

Summary
Novel therapies with unique new targets are needed for patients who are relapsed/refractory to current treatments for multiple myeloma. Ibrutinib is a first‐in‐class, once‐daily, oral covalent inhibitor of Bruton tyrosine kinase, which is overexpressed in the myeloma stem cell population. This study examined various doses of ibrutinib ± low‐dose dexamethasone in patients who received ≥2 prior lines of therapy, including an immunomodulatory agent. Daily ibrutinib ± weekly dexamethasone 40 mg was assessed in 4 cohorts using a Simon 2‐stage design. The primary objective was clinical benefit rate (CBR; ≥minimal response); secondary objectives included safety. Patients (n = 92) received a median of 4 prior regimens. Ibrutinib + dexamethasone produced the highest CBR (28%) in Cohort 4 (840 mg + dexamethasone; n = 43), with median duration of 9·2 months (range, 3·0–14·7). Progression‐free survival was 4·6 months (range, 0·4–17·3). Grade 3–4 haematological adverse events included anaemia (16%), thrombocytopenia (11%), and neutropenia (2%); grade 3–4 non‐haematological adverse events included pneumonia (7%), syncope (3%) and urinary tract infection (3%). Ibrutinib + dexamethasone produced notable responses in this heavily pre‐treated population. The encouraging efficacy, coupled with the favourable safety and tolerability profile of ibrutinib, supports its further evaluation as part of combination treatment.
Keywords: multiple myeloma, ibrutinib, dexamethasone, Bruton tyrosine kinase
The management of relapsed or relapsed and refractory multiple myeloma (RRMM) has changed considerably in the last decade, and there have been significant improvements with regards to patient outcomes. One reason behind these improvements is the incorporation of novel drugs with unique mechanisms of action into the treatment armamentarium, which until then, included the use of alkylating agents, corticosteroids, anthracyclines and autologous stem cell transplantation (Dimopoulos et al, 2015a). The most notable classes of new drugs include immunomodulatory agents (IMiDs) and proteasome inhibitors (PIs). Despite these improvements, most patients with multiple myeloma (MM) will ultimately relapse; therefore, agents with novel targets must be identified and explored to be used either as single agents or in combination with standard backbone therapies (Cottini & Anderson, 2015). Thus, MM remains one of the most significant areas of unmet medical need among lymphoid malignancies. In addition, the treatment of patients with RRMM is further complicated by advanced age, comorbidities (i.e., diabetes, cardiovascular and/or lung disease), residual toxicities from prior treatments (i.e., peripheral neuropathies, myelosuppression) and end‐organ damage resulting from the underlying disease (Dimopoulos et al, 2015a; Nooka et al, 2015).
The B‐cell receptor‐signalling pathway has emerged as a new therapeutic target in B‐cell malignancies, including MM. Within this pathway, Bruton tyrosine kinase (BTK) plays a central role in the activation of downstream pathways associated with survival and proliferation of B cells (Anderson et al, 1996; Craxton et al, 1999; Petro et al, 2000; Petro & Khan, 2001; Buggy & Elias, 2012). It is expressed in >85% of tumour cells from MM patients, may help regulate myeloma stemness in the bone marrow of MM patients (Yang et al, 2015), and may play a role in treatment‐resistant MM cells (Tai & Anderson, 2012). Ibrutinib is a first‐in‐class, potent, once‐daily, orally administered, covalently binding inhibitor of BTK and is indicated for the treatment of patients with mantle cell lymphoma who have received ≥1 prior therapy, marginal zone lymphoma who require systemic therapy and have received ≥1 prior anti‐CD20–based therapy, chronic lymphocytic leukaemia including patients with chronic lymphocytic leukaemia with 17p deletion, and Waldenström macroglobulinaemia (Pharmacyclics, 2017). Ibrutinib was designed as a selective inhibitor of the BTK protein and, in vitro, inhibits BTK activity and induces apoptosis in human B‐cell lymphoma cell lines (Pan et al, 2007; Honigberg et al, 2010). Furthermore, ibrutinib decreased MM‐induced bone destruction and suppressed tumour growth in an in vivo mouse model (Tai & Anderson, 2012). Myeloma regimens are commonly augmented with dexamethasone (dex), which often improves response rates to other agents (Sonneveld & Broijl, 2016). BTK expression was upregulated at both the protein and mRNA levels in MM1R dex‐resistant cells, suggesting a possible role of BTK in the mechanism of dex resistance (Chauhan et al, 2002).
This study was designed to assess the efficacy and safety of ibrutinib, a new agent with a novel mechanism of action, both alone and in combination with dex, in patients with RRMM.
Methods
Patients
This study was approved by the Institutional Review Board at all participating sites, and all patients provided written informed consent. Key inclusion criteria included measurable, symptomatic MM according to International Myeloma Working Group (IMWG) criteria (Durie et al, 2006); RRMM after receiving ≥2 lines of therapy (including a PI or IMiD), including lack of response or disease progression (according to IMWG response criteria; Rajkumar et al, 2014) to the most recent line of therapy; and Eastern Cooperative Oncology Group (ECOG) performance status ≤1. Key exclusion criteria were an absolute neutrophil count <0·75 × 109/l; platelet count <50 × 109/l; creatinine level >221 μmol/l; peripheral neuropathy grade ≥2; and a need for concomitant warfarin or other vitamin K antagonists or strong CYP3A4/5 inhibitors (Indiana University Department of Medicine 2016).
Study objectives
The primary objective was to determine the efficacy of ibrutinib, both as a single agent and in combination with dex, in patients with RRMM, as measured by the clinical benefit rate [CBR; ≥minimal response (MR)]. Secondary objectives included duration of clinical benefit, overall response rate (ORR), duration of response, and safety; exploratory objectives were progression‐free survival (PFS), time to progression and overall survival (OS).
Study design and treatment plan
PCYC‐1111 (NCT01478581) was a phase 2, open‐label, nonrandomized, multicohort, multicentre, Simon 2‐stage study of daily ibrutinib with or without weekly dex in RRMM. Protocol amendment 1 was designed to further explore the optimal regimen by increasing doses of ibrutinib and/or by combining it with low‐dose (LD) dex. Cohorts 1 and 3 assessed activity of ibrutinib monotherapy (420 and 840 mg/day, respectively, with 40 mg of dex once weekly allowed on disease progression at the discretion of the investigator); Cohorts 2 and 4 assessed ibrutinib (560 and 840 mg/day, respectively) in combination with 40 mg of oral dex once weekly (LD dex). The LD dex selected for this study corresponds to that typically used when given weekly in combination with other agents. The use of LD dex was established in combination with lenalidomide in a study that demonstrated superior safety and improved outcomes when compared with high‐dose dex (Rajkumar et al, 2010). An interim analysis was built into the study, if the enrolment targets were met after the pre‐specified Simon 2‐stage expansion criteria. In Stage 1, up to 18 patients could be enrolled into Cohorts 2, 3 and 4. If ≥3 patients with CBR were observed in Cohorts 2, 3 or 4 then, for Stage 2, the cohort(s) could be selected for expansion up to a total of 43 patients or until ≥8 patients with CBR were observed, whichever occurred first.
Study assessments
Patients had scheduled study visits on days 1, 2, 8 15, and 22 of Cycle 1. Thereafter, study visits occurred once per 28‐day cycle on day 1 (±2 days), continuing until Cycle 12 unless otherwise indicated. After Cycle 12, study visits occurred once every 2 cycles. Adverse events (AEs), ECOG performance status and blood samples for haematology and serum chemistry were collected at each study visit or as needed based on physician discretion. Myeloma‐specific assessments, including serum protein electrophoresis, urine protein electrophoresis, and/or serum free light‐chain assay and quantitative immunoglobulins, were obtained at the beginning of each treatment cycle to evaluate response.
CBR was defined as the proportion of patients who achieved stringent complete response, complete response, very good partial response, partial response (PR), or MR, as assessed by the modified IMWG response criteria (Rajkumar et al, 2011); patients were evaluated for response starting at Cycle 2 and required 2 consecutive assessments to confirm response. Assessments for response included M‐protein (serum protein electrophoresis, urine protein electrophoresis, serum immunofixation electrophoresis and serum free light‐chain assay), plasmacytoma evaluations and serum chemistries (e.g., calcium and albumin), with bone radiological examinations, bone marrow aspiration and biopsy performed when clinically indicated. Patients could continue to receive ibrutinib until progressive disease (PD), unacceptable toxicity or other protocol‐specified reason.
Treatment‐emergent AEs were coded using the MedDRA System Organ Class and Preferred Term (https://www.meddra.org. Last accessed 30 October 2017); the frequency, severity, and relationship to the study drug were assessed by the investigator. All reported treatment‐emergent AEs described are included regardless of investigator attribution. Study investigators assessed the occurrence of AEs and serious AEs at all patient evaluation time points during the study, and AEs were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 4·0 (https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf). All AEs and serious AEs were recorded, including duration, severity, suspected relationship to the study drug and any actions taken. The AE reporting period began with the first dose of study drug and ended 30 days after the last dose of study drug.
Patient follow‐up
For safety, each patient was followed for 30 (±7) days after the last dose of ibrutinib or until the start of a subsequent antineoplastic therapy, whichever came first. Patients who discontinued study treatment for reasons other than PD were followed approximately every 2 months until PD or the start of subsequent antineoplastic therapy. Once patients progressed or started use of a subsequent anticancer therapy, they were contacted for long‐term follow‐up every 3 months to assess survival, the use of subsequent antineoplastic therapy, and other malignancies, until last active patient discontinued study treatment.
Data analysis
The primary end point of the study was CBR, defined as the proportion of patients achieving an MR or better as assessed by investigator per modified IMWG criteria (Durie et al, 2006; Anderson et al, 2008). The primary efficacy analysis was performed on the all‐treated population. CBR and its corresponding 2‐sided 90% exact binomial confidence interval (CI) were calculated. Cohort 1 was planned to enrol with a target size of 35 patients if ≥2 patients with CBR were observed among the first 11 patients. This cohort was designed to test the null hypothesis that the CBR is ≤10% versus the alternative hypothesis of ≥30%, at a 1‐sided alpha of 5% with 85% power. Cohorts 2, 3 and 4 were planned with a sample size of up to 43 patients if ≥3 patients with CBRs were observed among the first 18 patients in the cohort, with 80% power and the alternative hypothesis of CBR rate ≥25%. The primary analysis was performed based on the assigned cohort treatment and does not include the response after the addition of dex for patients in Cohorts 1 and 3.
PFS was defined as the duration from start of the treatment to PD or death (regardless of cause of death), whichever came first. Kaplan‐Meier methods were used to estimate event‐free survival curves and corresponding quartiles (including the median). A 2‐sided 95% CI was provided for the median PFS.
Results
Patient characteristics
Ninety‐two patients with RRMM were enrolled between 20 March 2012 and 19 September 2014. Cohort 4 (ibrutinib 840 mg + LD dex) met the pre‐specified Simon 2‐stage expansion criteria, and enrolled a total of 43 patients. Median age was 62–66 years among the 4 cohorts; with the exception of cytogenic differences that were determined locally and difficult to interpret because of small sample size, patient baseline disease characteristics were similar between cohorts (Table 1). Prior treatment exposure patterns changed as subsequent cohorts were enrolled (Table 2). All patients had received prior steroid treatment, and prior treatment exposure changed between groups; for example, Cohorts 3 and 4 had more patients who received prior carfilzomib and pomalidomide given their increasing use in this setting since study initiation. In addition, 13% of patients were previously exposed to a monoclonal antibody. Overall, patients had received a median of 3·5 prior regimens (range, 2–14). In cohort 4, 70% were refractory to their last line of therapy; of these patients, 70% received either a PI or IMiD in that line, and 58% were dual refractory (PI and IMiD). Median follow‐up at the time of analysis was 23 months.
Table 1.
Patient characteristics
| Characteristic | Cohort 1 (n = 13) | Cohort 2 (n = 18) | Cohort 3 (n = 18) | Cohort 4 (n = 43) | Overall (N = 92) |
|---|---|---|---|---|---|
| Median age – years (range) | 62 (49–74) | 66 (46–77) | 66 (54–81) | 65 (43–81) | 65 (43–81) |
| Male – n (%) | 8 (62) | 9 (50) | 13 (72) | 26 (60) | 56 (61) |
| ECOG PS – % | |||||
| 0 | 54 | 33 | 44 | 47 | 45 |
| 1 | 46 | 67 | 56 | 53 | 55 |
| Median time since diagnosis – years | 3.9 | 5.0 | 6.3 | 6.5 | 5.9 |
| Measurable disease – n (%) | |||||
| SPEP/UPEP | 11 (85) | 14 (78) | 16 (89) | 33 (77) | 74 (80) |
| sFLC | 2 (15) | 4 (22) | 2 (11) | 10 (23) | 18 (20) |
| Disease status to last treatmenta – n (%) | |||||
| Relapsed | 4 (31) | 2 (11) | 4 (22) | 13 (30) | 23 (25) |
| Relapsed and refractory | 9 (69) | 16 (89) | 13 (72) | 30 (70) | 68 (74) |
| Last line of therapy – n (%) | |||||
| PI and/or IMiD | 11 (85) | 14 (78) | 13 (72) | 39 (91) | 77 (84) |
| No PI or IMiD | 2 (15) | 4 (22) | 5 (28) | 4 (9) | 15 (16) |
| Chromosomal abnormalities by FISH – n (%) | |||||
| t(11;14) | 1 (8) | 1 (6) | 5 (28) | 8 (19) | 15 (16) |
| del13q14 | 5 (38) | 3 (17) | 3 (17) | 4 (9) | 15 (16) |
| t(4;14) | 2 (15) | 5 (28) | 5 (28) | 1 (2) | 13 (14) |
| del17p | 3 (23) | 5 (28) | 0 (0) | 4 (9) | 12 (13) |
| High‐risk cytogeneticsb – n (%) | 5 (38) | 8 (44) | 5 (28) | 5 (12) | 23 (50) |
| ISS stage – n (%) | |||||
| I | 6 (46) | 6 (33) | 8 (44) | 23 (54) | 43 (47) |
| II | 6 (46) | 8 (44) | 8 (44) | 16 (37) | 38 (41) |
| III | 1 (8) | 4 (22) | 2 (11) | 4 (9) | 11 (12) |
ECOG PS, Eastern Cooperative Oncology Group performance status; FISH, fluorescence in situ hybridization; IMiD, immunomodulatory agent; ISS, International Staging System; PI, proteasome inhibitor; sFLC, serum free light chains; SPEP/UPEP, serum protein electrophoresis/urine protein electrophoresis.
The status of 1 patient in Cohort 3 was unknown.
High‐risk cytogenetics defined as those patients with del17p or t(4;14).
Table 2.
Prior treatment exposure
| Prior treatment regimen | Cohort 1 (n = 13) | Cohort 2 (n = 18) | Cohort 3 (n = 18) | Cohort 4 (n = 43) | Overall (N = 92) |
|---|---|---|---|---|---|
| Median prior treatments – n (range) | 3 (2–10) | 4 (2–11) | 3 (2–14) | 4 (2–10) | 3.5 (2–14) |
| Akylator – n (%) | 13 (100) | 17 (94) | 14 (78) | 40 (93) | 84 (91) |
| Refractory | 6 (46) | 10 (56) | 7 (39) | 17 (40) | 40 (43) |
| Thalidomide – n (%) | 3 (23) | 9 (50) | 11 (61) | 25 (58) | 48 (52) |
| Refractory | 1 (8) | 5 (28) | 5 (28) | 6 (14) | 17 (18) |
| Lenalidomide – n (%) | 13 (100) | 18 (100) | 16 (89) | 39 (91) | 86 (93) |
| Refractory | 9 (69) | 14 (78) | 9 (50) | 27 (63) | 59 (64) |
| Pomalidomide – n (%) | 1 (8) | 1 (6) | 3 (17) | 13 (30) | 18 (20) |
| Refractory | 1 (8) | 0 (0) | 3 (17) | 12 (28) | 16 (17) |
| Bortezomib – n (%) | 13 (100) | 18 (100) | 15 (83) | 39 (91) | 85 (92) |
| Refractory | 6 (46) | 14 (78) | 10 (56) | 22 (51) | 52 (57) |
| Carfilzomib – n (%) | 1 (8) | 5 (28) | 6 (33) | 14 (33) | 26 (28) |
| Refractory | 0 (0) | 5 (28) | 6 (33) | 12 (28) | 23 (25) |
| Monoclonal antibody – n (%) | 1 (8) | 4 (22) | 2 (11) | 4 (9) | 12 (13) |
| Elotuzumab | 0 (0) | 2 (11) | 1 (6) | 0 (0) | 3 (3) |
| Othera | 1 (8) | 2 (11) | 1 (6) | 5 (12) | 9 (10) |
| Autologous stem cell transplant – n (%) | 11 (85) | 14 (78) | 13 (72) | 33 (77) | 71 (77) |
| PI and IMiD – n (%) | 13 (100) | 18 (100) | 15 (83) | 40 (93) | 86 (93) |
IMiD, immunomodulatory agent; PI, proteasome inhibitor.
Other includes investigational (BB‐1091, BHQ880, BMS‐936564, BT062; n = 4), rituximab (n = 2), nivolumab (n = 1), tositumomab (n = 1), and SAR650984 (n = 1).
Overall response
The rate of stable disease (SD) stabilization or response increased with dose (Table 3). The highest CBR was observed in Cohort 4 (28%), including 2 PRs and 10 MRs, with median duration of 9·2 months (range, 3·0–14·7). An additional 23% of patients had SD ≥4 cycles, despite patients actively progressing at the time of enrolment. In the 25 patients (58%) who were dual refractory in Cohort 4, 4 patients (16%) achieved an MR, with an additional 5 patients (20%) maintaining SD ≥4 cycles. Cohort 3 had 33% SD and no responses.
Table 3.
Overall response by IMWG criteria
| Response | Cohort 1a (n = 13) | Cohort 2 (n = 18) | Cohort 3a (n = 18) | Cohort 4 (n = 43) |
|---|---|---|---|---|
| PR – n (%) | 0 (0) | 1 (6) | 0 (0) | 2 (5) |
| MR – n (%) | 1 (8) | 0 (0) | 0 (0) | 10 (23) |
| SD ≥4 cycles – n (%) | 1 (8) | 4 (22) | 6 (33) | 10 (23) |
| SD <4 cycles – n (%) | 5 (38) | 10 (56) | 5 (28) | 12 (28) |
| PD – n (%) | 5 (38) | 2 (11) | 4 (22) | 6 (14) |
| NE – n (%) | 0 (0) | 1 (6) | 1 (6) | 0 (0) |
| CBR (≥MR) – % | 8 | 6 | 0 | 28 |
| ORR (≥PR) – % | 0 | 6 | 0 | 5 |
CBR, clinical benefit rate; IMWG, International Myeloma Working Group; MR, minimal response; NE, not evaluable; ORR, overall response rate; PD, progressive disease; PR, partial response; SD, stable disease.
Cohort 1 has 1 patient with unconfirmed PD; Cohort 3 has 2 patients with unconfirmed PD; Cohort 4 has 3 patients with unconfirmed PD.
The primary analysis was performed based on the assigned cohort treatment and does not include the response after the addition of dexamethasone.
Progression‐free survival
Median time of PFS of each cohort is shown in Fig 1. Compared with Cohorts 1–3 [Cohort 1, 0·9 months (range, 0·5–36·0+); Cohort 2, 3·7 months (range, 0·8–8·3); Cohort 3, 2·8 months (range, 0·4–14·0)], Cohort 4 had the highest PFS at 4·6 months (range, 0·4+ to 17·3+). The PFS was higher in those cohorts containing dex and in cohorts with higher doses of ibrutinib.
Figure 1.
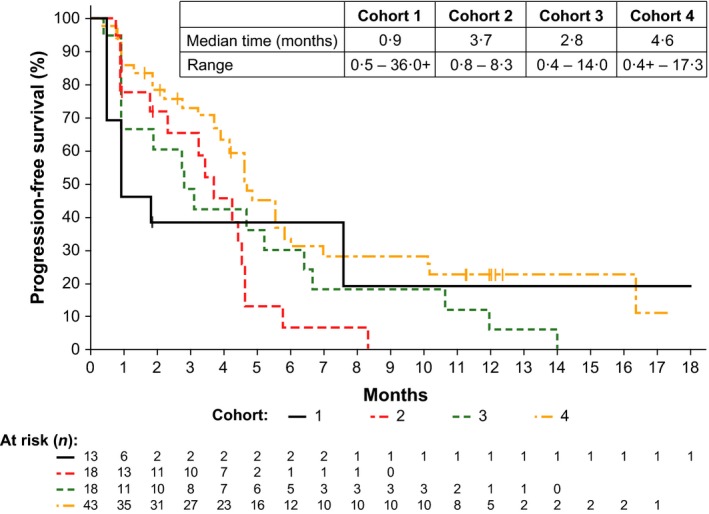
Progression‐free survival by treatment cohort. Cohort 4 showed a trend towards prolonged progression‐free survival at the highest dose of ibrutinib in combination with dexamethasone in a heavily pre‐treated population of patients with relapsed/refractory multiple myeloma. Tick mark indicates censored patients.
Safety
All treated patients received doses ranging from 420 to 840 mg of daily ibrutinib with median durations of ibrutinib treatment of 3·9, 2·6, 4·0, and 4·5 months in Cohort 1, 2, 3 and 4, respectively. No clinically significant differences in safety were observed across cohorts or in comparison with the established safety profile of ibrutinib (Table 4). Grade ≥3 treatment‐emergent AEs were experienced by 57% of all patients, and 29% experienced at least 1 serious AE. The most frequent AEs (all grades) included diarrhoea (53%), fatigue (43%), nausea (30%), anaemia (28%), thrombocytopenia (25%), muscle spasms (24%), cough (23%), insomnia (21%), upper respiratory tract infection (21%) and arthralgia (20%). The incidence of fatigue and diarrhoea were higher at the 840‐mg dose level of ibrutinib. However, these AEs were manageable with dose modification and/or concomitant therapy, and neither led to discontinuation of therapy. The rate of grade ≥3 haematological AEs was low, with 16% anaemia, 11% thrombocytopenia and 2% neutropenia. The most common grade ≥3 non‐haematological AE was pneumonia (7%). Rash was reported in 11 patients (12%), with no grade ≥3 events. Intervention for rash was required in 3 patients, with 1 patient requiring treatment delay and a subsequent dose reduction. Grade 3 atrial fibrillation occurred in 1 patient (1·1%), and grade 2 atrial flutter occurred in 1 patient (1·1%); neither of these patients had ongoing medical history of atrial fibrillation or atrial flutter at the time of study entry; no other atrial‐associated events of any grade were reported to date. Grade ≥3 acute kidney injury occurred in 1 patient after administration of intravenous (IV) contrast. Dose reductions due to AEs were reported in 9 patients (10%), with diarrhoea being the most frequent reason for dose reduction occurring in 4 patients. Of the 9 patients who had a dose reduction due to an AE, 3 patients subsequently discontinued treatment after the dose reduction due to grade 3 vision blurred, grade 1 renal impairment and grade 2 atrial flutter.
Table 4.
Treatment‐emergent adverse events
| Cohort 1 (n = 13) | Cohort 2 (n = 18) | Cohort 3 (n = 18) | Cohort 4 (n = 43) | Overall (N = 92) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Any grade | Grade 3/4 | Any grade | Grade 3/4 | Any grade | Grade 3/4 | Any grade | Grade 3/4 | Any grade | Grade 3/4 | |
| Haematological adverse events – n (%) | ||||||||||
| Anaemia | 31 | 23 | 33 | 28 | 33 | 17 | 23 | 9 | 28 | 16 |
| Thrombocytopenia | 38 | 0 | 28 | 22 | 22 | 11 | 21 | 9 | 25 | 11 |
| Neutropenia | 8 | 0 | 0 | 0 | 17 | 6 | 2 | 2 | 5 | 2 |
| Nonhaematological adverse events (>15%) – n (%) | ||||||||||
| Diarrhoea | 38 | 0 | 44 | 0 | 56 | 6 | 63 | 2 | 53 | 2 |
| Fatigue | 23 | 0 | 39 | 0 | 50 | 11 | 49 | 0 | 43 | 2 |
| Nausea | 46 | 0 | 28 | 0 | 39 | 0 | 23 | 0 | 30 | 0 |
| Muscle Spasms | 31 | 0 | 33 | 0 | 17 | 0 | 21 | 0 | 24 | 0 |
| Cough | 31 | 0 | 17 | 0 | 11 | 0 | 28 | 0 | 23 | 0 |
| Insomnia | 8 | 0 | 28 | 0 | 6 | 0 | 28 | 0 | 21 | 0 |
| Upper respiratory tract infection | 23 | 0 | 11 | 0 | 39 | 0 | 16 | 0 | 21 | 0 |
| Arthralgia | 23 | 0 | 22 | 0 | 22 | 0 | 16 | 0 | 20 | 0 |
| Dizziness | 15 | 0 | 22 | 0 | 11 | 0 | 21 | 0 | 18 | 0 |
| Back pain | 8 | 0 | 28 | 0 | 17 | 0 | 16 | 2 | 17 | 1 |
| Pain in the extremity | 8 | 0 | 28 | 6 | 0 | 0 | 23 | 2 | 17 | 2 |
| Pyrexia | 31 | 0 | 6 | 0 | 22 | 0 | 14 | 0 | 16 | 0 |
| Dyspnea | 15 | 0 | 22 | 0 | 11 | 6 | 14 | 2 | 15 | 2 |
Patient disposition
At the time of analysis, 8% of patients were still on treatment (1 patient in Cohort 1 and 6 in Cohort 4); all patients in Cohorts 2 and 3 had discontinued treatment. The most common reason for discontinuation across the cohorts was PD (60% overall). Other reasons for discontinuation across cohorts included investigator discretion (13%) and AE (10%; Table 5). AEs leading to treatment discontinuation included renal impairment (n = 2), with no other AEs occurring in more than 1 patient. One patient in Cohort 1 remains on study treatment (40 + cycles) at time of data cut‐off, having achieved an MR.
Table 5.
Patient disposition
| Disposition – n (%) | Cohort 1 (n = 13) | Cohort 2 (n = 18) | Cohort 3 (n = 18) | Cohort 4 (n = 43) | Overall (N = 92) |
|---|---|---|---|---|---|
| On treatment | 1 (8) | 0 (0) | 0 (0) | 6 (14) | 7 (8) |
| Discontinued treatment | 12 (92) | 18 (100) | 18 (100) | 37 (86) | 85 (92) |
| Progressive disease | 6 (46) | 11 (61) | 12 (67) | 26 (60) | 55 (60) |
| Investigator discretion | 3 (23) | 6 (33) | 2 (11) | 1 (2) | 12 (13) |
| Adverse event | 1 (8) | 0 (0) | 3 (17) | 5 (12) | 9 (10) |
| Othera | 2 (15) | 1 (6) | 1 (6) | 5 (12) | 9 (10) |
Includes patient withdrawal, noncompliance, and patient required prohibited concomitant medication.
Discussion
To date, no targeted kinase inhibitors have been approved for use in MM, despite the activity observed in other B‐cell malignancies. The BTK inhibitor ibrutinib, with or without weekly dex, demonstrated promising activity and was well tolerated in this heavily pre‐treated RRMM population with a median of 4 prior lines of therapy, including 74% of patients who had disease that was refractory to their most recent therapy. The AEs reported in this trial were consistent with the safety profile of ibrutinib observed across other B‐cell malignancies (Burger et al, 2015; Wang et al, 2015; Castillo et al, 2016; Falchi et al, 2016). The majority of AEs reported were grades 1–2 and were managed with supportive care. The incidence of grade ≥3 haematological AEs was consistent with that expected in this patient population, and no haematological AEs led to a dose reduction. Discontinuation of ibrutinib and dex due to AEs was uncommon (10%), suggesting that the treatment was generally well tolerated. Additionally, no increase in toxicity was observed with 840 mg of daily ibrutinib when compared with the other cohorts. The notable clinical activity demonstrated by the rate of sustained SD (≥4 cycles) or response observed in approximately half (51%) of the patients treated at 840 mg of daily ibrutinib in combination with dex is encouraging given the nature of the population evaluated. The highest activity was observed in Cohort 4 (at the highest dose of ibrutinib administered), with a 28% CBR, 5% ORR, 23% sustained SD and median PFS of 4·6 months. Of the 58% of patients who were double refractory to prior IMiDs and PIs and refractory to dex in this cohort, responses were seen in 16%, indicating the potential for benefit from ibrutinib and dex in this population.
Most patients with MM eventually relapse and the clinical benefit of treatment typically decreases with each subsequent line of therapy (Cottini & Anderson, 2015; Usmani et al, 2016). Treatment selection for MM should focus on improving long‐term outcomes. Median PFS with 840 mg of ibrutinib alone was 2·8 months compared with that for historical controls of 1·9 months with high‐dose dex alone (Dimopoulos et al, 2015b).The efficacy of ibrutinib appeared to be enhanced by the addition of LD dex, and compared with ibrutinib alone, increased the median PFS. Although no formal comparisons were made, increasing doses of ibrutinib to 560 to 840 mg was associated with longer PFS, with 3·7 months in the 560 mg + dex cohort (Cohort 3) and 4·6 months in 840 mg + dex cohort (Cohort 4), demonstrating the clinical activity of ibrutinib. In this setting, other agents have also shown to have enhanced activity in combination with dex. For example, carfilzomib + dex results in a PFS that ranges from 6·2 to 18·7 months, while carfilzomib alone ranged from 3·5 to 9 months; pomalidomide + dex increased PFS by 2·8 to 11·6 months, while pomalidomide alone was associated with a PFS of 4·6 to 9·5 months (Zou et al, 2017).
Although disease stabilization is not defined as a formal response to treatment, sustained SD in a heavily pretreated RRMM population with manageable toxicities compared with other treatment options suggests clinical benefit associated with ibrutinib treatment. Sustained disease stabilization for at least 4 cycles was observed in 10 of the 43 patients (23%) treated at the highest dose level of ibrutinib (840 mg) in combination with dex, with 2 of these patients receiving treatment and enjoying disease control beyond 1 year. In addition, 20% of patients who were double refractory to current backbone agents maintained disease stabilization for at least 4 cycles, further suggesting the potential activity of ibrutinib in this otherwise difficult‐to‐treat population.
Preclinical study results provided evidence that BTK plays a role in MM cell survival, with BTK overexpression observed in the majority of malignant plasma cells from patients with MM (Tai & Anderson, 2012; Bam et al, 2013). Moreover, the inhibition of BTK induces cytotoxicity in human MM cells (Rushworth et al, 2013), providing a scientific rationale for investigating ibrutinib as a therapeutic option for MM as a new combination partner with a novel mechanism of action. Preclinical studies have also demonstrated that ibrutinib could act synergistically with common backbone agents. Evidence of synergy between ibrutinib and the IMiD lenalidomide and the PI bortezomib has been observed in both MM patient cells and in MM cell lines, as evidenced by an increased cytotoxicity of malignant plasma cells (Rushworth et al, 2013). Additional preclinical data suggest that both BTK inhibitors and IMiDs target the clonogenic side populations of CD138neg cells, which are capable of clonogenic growth, self‐renewal and differentiation into myeloma plasma cells (Yang et al, 2006; Jakubikova et al, 2011; Beauvais et al, 2016). Increased BTK expression in the CD138neg side population cells is associated with clonogenic growth, increased expression of pluripotent/embryonic stem cell genes, and potential resistance to many standard myeloma treatments. In contrast, knockdown of BTK impeded these effects in vitro and was able to restore bortezomib sensitivity in BTK overexpressing cells (Yang et al, 2015).
The preliminary results of ibrutinib at 840 mg in combination with dex suggest clinical activity combined with a favourable safety/tolerability profile. Continued exploration of ibrutinib in triplet combinations with backbone agents, including pomalidomide and bortezomib, is warranted to potentially utilize synergistic effects observed preclinically. These investigations are underway in studies PCYC‐1138 (NCT02548962), PCYC‐1139 (NCT02902965), and PCYC‐1119 (NCT01962792). In PCYC‐1119, ibrutinib (840 mg) in combination with carfilzomib and dex has already shown early promising results with an initial ORR of 58%, a CBR of 67%, and no dose‐limiting toxicities during dose escalation (Chari et al, 2015). Additionally, augmenting the efficacy of immune therapies, such as monoclonal antibodies, with selective inhibitors, such as ibrutinib, is emerging as an area of great interest (Laubach & Richardson, 2015; van de Donk et al, 2016). Given the safety and activity of ibrutinib presented, future evaluation in combinations with next‐generation, small‐molecule inhibitors, monoclonal antibodies, and checkpoint inhibitors in patients with RRMM will allow evaluation of synergy across drug classes and with different targets with the hope to identify regimens that will provide greatest benefit to patients (Bianchi et al, 2015).
Author contributions
Contributions: All authors had access to primary clinical trial data. LE, FC, TG, and EB designed the study; PGR, WIB, CAH, CLC, NL, JGB, LDA, DSS, DL, SJ, JPL, KES‐G, and RV collected data; PGR, LK, FC, LE, ZS, TG, and EB analysed and interpreted the data; LK performed statistical analysis; and all authors were involved in manuscript preparation and approved the final version of the manuscript.
Conflict of interest
PGR has had a consultancy/advisory role with Celgene, Millenium/Takeda, Janssen, Novartis, Bristol‐Meyers Squibb (BMS) and Genmab. WIB has received honoraria from Celgene, Amgen and Takeda; has a consultancy/advisory role from Celgene, Sanofi and BMS; has received research funding from Celgene, Sanofi, Acetylon, BMS and Takeda; has been on the speakers' bureau of Celgene, Amgen and Takeda; and has provided expert testimony from Celgene and Takeda. CAH has had a consultancy/advisory role with Celgene, Glenmark, Takeda and Karyopharm; and has received research funding from Pharmacyclics LLC, an AbbVie Company, Karyopharm and Medimmune. CC has had a consultancy/advisory role with Celgene and Millennium/Takeda; has received honoraria from Celgene and Takeda; and has been on the speakers' bureau from Janssen. NL has employment with Janssen and has received research funding from Millennium, Karyopharm, GlaxoSmithKline and Sanofi. JGB has received research funding from Pharmacyclics LLC, an AbbVie company, Amgen, Teva, Constellation, Vivolux, Bluebird, Takeda, Celgene, BMS, AbbVie, Janssen and Novartis and has travelled with BMS, Celgene and Takeda. LDA has been on the speakers' bureau for Takeda, Onyx and Celgene. DSS has received honoraria, has had a consultancy/advisory role, and has been on the speaker's bureau for Celgene, Takeda, BMS, Amgen, Novartis and Merck. DL has received research funding from Celgene; has been on the speakers’ bureau for Amgen, Celgene and Takeda; and has received travel expenses from Amgen, Celgene and Takeda. SJ has received honoraria, has had a consultancy/advisory role, and has been on the speakers’ bureau for Celgene, Janssen, BMS, Medicom and Oncotracker; and has received research funding from Celgene, Janssen and BMS. JPL has received research funding from Onyx, Novartis, Celgene and Millennium. KES‐G has equity ownership with Abbot and AbbVie and has been on the speakers’ bureau for Janssen. LK is an employee of Pharmacyclics LLC, an AbbVie Company, and has equity ownership with AbbVie and Abbott. FC has had employment from Pharmacyclics LLC, an AbbVie Company; and has equity ownership with AbbVie. LE has had employment with Pharmacyclics LLC, an AbbVie Company; and has equity ownership with AbbVie, Gilead, Ziopharm, Exelixis and Karyopharm. ZS has employment with Pharmacyclics LLC, an AbbVie Company, and has equity ownership with AbbVie. TG has employment and patents/royalties/other intellectual property with Pharmacyclics LLC, an AbbVie Company, and has equity ownership with AbbVie. EB has employment with Pharmacyclics LLC, an Abbvie Company, and has equity ownership in AbbVie. RV has received honoraria from Takeda, Celgene, Amgen, BMS, Janssen, AbbVie, Karyopharm and Sanofi and research funding from Takeda and Amgen.
Acknowledgements
This study was supported by funding from Pharmacyclics LLC, an AbbVie Company and by a grant from the NIH/NCI Cancer Center Support (Grant P30 CA008748). We would like to thank Michelle Maglio, BS, of Dana‐Farber Cancer Institute, funded by the RJ Corman Multiple Myeloma Research Fund, and Brian Haas, PhD, a medical writer supported by funding from Pharmacyclics LLC, an AbbVie Company, for editorial assistance to the authors during preparation of this manuscript.
Presented in part as an oral presentation at the 2014 American Society of Hematology Annual Meeting and the 2015 Lymphoma & Myeloma Congress and as a poster presentation at the 2015 International Myeloma Workshop.
References
- Anderson, J.S. , Teutsch, M. , Dong, Z. & Wortis, H.H. (1996) An essential role for Bruton's tyrosine kinase in the regulation of B‐cell apoptosis. Proceedings of the National Academy of Science U S A, 93, 10966–10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, K.C. , Kyle, R.A. , Rajkumar, S.V. , Stewart, A.K. , Weber, D. & Richardson, P. ; ASH/FDA Panel on Clinical Endpoints in Multiple Myeloma (2008) Clinically relevant end points and new drug approvals for myeloma. Leukemia, 22, 231–239. [DOI] [PubMed] [Google Scholar]
- Bam, R. , Ling, W. , Khan, S. , Pennisi, A. , Venkateshaiah, S.U. , Li, X. , van Rhee, F. , Usmani, S. , Barlogie, B. , Shaughnessy, J. , Epstein, J. & Yaccoby, S. (2013) Role of Bruton's tyrosine kinase in myeloma cell migration and induction of bone disease. American Journal of Hematology, 88, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvais, D.M. , Jung, O. , Yang, Y. , Sanderson, R.D. & Rapraeger, A.C. (2016) Syndecan‐1 (CD138) suppresses apoptosis in multiple myeloma by activating IGF1 receptor: prevention by synstatinIGF1R inhibits tumor growth. Cancer Research, 76, 4981–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi, G. , Richardson, P.G. & Anderson, K.C. (2015) Promising therapies in multiple myeloma. Blood, 126, 300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggy, J.J. & Elias, L. (2012) Bruton tyrosine kinase (BTK) and its role in B‐cell malignancy. International Review of Immunology, 31, 119–132. [DOI] [PubMed] [Google Scholar]
- Burger, J.A. , Tedeschi, A. , Barr, P.M. , Robak, T. , Owen, C. , Ghia, P. , Bairey, O. , Hillmen, P. , Bartlett, N.L. , Li, J. , Simpson, D. , Grosicki, S. , Devereux, S. , McCarthy, H. , Coutre, S. , Quach, H. , Gaidano, G. , Maslyak, Z. , Stevens, D.A. , Janssens, A. , Offner, F. , Mayer, J. , O'Dwyer, M. , Hellmann, A. , Schuh, A. , Siddiqi, T. , Polliack, A. , Tam, C.S. , Suri, D. , Cheng, M. , Clow, F. , Styles, L. , James, D.F. & Kipps, T.J. ; RESONATE‐2 Investigators . (2015) Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. New England Journal of Medicine, 373, 2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo, J.J. , Palomba, M.L. , Advani, R. & Treon, S.P. (2016) Ibrutinib in Waldenström macroglobulinemia: latest evidence and clinical experience. Therapeutic Advances in Hematology, 7, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari, A.C.S. , Usmani, S. , Larson, S. , Niesvizky, R. , Matous, J. , Gasparetto, C. , Holkova, B. , Lunning, M. , Valent, J. , Anderson, L.D. , Karanes, C. , Kwei, L. , Chang, L. , Graef, T. , Bilotti, E. & McDonagh, K. (2015) The Bruton's tyrosine kinase inhibitor ibrutinib in combination with carfilzomib in patients with relapsed or relapsed and refractory multiple myeloma: initial results from a multicenter phase 1/2b study. Clinical Lymphoma, Myeloma, Leukemia, 15(Suppl. 3), e275–e276. [Google Scholar]
- Chauhan, D. , Auclair, D. , Robinson, E.K. , Hideshima, T. , Li, G. , Podar, K. , Gupta, D. , Richardson, P. , Schlossman, R.L. , Krett, N. , Chen, L.B. , Munshi, N.C. & Anderson, K.C. (2002) Identification of genes regulated by dexamethasone in multiple myeloma cells using oligonucleotide arrays. Oncogene, 21, 1346–1358. [DOI] [PubMed] [Google Scholar]
- Cottini, F. & Anderson, K. (2015) Novel therapeutic targets in multiple myeloma. Clinical Advances in Hematology and Oncology, 13, 236–248. [PubMed] [Google Scholar]
- Craxton, A. , Jiang, A. , Kurosaki, T. & Clark, E.A. (1999) Syk and Bruton's tyrosine kinase are required for B cell antigen receptor‐mediated activation of the kinase Akt. Journal of Biology and Chemistry, 274, 30644–30650. [DOI] [PubMed] [Google Scholar]
- Dimopoulos, M.A. , Richardson, P.G. , Moreau, P. & Anderson, K.C. (2015a) Current treatment landscape for relapsed and/or refractory multiple myeloma. National Review of Clinical Oncology, 12, 42–54. [DOI] [PubMed] [Google Scholar]
- Dimopoulos, M.A. , Weisel, K.C. , Song, K.W. , Delforge, M. , Karlin, L. , Goldschmidt, H. , Moreau, P. , Banos, A. , Oriol, A. , Garderet, L. , Cavo, M. , Ivanova, V. , Alegre, A. , Martinez‐Lopez, J. , Chen, C. , Spencer, A. , Knop, S. , Bahlis, N.J. , Renner, C. , Yu, X. , Hong, K. , Sternas, L. , Jacques, C. , Zaki, M.H. & San Miguel, J.F. (2015b) Cytogenetics and long‐term survival of patients with refractory or relapsed and refractory multiple myeloma treated with pomalidomide and low‐dose dexamethasone. Haematologica, 100, 1327–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Donk, N.W. , Moreau, P. , Plesner, T. , Palumbo, A. , Gay, F. , Laubach, J.P. , Malavasi, F. , Avet‐Loiseau, H. , Mateos, M.V. , Sonneveld, P. , Lokhorst, H.M. & Richardson, P.G. (2016) Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood, 127, 681–695. [DOI] [PubMed] [Google Scholar]
- Durie, B.G. , Harousseau, J.L. , Miguel, J.S. , Bladé, J. , Barlogie, B. , Anderson, K. , Gertz, M. , Dimopoulos, M. , Westin, J. , Sonneveld, P. , Ludwig, H. , Gahrton, G. , Beksac, M. , Crowley, J. , Belch, A. , Boccadaro, M. , Cavo, M. , Turesson, I. , Joshua, D. , Vesole, D. , Kyle, R. , Alexanian, R. , Tricot, G. , Attal, M. , Merlini, G. , Powles, R. , Richardson, P. , Shimizu, K. , Tosi, P. , Morgan, G. & Rajkumar, S.V. for the International Myeloma Working Group (2006) International uniform response criteria for multiple myeloma. Leukemia, 20, 1467–1473. [DOI] [PubMed] [Google Scholar]
- Falchi, L. , Baron, J.M. , Orlikowski, C.A. & Ferrajoli, A. (2016) BCR Signaling inhibitors: an overview of toxicities associated with ibrutinib and idelalisib in patients with chronic lymphocytic leukemia. Mediterranean Journal of Hematology and Infectious Diseases, 8, e2016011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigberg, L.A. , Smith, A.M. , Sirisawad, M. , Verner, E. , Loury, D. , Chang, B. , Li, S. , Pan, Z. , Thamm, D.H. , Miller, R.A. & Buggy, J.J. (2010) The Bruton tyrosine kinase inhibitor PCI‐32765 blocks B‐cell activation and is efficacious in models of autoimmune disease and B‐cell malignancy. Proceedings of the National Academy of Science U S A, 107, 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indiana University Department of Medicine (2016) The Flockhart Table™ P450 Drug Interaction Table. Available at: http://medicine.iupui.edu/clinpharm/ddis/main-table. (accessed 30 October 2017).
- Jakubikova, J. , Adamia, S. , Kost‐Alimova, M. , Klippel, S. , Cervi, D. , Daley, J.F. , Cholujova, D. , Kong, S.Y. , Leiba, M. , Blotta, S. , Ooi, M. , Delmore, J. , Laubach, J. , Richardson, P.G. , Sedlak, J. , Anderson, K.C. & Mitsiades, C.S. (2011) Lenalidomide targets clonogenic side population in multiple myeloma: pathophysiologic and clinical implications. Blood, 117, 4409–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubach, J.P. & Richardson, P.G. (2015) CD38‐targeted immunochemotherapy in refractory multiple myeloma: a new horizon. Clinical Cancer Research, 21, 2660–2662. [DOI] [PubMed] [Google Scholar]
- Nooka, A.K. , Kastritis, E. , Dimopoulos, M.A. & Lonial, S. (2015) Treatment options for relapsed and refractory multiple myeloma. Blood, 125, 3085–3099. [DOI] [PubMed] [Google Scholar]
- Pan, Z. , Scheerens, H. , Li, S.J. , Schultz, B.E. , Sprengeler, P.A. , Burrill, L.C. , Mendonca, R.V. , Sweeney, M.D. , Scott, K.C. , Grothaus, P.G. , Jeffery, D.A. , Spoerke, J.M. , Honigberg, L.A. , Young, P.R. , Dalrymple, S.A. & Palmer, J.T. (2007) Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem, 2, 58–61. [DOI] [PubMed] [Google Scholar]
- Petro, J.B. & Khan, W.N. (2001) Phospholipase C‐gamma 2 couples Bruton's tyrosine kinase to the NF‐kappaB signaling pathway in B lymphocytes. Journal of Biological Chemistry, 276, 1715–1719. [DOI] [PubMed] [Google Scholar]
- Petro, J.B. , Rahman, S.M. , Ballard, D.W. & Khan, W.N. (2000) Bruton's tyrosine kinase is required for activation of IkappaB kinase and nuclear factor kappaB in response to B cell receptor engagement. Journal of Experimantal Medicine, 191, 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharmacyclics, I. (2017) Imbruvica: Full Prescribing Information.
- Rajkumar, S.V. , Jacobus, S. , Callander, N.S. , Fonseca, R. , Vesole, D.H. , Williams, M.E. , Abonour, R. , Siegel, D.S. , Katz, M. & Greipp, P.R. ; Eastern Cooperative Oncology Group . (2010) Lenalidomide plus high‐dose dexamethasone versus lenalidomide plus low‐dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: an open‐label randomised controlled trial. Lancet Oncology, 11, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar, S.V. , Harousseau, J.L. , Durie, B.G. , Anderson, K.C. , Dimopoulos, M.A. , Kyle, R. , Blade, J. , Richardson, P. & Orlowski, R. (2011) Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood, 117, 4691–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar, S.V. , Dimopoulos, M.A. , Palumbo, A. , Blade, J. , Merlini, G. , Mateos, M.V. , Kumar, S. , Hillengass, J. , Kastritis, E. , Richardson, P. , Landgren, O. , Paiva, B. , Dispenzieri, A. , Weiss, B. , LeLeu, X. , Zweegman, S. , Lonial, S. , Rosinol, L. , Zamagni, E. , Jagannath, S. , Sezer, O. , Kristinsson, S.Y. , Caers, J. , Usmani, S.Z. , Lahuerta, J.J. , Johnsen, H.E. , Beksac, M. , Cavo, M. , Goldschmidt, H. , Terpos, E. , Kyle, R.A. , Anderson, K.C. , Durie, B.G. & Miguel, J.F. (2014) International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncology, 15, e538–e548. [DOI] [PubMed] [Google Scholar]
- Rushworth, S.A. , Bowles, K.M. , Barrera, L.N. , Murray, M.Y. , Zaitseva, L. & MacEwan, D.J. (2013) BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF‐κB. Cell Signalling, 25, 106–112. [DOI] [PubMed] [Google Scholar]
- Sonneveld, P. & Broijl, A. (2016) Treatment of relapsed and refractory multiple myeloma. Haematologica, 101, 995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai, Y.T. & Anderson, K.C. (2012) Bruton's tyrosine kinase: oncotarget in myeloma. Oncotarget, 3, 913–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usmani, S. , Ahmadi, T. , Ng, Y. , Lam, A. , Desai, A. , Potluri, R. & Mehra, M. (2016) Analysis of real‐world data on overall survival in multiple myeloma patients with ≥3 prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory drug (IMiD), or double refractory to a PI and an IMiD. Oncologist, 21, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M.L. , Blum, K.A. , Martin, P. , Goy, A. , Auer, R. , Kahl, B.S. , Jurczak, W. , Advani, R.H. , Romaguera, J.E. , Williams, M.E. , Barrientos, J.C. , Chmielowska, E. , Radford, J. , Stilgenbauer, S. , Dreyling, M. , Jedrzejczak, W.W. , Johnson, P. , Spurgeon, S.E. , Zhang, L. , Baher, L. , Cheng, M. , Lee, D. , Beaupre, D.M. & Rule, S. (2015) Long‐term follow‐up of MCL patients treated with single‐agent ibrutinib: Updated safety and efficacy results. Blood, 126, 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z.Z. , Novak, A.J. , Stenson, M.J. , Witzig, T.E. & Ansell, S.M. (2006) Intratumoral CD4 + CD25 + regulatory T‐cell‐mediated suppression of infiltrating CD4 + T cells in B‐cell non‐Hodgkin lymphoma. Blood, 107, 3639–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Shi, J. , Gu, Z. , Salama, M.E. , Das, S. , Wendlandt, E. , Xu, H. , Huang, J. , Tao, Y. , Hao, M. , Franqui, R. , Levasseur, D. , Janz, S. , Tricot, G. & Zhan, F. (2015) Bruton tyrosine kinase is a therapeutic target in stem‐like cells from multiple myeloma. Cancer Research, 75, 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, Y. , Ma, X. , Yu, H. , Hu, C. , Fan, L. & Ran, X. (2017) Carfilzomib/pomalidomide single‐agent or in combination with other agents for the management of relapsed/refractory multiple myeloma: a meta‐analysis of 37 trials. Oncotarget, 8, 39805–39817. [DOI] [PMC free article] [PubMed] [Google Scholar]