at scheduled visits over 104 weeks based on (i) skin symptoms, as assessed using PASI improvement from baseline, achievement of PASI‐75 response and achievement of response on the 5‐point static Physician Global Assessment (sPGA; response = score of 0 [clear] or 1 [almost clear]); (ii) change from baseline in pruritus, as assessed using a 100‐mm visual analog scale (VAS; 0 = no pruritus, 100 = worst possible pruritus); (iii) scalp symptoms, as assessed based on achievement of response on the 6‐point Scalp Physician Global Assessment (ScPGA; response = score of 0 [clear] or 1 [minimal]); (iv) nail symptoms, as assessed based on improvement from baseline in Nail Psoriasis Severity Index (NAPSI) score in the target nail and achievement of NAPSI‐50 (≥50% reduction from baseline in NAPSI score) among patients with nail symptoms (i.e. NAPSI score ≥1); and (v) impact of psoriasis on quality of life (QOL), as assessed by improvement from baseline in Dermatology Life Quality Index (DLQI) total score, achievement of DLQI total score ≤5 (minimal impairment) and achievement of DLQI total score of 0 or 1. Safety was assessed based on adverse events (AEs), vital signs, clinical laboratory assessments and physical examinations.
Statistical analysis
Efficacy analyses were performed in the modified intent‐to‐treat population (all randomized patients who received ≥1 dose of study medication and had baseline PASI and ≥1 post‐treatment PASI assessments). Statistical analyses for between‐group comparisons of efficacy assessments at Week 16 included Cochran–Mantel–Haenszel tests for categorical variables and an analysis of covariance model for continuous variables. Efficacy assessments during the apremilast‐extension phase were summarized using descriptive statistics at visits after treatment initiation. Missing values were imputed using last‐observation‐carried‐forward (LOCF) methodology. Responder analyses of DLQI total score ≤5 and DLQI score of 0 or 1 were performed using data from completers without imputation. Safety was summarized using descriptive statistics.1
Results
Patients
There were 233 patients who completed the placebo‐controlled phase among 250 randomized patients. There were 226 patients who entered the apremilast‐extension phase (placebo/apremilast, n = 73; apremilast/apremilast, n = 74; etanercept/apremilast, n = 79); 138 (60.8%) completed the Week 104 visit. The most common reasons for withdrawal from the apremilast‐extension phase were lack of efficacy (n = 27, 11.9%), lost to follow‐up (n = 25, 11.0%) and withdrawal by patient (n = 24, 10.6%). Baseline characteristics were balanced between groups.1 Mean psoriasis duration was 18.2 years, and mean PASI score was 19.6.1
Efficacy
At Week 16, significantly greater proportions of patients receiving apremilast vs. placebo achieved PASI‐75 (39.8% vs. 11.9%, P < 0.0001). In patients who continued or were switched from placebo or etanercept to apremilast during the apremilast‐extension phase, PASI‐75 response rate was maintained across treatment groups at Week 52 (range, 52.7%–57.0%)1 and Week 104 (45.9%–51.9%; Table 1; Fig. 1) and was similar between treatment groups. At Week 16, mean percentage improvement in PASI score was significantly greater with apremilast compared with placebo (P < 0.0001). Mean percentage improvement in PASI score was maintained across treatment groups at Week 52 (range, 66.2%–75.1%)1 and continued through Week 104 (63.3%–70.1%; Table 1). Likewise, achievement of sPGA response was significantly greater with apremilast vs. placebo at Week 16 (21.7% vs. 3.6%, P = 0.0005)1; achievement was 24.3%–35.6% across treatment groups at Week 521 and 18.9%–27.4% at Week 104 (Table 1).
Table 1.
Clinical response across efficacy end points at Week 104 (LOCF)
| Placebo/ Apremilast Patient‐Years = 95.6 | Apremilast/ Apremilast Patient‐Years = 89.4 | Etanercept/ Apremilast Patient‐Years = 102.3 | |
|---|---|---|---|
| PASI‐75, n/m (%) | 37/73 (50.7) | 34/74 (45.9) | 41/79 (51.9) |
| PASI percentage change from baseline, mean (SD) | −63.3 (33.4) | −63.7 (28.1) | −70.1 (25.5) |
| sPGA 0 or 1, n/m (%) | 20/73 (27.4) | 14/74 (18.9) | 21/79 (26.6) |
| ScPGA 0 or 1*, n/m (%) | 25/50 (50.0) | 29/49 (59.2) | 30/53 (56.6) |
| NAPSI‐50, n/m (%) | 18/37 (48.6) | 29/48 (60.4) | 30/46 (65.2) |
| NAPSI percentage change from baseline*, mean (SD) |
−48.1 (49.6) (n = 33) |
−48.2 (48.4) (n = 48) |
−51.1 (72.2) (n = 45) |
| Pruritus VAS change from baseline, mean (SD), mm |
−32.3 (33.4) (n = 70) |
−26.6 (29.1) (n = 71) |
−24.4 (31.2) (n = 73) |
| DLQI ≤5, n † (%) | 34/51 (66.7) | 29/40 (72.5) | 33/50 (66.0) |
| DLQI change from baseline, mean (SD) |
−5.6 (6.3) (n = 69) |
−7.5 (7.0) (n = 73) |
−5.2 (7.3) (n = 73) |
Data are from patients who entered the apremilast‐extension phase and were treated in the phase; missing values were imputed using LOCF. *For ScPGA 0 or 1, data are from patients with ScPGA ≥3 (moderate or greater) at baseline and for NAPSI mean percentage change, among patients with NAPSI ≥1 at baseline. †For DLQI ≤5, data are from patients with data at Week 104. n/m, number of responders/number of patients who entered and were treated in the apremilast‐extension phase. DLQI, Dermatology Life Quality Index; NAPSI, Nail Psoriasis Severity Index; NAPSI‐50, ≥50% reduction from baseline in NAPSI score; PASI, Psoriasis Area and Severity Index; PASI‐75, ≥75% reduction from baseline in PASI score; sPGA, static Physician Global Assessment; ScPGA, Scalp Physician Global Assessment.
Figure 1.
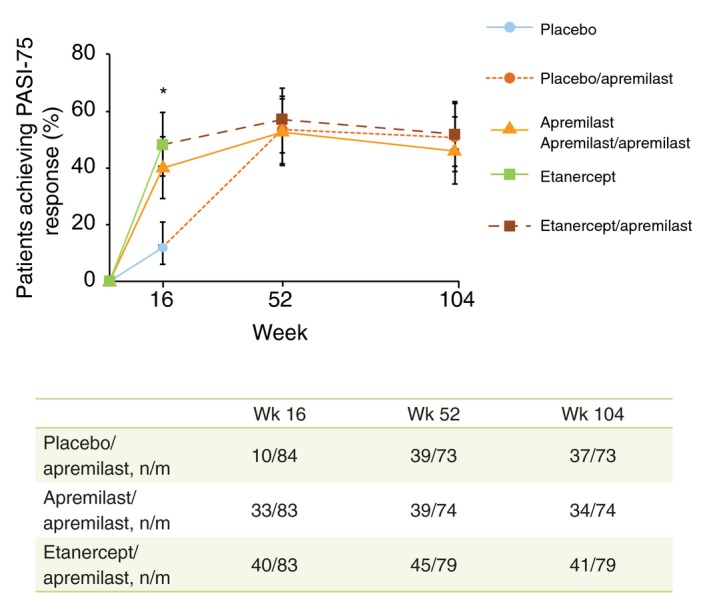
PASI‐75 response over 104 weeks with apremilast treatment (LOCF). *P < 0.0001, apremilast and etanercept vs. placebo. The vertical lines indicate two‐sided 95% confidence intervals. Missing values were imputed using LOCF methodology. n/m, number of patients with PASI‐75 response/number of patients included in the LOCF analysis at the time point (mITT population for Week 16 [patients who entered and were treated in the apremilast‐extension phase for Weeks 52 and 104]). Numbers of patients in each group with an observation at each time point were: Week 16: placebo n = 73, apremilast n = 74, etanercept n = 81; Weeks 52 and 104 (respectively): placebo/apremilast n = 61 and n = 49, apremilast/apremilast n = 57 and n = 42, etanercept/apremilast n = 67 and n = 52. LOCF, last observation carried forward; mITT, modified intent to treat; PASI, Psoriasis Area and Severity Index; PASI‐75, ≥75% reduction from baseline in PASI score.
Other signs and symptoms of psoriasis that were significantly improved at Week 16 with apremilast vs. placebo included pruritus, scalp involvement and nail involvement (Fig. 2a and 2b). Improvements were maintained at Week 521 and Week 104 in patients who continued or were switched to apremilast during the apremilast‐extension phase. Mean change from baseline in pruritus VAS score across treatment groups was −31.7 to −35.9 mm at Week 521 and −24.4 to −32.3 mm at Week 104 (Table 1). At Weeks 52 and 104, ScPGA response of 0 (clear) or 1 (minimal) was achieved by 52.0%–60.4%1 and 50.0%–59.2% of patients, respectively, across treatment groups (Table 1; Fig. 2a). Mean percentage change from baseline in NAPSI score ranged from −44.6% to −60.7% at Week 521 and −48.1% to −51.1% at Week 104 (Table 1; Fig. 2b). At Weeks 52 and 104, NAPSI‐50 was achieved by 50.0%–67.4% and 48.6%–65.2% of patients, respectively, across treatment groups (Table 1). At baseline, mean (SD) DLQI scores were 11.4 (6.3), 13.8 (6.6) and 12.5 (7.0) in the placebo, apremilast and etanercept groups, respectively. At Week 16, there was a significant improvement with apremilast vs. placebo in total DLQI score (mean change from baseline, −8.3 vs. −3.8, P < 0.0001).1 At Weeks 52 and 104, mean change from baseline in DLQI score was −6.7 to −8.21 and −5.2 to −7.5 points, respectively (Table 1). At Week 16, across treatment groups, 53.4%–65.0% of patients achieved a DLQI score ≤5 (P = NS vs. placebo) (Fig. 2c). At Weeks 52 and 104, DLQI ≤5 was achieved by 65.2%–74.6% and 66.0%–72.5% of patients, respectively, across treatment groups (Table 1; Fig. 2c). Among patients remaining in the study at Week 104, 19/51 (37.3%), 12/40 (30.0%) and 13/50 (26.0%) achieved a DLQI score of 0 or 1 at Week 104 in the placebo/apremilast, apremilast/apremilast and etanercept/apremilast groups, respectively.
Figure 2.
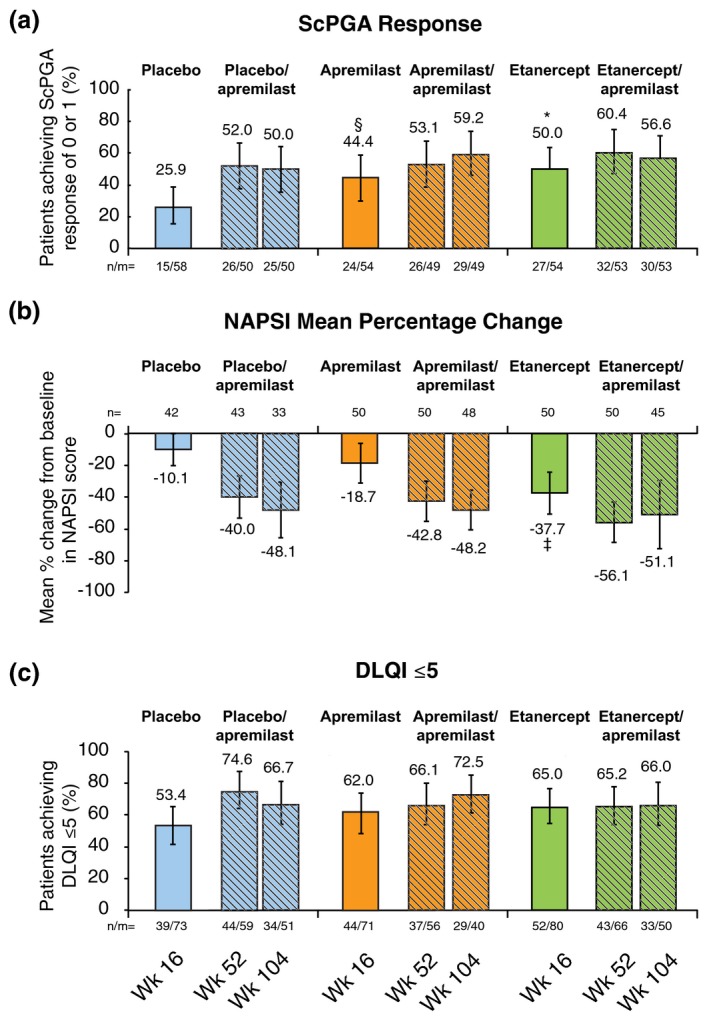
Improvement in scalp (a) and nail (b) psoriasis and quality of life (c) over 104 weeks with apremilast treatment. *P = 0.0083 vs. placebo. † P = 0.0458 vs. placebo. & P = 0.0024 vs. placebo. n/m, number of responders/number of patients with data at time of observation. Week 16 group comparisons based on Cochran–Mantel–Haenszel test stratified by baseline body mass index (<30 kg/m2 and ≥30 kg/m2). Missing values for improvements in scalp and nail psoriasis were imputed using LOCF methodology. The vertical lines indicate two‐sided 95% confidence intervals. ScPGA response (0 [clear] or 1 [minimal]) examined among patients with ScPGA ≥3 (moderate or greater) at baseline; NAPSI examined among patients with NAPSI ≥1 at baseline; DLQI ≤5 examined among all patients who had data at time of observation (i.e. completers, without imputation; both P=NS vs. placebo at Week 16). DLQI, Dermatology Life Quality Index; LOCF, last observation carried forward; mITT, modified intent to treat; NAPSI, Nail Psoriasis Severity Index; ScPGA, Scalp Physician Global Assessment.
Safety
During the apremilast‐extension phase, most AEs were mild or moderate in severity, and incidence of serious adverse events (SAEs) was similar across groups (Table 2). Discontinuation rates due to AEs were generally low across groups during the apremilast‐extension phase. No deaths occurred during the study. For AEs that occurred in ≥5% of patients during the placebo‐controlled phase (diarrhoea, nausea, upper respiratory tract infection, nasopharyngitis, headache and tension headache1), no increase in incidence was observed among patients in the apremilast/apremilast group with long‐term exposure (Table 2). No psychiatric AEs occurred in ≥5% of patients during the placebo‐controlled phase or with long‐term exposure in patients who continued or were switched to apremilast.1 During the apremilast‐extension phase, four patients in the apremilast/apremilast group reported five psychiatric AEs (n = 1 [1.4%] each AE): anxiety, depression, insomnia, depressed mood and stress. Among placebo/apremilast or etanercept/apremilast patients, one in each group (1.4%, 1.3%, respectively) experienced an AE of depression, anxiety and insomnia. Among placebo/apremilast patients, psychotic disorder and suicidal ideation were experienced by one (1.4%) patient each; among etanercept/apremilast patients, two patients (2.5%) experienced altered mood. In patients reporting AEs of rebound psoriasis in the etanercept/apremilast group, the majority of events (five of seven) involved loss of PASI response after treatment was discontinued or completed, and no patients experienced worsening of PASI score ≥125% of baseline.
Table 2.
Summary of adverse events during the apremilast‐extension phase (Weeks 16–104)
| Placebo/Apremilast* n = 73 Patient‐years = 95.6 | Apremilast/Apremilast n = 74 Patient‐years = 89.4 | Etanercept/Apremilast † n = 79 Patient‐years = 102.3 | |
|---|---|---|---|
| Patients, n (%) | |||
| ≥1 AE | 45 (61.6) | 49 (66.2) | 54 (68.4) |
| ≥1 Severe AE | 4 (5.5) | 4 (5.4) | 7 (8.9) |
| ≥1 SAE | 5 (6.8) | 3 (4.1) | 4 (5.1) |
| AE leading to withdrawal | 3 (4.1) | 4 (5.4) | 2 (2.5) |
| AE leading to death | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| AEs occurring in ≥5% of patients in any treatment group, n (%) & | |||
| Diarrhoea | 13 (17.8) | 4 (5.4) | 6 (7.6) |
| Nausea | 5 (6.8) | 3 (4.1) | 5 (6.3) |
| URTI | 5 (6.8) | 5 (6.8) | 1 (1.3) |
| Bronchitis | 1 (1.4) | 4 (5.4) | 1 (1.3) |
| Nasopharyngitis | 4 (5.5) | 2 (2.7) | 5 (6.3) |
| Headache | 5 (6.8) | 2 (2.7) | 3 (3.8) |
| Sinusitis | 0 (0.0) | 1 (1.4) | 5 (6.3) |
| Pain in extremity | 1 (1.4) | 3 (4.1) | 4 (5.1) |
| Arthralgia | 4 (5.5) | 4 (5.4) | 3 (3.8) |
| Rebound psoriasis | 1 (1.4) | 2 (2.7) | 7 (8.9) |
| Psoriasis | 2 (2.7) | 4 (5.4) | 0 (0.0) |
Data are from patients who entered the apremilast‐extension phase and were treated in the phase.
*No dose titration for apremilast. †Dose titration for apremilast. &Each patient is counted once for each applicable category. AE, adverse event; SAE, serious adverse event; URTI, upper respiratory tract infection.
All cases of diarrhoea and nausea occurring in the apremilast‐extension phase were mild or moderate in severity and generally resolved within 1 month. Of note, patients in the placebo/apremilast group had no dose titration with apremilast at Week 16, which likely explains the higher incidence of diarrhoea in the placebo/apremilast group (17.8%) compared with incidence of diarrhoea with apremilast during the placebo‐controlled phase (10.8%).1 Marked laboratory abnormalities were infrequent and transient; incidence remained low across groups with continued apremilast treatment through 104 weeks. At Week 104, mean percentage change from baseline in bodyweight was −1.1% (placebo/apremilast), 0.21% (apremilast/apremilast) and −1.9% (etanercept/apremilast).
Discussion
Apremilast treatment in biologic‐naive patients with moderate to severe psoriasis was effective and safe for up to 104 weeks, with improvements in skin, scalp, nails, pruritus and QOL. Efficacy and safety were maintained in patients who switched from etanercept to apremilast and remained in the study at Week 104. Patients also experienced minimal impact of psoriasis on QOL, with ≥66% of patients achieving DLQI ≤5 at Week 104. Improved QOL may help motivate patients to maintain treatment over time, which may lead to better disease control. The safety profile of apremilast during the apremilast‐extension phase was consistent with the ESTEEM and PALACE trials.2, 3, 4, 5, 6, 7
Conflicts of interest
K. Reich has received honoraria as a consultant and/or advisory board member and/or acted as a paid speaker and/or participated in clinical trials sponsored by AbbVie, Amgen, Biogen, Boehringer Ingelheim, Celgene Corporation, Centocor, Covagen, Eli Lilly, Forward Pharma, GlaxoSmithKline, Janssen‐Cilag, LEO Pharma, Medac, Merck Sharp & Dohme Corp., Novartis, Ocean Pharma, Pfizer, Regeneron, Takeda, UCB Pharma and XenoPort. M. Gooderham has received honoraria, grants and/or research funding as a speaker, investigator, advisory board member, data safety monitoring board member and/or consultant for AbbVie, Actelion, Amgen, Astellas Pharma US, Boehringer Ingelheim, Celgene Corporation, Dermira, Eli Lilly, Galderma, Janssen, Kyowa Hakko Kirin Pharma, LEO Pharma, MedImmune, Merck & Co., Inc., Novartis, Pfizer, Regeneron, Roche Laboratories, Sanofi‐Genzyme, Takeda Pharmaceuticals USA Inc, UCB and Valeant.
A. Bewley has had ad hoc consultancy agreements with AbbVie, Celgene Corporation, Galderma, Janssen Pharmaceuticals, LEO Pharma, Novartis and Stiefel (a GSK company).
L. Green has been a speaker, advisory board member and/or investigator for Amgen, AbbVie, Celgene Corporation, LEO Pharma, Novartis, Pfizer and Valeant.
J. Soung has received honoraria and/or grants as a speaker, advisory board member and/or investigator for AbbVie, Actelion, Allergan, Amgen, Cassiopeia, Celgene Corporation, Cutanea, Eli Lilly, Galderma, Genentech, GenZum, GlaxoSmithKline, Janssen, Kadmon, LEO Pharma, Merz, Pfizer, Regeneron and/or Valeant.
R. Petric, J. Marcsisin and J. Cirulli are employed by Celgene Corporation.
R. Chen was employed by Celgene Corporation at the time of study conduct.
V. Piguet has been a principal investigator in clinical trials sponsored by Celgene Corporation and has received honoraria and/or grants as a speaker, advisory board member and/or investigator for AbbVie, Celgene Corporation, Almirall, Galderma, GlaxoSmithKline, Janssen and Novartis.
Funding sources
The authors acknowledge financial support for this study from Celgene Corporation. The authors received editorial support in the preparation of this manuscript from Amy Shaberman, PhD, of Peloton Advantage, LLC, Parsippany, NJ, USA, funded by Celgene Corporation, Summit, NJ, USA. The authors, however, directed and are fully responsible for all content and editorial decisions for this manuscript. All authors had full access to all of the data, and data are available upon request.
References
- 1. Reich K, Gooderham M, Green L et al The efficacy and safety of apremilast, etanercept, and placebo in patients with moderate‐to‐severe plaque psoriasis: 52‐week results from a phase IIIb, randomized, placebo‐controlled trial (LIBERATE). J Eur Acad Dermatol Venereol 2017; 31: 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papp K, Reich K, Leonardi CL et al Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM 1]). J Am Acad Dermatol 2015; 73: 37–49. [DOI] [PubMed] [Google Scholar]
- 3. Paul C, Cather J, Gooderham M et al Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate to severe plaque psoriasis over 52 weeks: a phase III, randomized, controlled trial (ESTEEM 2). Br J Dermatol 2015; 173: 1387–1399. [DOI] [PubMed] [Google Scholar]
- 4. Kavanaugh A, Mease PJ, Gomez‐Reino JJ et al Longterm (52‐week) results of a phase III randomized, controlled trial of apremilast in patients with psoriatic arthritis. J Rheumatol 2015; 42: 479–488. [DOI] [PubMed] [Google Scholar]
- 5. Cutolo M, Myerson GE, Fleischmann R et al A phase III, randomized, controlled trial of apremilast in patients with psoriatic arthritis: results of the PALACE 2 trial. J Rheumatol 2016; 43: 1724–1734. [DOI] [PubMed] [Google Scholar]
- 6. Edwards CJ, Blanco FJ, Crowley J et al Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis and current skin involvement: a phase III, randomised, controlled trial (PALACE 3). Ann Rheum Dis 2016; 75: 1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wells AF, Edwards CJ, Kivitz AJ et al Apremilast monotherapy as the first systemic treatment in DMARD‐naive patients with active psoriatic arthritis: 3‐year treatment results [abstract 1680]. Arthritis Rheumatol 2016; 68(suppl 10): 1680. [Google Scholar]