

Abstract
Secreted noggin protein regulates bone morphogenetic protein activity during development. In mice, a complete loss of noggin protein leads to multiple malformations including joint fusion, whereas mice heterozygous for Nog loss-of-function mutations are normal. In humans, heterozygous NOG missense mutations have been found in patients with two autosomal dominant disorders of joint development, multiple synostosis syndrome (SYNS1) and a milder disorder proximal symphalangism (SYM1). This study investigated the effect of one SYNS1 and two SYM1 disease-causing missense mutations on the structure and function of noggin. The SYNS1 mutation abolished, and the SYM1 mutations reduced, the secretion of functional noggin dimers in transiently transfected COS-7 cells. Coexpression of mutant noggin with wild-type noggin, to resemble the heterozygous state, did not interfere with wild-type noggin secretion. These data indicate that the human disease-causing mutations are hypomorphic alleles that reduce secretion of functional dimeric noggin. Therefore, we conclude that noggin has both species-specific and joint-specific dosage-dependent roles during joint formation. Surprisingly, in contrast to the COS-7 cell studies, the SYNS1 mutant was able to form dimers in Xenopus laevis oocytes. This finding indicates that there also exist species-specific differences in the ability to process mutant noggin polypeptides.
Structures of the appendicular skeleton arise from the condensation and differentiation of mesenchymal cells. The bone morphogenetic protein (BMP) family of secreted signaling molecules comprises a large fraction of the transforming growth factor β superfamily and has important roles in this process (1). BMPs were first purified from demineralized bone matrix as proteins capable of causing ectopic bone formation after s.c. or i.m. administration in rodents (2); the BMP protein family has grown to include 15 distinct members. The essential roles that BMPs play in skeletogenesis include recruiting mesenchymal cells into future skeletal anlagen, promoting mesenchymal cell proliferation and differentiation into chondroblasts and osteoblasts, and inducing apoptosis at sites of future joints (3–5). An individual BMP may play each of these roles in a temporally dependent, site-specific manner. Regulation of BMP activity occurs in several different ways including the direct inhibition of BMP signaling by secreted antagonists.
Noggin was the first identified BMP antagonist (6). In Xenopus laevis embryos, noggin can bind and inhibit BMP-4 and thereby induce the formation of the head and other dorsal structures (7). Noggin is posttranslationally modified and is secreted as a disulfide-bonded homodimer (8). In addition to binding to BMP-4, it binds to several other BMP family members including BMPs 2, 7, 13 (growth/differentiation factor-6), and 14 (growth/differentiation factor-5) (7, 9, 10). Roles for noggin during mammalian development were demonstrated by knocking out the gene in transgenic mice (11, 12). Mice heterozygous for a null mutation in the noggin gene (Nog) appear normal, whereas mice homozygous for Nog-null alleles display several developmental defects, including a severely malformed skeletal system (11, 12). Noggin-null mice have shorter bones, are missing skeletal elements, and lack multiple articulating joints (11).
We have previously shown that heterozygous missense mutations in the human noggin gene (NOG) cause two similar, but clinically distinct, skeletal dysplasias, proximal symphalangism (SYM1) and multiple synostoses syndrome (SYNS1) (13). SYM1 and SYNS1 have apical joint fusions as their principal feature (13), but SYNS1 is distinguished from SYM1 by also having hip and vertebral fusions (14). The absence of a phenotype in mice that are heterozygous for noggin-null mutations contrasts with the autosomal dominant skeletal phenotype observed in humans. Furthermore, NOG missense mutations appear capable of causing a spectrum of joint involvement (i.e., SYM1 and SYNS1). These observations led us to study the synthesis, secretion, and BMP binding activity of wild-type and mutant noggin to explore the mechanism by which human NOG missense mutations exert their effect.
Materials and Methods
Noggin and BMP Expression Vectors.
The coding sequence for human noggin, a 232-aa residue polypeptide, is contained within a single exon (13). The noggin coding sequence from the genomic DNA of two patients with SYM1 and one patient with SYNS1 was amplified by using the previously described primers, NOG1 and NOG5 (13). The SYM1-derived mutants were a glycine to cysteine substitution at residue 189 (G189C) and a proline to leucine substitution at residue 223 (P223L); the SYNS1-derived mutant was a tryptophan to glycine substitution at residue 217 (W217G) (13). The PCR amplimers were cloned into the pCR2.1 vector (Invitrogen). Plasmids containing mutant alleles were identified by DNA sequencing. Wild-type NOG sequence and the three mutant NOG sequences were moved from pCR2.1 into the Xenopus expression vector pT7TS (gift of P. Krieg, Univ. of Texas, Austin) by using EcoRV and SpeI cloning sites. Wild-type and mutant NOG sequence was moved from pT7TS into the mammalian expression vector pcDNA3 (Invitrogen) by using HindIII and XbaI cloning sites. A wild-type noggin construct containing three tandem-myc epitopes (EQKLISEEDLGG) at its C-terminal end was obtained from Regeneron Pharmaceuticals. This tagged version of noggin was subsequently moved to pcDNA3 to maintain vector uniformity. A BMP-14 (also known as CDMP1 and GDF5) expression construct in which the active domain of BMP-14 is myc-tagged and fused to the pro-domain of dorsalin has been described (15), as has a myc-tagged BMP-4 expression construct (16).
Transfections.
COS-7 cells were cultured in growth medium (DMEM with 10% FBS/100 units/ml penicillin/100 μg/ml streptomycin/250 ng/ml amphotericin B; GIBCO/BRL) at 37°C in an atmosphere containing 5% CO2. Cells at ≈80% confluence in 100-mm dishes were transfected with 40 μl of FuGENE 6 (Roche Molecular Biochemicals) and 5 μg of mammalian expression plasmid DNA in Opti-MEM I according to manufacturer's recommendations. When cotransfection of noggin wild-type/noggin mutant or noggin/BMP was performed, 5 μg of each plasmid was used. Titration experiments were also performed by cotransfecting 1 μg of untagged noggin (wild-type and mutants) with decreasing amounts (1.0, 0.5, and 0.1 μg) of myc-tagged noggin (wild type). Transfected COS-7 cells were maintained for 24 h in growth medium and then switched to 10 ml of serum-free DMEM/plate. Transfection efficiency (≈50%) was assessed by cotransfection with a green fluorescent protein construct. After 24 h, the culture media and the cell layer were separately harvested.
Coculture experiments of wild-type or mutant noggin (1 μg)-transfected COS-7 cells and myc-tagged BMP-14 (1 μg)-transfected COS-7 cells were performed by using cell culture inserts (6-well plate, Falcon). BMP-14-transfected COS-7 cells were maintained for 24 h in growth medium and then switched to serum-free DMEM. Twenty-four hours later, inserts containing noggin transfected COS-7 cells were added to this system. Total conditioned media (5 ml) were collected after 24 h of coculture.
Oocyte Isolation and Injection.
X. laevis were purchased from Xenopus Express (Beverly Hills, FL). Oocytes were removed, and collagenase dissociated (2 mg/ml) in Ca2+-free Ringer's solution for 35–40 min as described (17). Capped RNA was synthesized by using the linearized pT7TS expression constructs and the T7 mMessage mMachine kit according to manufacturer's instructions (Ambion, Austin, TX). Oocytes were injected with 50 nl of 1 μg/μl (50 ng) noggin-capped RNA or water and maintained at 18°C in OR3 (modified Leibovitz media, 5% Pen/Strep, pH 7.5). Three to four oocytes were incubated in 200 μl of OR3 solution for 72 h after injection.
Western Blot Analysis.
Each injected Xenopus oocyte was dissolved in 20 μl of 2× sample loading buffer, boiled for 5 min, and then centrifuged at 12,000 × g for 5 min at room temperature; 30 μl was loaded per lane. Conditioned media from the COS-7 cells were cleared by centrifugation at 1,800 × g for 10 min at room temperature. After centrifugation, equal aliquots of conditioned media supernatant and 2× sample loading buffer were mixed and boiled for 5 min; 20 μl was loaded per lane. The COS-7 cell layer was scraped and extracted with 1 ml of RIPA buffer (150 mM NaCl/1% Nonidet P-40/0.5% deoxycholate/0.1% SDS/50 mM Tris, pH 8) and centrifuged at 12,000 × g for 10 min at room temperature. Equal aliquots of the cell lysis supernatant and 2× sample loading buffer were mixed and boiled for 5 min; 20 μl was loaded per lane.
Samples were separated by SDS/PAGE under nonreducing or reducing (5% β-mercaptoethanol) conditions and transferred onto Immobilon-P (Millipore). Blots were blocked for 1 h with 5% nonfat dry milk in TBS (150 mM NaCl/20 mM Tris, pH 7.5) and then incubated with either 50 ng/ml anti-noggin antibody (rat anti-noggin RP57–16, Regeneron Pharmaceuticals) or a 1:250 dilution anti-myc antibody (Santa Cruz Biotechnology) in antibody buffer (2.5% nonfat dry milk/150 mM NaCl/20 mM Tris/0.05% Tween 20, pH 7.5) for 1 h. After three washes with TBST (150 mM NaCl/20 mM Tris/0.05% Tween 20, pH 7.5), blots were incubated with a 1:8,000 dilution of horseradish-peroxidase secondary antibody (goat anti-rat, Santa Cruz Biotechnology) in antibody buffer for 1 h and then washed again three times with TBST. Immunoreactive proteins were detected with the ECL Plus chemiluminescent detection system (Amersham Pharmacia).
Coimmunoprecipitation.
Conditioned media (0.5 ml) from COS-7 cells that had been transfected with the pcDNA3 constructs (wild type or mutants) were mixed with conditioned media (0.5 ml) from cells that had been transfected with myc-tagged BMP (BMP-4 or BMP-14) constructs. Media were incubated for 16 h at 4°C and then immunoprecipitated by using anti-myc antibody (1:100) and 40 μl of protein G (Amersham Pharmacia) for 16 h at 4°C. Conditioned media (1 ml) from COS-7 cells that had been cotransfected with pcDNA3 noggin constructs (wild type or mutants) and myc-tagged BMP constructs (BMP-4 or BMP-14) were similarly immunoprecipitated. Immunoprecipitates were washed three times with RIPA buffer, dissolved in 40 μl of 1× sample loading buffer, boiled for 5 min, briefly vortexed, and centrifuged at 12,000 × g for 5 min at room temperature; 10 μl of sample was loaded per lane. Western analysis of coimmunoprecipitated noggin was performed as described above.
Noggin Functional Assay.
Wild-type and mutant NOG sequences were moved from pT7TS into pCS107 (18) Xenopus expression vectors by EcoRI digest. Capped synthetic mRNAs were transcribed by using linearized expression constructs and the mMessage mMachine kit (Ambion); 50 pg of synthetic mRNA was injected into the marginal zone of a single ventral blastomere of a four-cell embryo. All injections were in volumes of about 10 nl. Embryos were allowed to develop to tailbud stage when they were assayed for the presence of secondary axes.
Results
Noggin Missense Mutations Affect the Protein's Ability to Form Secreted Disulfide-Linked Dimers in COS-7 Cells.
SDS/PAGE performed under reducing conditions indicated that the transiently transfected COS-7 cells transcribed and translated mutant noggin similar to wild-type noggin, although some mutant protein had altered posttranslational processing (Fig. 1B). Because wild-type noggin polypeptides normally form disulfide-linked homodimers, the polypeptides were analyzed under nonreducing conditions (Fig. 1A). Mutant noggin polypeptides formed disulfide-linked dimers less efficiently than wild-type noggin. Furthermore, mutant polypeptides formed a large molecular weight aggregate that is not found in wild-type noggin, indicative of abnormally disulfide-bonded protein.
Figure 1.

Noggin mutant proteins synthesized by COS-7 cells. Western blot analysis of cell lysates of COS-7 cells transfected with pcDNA3 containing wild-type sequence (WT), no insert (− control), SYM1-derived sequences (P223L, G189C), or SYNS1-derived sequence (W217G). (A) Nonreducing SDS/8% PAGE and immunodetection using an anti-noggin mAb. Wild-type noggin forms disulfide-bonded dimers and a higher molecular weight species (denoted by arrow) that is an artifact of the electrophoresis method. SYM1-derived mutant proteins are present as monomer, disulfide-bonded dimer, and as higher molecular weight species (denoted by *) that do not penetrate the separating or stacking gel. SYNS1-derived mutant protein is present as monomer and as higher molecular weight species that do not penetrate the separating or stacking gel. (B) Reducing SDS/10% PAGE and immunodetection using anti-noggin antibody. All higher molecular weight species resolve to monomeric noggin. All three mutant noggins are synthesized in amounts similar to wild-type noggin. A second, slightly slower migrating band in the mutants indicates additional posttranslational modifications that are not present in wild-type protein.
Because noggin is a secreted protein, wild-type and mutant noggin expression was also analyzed by using the conditioned media from the transfected COS-7 cells. Western blots of conditioned media electrophoresed under nonreducing conditions revealed that the SYM1 noggin mutants (P223L and G189C) showed decreased levels of disulfide-linked dimeric noggin compared with wild type; in fact, the G189C mutant protein is barely able to form dimers (Fig. 2A). Some mutant noggin also seems to be secreted in either monomeric or at least nondisulfide bonded form (Fig. 2A). The SYNS1 noggin mutant (W217G) did not appear secreted as either a monomer or a dimer. As with the cell extracts, all three mutant noggin polypeptides were present as high molecular weight disulfide-linked aggregates. When conditioned medium was run in reducing conditions to assess the apparent levels of secreted noggin, the SYNS1 mutant (W217G) showed the least amount of immunodetectable noggin (Fig. 2B). However, as indicated by Fig. 1B, this decrease is not caused by altered epitope recognition of this particular mutant protein or a decrease in protein synthesis. These results suggest that there are differences in each missense mutation's ability to form disulfide-linked dimers and to be secreted.
Figure 2.

Noggin mutant proteins secreted by COS-7 cells. Western blot analysis of conditioned media from COS-7 cells transfected with pcDNA3 containing NOG wild-type sequence (WT), SYM1-derived sequences (P223L and G189C), or SYNS1-derived sequence (W217G). (A) Nonreducing SDS/8% PAGE and immunodetection using anti-noggin antibody. Wild-type noggin forms disulfide-bonded dimers and a higher molecular weight species (denoted by arrow) that is an artifact of the electrophoresis method. The SYM1-derived mutant (P223L) is secreted as a dimer, but less efficiently than wild type. The SYM1-derived mutant (G189C) is secreted predominantly as a monomer; a small amount of disulfide-bonded dimer is also secreted. The SYNS1 mutant does not appear to be secreted as either a monomer or dimer. All three mutant proteins are also present as higher molecular weight species (denoted by *), which do not penetrate the separating or stacking gel. (B) Reducing SDS/10% PAGE and immunodetection using anti-noggin antibody. All higher molecular weight species resolve to monomeric noggin. Less mutant protein is secreted for the mutants (P223L and W219G) when compared with wild type.
Mutant Noggin Polypeptides Do Not Affect Wild-Type Noggin Expression.
Because SYM1 and SYNS1 are associated with heterozygous NOG missense mutations, it is possible that the product of the mutant allele could interfere with the product of the wild-type allele. To test this hypothesis, a myc-tagged, wild-type noggin construct was cotransfected with wild-type and mutant constructs. Western blot analysis of conditioned media from the different cotransfectants showed that the synthesis, dimerization, and secretion of myc-tagged, wild-type noggin were not significantly affected by the mutant allele (Fig. 3A). Specifically, myc-tagged wild-type noggin was not found in monomeric form or in high molecular weight species. Furthermore, altering the relative amount of mutant to wild-type noggin during the cotransfection experiments did not affect the ability of wild-type protein to be synthesized or secreted (data not shown), indicating that the synthesis of mutant polypeptide does not interfere with the synthesis of wild-type noggin.
Figure 3.

Coexpression of mutant and wild-type noggin. Western blot analysis of conditioned media from COS-7 cells that had been cotransfected with pcDNA3 containing myc-tagged, wild-type NOG sequence (WT-myc) and pcDNA3 containing wild-type sequence (WT), SYM1-derived sequences (P223L, G189C), or SYNS1-derived sequence (W217G). (A) Nonreducing SDS/PAGE and immunodetection using anti-myc antibody. Myc-tagged, wild-type noggin is present only in dimeric form. (B) Nonreducing SDS/PAGE and immunodetection using anti-noggin antibody. Most noggin is present as a disulfide-linked dimer, indicating that myc-tagged wild-type noggin may be more efficiently expressed than mutant noggin. It is possible that some heterodimeric wild-type/mutant noggin may also be present. (C) Reducing 10% SDS/PAGE and immunodetection using anti-noggin antibody. Slightly faster migrating bands in the WT and G189C lanes probably represent untagged protein, which is 36 aa residues smaller than the myc-tagged noggin.
Both Wild-Type and Mutant Noggin Dimers Can Bind BMPs.
Noggin can bind to several BMPs, including BMP-14 (also known as GDF5 for growth/differentiation factor 5 or CDMP1 for cartilage-derived morphogenetic protein 1) (10). To determine whether the secreted noggin mutants are able to bind BMPs, conditioned media from noggin transfectants and BMP transfectants were mixed to allow the interaction of noggin proteins with myc-tagged BMP-14. Immunoprecipitation with an anti-myc antibody directed against myc-tagged BMP-14 protein resulted in the marked coprecipitation of wild-type noggin and the two SYM1 mutants (Fig. 4). Coimmunoprecipitation experiments using myc-tagged BMP-4 were also performed, and similar results were obtained (data not shown). Only disulfide-stabilized dimeric noggin was coprecipitated with BMP-14 and BMP-4, suggesting that free noggin monomer cannot bind to BMPs (data not shown). Because cells may coexpress both noggin and BMPs during development, coimmunoprecipitation of noggin mutant proteins with BMP-14 was also performed on conditioned media from COS-7 cells that had been cotransfected with noggin and BMP-14 constructs. Wild-type noggin and both SYM1 mutant noggins were coprecipitated as disulfide-linked dimers (Fig. 5). SYNS1-derived mutant noggin (W217G) coimmunoprecipitated with BMP-14 as a high molecular aggregate (Fig. 5). It is interesting that dimeric SYM1-derived mutant protein (G189C) is strongly coimmunoprecipitated when coexpressed with BMP-14, despite its predominant expression as a monomer when solely expressed (see Figs. 2 and 5). This result suggests that BMP-14 coexpression might facilitate formation of mutant noggin dimers.
Figure 4.

Interaction of noggin with BMP-14. Western blot analysis of noggin after precipitation with anti-myc antibody. Conditioned media from myc-tagged BMP-14 transfected COS-7 cells (+BMP-14) was mixed with conditioned media from COS-7 cells that had been transfected with pcDNA3 containing NOG wild-type sequence (WT), SYM1-derived sequences (P223L, G189C), or SYNS1-derived sequence (W217G). As negative controls, anti-myc immunoprecipitation was performed on the conditioned media from the different noggin transfectants not mixed with BMP-14 conditioned media (−BMP-14). Samples were subjected to nonreducing SDS/8% PAGE and immunodetected using an anti-noggin antibody. Arrow denotes artifact band of noggin. Band migrating above the arrow is because of cross reaction with the anti-myc antibody. Wild-type noggin dimer and the two SYM1 mutant dimers coprecipitate with BMP-14.
Figure 5.

Interaction of noggin mutant proteins and coexpressed BMP-14. Conditioned media from COS-7 cells cotransfected with pcDNA3 containing myc-tagged BMP-14 (+BMP-14) and pcDNA3 containing NOG wild-type sequence (WT), SYM1-derived sequences (P223L, G189C), or SYNS1-derived sequence (W217G). As negative controls, anti-myc immunoprecipitation was performed on the conditioned media from the noggin constructs expressed alone (−BMP-14). Samples were subjected to nonreducing SDS/8% PAGE and immunodetected using an anti-noggin antibody. Arrow denotes artifact band of noggin, and asterisks denote high molecular weight species of noggin. Band migrating between * and arrow is the anti-myc antibody. Wild-type noggin dimer and the two SYM1 mutant dimers coprecipitate with BMP14. Monomeric noggin does not coprecipitate with BMP-14. A much larger amount of dimeric noggin mutant (G189C) coprecipitates than might be expected based on this mutant's predominant secretion as a monomer when expressed alone (see Fig. 2).
Intracellular BMP-14 Facilitates Formation of Mutant Noggin Dimers.
BMP-14 and the different noggin constructs were coexpressed in COS-7 cells, and the conditioned media were analyzed by Western blot. Cotransfection with BMP-14 leads to a reproducible increase in the secretion of dimeric noggin species for both SYM1-derived mutants (Fig. 6). The most striking effect occurs in the SYM1 mutant (G189C), where the increase in dimeric noggin is accompanied by a decrease in monomeric noggin. Coculture experiments indicated that BMP-14 acts intracellularly, rather than by facilitating or stabilizing noggin dimer formation extracellularly, as coculturing noggin-expressing cells with BMP-14-expressing cells did not enhance noggin dimer formation (data not shown).
Figure 6.

BMP-14 coexpression facilitates dimerization of noggin. Western blot analysis of conditioned media from COS-7 cells transfected with pcDNA3 containing NOG wild-type sequence (WT), SYM1-derived sequences (P223L, G189C), or SYNS1-derived sequence (W217G), with (+) or without (−) cotransfection of pcDNA3 containing myc-tagged BMP-14. Samples were subjected to nonreducing SDS/8% PAGE and immunodetected using an anti-noggin antibody. The SYM1 mutant proteins have increased disulfide-linked dimer formation. The SYM1 mutant (G189C) has the most striking change in relative abundance of secreted monomer and dimer when coexpressed with BMP-14.
Mutant Noggin Proteins Can Form Dimers in Xenopus Oocytes.
The X. laevis oocyte system has previously been used to study wild-type noggin protein expression (8). In this system, all three mutant noggin proteins were able to form disulfide-stabilized dimers; however, they did so with varying efficiency (Fig. 7). Surprisingly, the SYNS1 mutant, which did not form dimers in COS-7 cells, dimerized more efficiently than one of the SYM1 mutants in the Xenopus oocyte (Fig. 7).
Figure 7.

Noggin mutant proteins synthesized by Xenopus oocytes. Western blot analysis of Xenopus oocytes that were injected with capped RNA from pT7TS constructs containing SYM1-derived sequences (P223L and G189C) and SYNS1-derived sequence (W217G). Samples were subjected to nonreducing SDS/12% PAGE and immunodetected using an anti-noggin antibody. All noggin mutants form disulfide-bonded dimers, albeit with varying efficiency. Monomeric noggin migrates at 32 kDa, disulfide-bonded dimeric noggin migrates at 52 kDa, and a high molecular weight noggin aggregate (denoted by *) does not penetrate the separating gel.
Noggin Mutant Proteins Are Functional in X. laevis Embryos.
Having found that SYM1 mutant noggin protein, although secreted less efficiently than wild-type protein by COS-7 cells, still retains the ability to bind BMP, and that SYNS1 mutant protein can form dimers in Xenopus oocytes, we sought to determine whether these mutant noggin dimers retain function. Fifty picograms of mRNA encoding each mutant was tested for dorsalizing activity by injection into a single ventral blastomere of a four-cell Xenopus embryo. Injected embryos were allowed to grow to the tailbud stage and were scored for secondary axes. As shown in Fig. 8, embryos ectopically expressing wild-type human noggin on the ventral side display a partial secondary axis as expected for a secreted BMP antagonist (19). Interestingly, injection of the same amount of P223L noggin mutant mRNA also elicited a secondary axis in Xenopus embryos. Similar results were obtained for W217G and G189C mutants (data not shown). These results indicate that the noggin mutant proteins retain their ability to antagonize BMP signaling in an intact embryo.
Figure 8.
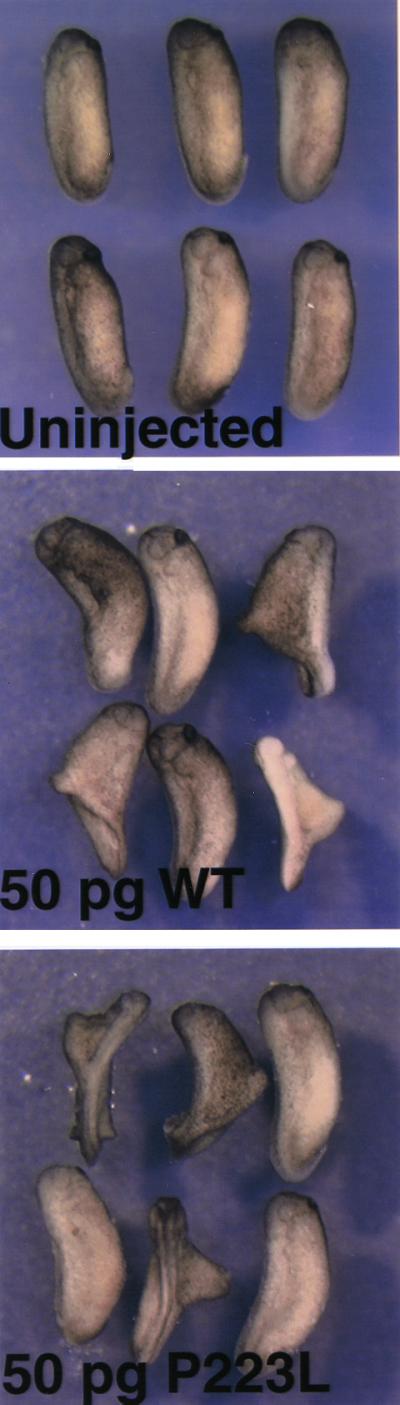
Noggin mutant proteins are capable of inducing secondary axis formation in Xenopus embryos. Capped RNA (50 pg) synthesized from NOG constructs (WT or P223L) were injected into the marginal zone of the ventral blastomere of a four-cell embryo. Secondary axis formation is observed in both wild-type noggin (WT) and mutant noggin (P223L) capped RNA-injected embryos but not in uninjected embryos.
Discussion
Noggin can bind to BMPs and inhibit their BMP signaling. Analysis of mouse mutants shows that this interaction is essential to modulate the activities of BMPs during development (11). In contrast to the Nog-null allele in mice, which is asymptomatic in the heterozygous state but causes a perinatal lethal, recessive phenotype when homozygous, several heterozygous NOG missense mutations have been identified in human families with the autosomal dominant phenotypes SYM1 and SYNS1 (13). To explore the mechanism of mutational effect of the human missense mutations in the noggin polypeptide, we studied their expression using transiently transfected mammalian COS-7 cells and noggin mRNA-injected Xenopus oocytes.
We found differences in the ability of COS-7 cells and Xenopus oocytes to handle mutant protein. Each missense mutation was able to form disulfide-stabilized dimers in the Xenopus system and to dorsalize developing Xenopus embryos (Figs. 7 and 8), suggesting that the mutant proteins retain their ability to bind to and antagonize BMPs. Using expression in COS-7 cells to analyze synthesis and secretion of protein, the two SYM1 mutant proteins, but not the SYNS1 mutant protein, were able to form dimers and to bind BMP. Importantly, however, dimer formation for the two SYM1 mutants was consistently reduced in comparison with wild type (Figs. 1 and 2). Cotransfection studies in COS-7 cells indicated that mutant polypeptide did not interfere with the dimerization and secretion of wild-type protein (Fig. 3). Taken together, these results suggest that reduction in the amount of secreted dimeric noggin accounts for the two human skeletal phenotypes and that alleles that produce less protein will cause more severe phenotypes. This study also highlights the difference between humans and mice in their dosage requirements for secreted growth regulators during skeletal development, as mice that are heterozygous for Nog-null alleles have normal joints. Why humans and mice differ in their dosage requirement for noggin is not known. Skeletal elements in humans are larger and take longer to form than do the comparable elements in mice. Therefore, humans may require more noggin protein to form a gradient over larger distances or to maintain a gradient over longer duration. Alternatively, cells that comprise the human and mouse skeletal anlages may differ in their absolute threshold requirements for BMP signals. At present, noggin is the only secreted inhibitor of BMP signaling that has been associated with a human disease phenotype. Therefore, it is not yet possible to determine whether interspecific differences in dosage requirements for inhibiting BMP signaling will be unique to noggin or more generalized. However, support for the latter hypothesis derives from two other autosomal dominant human phenotypes, brachydactyly type C and situs ambiguus, which have been attributed to heterozygous loss-of-function mutations that affect the secreted transforming growth factor β superfamily members BMP-14 and Lefty-1, respectively (20, 21); comparable mutations in the mouse orthologs of these genes act as recessive alleles (22, 23).
Among the surprising findings in our study was that the SYNS1 mutant (W217G) was able to form dimers in Xenopus oocytes but not in COS-7 cells. This observation could reflect Xenopus oocytes having factors that facilitate efficient noggin dimerization, or COS-7 cells having a more sophisticated system for detecting and not secreting mutant noggin polypeptides. Although we do not know the precise reason for this difference, the observation indicates a need to be cautious when exploring the consequences of mutations in different model systems. Even within the same organism, diverse cell types differ in their abilities to process mutant polypeptides. For example, osteoblasts obtained from patients with osteogenesis imperfecta are less able to identify and sequester mutant collagen molecules than are the patients' skin fibroblasts (24, 25). A second unexpected finding was that the SYM1 mutants had enhanced dimer secretion from COS-7 cells when they were coexpressed with BMP-14. This suggests that noggin may normally interact intracellularly with its binding partner and that such interactions could modify the consequences of some missense mutations by improving their ability to dimerize and be secreted. Because noggin and BMPs have overlapping patterns during development, it is intriguing to speculate that endogenous intracellular interactions between noggin and BMPs may be a particularly potent regulator of BMP function during development.
Acknowledgments
We thank Dr. J. Terrig Thomas for providing the BMP-14 expression construct and Dr. Daniel Constam for providing the BMP-4 expression construct. This work was supported by National Institutes of Health Developmental Biology Grant HD 07104 (to J.M.), a biomedical research grant from the Arthritis Foundation (to M.L.W.), and National Institutes of Health Grants AR 43527 (to M.L.W.) and GM 49346 (to R.M.H). M.L.W. is an Assistant Investigator with the Howard Hughes Medical Institute and a recipient of a Burroughs Wellcome Fund Clinical Scientist Award in Translational Research.
Abbreviations
- BMP
bone morphogenetic protein
- SYM1
proximal symphalangism
- SYNS1
multiple synostosis syndrome
References
- 1.Hogan B L. Curr Opin Genet Dev. 1996;6:432–438. doi: 10.1016/s0959-437x(96)80064-5. [DOI] [PubMed] [Google Scholar]
- 2.Van de Putte K A, Urist M R. Clin Orthop. 1965;43:257–270. doi: 10.1097/00003086-196500430-00026. [DOI] [PubMed] [Google Scholar]
- 3.Groeneveld E H, Burger E H. Eur J Endocrinol. 2000;142:9–21. doi: 10.1530/eje.0.1420009. [DOI] [PubMed] [Google Scholar]
- 4.Wozney J M, Rosen V. Clin Orthop. 1998;346:26–37. [PubMed] [Google Scholar]
- 5.Zou H, Niswander L. Science. 1996;272:738–741. doi: 10.1126/science.272.5262.738. [DOI] [PubMed] [Google Scholar]
- 6.Smith W C, Harland R M. Cell. 1992;70:829–840. doi: 10.1016/0092-8674(92)90316-5. [DOI] [PubMed] [Google Scholar]
- 7.Zimmerman L B, De Jesus-Escobar J M, Harland R M. Cell. 1996;86:599–606. doi: 10.1016/s0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]
- 8.Smith W C, Knecht A K, Wu M, Harland R M. Nature (London) 1993;361:547–549. doi: 10.1038/361547a0. [DOI] [PubMed] [Google Scholar]
- 9.Chang C, Hemmati-Brivanlou A. Development (Cambridge, UK) 1999;126:3347–3357. doi: 10.1242/dev.126.15.3347. [DOI] [PubMed] [Google Scholar]
- 10.Merino R, Macias D, Ganan Y, Economides A N, Wang X, Wu Q, Stahl N, Sampath K T, Varona P, Hurle J M. Dev Biol. 1999;206:33–45. doi: 10.1006/dbio.1998.9129. [DOI] [PubMed] [Google Scholar]
- 11.Brunet L J, McMahon J A, McMahon A P, Harland R M. Science. 1998;280:1455–1457. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- 12.McMahon J A, Takada S, Zimmerman L B, Fan C M, Harland R M, McMahon A P. Genes Dev. 1998;12:1438–1452. doi: 10.1101/gad.12.10.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong Y, Krakow D, Marcelino J, Wilkin D, Chitayat D, Babul-Hirji R, Hudgins L, Cremers C W, Cremers F P, Brunner H G, et al. Nat Genet. 1999;21:302–304. doi: 10.1038/6821. [DOI] [PubMed] [Google Scholar]
- 14.Krakow D, Reinker K, Powell B, Cantor R, Priore M A, Garber A, Lachman R S, Rimoin D L, Cohn D H. Am J Hum Genet. 1998;63:120–124. doi: 10.1086/301921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas J T, Kilpatrick M W, Lin K, Erlacher L, Lembessis P, Costa T, Tsipouras P, Luyten F P. Nat Genet. 1997;17:58–64. doi: 10.1038/ng0997-58. [DOI] [PubMed] [Google Scholar]
- 16.Constam D B, Robertson E J. J Cell Biol. 1999;144:139–149. doi: 10.1083/jcb.144.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sciortino C M, Romero M F. Am J Physiol. 1999;277:F611–F623. doi: 10.1152/ajprenal.1999.277.4.F611. [DOI] [PubMed] [Google Scholar]
- 18.Baker J C, Beddington R S, Harland R M. Genes Dev. 1999;13:3149–3159. doi: 10.1101/gad.13.23.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glinka A, Wu W, Onichtchouk D, Blumenstock C, Niehrs C. Nature (London) 1997;389:517–519. doi: 10.1038/39092. [DOI] [PubMed] [Google Scholar]
- 20.Polinkovsky A, Robin N H, Thomas J T, Irons M, Lynn A, Goodman F R, Reardon W, Kant S G, Brunner H G, van der Burgt I, et al. Nat Genet. 1997;17:18–19. doi: 10.1038/ng0997-18. [DOI] [PubMed] [Google Scholar]
- 21.Kosaki K, Bassi M T, Kosaki R, Lewin M, Belmont J, Schauer G, Casey B. Am J Hum Genet. 1999;64:712–721. doi: 10.1086/302289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Storm E E, Huynh T V, Copeland N G, Jenkins N A, Kingsley D M, Lee S J. Nature (London) 1994;368:639–643. doi: 10.1038/368639a0. [DOI] [PubMed] [Google Scholar]
- 23.Meno C, Shimono A, Saijoh Y, Yashiro K, Mochida K, Ohishi S, Noji S, Kondoh H, Hamada H. Cell. 1998;94:287–297. doi: 10.1016/s0092-8674(00)81472-5. [DOI] [PubMed] [Google Scholar]
- 24.Mundlos S, Chan D, Weng Y M, Sillence D O, Cole W G, Bateman J F. J Biol Chem. 1996;271:21068–21074. doi: 10.1074/jbc.271.35.21068. [DOI] [PubMed] [Google Scholar]
- 25.Sarafova A P, Choi H, Forlino A, Gajko A, Cabral W A, Tosi L, Reing C M, Marini J C. Hum Mutat. 1998;11:395–403. doi: 10.1002/(SICI)1098-1004(1998)11:5<395::AID-HUMU7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]