

Abstract
Scope
The renin‐angiotensin system (RAS) is a major contributor to the development of insulin resistance and its related complications. Egg white ovotransferrin‐derived tripeptides, IRW (Ile‐Arg‐Trp), IQW (Ile‐Gln‐Trp), or LKP (Leu‐Lys‐Pro) are previously identified as the inhibitors of angiotensin‐converting enzyme (ACE), a key enzyme in the RAS. This study aims at determining whether these peptides are effective in improving insulin resistance, and their mechanisms of action, in a rat derived skeletal muscle cell line (L6 cells).
Methods and results
Insulin resistance is induced by treating L6 cells with 1 μm angiotensin II (Ang II) for 24 h. Effects of peptides on glucose uptake are determined using glucose uptake assay, glucose transporter 4 (GLUT4) translocation by immunofluorescence, reactive oxygen species (ROS) by dihydroethidium (DHE) staining, while insulin signaling pathway, Ang II receptor (AT1R or AT2R) levels, and NADPH oxidase activation are measured using Western Blot. Only IRW treatment significantly improves insulin resistance in L6 cells via stimulation of insulin signaling. IRW decreases Ang II‐stimulated AT1R expression, ROS formation, and NADPH oxidase activation.
Conclusions
Of three ACE inhibitory peptides studied, only IRW improves insulin resistance in L6 cells, at least partially via reduced AT1R expression and its anti‐oxidative activity.
Keywords: ACE inhibitory peptides, egg white protein ovotransferrin, insulin resistance, renin angiotensin system, skeletal muscle cells
1. Introduction
According to the International Diabetes Federation, Metabolic Syndrome is defined as an array of cardiovascular risk factors including type 2 diabetes (T2D), obesity, elevated triglyceride and cholesterol, and hypertension.1 Insulin resistance precedes the onset of metabolic syndrome and is unequivocally associated with an increased risk of T2D, obesity, hypertension, and cardiovascular diseases.2, 3 The renin‐angiotensin system (RAS) is classically known as a regulator of blood pressure, cardiac function, and fluid homeostasis.4 The major biologically active component generated by this system is angiotensin II (Ang II), which binds to type 1 (AT1R) and type 2 (AT2R) receptors, triggering various biological actions in the body that maintain circulation and body fluid volume.4, 5 While the role of RAS in blood pressure is well established, evidence from both clinical and rodent studies strongly suggests that abnormal activation of RAS is associated with development of insulin resistance and obesity.6, 7, 8, 9, 10 Recent evidence also indicates a critical role for RAS inhibition in obesity and T2D. Several clinical studies have shown that treating hypertensive patients with angiotensin converting enzyme (ACE) inhibitors or AT1R blockers results in a lower risk of developing T2D when compared with other antihypertensive treatments.11, 12, 13, 14 In rodent models of obesity and insulin resistance, RAS blockade via either pharmacologically active compounds or genetic deletion also improves insulin sensitivity.15, 16, 17, 18, 19 Therefore, inhibition of RAS might represent a promising approach to alleviate insulin resistance in metabolic syndrome. In addition to the systemic RAS, there are several local RAS presenting in brain, pancreas, heart, adipose tissue, and skeletal muscle.20, 21, 22 Skeletal muscle is a crucial tissue for development of insulin resistance because it accounts for the majority of insulin‐stimulated glucose utilization. In skeletal muscle, insulin increases glucose uptake through a signaling pathway that leads to tyrosine phosphorylation of insulin receptor substrate‐1 (IRS‐1), with subsequent phosphorylation of phosphatidylinositol‐3 kinase and protein kinase B (PKB), also known as Akt. As a result, translocation of glucose transporter 4 (GLUT4) from the cytosol to the plasma membrane removes glucose from the circulation to maintain whole‐body glucose homeostasis.23 The role of Ang II in insulin resistance of skeletal muscle has been well established.24, 25, 26, 27 Exposure of skeletal muscle to Ang II impairs GLUT4 translocation as well as glucose utilization via inhibition of the insulin signaling pathway.24, 26 Binding of Ang II to AT1R leads to increased nicotinamide adenine dinucleotide phosphate‐oxidase (NADPH) activation and thus reactive oxygen species (ROS) production.28 Increased ROS production leads to impaired insulin signaling pathway by inducing IRS serine phosphorylation, followed by decreasing GLUT4 translocation.29 Although several reports suggested that food‐derived peptides could improve insulin resistance,30, 31 effect of peptides on RAS stimulated insulin resistance in skeletal muscle and their mechanism have yet to be investigated. Egg white is a rich source of various bioactive peptides.32 In our previous research, three egg white ovotransferrin‐derived ACE inhibitory peptides, IRW (Ile‐Arg‐Trp), IQW (Ile‐Gln‐Trp), and LKP (Leu‐Lys‐Pro) were identified.33, 34 Our recent research demonstrated that IRW, IQW, and LKP significantly decreased blood pressure and plasma Ang II level in spontaneously hypertensive rats.35, 36 Interestingly, the mechanisms of their antihypertensitve action differed; IRW and IQW but not LKP treatment decreased proinflammatory/oxidative stress markers.35, 36, 37, 38, 39 Using human umbilical vein endothelial cells, both IRW and IQW exhibited antiinflammatory and anti‐antioxidant effects as shown by reduction of tumor necrosis factor‐alpha (TNF‐α) induced superoxide generation.37, 38 The actions were mediated by the nuclear factor‐κB (NF‐κB) pathway, which could be completely inhibited by IRW while only partially suppressed by IQW.38 Our recent work also showed that IRW can downregulate AT1R expression and NF‐κB signaling in Ang II treated vascular smooth muscle cells.40 Since inhibition of RAS overactivation can ameliorate insulin resistance and metabolic syndrome, our recent research findings prompted us to hypothesize that egg white ovotransferrin‐derived tripeptides may impart a beneficial effect in modulating RAS induced insulin resistance. The objective of this study was to determine effects of three ACE inhibitory peptides, derived from egg white ovotransferrin, on insulin resistance, and their mechanisms of action, in a rat derived skeletal muscle cell line (L6 cells).
2. Experimental Section
2.1. Materials
Ang II, insulin, dithiothreitol (DTT), Triton X‐100, NADPH, and Lucigenin were from Sigma–Aldrich (St Louis, MO, USA). DMEM, fetal bovine serum (FBS), Antibiotic‐antimycotic solution, and horse serum were purchased from Gibco/Invitrogen (Carlsbad, CA, USA). Dihydroethidium (DHE) and Hoechst 33342 were purchased from Thermo Fisher Scientific (Thermo Fisher Scientific, Burlington, Canada). The tripeptide IRW, IQW, and LKP were synthesized by Genscript (Piscataway, NJ, USA). Peptide sequence and purity (99.8%) were validated by HPLC–MS/MS.
2.2. Antibodies
Rabbit monoclonal primary antibodies against AT1R, phospho‐insulin receptor 1 (Tyr632, Ser307), insulin receptor 1, p67 phox, and p47phox were obtained from Santa Cruz Biotechnology Inc (SantaCruz, CA, USA). Rabbit monoclonal primary antibodies against AT2R, rabbit monoclonal primary antibody against GLUT4, and mouse monoclonal primary antibody against α‐tubulin were bought from Abcam (Cambridge, MA, USA). Rabbit monoclonal primary antibody against phospho‐Akt (Ser473), Akt was bought from Cell Signaling Technology Inc. (Danvers, MA, USA). Goat anti‐rabbit IRDye 680RD secondary antibody or Donkey anti‐mouse 800CW secondary antibody was purchased from Licor Biosciences (Lincoln, NE, USA).
2.3. Cell Culture
Rat‐derived L6 myoblasts were obtained from American Type Culture Collection (Manassas, VA; ATCC_ numbers: CRL‐1458). The cells were grown in DMEM supplemented with 10% FBS and 1% v/v antibiotic‐antimycotic solution (10 000 units mL–1 penicillin G, 10 mg mL–1 streptomycin, and 25 mg mL–1 amphotericin B) at 5% CO2 and 37 °C until they reached 80% confluence. For further differentiation, the cells were cultured in DMEM containing 2% horse serum for 6–7 days. The media were changed every 48 h and cells were used at the stage of myotubes (60–70%) when GLUT4 expression is the highest.41
2.4. Cell Viability Test
After termination of cell culture, the fully differentiated myotubes were incubated with 100 μm of IRW, IQW, or LKP in serum free DMEM for 2 h (vehicle), followed by treatment with 1 μm Ang II for 24 h. Next, 5 mg mL–1 3‐(4,5‐dimethythiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) (Manassas, VA, USA) was added and the plate reincubated at 5% CO2 and 37 °C for a further 3 h. The MTT mixture was then removed and 100 μL of DMSO was added to dissolve the formazan crystals. The plate was gently mixed on a shaker for 10 min, and then read on a microplate reader (SpectraMax M3, Molecular devices, CA, USA) at 570 nm and expressed as fold change over the vehicle control group.
2.5. Glucose Uptake Assay
Glucose uptake assay was examined by the procedure described previously42, 43 with slight modifications. Briefly, L6 myoblasts (5 × 104 cells per well) were subcultured into Nunc 24‐place multiwell plates and grown for 10–11 days until they formed myotubes. The fully differentiated myotubes were incubated with 100 μm of IRW, IQW, or LKP in serum free DMEM for 2 h, followed by treatment with 1 μm Ang II for 24 h. Next, the myotubes were kept for 2 h in Krebs–Henseleit buffer (pH 7.4) containing 0.1% BSA, 10 mm Hepes, and 2 mm sodium pyruvate (Krebs–Henseleit buffer, KHH buffer). The myotubes were then cultured in KHH buffer containing 11 mm glucose in the absence or presence of 1 μm of Insulin for another 4 h. Glucose concentrations in KHH buffer were determined with Glucose CII‐Test kit (Wako Pure Chemical Industries, Ltd., Osaka, Japan) and the amounts of glucose consumed were calculated from the differences in glucose concentrations between before and after culture.
2.6. Western Blot
The fully differentiated L6 myotubes were incubated with 100 μm of IRW, IQW, or LKP in serum free DMEM for 2 h, followed by treatment with 1 μm Ang II for different time conditions. To detect the total and phosphorylated proteins of the insulin signaling pathway, the myotubes were kept for 2 h in KHH buffer (pH 7.4) and then incubated in KHH buffer containing 11 mm glucose in the absence or presence of 1 μm insulin for 30 min. At the end of incubation, L6 myotubes were lysed in boiling hot Laemmle's buffer containing 50 mm DTT and 0.2% Triton‐X‐100 to prepare samples for Western blotting. To extract the protein from cell membrane and cytosol respectively, a Mem‐PER Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific) was used. Briefly, cells were washed with Cell Wash Solution and centrifuged at 300 × g for 5 min. After discarding the supernatant, cells were resuspended and incubated with Permeabilization Buffer at 4 °C. Next, permeabilized cells were centrifuged for 15 min at 16 000 × g. The supernatant containing cytosolic proteins was transferred into a new tube and the pellet was incubated with solubilization buffer at 4 °C for 30 min. After centrifuging for 15 min at 16 000 × g, the supernatant containing solubilized membrane and membrane‐associated proteins was transferred to a new tube for western blotting analysis. The cell lysates were then run in 9% SDS‐PAGE, transferred to nitrocellulose membranes, and immunoblotted with primary antibodies. After incubating with secondary antibodies, protein bands were detected by Licor Odyssey BioImager (Licor Biosciences, NE, USA) and quantified by densitometry using Image Studio Lite 5.2.
2.7. Superoxide Detection
Cellular superoxide generation was detected by DHE staining. ROS react with DHE to form ethidium, which then binds to nuclear DNA and releases nuclear fluorescence. Briefly, L6 myotubes were incubated with 100 μm of IRW, IQW, or LKP for 2 h, followed by treatment with Ang II (1 μm) for 30 min. Cells were then treated with 20 μm of DHE and incubated in the dark for 30 min. After three washings, the fluorescence signal was detected by an EVOS FL Auto Cell Imaging System (Thermo Fisher Scientific). For each data point, images from four random fields were taken. The total fluorescence intensity in each field was quantified by ImageJ software (http://imagej.net/Welcome), and the mean fluorescence intensity was determined.
2.8. Immunofluorescence
Fully differentiated L6 myotubes were incubated with 100 μm of IRW, IQW, or LKP 2 h prior to treatment with 1 μm of Ang II for 24 h. The myotubes were then incubated for 2 h in KHH buffer and then kept in KHH buffer containing 11 mm glucose in the absence or presence of 1 μg of insulin for 30 min. The myotubes were fixed in 3.75% paraformaldehyde. To selectively stain the proteins on cell membranes, the cells were not permeabilized. After washing twice with PBS, cells were blocked with 1% BSA in PBS for 1 h and incubated with anti‐GLUT4 primary antibody overnight at 4 °C. After washing three times with PBS, cells were treated with Alexa Fluor 546 conjugated goat anti‐rabbit secondary antibody for 1 h at room temperature. The cell nuclei were stained with Hoechst 33342 dye from Molecular Probes. Finally, after washing three times with PBS, cells were examined with an EVOS FL Auto Cell Imaging System (Thermo Fisher Scientific). All images presented are ×100 magnification.
2.9. Statistical Analysis
All data are presented as the mean ± SEM. The data were evaluated by IBM SPSS version 22. Differences between the mean values were assessed using one‐way analysis of variance, followed by Tukey multiple comparisons test if applicable. Statistical significance was considered for values of p < 0.05.
3. Results
3.1. Effect of Egg White Ovotransferrin‐Derived Tripeptides on Cell Viability and Insulin Stimulated Glucose Uptake in Ang II Treated L6 Myotubes
Cell viability was performed by MTT assay. At concentrations previously shown to have biological activity, the cell viability was not affected by any of the peptides (IRW, IQW, or LKP) (Figure 1A). To investigate the effect of peptides on insulin‐independent glucose uptake in L6 myotubes, the glucose uptake test was performed in comparison with insulin. IRW, IQW, or LKP were applied to L6 myotubes for 4 h. None of the peptides (IRW, IQW, or LKP) stimulated basal glucose uptake (Figure 1B). Adding insulin (1μm) however stimulated glucose uptake by 25%. Previous research has shown that Ang II decreases insulin‐stimulated glucose uptake in skeletal muscle.26, 45 Thus, we examined whether ovotransferrin‐derived peptides could reverse decreased glucose uptake in Ang II treated L6 myotubes. Cells were incubated with IRW, IQW, or LKP for 2 h prior to 24 h of treatment with Ang II. As shown in Figure 1C, adding Ang II significantly inhibited insulin‐stimulated glucose uptake in comparison with insulin alone. However, this decrease was significantly prevented by IRW treatment in Ang II treated L6 myotubes. IRW treatment (50, 100, and 150 μm) dose dependently increased glucose uptake (data not shown). On the other hand, treatment with IQW and LKP did not enhance insulin stimulated glucose uptake.
Figure 1.
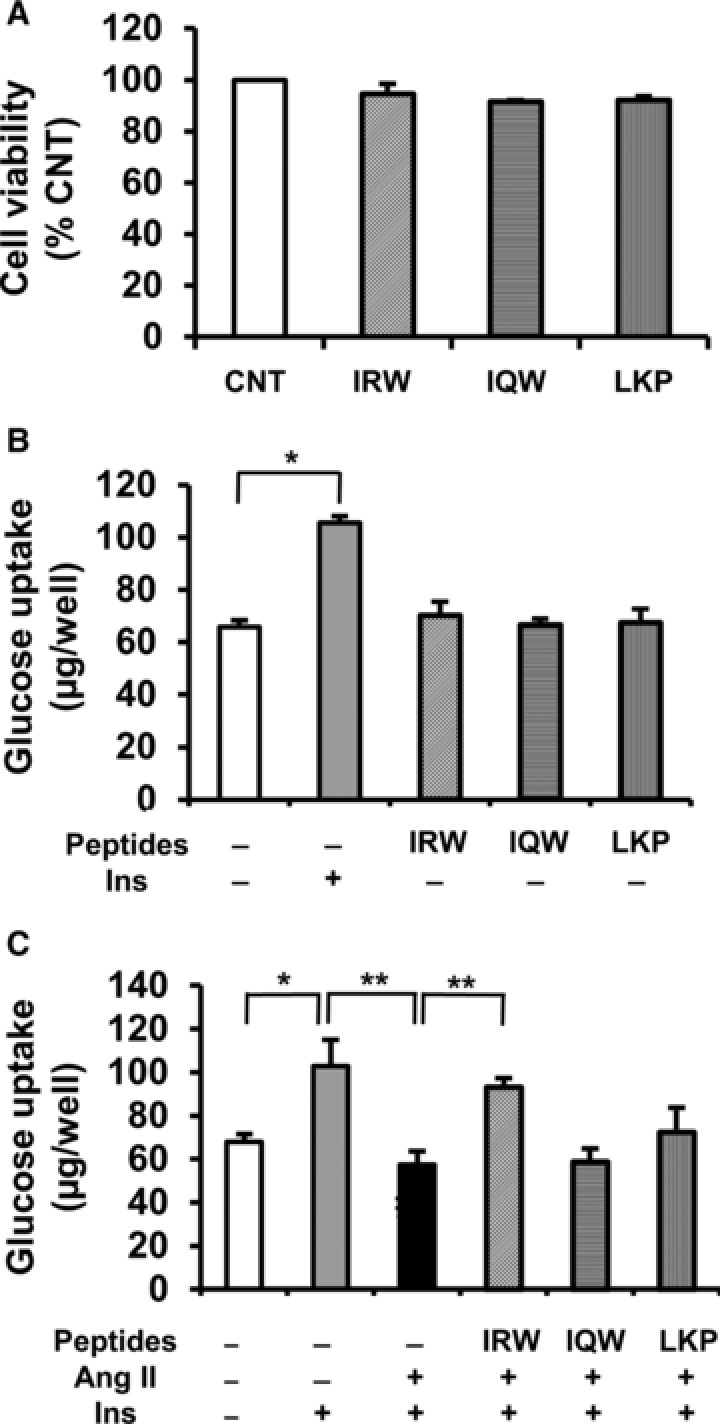
Effects of egg white ovotransferrin‐derived tripeptides on cell viability and glucose uptake in L6 myotubes. A) Effects of IRW, IQW, or LKP on cell viability. Fully differentiated myotubes were incubated with 100 μm of IRW, IQW, or LKP 2 h prior to the treatment of 1 μm of Ang II for 24 h. Cell viability was examined using MTT assay. B) Effects of IRW, IQW, or LKP on insulin‐independent glucose uptake. Fully differentiated L6 myotubes were preincubated in 24‐place multiwell plates in Krebs‐Henseleit‐Hepes buffer (KHH buffer) without glucose for 2 h. They were then incubated in KHH buffer containing 11 mm glucose without or with insulin and 100 μm of peptides for 4 h. C) Effect of peptides on insulin stimulated glucose uptake in Ang II‐treated L6 myotubes. Fully differentiated myotubes were incubated with 100 μm of IRW, IQW, or LKP 2 h prior to the treatment of 1 μm of Ang II for 24 h. Next, the myotubes were kept for 2 h in KHH buffer. The myotubes were then cultured in KHH buffer containing 11 mm glucose in the absence or presence of 100 nm of insulin for another 4 h, and then the glucose uptake was measured using a Glucose CII Test Kit. Each value represents the mean ± SEM of five independent experiments. **indicates p < 0.05 as compared to the Ang II‐treated group. *indicates p < 0.05 as compared to the untreated group.
3.2. Effect of Egg White Ovotransferrin‐Derived Tripeptides on Insulin Signaling Pathway on Ang II Treated L6 Myotubes
Previous studies in L6 skeletal muscle cells showed that Ang II impairs the insulin signaling pathway by inhibiting tyrosine residue phosphorylation of IRS‐1.24 To assess whether improved glucose uptake by IRW is due to improved insulin signaling, we investigated the effect of peptides on IRS‐1 and Akt phosphorylation in Ang II treated L6 myotubes. Ang II treatment induced serine residue phosphorylation of IRS‐1 (Figure 2A) but reduced tyrosine residue phosphorylation of IRS‐1 (Figure 2B) in comparison with insulin‐treated myotubes, consistent with a state of insulin resistance (Figure 2A, B). IRW but no other peptides normalized serine residue phosphorylation whereas the phosphorylation of tyrosine residue remained unaffected by any peptide (Figure 2B). In addition, phosphorylation of Akt was also significantly decreased by Ang II compared to that in insulin‐treated myotubes, whereas it was improved by IRW (Figure 2C). However, IQW and LKP did not exert any significant effect on impaired insulin signaling caused by Ang II, which indicates IRW was more competent in improving Ang II induced insulin resistance than IQW and LKP.
Figure 2.
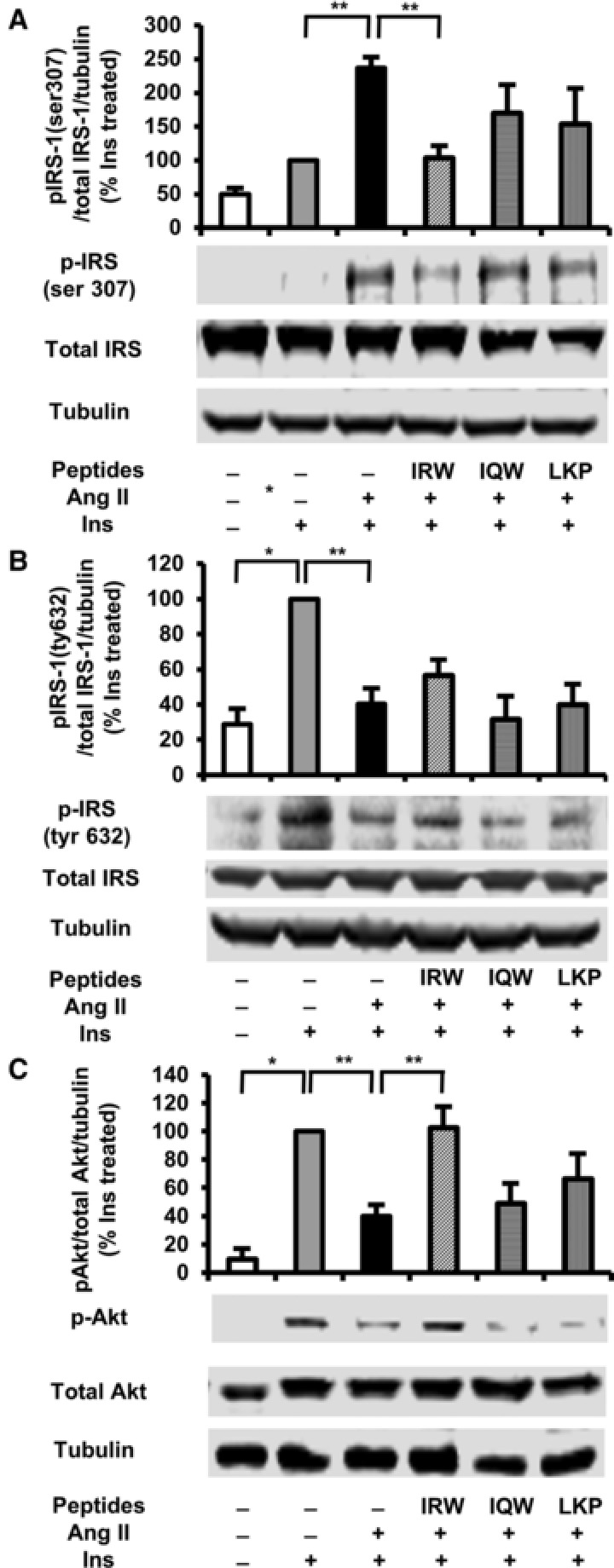
Effects of egg white ovotransferrin‐derived tripeptides on insulin signaling pathway in Ang II treated L6 myotubes. Fully differentiated myotubes were treated with 100 μm of peptides for 2 h followed by treatment with 1 μm of Ang 2 for 24 h. L6 myotubes were preincubated in KHH buffer for 2 h. They were then incubated in KHH buffer containing 11 mm glucose without or with 100 nm of insulin for 30 min. The cells were lysed and western blotting of the lysates was performed with antibodies against p‐IRS‐1 Ser 307 (A), p‐IRS‐1 tyr 632 (B), total IRS‐1 (A,B), p‐Akt (C), total Akt (C), and α‐tubulin (loading control). A set of representative images is shown. Data are presented as mean ± SEM of three independent experiments. **indicates p < 0.05 as compared to the Ang II‐treated group. *indicates p < 0.05 as compared to the untreated group.
3.3. Effect of Egg White Ovotransferrin Derived Tripeptides on GLUT4 Translocation in L6 Myotubes
To visually observe the translocation of GLUT4 in L6 myotubes, we detected GLUT4 abundance in L6 myotubes using immunofluorescence. As shown in Figure 3A, red fluorescence (left) shows cellular localization of GLUT4 while DAPI+GLUT4 (right) shows merging of GLUT4 and nuclei. In the presence of Ang II, GLUT4 abundance (left) was diminished as compared with that in insulin treated group. In the cells treated with Ang II and IRW, GLUT4 presence in the plasma membrane was higher than in Ang II‐treated cells indicating IRW treatment prevents the decreased GLUT4 translocation by Ang II in L6 myotubes. However, IQW and LKP did not alter GLUT4 abundance on the cell membrane. This result was verified using Western Blot (Figure 3B). The treatment of Ang II significantly decreased GLUT4 level in cell membrane as compared with insulin treated cells. This decrease was significantly recovered by IRW but not IQW and LKP (Figure 3B).
Figure 3.
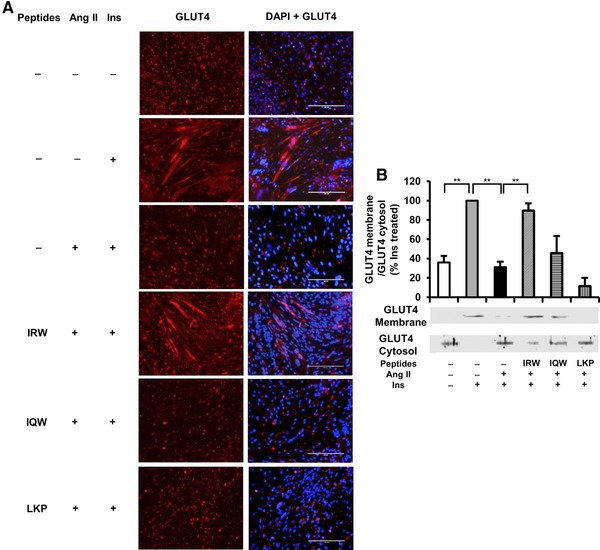
Effects of egg white ovotransferrin‐derived tripeptides (peptides) on GLUT4 translocation in L6 myotubes. Fully differentiated myotubes were treated with 100 μm of peptides for 2 h followed by treatment with 1 μm of Ang II for 24 h. L6 myotubes were preincubated in KHH buffer for 2 h. They were then incubated in KHH buffer containing 11 mm glucose without or with 100 nm of Insulin for 30 min. A) GLUT4 localization was detected using immunofluorescence technique and western blotting. Cellular localization of GLUT4 proteins is shown in red fluorescence (left) and merged image is also shown (right). A representative set of images from three independent experiments is shown. B) The membrane and cytosol were separated from the cells. The expression of GLUT4 was tested by western blot. Each value represents the mean ± SEM of three independent experiments. **indicates p < 0.05 as compared to the Ang II‐treated group. *indicates p < 0.05 as compared to the untreated group.
3.4. Effect of Egg White Ovotransferrin‐Derived Tripeptides on Ang II Receptor Abundance in Ang II Treated L6 Myotubes
To elucidate the molecular mechanisms underlying the effects of peptides on Ang II‐treated L6 myotubes, we detected AT1R protein in membranes by western blotting under basal conditions and in the presence of Ang II or Ang II plus insulin. None of the peptides influenced basal AT1R expression in L6 myotubes without Ang II stimulation (Figure 4A–C). Interestingly, treatment with IRW (Figure 4A, D), but not IQW and LKP (Figure 4B, C, D), significantly decreased the level of AT1R expression in L6 myotubes compared to that in Ang II treated control. However, the expression of AT2R was not affected by the incubation with the peptides (Figure 4E).
Figure 4.
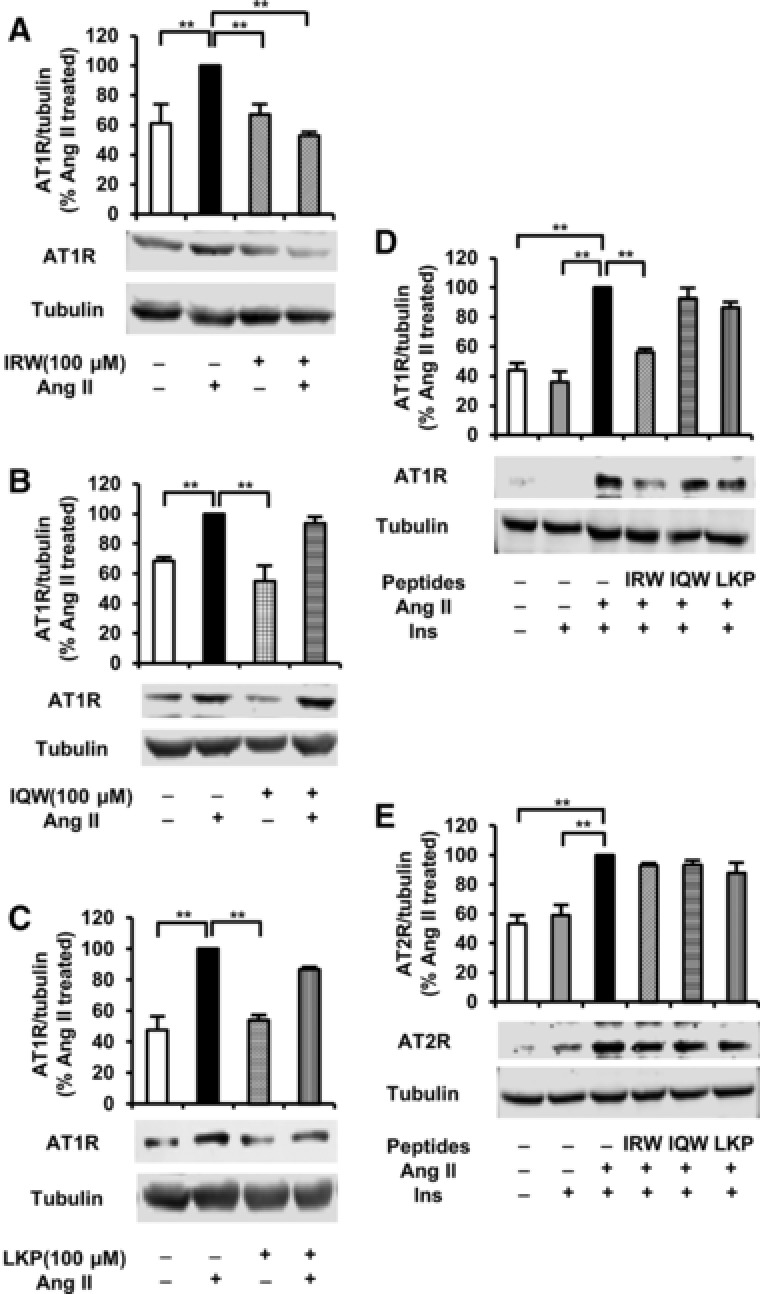
Effects of egg white ovotransferrin‐derived tripeptides on AT1R and AT2R expression in Ang II treated L6 myotubes. Fully differentiated myotubes were treated with 100 μm of peptides for 2 h followed by the treatment with 1 μm of Ang II for 24 h. The cells were lysed and western blotting of the lysates was performed with antibodies against AT1R A–D), AT2R E), and α‐tubulin (loading control). A set of representative images is shown. Data are presented as mean ± SEM of three independent experiments. **indicates p < 0.05 as compared to the Ang II‐treated group.
3.5. Effect of Egg White Ovotransferrin‐Derived Tripeptides on Ang II‐Induced ROS in L6 Myotubes
Binding of Ang II to AT1R increases the generation of ROS, which impairs insulin signaling pathway and GLUT4 translocation.28, 29 We investigated the effect of peptides on Ang II‐induced ROS in L6 myotubes. Compared with untreated cells, Ang II treatment increased the ROS level by 60% (Figure 5). Pretreatment with IRW significantly decreased the levels of ROS level suggesting that improvement of insulin resistance by IRW is at least partially mediating Ang II induced oxidative stress.
Figure 5.
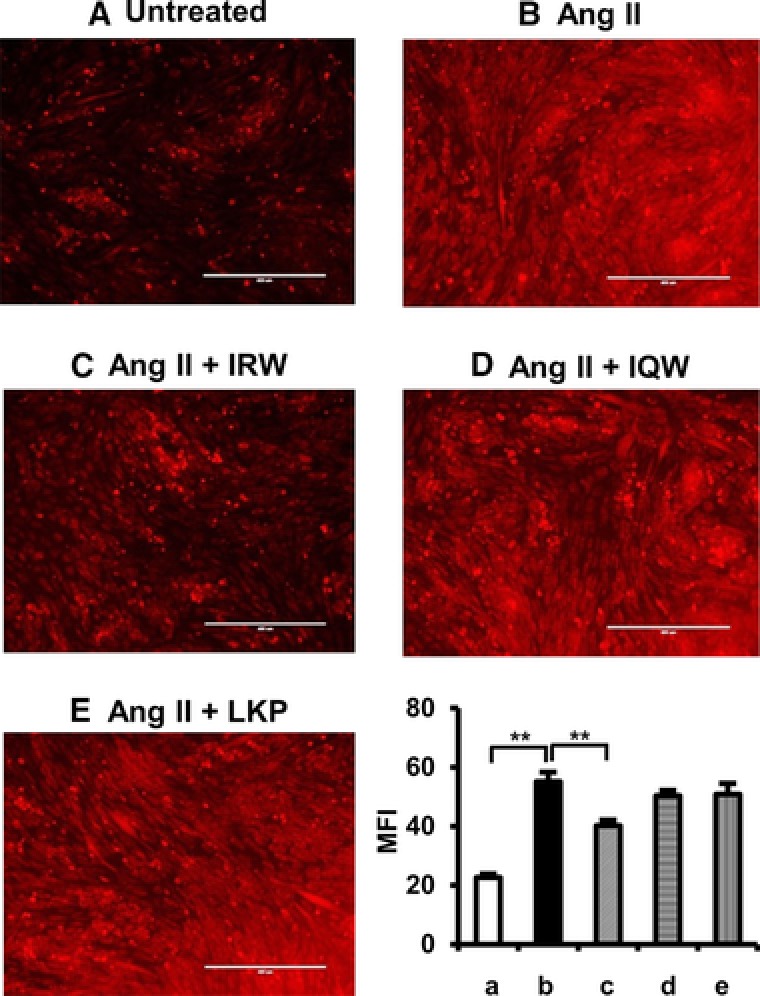
Effects of egg white ovotransferrin derived tripeptides on Ang II induced ROS in L6 myotubes. L6 myotubes were pretreated with 100 μm of Peptides for 2 h prior to 30 min stimulation with 1 μm of Ang II. Cells were treated with 10 μm of DHE for 30 min, and then visualized by fluorescent microscopy. The mean fluorescence intensity was calculated, and the data were expressed as % of the Ang II‐treat group from four independent experiments. Mean ± SEM are shown. **indicates p < 0.05 as compared to the Ang II‐treated group. A representative set of images from three independent experiments is shown.
3.6. Effect of Egg White Ovotransferrin‐Derived Tripeptides on the Translocation of P67phox and p47phox, Subunits of NADPH Oxidase, in L6 Myotubes
NADPH oxidase is a major source of superoxide formation and its activity is highly correlated with cytosolic regulatory subunits, p67phox and p47phox upon translocation46, 47 to the plasma membrane. Ang II treatment significantly increased translocation of both p67phox and p47phox as compared with those in untreated cells (Figure 6A, B). However, pretreatment with IRW abolished Ang II stimulated translocation of both p67phox and p47phox. Similarly, pretreatment with IQW and LKP also inhibited the translocation of p47phox and p67phox. These results indicate that the antioxidant ability of IRW may stem from decreasing NADPH oxidase activity.
Figure 6.
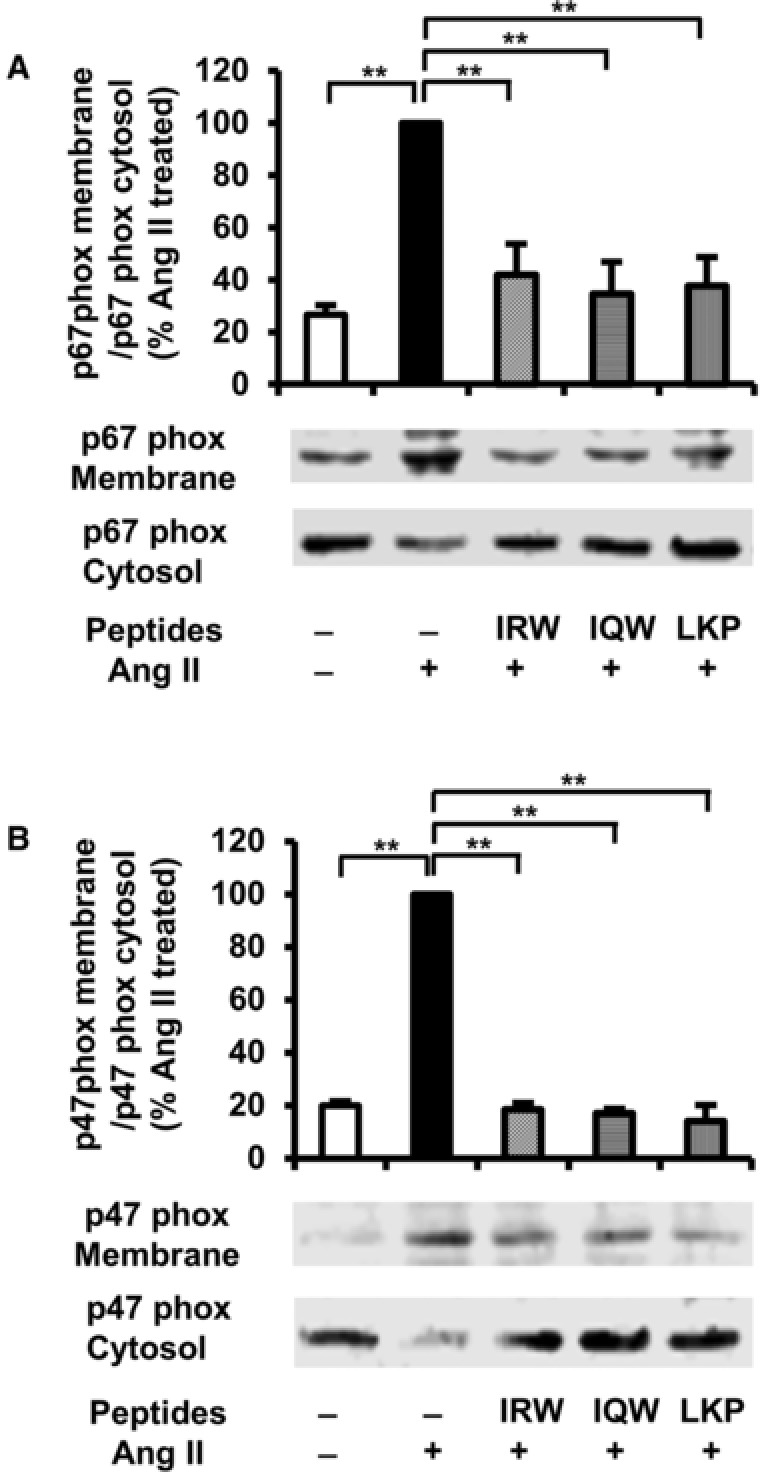
Effects of egg white ovotransferrin‐derived tripeptides on p67phox and p47phox translocation in L6 myotubes. L6 myotubes were pretreated with 100 μm of Peptides for 2 h followed by 30 min stimulation with 1 μm of Ang II. The membrane and cytosol were separated from the cells. The expression of p67phox (A) and p47phox (B) were tested by western blotting. The data are expressed as % of the Ang II‐treated group from four independent experiments. Mean ± SEM are shown. **indicates p < 0.05 as compared to the Ang II‐treated group.
4. Discussion
In this study, effects of three egg white ovotransferrin‐derived ACE inhibitory peptides on Ang II‐induced insulin resistance were studied in rat‐derived L6 skeletal muscle cells. Ang II treatment significantly decreased insulin‐stimulated glucose uptake, impaired insulin signaling pathway and GLUT4 translocation, while adding IRW, but not IQW or LKP, significantly reversed these outcomes. The improvement in insulin sensitivity by IRW was mediated by downregulation of Ang II stimulated AT1R expression.
Ang II is the major bioactive peptide of RAS playing a role in the regulation of vascular function and structure.48 In pathological conditions, overexpression of Ang II increases the generation of ROS leading to endothelial dysfunction, vascular injury, and inflammation underlying cardiovascular disease.49 Growing evidence supports an important role of local RAS and Ang II in the pathogenesis of insulin resistance and metabolic syndrome. In skeletal muscle, the major insulin sensitive tissue, Ang II decreases glucose utilization and insulin sensitivity.24, 26, 44 Thus, inhibiting the action of Ang II in skeletal muscle will contribute to the prevention and treatment of insulin resistance. Ang II was reported to inhibit insulin‐stimulated Akt activation and decrease insulin‐stimulated GLUT4 abundance on cell membrane in L6 myotubes.24 In isolated mammalian skeletal muscle, exposure of Ang II for 2 h impaired the phosphorylation of Akt in insulin signaling and directly decreased insulin‐stimulated glucose uptake,26 which was in alignment with our results. Interestingly, we found that incubation of IRW for 2 h but not IQW or LKP ameliorated Ang II‐induced decrease in insulin‐stimulated glucose uptake and GLUT4 abundance on cell membrane. In addition, IRW decreased IRS‐1 phosphorylation at Ser residue but normalized Akt phosphorylation, suggested a positive effect of IRW on Ang II‐inhibited insulin signaling.
This preventative effect of IRW observed in Ang II‐induced insulin resistance may be mediated through AT1R inhibition. A large body of data from cellular and animal studies showed that the inhibition of AT1R improved insulin resistance in diabetic model animals.15, 16, 17, 18, 19, 24, 25, 26, 45 Wei et al.24 found that AT1R blocker losartan improved insulin signaling and GLUT4 abundance in cell membrane of L6 myotubes. Shiuchi et al.45 reported that AT1R blocker valsartan increased insulin sensitivity and glucose uptake in skeletal muscle of diabetic KK‐Ay mice via stimulating the insulin signaling pathway and GLUT4 translocation. In our recent work, IRW inhibited AT1R overexpression in vascular smooth muscle cells.40 In agreement with these results, cotreatment of IRW but not IQW or LKP with Ang II decreased AT1R overexpression in L6 myotubes. These results suggest that the effects of IRW on Ang II‐stimulated insulin resistance are at least partially due to reduced AT1R expression. Our previous study using transcriptome analysis showed that IRW affected the expression of a number of genes related to cardiovascular disease in spontaneously hypertensive rats, including ACE2,39 a recently identified member of RAS that catalyzes the conversion of Ang II to Ang (1‐7).50, 51 Ang (1‐7) was reported to improve insulin resistance by inducing insulin signaling pathway in vitro and in vivo.52, 53 Thus, the reduction in Ang II induced AT1R expression might be associated with increased ACE2 activation, awaiting further investigation.
Ang II induced‐ROS is involved in the inhibition of insulin signaling in skeletal muscle.25, 26 Ang II stimulation of NADPH oxidase and ROS production is mediated by AT1R.28, 46 ROS impairs insulin signaling pathway and GLUT4 translocation in skeletal muscle.29 We previously identified that IRW had antioxidant activity in vascular cells and in vivo.37, 38, 40 Herein, we found that Ang II exhibited increased ROS in skeletal muscle cells, but was attenuated by the presence of IRW. The antioxidant activity of peptides was also accompanied by decreased NADPH oxidase activity. The activation of NADPH is dependent on stable formation of membrane bound cytochrome b558 by its association with cytosolic regulatory subunits, p67phox and p47phox upon translocation. We observed that the translocation of both p67phox and p47phox was increased in response to Ang II, which was reversed by coincubation with IRW, IQW, or LKP. Interestingly, while only IRW showed significant ability to reduce ROS generation after Ang II treatment for 30 min, all three tripeptides were capable of diminishing the translocation of p67phox and p47 phox. There are various intracellular sources of ROS such as mitochondria, the ER, peroxisomes, and oxidases/oxygenases including NAD(P)H oxidases, xanthine oxidase, and nitric oxide synthases.54 Our results suggest the possibility that antioxidant ability of IRW might mediate other probable sources of ROS generation responsible for Ang II induced insulin resistance.
ROS has been proposed to be involved in the activation of NF‐κB.55, 56 Furthermore, several studies on rodents demonstrated that high fat diet or acute hyperlipidemia activated NF‐κB in skeletal muscle, which was associated with impaired insulin signaling.57, 58 Wei et al.25 previously reported the involvement of NF‐κB pathway via NADPH oxidase in Ang II‐induced insulin resistance. Ang II significantly induced NF‐κB p65 nuclear translocation in L6 cells. Our research group previously demonstrated that pretreatment with IRW completely abolished TNF‐α induced translocation of both p65 and p50, while IQW partially restricted only the translocation of p50, which suggests differential regulation of NF‐κB by egg peptides.38 LKP did not exert any effect in modulating inflammatory pathway.36 To test the possibility that IRW inactivates NF‐κB in skeletal muscle to alleviate insulin resistance, further studies are required. In addition, future research in vivo is necessary to confirm the efficacy of IRW in an insulin resistant animal model.
Although there is only one amino acid residue difference between IRW and IQW, they functioned differently with regard to insulin resistance, similar to our previous studies on vascular functions in cells and in animals.35, 36, 38 It is not known if this difference is due to the presence of l‐arginine within IRW, as l‐arginine is a known precursor of nitric oxide (NO), and l‐arginine supplementation improves insulin sensitivity by altering nitric oxide pathway.59 In vitro study showed, however, that NO stimulates GLUT4 translocation in skeletal muscle independent of insulin signaling pathway.60 Hence, IRW might have different mechanism from l‐arginine alone, which awaits further research.
In conclusion, IRW improves Ang II‐induced insulin resistance, at least partially via reduced AT1R expression and its antioxidant activity. We provided the evidence that specific food derived ACE inhibitory peptides could improve insulin resistance. Our studies indicate that RAS system may become a complementary therapeutic target for studying the potential beneficial effect of food‐derived bioactive peptides for the treatment of metabolic syndrome and its related complications.
Abbreviations
- ACE
angiotensin converting enzyme
- Akt
protein kinase B
- Ang II
angiotensin II
- AT1R
angiotensin II type 1 receptor
- AT2R
angiotensin II type 2 receptor
- DHE
dihydroethidium
- GLUT4
glucose transporter 4
- IRS‐1
insulin receptor substrate‐1
- KHH buffer
Krebs–Henseleit buffer
- MTT
3‐(4,5‐dimethythiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- NADPH
nicotinamide adenine dinucleotide phosphate‐oxidase
- NF‐κB
nuclear factor‐κB
- PI3K
phosphatidylinositol‐3 kinase
- RAS
the renin‐angiotensin system
- ROS
reactive oxygen species
- T2D
type 2 diabetes
- VSMCs
vascular smooth muscle cells
Conflict of Interest
The authors have declared no conflict of interest.
Acknowledgments
This work is supported by research grants from the Alberta Livestock and Meat Agency, (ALMA), Egg Farmers of Canada, and the Natural Sciences and Engineering Research Council of Canada (NSERC).
M.S. and J.W. conceived the study concept. M.S. performed the experiments, analyzed and interpreted the data, and drafted the first version of this manuscript. C.B.C. and J.W. interpreted data and reviewed the manuscript. All authors read and approved the final manuscript.
Son M., Chan C. B., Wu J., Mol. Nutr. Food Res. 2018, 62, 1700602 https://doi.org/10.1002/mnfr.201700602
References
- 1. International Diabetes Federation . https://www.idf.org/our-activities/advocacy-awareness/resources-and-tools/60:idfconsensus-worldwide-definitionof-the-metabolic-syndrome.html. (accessed: June 2017).
- 2. Sowers J. R., Am. J. Physiol. Heart. Circ. Physiol. 2004, 286, H1597. [DOI] [PubMed] [Google Scholar]
- 3. McFarlane S. I., Banerji M., Sowers J. R., J. Clin. Endocrinol. Metab. 2001, 86, 713. [DOI] [PubMed] [Google Scholar]
- 4. Schmieder R. E., Hilgers K. F., Schlaich M. P., Schmidt B. M., Lancet 2007, 369, 1208. [DOI] [PubMed] [Google Scholar]
- 5. Matthew A. S., Steven D. C., Susan B. G., Maria M., Thomas M. C., Compr. Physiol. 2014, 4, 1201.24944035 [Google Scholar]
- 6. Goossens G. H., Jocken J. W., Blaak E. E., Schiffers P. M., Saris W. H., van Baak M. A., Hypertension 2007, 49, 542. [DOI] [PubMed] [Google Scholar]
- 7. Gorzelniak K., Engeli S., Janke J., Luft F. C., Sharma A. M., J. Hypertension 2002, 20, 965. [DOI] [PubMed] [Google Scholar]
- 8. Boustany C. M., Bharadwaj K., Daugherty A., Brown D. R., Randall D. C., Cassis L. A., Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R943. [DOI] [PubMed] [Google Scholar]
- 9. Ogihara T., Asano T., Ando K., Chiba Y., Sakoda H., Anai M., Shojima N., Ono H., Onishi Y., Fujishiro M., Katagiri H., Fukushima Y., Kikuchi M., Noguchi N., Aburatani H., Komuro I., Fujita T., Hypertension 2002, 40, 872. [DOI] [PubMed] [Google Scholar]
- 10. Sloniger J. A., Saengsirisuwan V., Diehl C. J., Dokken B. B., Lailerd N., Lemieux A. M., Kim J. S., Henriksen E. J., Am. J. Physio. Endocrinol. Metab. 2005, 288, E1074. [DOI] [PubMed] [Google Scholar]
- 11. Hansson L., Lindholm L. H., Niskanen L., Lanke J., Hedner T., Niklason A., Luomanmäki K., Dahlöf B., de Faire U., Mörlin C., Karlberg B. E., Wester P. O., Björck J. E., Lancet 1999, 353, 611. [DOI] [PubMed] [Google Scholar]
- 12. Kjeldsen S. E., Dahlöf B., Devereux R. B., Julius S., Aurup P., Edelman J., Beevers G., de Faire U., Fyhrquist F., Ibsen H., Kristianson K., Lederballe‐Pedersen O., Lindholm L. H., Nieminen M. S., Omvik P., Oparil S., Snapinn S., Wedel H., J. Am. Med. Assoc. 2002, 288, 1491. [DOI] [PubMed] [Google Scholar]
- 13. Yusuf S., Sleight P., Pogue J., Bosch J., Davies R., Dagenais G., N. Engl. J. Med. 2000, 342, 145. [DOI] [PubMed] [Google Scholar]
- 14. Yusuf S., Gerstein H., Hoogwerf B., Pogue J., Bosch J., Dagenais G., Wolffenbuttel B. H., Zinman B., HOPE Study Investigators . JAMA 2001, 286, 1882. [DOI] [PubMed] [Google Scholar]
- 15. Henriksen E. J., Jacob S., Kinnick T. R., Teachey M. K., Krekler M., Hypertension 2001, 38, 884. [DOI] [PubMed] [Google Scholar]
- 16. Iwai M., Kanno H., Tomono Y., Inaba S., Senba I., Furuno M., Mogi M., Horiuchi M., J. Hypertens. 2010, 28, 1471. [DOI] [PubMed] [Google Scholar]
- 17. Takahashi N., Li F., Hua K., Deng J., Wang C. H., Bowers R. R., Bartness T. J., Kim H. S., Harp J. B., Cell. Metab. 2007, 6, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jayasooriya A. P., Mathai M. L., Walker L. L., Begg D. P., Denton D. A., Cameron‐Smith D., Egan G. F., McKinley M. J., Rodger P. D., Sinclair A. J., Wark J. D., Weisinger H. S., Jois M., Weisinger R. S., Proc. Natl. Acad. Sci. USA 2008, 105, 6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kouyama R., Suganami T., Nishida J., Tanaka M., Toyoda T., Kiso M., Chiwata T., Miyamoto Y., Yoshimasa Y., Fukamizu A., Horiuchi M., Hirata Y., Ogawa Y., Endocrinology 2005, 146, 3481. [DOI] [PubMed] [Google Scholar]
- 20. Jones B. H., Standridge M. K., Taylor J. W., Moustaid N., Am. J. Physiol. 1997, 273, R236. [DOI] [PubMed] [Google Scholar]
- 21. Paul M., Poyan Mehr A., Kreutz R., Physiol. Rev. 2006, 86, 747. [DOI] [PubMed] [Google Scholar]
- 22. Johnston A. P., Baker J., De Lisio M., Parise G., J. Renin Angiotensin Aldosterone Syst. 2011, 12, 75. [DOI] [PubMed] [Google Scholar]
- 23. Saltiel A. R., Kahn C. R., Nature 2011, 414, 799. [DOI] [PubMed] [Google Scholar]
- 24. Wei Y., Sowers J. R., Nistala R., Gong H., Uptergrove G. M., Clark S. E., Morris E. M., Szary N., Manrique C., Stump C. S., J. Biol. Chem. 2006, 281, 35137. [DOI] [PubMed] [Google Scholar]
- 25. Wei Y., Sowers J. R., Clark S. E., Li W., Ferrario C. M., Stump C. S., Am. J. Physiol. Endocrinol. Metab. 2008, 294, E345. [DOI] [PubMed] [Google Scholar]
- 26. Diamond‐Stanic M. K., Henriksen E. J., Arch. Physiol. Biochem. 2010, 116, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mitsuishi M., Miyashita K., Muraki A., Itoh H., Diabetes 2009, 58, 710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Griendling K. K., Minieri C. A., Ollerenshaw J. D., Alexander R. W., Circ. Res. 1994, 74, 1141. [DOI] [PubMed] [Google Scholar]
- 29. Bloch‐Damti A., Bashan N., Antioxid. Redox Signal. 2005, 7, 1553. [DOI] [PubMed] [Google Scholar]
- 30. Song J. J., Wang Q., Du M., Li T. G., Chen B., Mao X. Y., Mol. Nutr. Food Res. 2017, 61, 1. [DOI] [PubMed] [Google Scholar]
- 31. Sawada Y., Sakamoto Y., Toh M., Ohara N., Hatanaka Y., Naka A., Kishimoto Y., Kondo K., Iida K., Mol. Nutr. Food Res. 2015, 59, 2502. [DOI] [PubMed] [Google Scholar]
- 32. Elizabeth A., J. Am. Coll. Nutr. 2000, 19, 495S. [Google Scholar]
- 33. Majumder K., Wu J., Food Res. Int. 2010, 43, 1371. [Google Scholar]
- 34. Majumder K., Wu J., Food Chem. 2011, 126, 1614. [DOI] [PubMed] [Google Scholar]
- 35. Majumder K., Chakrabarti S., Morton J. S., Panahi S., Kaufman S., Davidge S. T., Wu J., PLoS One 2013, 8, e82829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Majumder K., Chakrabarti S., Jude S. M., Sareh P., Susan K., Davidge S. T., Wu J., J. Funct. Foods 2015, 13, 50. [Google Scholar]
- 37. Huang W., Chakrabarti S., Majumder K., Jiang Y., Davidge S. T., Wu J., J. Agric. Food Chem. 2010, 58, 10840. [DOI] [PubMed] [Google Scholar]
- 38. Majumder K., Chakrabarti S., Davidge S. T., Wu J., J. Agric. Food Chem. 2013, 61, 2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Majumder K., Liang G., Chen Y., Guan L., Davidge S. T., Wu J., Mol. Nutr. Food Res. 2015, 59, 1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liao W., Chakrabarti S., Davidge S. T., Wu J., J. Agric. Food Chem. 2016, 64, 7342. [DOI] [PubMed] [Google Scholar]
- 41. Mitsumoto Y., Burdett E., Grant A., Klip A., Biochem. Biophys. Res. Commun. 1991, 175, 652. [DOI] [PubMed] [Google Scholar]
- 42. Son M. J., Minakawa M., Miura Y., Yagasaki K., Eur. J. Nutr. 2013, 52, 1607. [DOI] [PubMed] [Google Scholar]
- 43. Ha B. G., Nagaoka M., Yonezawa T., Tanabe R., Woo J. T., Kato H., Chung U. I., Yagasaki K., J. Nutr. Biochem. 2012, 23, 501. [DOI] [PubMed] [Google Scholar]
- 44. Shinshi Y., Higashiura K., Yoshida D., Togashi N., Yoshida H., Miyazaki Y., Ura N., Shimamoto K., J. Am. Soc. Hypertens. 2007, 1, 251. [DOI] [PubMed] [Google Scholar]
- 45. Shiuchi T., Iwai M., Li H. S., Wu L., Min L. J., Li J. M., Okumura M., Cui T. X., Horiuchi M., Hypertension 2004, 43, 1003. [DOI] [PubMed] [Google Scholar]
- 46. Choi H., Leto T. L., Hunyady L., Catt K. J., Bae Y. S., Rhee S. G., J. Biol. Chem. 2008, 283, 255. [DOI] [PubMed] [Google Scholar]
- 47. Aljada A., Ghanim H., Dandona P., Methods Mol. Biol. 2002, 196, 99. [DOI] [PubMed] [Google Scholar]
- 48. Nguyen‐Dinh‐Cat A., Montezano A. C., Burger D., Touyz R. M., Antioxid. Redox Signal. 2013, 19, 1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sergey I. D., Rafal R. N., Antioxid. Redox Signal. 2013, 19, 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mohammed A. R., Chamsi P., Zhili S., Wilson Tang W. H., Curr. Heart Fail. Rep. 2014, 11, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vickers C., Hales P., Kaushik V., Dick L., Gavin J., Tang J., Godbout K., Parsons T., Baronas E., Hsieh F., Acton S., Patane M., Nichols A., Tummino P., J. Biol. Chem. 2002, 277, 14838. [DOI] [PubMed] [Google Scholar]
- 52. Takeda M., Yamamoto K., Takemura Y., Takeshita H., Hongyo K., Kawai T., Hanasaki‐Yamamoto H., Oguro R., Takami Y., Tatara Y., Takeya Y., Sugimoto K., Kamide K., Ohishi M., Rakugi H., Diabetes 2013, 62, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Giani J. F., Mayer M. A., Muñoz M. C., Silberman E. A., Höcht C., Am. J. Physiol. Endocrinol. Metab. 2009, 296, 262. [DOI] [PubMed] [Google Scholar]
- 54. Wei Y., Chen K., Whaley‐Connell A. T., Stump C. S., Ibdah J. A., Sowers J. R., Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R673. [DOI] [PubMed] [Google Scholar]
- 55. Anderson M. T., Staal F. J., Gitler C., Herzenberg L. A., Herzenberg L. A., Proc. Natl. Acad. Sci. USA 1994, 91, 11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Flohé L., Brigelius‐Flohé R., Saliou C., Traber M. G., Packer L., Free Radic. Biol. Med. 1997, 22, 1115. [DOI] [PubMed] [Google Scholar]
- 57. Bhatt B. A., Dube J. J., Dedousis N., Reider J. A., O'Doherty R. M., Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R233. [DOI] [PubMed] [Google Scholar]
- 58. Ropelle E. R., Pauli J. R., Prada P. O., de Souza C. T., Picardi P. K., Faria M. C., Cintra D. E., Fernandes M. F., Flores M. B., Velloso L. A., Saad M. J., Carvalheira J. B., J. Physiol. 2006, 577, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Claybaugh T., Decker S., McCall K., Slyvka Y., Steimle J., Wood A., Schaefer M., Thuma J., Inman S., Int. J. Endocrinol. 2014, 2014, 171546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Higaki Y., Hirshman M.‐F., Fujii N., Goodyear L. J., Diabetes 2001, 50, 241. [DOI] [PubMed] [Google Scholar]