

ABSTRACT
The analysis of DNA methylation has become routine in the pipeline for diagnosis of imprinting disorders, with many publications reporting aberrant methylation associated with imprinted differentially methylated regions (DMRs). However, comparisons between these studies are routinely hampered by the lack of consistency in reporting sites of methylation evaluated. To avoid confusion surrounding nomenclature, special care is needed to communicate results accurately, especially between scientists and other health care professionals. Within the European Network for Human Congenital Imprinting Disorders we have discussed these issues and designed a nomenclature for naming imprinted DMRs as well as for reporting methylation values. We apply these recommendations for imprinted DMRs that are commonly assayed in clinical laboratories and show how they support standardized database submission. The recommendations are in line with existing recommendations, most importantly the Human Genome Variation Society nomenclature, and should facilitate accurate reporting and data exchange among laboratories and thereby help to avoid future confusion.
KEYWORDS: Imprinting, imprinting disorders, methylation, nomenclature
Mammalian genomic imprinting is an epigenetic regulatory mechanism that results in parent-of-origin specific gene expression in diploid somatic cells (for a review, see ref1). Several features of the imprinting mechanism have been identified including allelic DNA methylation, histone modifications, and noncoding RNAs.2 Clustering and coordinate regulation is a key feature of imprinted domains with much effort invested in understanding how multiple genes are regulated by long-range cis-acting differentially methylated regions (DMRs).
In 1993, two publications reported parent-of-origin specific methylation associated with imprinted domains. Both of these studies were in mouse, the first described the paternally methylated regions associated with the H19-Igf2 gene cluster,3 the second identified a region of methylation on the maternal allele within the Igf2r gene associated with the T-associated maternal effect (Tme) deletion.4 Since these first pivotal reports, with the advent of genome-wide methylation screening technologies, the number of imprinted DMRs in mammalian species has steadily increased, including those originating from the respective germlines and those that are somatically acquired.
Primary methylation defects of some well characterized imprinted DMRs are directly responsible for developmental disorders, including Beckwith-Wiedemann syndrome (OMIM 130,650) (BWS), Silver-Russell syndrome (OMIM 180,860) (SRS), Transient Neonatal Diabetes Mellitus (OMIM 601,410) (TNDM), Kagami-Ogata syndrome (OMIM 608,149) (KOS14), Temple syndrome (OMIM 616,222) (TS14), Prader-Willi syndrome (OMIM 176,270) (PWS), Angelman syndrome (OMIM 105,830) (AS), and Pseudohypoparathyroidism Ib (OMIM 103,580) (PHP1b) (a detailed description of human imprinting disorders is available at the webpage of the European Network for Human Congenital Imprinting Disorders, EUCID.net). The aberrant methylation often affects solely the disease-associated locus but, recently, some individuals have been reported to have additional imprinted DMRs affected, leading to a scenario termed multi-locus imprinting disturbance (MLID) (for a review, see ref5). Reporting these epigenomic data from molecular tests in laboratory reports or for publication is troubled by the lack of a uniform nomenclature. In this article we recommend unified names for imprinted DMRs and give details of their precise locations and suggest nomenclature for describing the results similar to those routinely used for DNA sequence variants.
Consensus for names of imprinted DMR
From the earliest days of the molecular descriptions of imprinting aberrations it became obvious that recording methylation defects would be challenging. This was evident simply because some imprinted genes have historically had multiple names, since many were identified simultaneously by independent groups who have termed the transcripts and DMRs differently. This is exemplified by the maternally methylated region overlapping the promoter of KCNQ1OT1 within intron 10 of KCNQ1 on chromosome 11, which has more than five aliases (Table 1). Ultimately, this causes confusion when cross-referencing original literature and modern databases. To improve this situation, the 41 EUCID members from 22 countries, have developed a uniform nomenclature system for reporting methylation aberrations. The final consensus after careful consideration was that the name of an imprinted DMR should be attributed to the nearest transcript with an approved symbol from the Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC).
Table 1.
The extent of imprinted methylation defined by methyl-seq data sets with the commonly used name for each imprinted DMR, those proposed by of EUCID using HGNC approved gene names, previous aliases and LRG identifiers. For completeness, origin of the allelic methylation is given, as are any associated disorders and information whether the methylation is germline or somatically derived. Secondary DMRs are regions of differential methylation, the establishment of which is often somatically acquired and dependent on hierarchical interactions with a neighboring germline DMR. All coordinates are given as GRCh37/hg19. M, maternally derived methylation; P, paternally derived methylation; gDMR, germline DMR; PHP1b, Pseudohypoparathyroidism; SRS, Silver Russell syndrome; BWS, Beckwith Wiedemann syndrome; AS, Angelman syndrome; PWS, Prader Willi syndrome; MLID, Mutlilocus imprinting disturbance; TS14, Temple syndrome; KOS14, Kagami-Ogata syndrome; TNDM, Transient Neonatal Diabetes Mellitus. All relevant DMR information and aliases can also be found in the “community” section of each LRG record.
| Extent of DMR (methyl-seq) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Imprinted DMR name |
Chr |
Start |
Finish |
Number CpGs |
Methylation origin |
Germline derived |
LRG identifier |
Aliases |
| PPIEL:Ex1-DMR | 1 | 40,024,626 | 40,025,540 | 39 | M | Oocyte gDMR | ||
| DIRAS3:TSS-DMR | 1 | 68,515,433 | 68,517,545 | 88 | M | Oocyte gDMR | LRG_1,034 | NOEY2, ARH1 |
| DIRAS3:Ex2-DMR | 1 | 68,512,505 | 68,513,486 | 39 | M | Oocyte gDMR | LRG_1,034 | |
| GPR1-AS:TSS-DMR | 2 | 207,066,967 | 207,069,445 | 86 | M | Oocyte gDMR | ||
| ZDBF2/GPR1:IG-DMR | 2 | 207,114,583 | 207,136,544 | 439 | P | Sperm gDMR-secondary DMR | LRG_1,065 | |
| NAP1L5:TSS-DMR | 4 | 89,618,184 | 89,619,237 | 57 | M | Oocyte gDMR | LRG_1,072 | |
| VTRNA2–1:DMR | 5 | 135,414,802 | 135,416,645 | 76 | M | Oocyte gDMR | nc886 | |
| FAM50B:TSS-DMR | 6 | 3,849,082 | 3,850,359 | 90 | M | Oocyte gDMR | LRG_1,062 | |
| PLAGL1:alt-TSS-DMR | 6 | 144,328,078 | 144,329,888 | 143 | M | Oocyte gDMR | LRG_1,101 | LOT1, ZAC1 |
| IGF2R:Int2-DMR | 6 | 160,426,558 | 160,427,561 | 74 | M | Oocyte gDMR | LRG_1,036 | |
| WDR27:Int13-DMR | 6 | 170,054,504 | 170,055,618 | 58 | M | Oocyte gDMR | LRG_1,071 | |
| GRB10:alt-TSS-DMR | 7 | 50,848,726 | 50,851,312 | 171 | M | Oocyte gDMR | LRG_1,032 | |
| PEG10:TSS-DMR | 7 | 94,285,537 | 94,287,960 | 119 | M | Oocyte gDMR | LRG_1,097 | |
| MEST:alt-TSS-DMR | 7 | 130,130,122 | 130,134,388 | 226 | M | Oocyte gDMR | LRG_1,033 | PEG1 |
| SVOPL:alt-TSS-DMR | 7 | 138,348,118 | 138,349,069 | 31 | M | Oocyte gDMR | LRG_1,103 | |
| HTR5A:TSS-DMR | 7 | 154,862,719 | 154,863,382 | 55 | M | Oocyte gDMR | LRG_1,057 | |
| ERLIN2:Int6-DMR | 8 | 37,604,992 | 37,606,088 | 37 | M | Oocyte gDMR | LRG_1,040 | |
| PEG13:TSS-DMR | 8 | 141,108,147 | 141,111,081 | 193 | M | Oocyte gDMR | LRG_1,041 | TRAPPC9 intronic DMR |
| FANCC:Int1-DMR | 9 | 98,075,400 | 98,075,744 | 26 | M | Oocyte gDMR | LRG_497 | |
| INPP5F:Int2-DMR | 10 | 121,578,046 | 121,578,727 | 52 | M | Oocyte gDMR | LRG_1,061 | |
| H19/IGF2:IG-DMR | 11 | 2,018,812 | 2,024,740 | 250 | P | Sperm gDMR | LRG_1,030 | ICR1, IC1, H19 DMR, H19 DMD,IGF2-H19 DMR |
| IGF2:Ex9-DMR | 11 | 2,153,991 | 2,155,112 | 63 | P | No-secondary DMR | LRG_1,031 | |
| IGF2:alt-TSS-DMR | 11 | 2,168,333 | 2,169,768 | 33 | P | Sperm gDMR | LRG_1,031 | |
| KCNQ1OT1:TSS-DMR | 11 | 2,719,948 | 2,722,259 | 192 | M | Oocyte gDMR | LRG_1,052 | ICR2, IC2, KvDMR1, KvLQT1-A, LIT1 |
| RB1:Int2-DMR | 13 | 48,892,341 | 48,895,763 | 195 | M | Oocyte gDMR | LRG_517 | |
| MEG3/DLK1:IG-DMR | 14 | 101,275,427 | 101,278,058 | 64 | P | Sperm gDMR | LRG_1,044 | GLT2-DLK1 |
| MEG3:TSS-DMR | 14 | 101,290,524 | 101,293,978 | 188 | P | No-secondary DMR | LRG_1,098 | GTL2 |
| MEG8:Int2-DMR | 14 | 101,370,741 | 101,371,419 | 43 | M | No-secondary DMR | LRG_1,058 | |
| MKRN3:TSS-DMR | 15 | 23,807,086 | 23,812,495 | 109 | M | Oocyte gDMR-secondary DMR | LRG_1,045 | |
| MAGEL2:TSS-DMR | 15 | 23,892,425 | 23,894,029 | 51 | M | No-secondary DMR | LRG_1,046 | |
| NDN:TSS-DMR | 15 | 23,931,451 | 23,932,759 | 108 | M | No-secondary DMR | LRG_1,047 | |
| SNRPN:alt-TSS-DMR | 15 | 25,068,564 | 25,069,481 | 19 | M | No-secondary DMR | ||
| SNRPN:Int1-DMR1 | 15 | 25,093,008 | 25,193,829 | 44 | M | No-secondary DMR | ||
| SNRPN:Int1-DMR2 | 15 | 25,123,027 | 25,123,905 | 45 | M | No-secondary DMR | ||
| SNURF:TSS-DMR | 15 | 25,200,004 | 25,201,976 | 113 | M | Oocyte gDMR | ||
| IGF1R:Int2-DMR | 15 | 99,408,496 | 99,409,650 | 55 | M | Oocyte gDMR | LRG_1,055 | IRAIN |
| ZNF597:3′ DMR | 16 | 3,481,801 | 3,482,388 | 29 | M | Oocyte gDMR | LRG_1,066 | |
| ZNF597:TSS-DMR | 16 | 3,492,828 | 3,494,463 | 76 | P | No-secondary DMR | LRG_1,066 | NAT15 |
| ZNF331:alt-TSS-DMR1 | 19 | 54,040,510 | 54,042,212 | 125 | M | Oocyte gDMR | LRG_1,068 | |
| ZNF331:alt-TSS-DMR2 | 19 | 54,057,086 | 54,058,425 | 102 | M | Oocyte gDMR | LRG_1,068 | |
| PEG3:TSS-DMR | 19 | 57,348,493 | 57,353,271 | 221 | M | Oocyte gDMR | ZIM2, ZNF904 | |
| MCTS2P:TSS-DMR | 20 | 30,134,663 | 30,135,933 | 47 | M | Oocyte gDMR | psiMCT-1, MCTS2 | |
| NNAT:TSS-DMR | 20 | 36,148,604 | 36,150,528 | 135 | M | Oocyte gDMR | LRG_1,048 | PEG5 |
| L3MBTL1:alt-TSS-DMR | 20 | 42,142,365 | 42,144,040 | 84 | M | Oocyte gDMR | LRG_1,049 | ZC2HC3,KIAA0681 |
| GNAS-NESP:TSS-DMR | 20 | 57,414,039 | 57,418,612 | 257 | P | No-secondary DMR | LRG_1,051 | NESP55 |
| GNAS-AS1:TSS-DMR | 20 | 57,425,649 | 57,428,033 | 128 | M | Oocyte gDMR | LRG_1,051 | NESP-AS |
| GNAS-XL:Ex1-DMR | 20 | 57,428,905 | 57,431,463 | 200 | M | Oocyte gDMR | LRG_1,051 | secretogranin VI |
| GNAS A/B:TSS-DMR | 20 | 57,463,265 | 57,465,201 | 198 | M | No-secondary DMR | LRG_1,051 | secretogranin VI |
| WRB:alt-TSS-DMR | 21 | 40,757,510 | 40,758,276 | 43 | M | Oocyte gDMR | LRG_1,060 | |
| SNU13:alt-TSS-DMR | 22 | 42,077,774 | 42,078,873 | 63 | M | Oocyte gDMR | LRG_1,102 | NHP2L1 |
Furthermore, we named a DMR in such a way that it gives basic information regarding its localization in relation to the nearest RefSeq transcript with the use of several prefixes outlined in Fig. 1 (e.g., TSS for transcription start site, IG for intergenic, Int for intronic and alt-TSS for alternative transcription start site). The precise location of each imprinted DMR is derived from methyl-seq data from whole blood samples as described by Court et al.,6 which has base-pair resolution. To ensure that the same genomic regions are identifiable in subsequent genome builds, all imprinted domains, including their corresponding DMRs, have been submitted to obtain Locus Reference Genomic (LRG) identifiers.7 LRGs are manually curated reference sequence records specifically designed for the reporting of variants with clinical implications. The inclusion of stable and unique genomic, transcript, and protein reference sequences ensures that variants are unambiguously and consistently reported over time (www.lrg-sequence.org). The records will contain all relevant DMR annotations. Information regarding the recommended naming, localization, and sizes of each DMR are given in Table 1.
Figure 1.
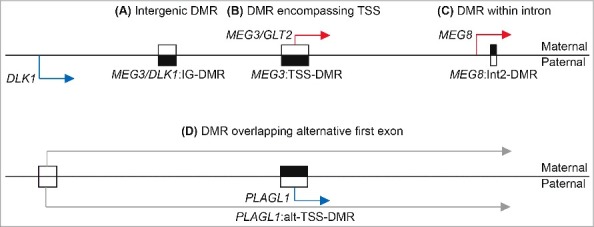
Schematic overview of the naming system for imprinted DMRs. The upper panel shows the three imprinted DMRs located within the chromosome 14q32.2 domain depicting (A) an intergenic DMR, (B) a DMR overlapping a transcription start site, and (C) an intronic DMR. The lower panel illustrates a DMR associated with (D) an alternative first exon as found within the PLAGL1 locus on chromosome 6q24. Black boxes represent methylated regions while open boxes are unmethylated. Blue arrows signify paternally expressed genes, red arrows denote maternally expressed genes, and gray arrows are biallelically expressed transcripts.Webpage URLsEUCID.net webpage. www.imprinting-disorders.eu285 Locus Reference Genomic identifier. www.lrg-sequence.orgHGVS nomenclature webpage. www.HGVS.org/varnomen
Standardization of reporting exact sites of imprinted methylation
It has previously been discussed that in order to allow correct identification and eventual reproduction of published observations, a universal system for the description of specific sites of DNA methylation tested needs to be employed.8 In the case of imprinted DMRs, this is reasonably straightforward if laboratories use commercially available methods to analyze methylation, such as methylation-sensitive multiplex ligation-dependent probe amplifications (MLPA) or high-density methylation arrays. In such cases, the precise location of the probe identifier, restriction site, or the interrogated CpG probes found on commonly used methylation profiling platforms can easily be identified, the genomic nucleotide tested accurately described, and methylation values reported. As an initial step to assist in this standardization, we have provided a resource listing all probes mapping to imprinted DMRs on the popular Infinium HumanMethylation450 BeadChips (Illumina, USA)(Suppl. Table 1), as well as the CpG dinucleotides interrogated by commonly used methylation-sensitive MLPA kits (MRC Holland, Netherlands)(Suppl. Table 2). For custom technologies, such as in-house pyrosequencing, for example, different CpG positions within the imprinted DMRs may be examined. In such cases, we recommend that the genomic coordinates targeted by the assays be listed and the methylation status described as an average percentage of all CpGs analyzed. However, such a description lacks resolution at the individual CpG level and, for future standardized reporting, it would be advantageous to have this information, not only for methylation at imprinted DMRs but also for all CpG positions in the genome. Such an approach could be based upon the current annotation of genomic locations as recommended by the Human Genome Variation Society (HGVS), allowing methylation values to be paired to each CpG position.9,10
Use of the suggested nomenclature
Following HGVS recommendations, methylation values at a specific region are described with (A) the chromosome number or LRG followed by (B) a colon :“;” (C) prefix “g.” for genomic DNA; (D) the position of the cytosine nucleotide or the range of nucleotides tested for the CpG containing interval; (E) the “|” character to indicate that it is a modification of the sequence not a sequence variant; (F) prefix describing the specific modification.
In collaboration with the HGVS' Sequence Variant Description Working Group (SVD-WG), it was decided to use the abbreviation “gom” to report a gain of methylation and “lom” for a loss. For non-specific methylation resistant to bisulphite conversion we suggest “bis” followed by a methylation value in brackets. If the molecular assay differentiates between 5-methylcytosine and its oxidative derivative 5-hydroxymethylcytosine, we propose the use of the “met” and “hmt,” respectively. This is consistent with HGVS standards to use three-letter abbreviations that do not include the nucleotide so that the modification can be added to any DNA base. When utilizing this format, it is important to mention the correct imprinted DMR name, the genome build used, and the technique used to measure the methylation status. This is because the EUCID COST action has previously reported that different methods targeting subtly different locations within the same imprinted DMR having different sensitivities.11,12 Furthermore, to help characterize variation due to tissue mosaicism, the tissue source from which the DNA is derived should be stated in any report because the methylation levels can be different in different tissues.13
For example, the nomenclature for a bisulphite PCR targeting the KCNQ1OT1:TSS-DMR negative DNA strand:
GRCh37/hg19 chr11:g. 2,722,063_ 2,722,087, in detail chr11:g. 2,722,063_ 2,722,087|met {0.52} indicates that the CpGs within this 24 bp interval have an average methylation of 52%. Using the LRGs, the nomenclature would be LRG_1,052:g.94506_94,530; LRG_1,052:g.94506_94,530|met {0.52}. For an example with loss of methylation of the same interval, the nomenclature would be GRCh37/hg19 chr11:g.2722063_ 2,722,087|lom, in detail chr11:g. 2,722,063_ 2,722,087|met {0.22} or LRG_1,052:g.94506_94,530; LRG_1,052:g.94506_94,530|met {0.22}.
An example of the same region reporting methylation at individual cytosines of CpG dinucleotides would be:
GRCh37/hg19 chr11:g. 2722063C|met {0.50}; 2722072C|met {0.54}; 2722074C|met {0.52}; 2722077C|met {0.53}; 2722083C|met {0.50}; 2722085C|met {0.51}; 2722087C|met {0.55} or LRG_1,052:g.94506C|met {0.50}; 94515C|met {0.54}; 94517C|met {0.52}; 94520C|met {0.53}; 94526C|met {0.50}; 94528C|met {0.51}; 94530C|met {0.55} in the sample with normal methylation. For the sample with loss of methylation, the nomenclature would be GRCh37/hg19 chr11:g. 2722063C|met {0.22}; 2722072C|met {0.26}; 2722074C|met {0.23}; 2722077C|met {0.17}; 27220783C|met {0.16}; 2722085C|met {0.26}; 2722087C|met {0.26} or LRG_1,052: g.94506C|met {0.22}; 94515C|met {0.26}; 94517C|met {0.23}; 94520C|met {0.17}; 94526C|met {0.16}; 94528C|met {0.26}; 94530C|met {0.26}.
In light of our suggestions, we encourage comments and discussion from clinical geneticists, molecular geneticists, and researchers from the epigenomics community and trust that the recommendations we have made for standardized reporting format will be useful for accurately communicating results. To give the wider epigenetics community the opportunity to be involved in the final discussions the proposed gom/lom nomenclature is open for community consultation on the HGVS webpage (see http://varnomen.hgvs.org/bg-material/consultation/svd-wg005/). We hope that by giving precise methylation values as percentages, it will overcome issues of comparing results between laboratories who often describe abnormalities using different methylation indexes.
The next issue that needs a consensus is defining the criteria to allow the description ‘lom’ or ‘gom’. This is complicated as not only statistical cut-offs need to be discussed (i.e., using mean ± standard deviation), but also the number of controls analyzed to define the normal range. Furthermore, utilizing fixed statistical criteria will be complicated in cases with mosaic epimutations, as methylation variance at different CpGs within a DMR need to be taken into account, as does the reproducibility of the molecular techniques used. 11
Supplementary Material
**Author appendix
Members of the Nomenclature group of the European Network for Human Congenital Imprinting Disorders
Frederic Brioude, Irene Netchine, Deborah Mackay, Pablo Lapunzina, Eammon Maher, Anne Rochtus, Guiomar Perez de Nanclares, Matthias Begemann, Lukas Soellner, Karen Grønskov, Jet Bliek, Maria Paola Lombardi, Lidia Larizza, Dorota Jurkiewicz, Francesca Elli, Serap Turan, Alessandra Baumer, Tiina Kehre, O'hlci Bronagh
AP-HP, Hospitaux Universitaires Paris Est (AP-HP) Hopital des Enfants Armand Trousseau, Service d'Eplorations Fonctionnalles Endocrtiniennes, 26 avenue du Dr Arnold Netter, 75012 Paris, France.
Human Genetics and Genomic Medicine, Faculty of Medicine University of Southampton, UK.
Instituto de Genética Médica y Molecular (INGEMM)-IdiPAZ, Hospital Universitario La Paz, Madrid, Spain; CIBERER, Centro de Investigación Biomédica en Red de Enfermedades Raras, ISCIII, Madrid, Spain.
Department of Medical Genetics, University of Cambridge and NIHR Cambridge Biomedical Research Center, Cambridge, UK.
Department of Pediatrics, University Hospitals Leuven, 3000 Leuven, Belgium.
Molecular (Epi)Genetics Laboratory, BioAraba National Health Institute, OSI Araba-Txagorritxu, E-01009 Vitoria-Gasteiz, Spain.
Department of Human Genetics, RWTH Aachen, Aachen, Germany.
Clinical Genetic Clinic, Kennedy Center, Rigshospitalet, Copenhagen University Hospital, Glostrup, Denmark.
Academic Medical Center, Department of Clinical Genetics, Laboratory for Genome Diagnostics, Meibergdreef 15, 1105AZ Amsterdam, Netherlands.
Laboratory of Cytogenetics and Molecular Genetics, Istituto Auxologico Italiano, Milan, Italy.
Department of Medical Genetics, Children's Memorial Health Institute, Warsaw, Poland.
Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Department of Clinical Sciences and Community Health, University of Milan, Endocrinology and Diabetology Unit, Milan, Italy.
Pediatric Endocrinology and Diabetes, Marmara University Hospital, Istanbul, Turkey.
Institute of Medical Genetics, University of Zürich, Schlieren-Zürich, Switzerland.
Department of Genetics, Tartu University Hospital, Tartu, Estonia; Department of Pediatrics, Institute of Molecular and Cell Biology, University of Tartu, Tartu, Estonia.
National Center for Medical Genetics, Ireland.
Funding Statement
This work was supported by the European COST action under grant number BM1208, the Bundesministerium für Bildung und Forschung (BMBF) under grant number 01GM1513A, B, C and D, the Spanish Ministerio de Educación y Competitividad (MINECO) under grant number BFU2014–53,093 co-funded with the European Union Regional Development Fund (FEDER), the Wellcome Trust under grant number WT200990/Z/16/Z and by the European Molecular Biology Laboratory.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Monk D. Germline-derived DNA methylation and early embryo epigenetic reprogramming: The selected survival of imprints. Int J Biochem Cell Biol 2015; 67:128-38; PMID:; https://doi.org/ 10.1016/j.biocel.2015.04.014 [DOI] [PubMed] [Google Scholar]
- 2.Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet 2011; 12(8):565-75; PMID:; https://doi.org/ 10.1038/nrg3032 [DOI] [PubMed] [Google Scholar]
- 3.Ferguson-Smith AC, Sasaki H, Cattanach BM, Surani MA. Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature 1993; 362(6422):751-5; PMID:; https://doi.org/ 10.1038/362751a0 [DOI] [PubMed] [Google Scholar]
- 4.Stöger R, Kubicka P, Liu CG, Kafri T, Razin A, Cedar H, Barlow DP. Maternal-specific methylation of the imprinted mouse Igf2r locus identifies the expressed locus as carrying the imprinting signal. Cell 1993; 73(1):61-71; PMID:; https://doi.org/ 10.1016/0092-8674(93)90160-R [DOI] [PubMed] [Google Scholar]
- 5.Sanchez-Delgado M, Riccio A, Eggermann T, Maher ER, Lapunzina P, Mackay D, Monk D. Causes and Consequences of Multi-Locus Imprinting Disturbances in Humans. Trends Genet 2016; 32(7):444-55; PMID:; https://doi.org/ 10.1016/j.tig.2016.05.001 [DOI] [PubMed] [Google Scholar]
- 6.Court F, Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, Sugahara N, Simón C, Moore H, Harness JV, et al.. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res 2014; 24(4):554-69; PMID:; https://doi.org/ 10.1101/gr.164913.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacArthur JA, Morales J, Tully RE, Astashyn A, Gil L, Bruford EA, Larsson P, Kileck P, Dalgleish R, Maglott DR, Cunningham F. Locus Reference Genomic: reference sequences for the reporting of clinically relevant sequence variants. Nucl Acid Res 2014; 42:D873-D878; PMID:; https://doi.org/ 10.1093/nar/gkt1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saffery R, Gordon L. Time for a standardized system of reporting sites of genomic methylation. Genome Biol 2015; 16:85; PMID:; https://doi.org/ 10.1186/s13059-015-0654-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum.Mutat 2016; 37(6):564-569; PMID:; https://doi.org/ 10.1002/humu.22981 [DOI] [PubMed] [Google Scholar]
- 10.Ogino S, Gulley ML, den Dunnen JT, Wilson RB, Association for Molecular Patholpogy Training and Education Committtee . Standard mutation nomenclature in molecular diagnostics: practical and educational challenges. J Mol Diagn 2007; 9(1):1-6; PMID:; https://doi.org/ 10.2353/jmoldx.2007.060081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soellner L, Monk D, Rezwan FI, Begemann M, Mackay D, Eggermann T. Congenital imprinting disorders: Application of multilocus and high throughput methods to decipher new pathomechanisms and improve their management. Mol Cell Probes 2015; 29(5):282-90; PMID:; https://doi.org/ 10.1016/j.mcp.2015.05.003 [DOI] [PubMed] [Google Scholar]
- 12.Garin I, Mantovani G, Aguirre U, Barlier A, Brix B, Elli FM, Freson K, Grybek V, Izzi B, Linglart A, et al.. European guidance for the molecular diagnosis of pseudohypoparathyroidism not caused by point genetic variants at GNAS: an EQA study. Eur J Hum Genet 2015; 23(4):560; PMID:; https://doi.org/ 10.1038/ejhg.2014.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azzi S, Blaise A, Steunou V, Harbison MD, Salem J, Brioude F, Rossignol S, Habib WA, Thibaud N, Neves CD, et al.. Complex tissue-specific epigenotypes in Russell-Silver syndrome associated with 11p15 ICR1 hypomethylation. Hum Mut 2014; 35(10):1211-20; PMID:; https://doi.org/ 10.1002/humu.22623 [DOI] [PubMed] [Google Scholar]
Webpage URLs
- EUCID.net webpage www.imprinting-disorders.eu
- Locus Reference Genomic identifier www.lrg-sequence.org
- HGVS nomenclature webpage www.HGVS.org/varnomen
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.