

Abstract
Mutations in fused in sarcoma (FUS) are linked to amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease affecting both upper and lower motor neurons. While it is established that astrocytes contribute to the death of motor neurons in ALS, the specific contribution of mutant FUS (mutFUS) through astrocytes has not yet been studied. Here, we used primary astrocytes expressing a N‐terminally GFP tagged R521G mutant or wild‐type FUS (WTFUS) and show that mutFUS‐expressing astrocytes undergo astrogliosis, damage co‐cultured motor neurons via activation of an inflammatory response and produce conditioned medium (ACM) that is toxic to motor neurons in isolation. Time lapse imaging shows that motor neuron cultures exposed to mutFUS ACM, but not WTFUS ACM, undergo significant cell loss, which is preceded by progressive degeneration of neurites. We found that Tumor Necrosis Factor‐Alpha (TNFα) is secreted into ACM of mutFUS‐expressing astrocytes. Accordingly, mutFUS astrocyte‐mediated motor neuron toxicity is blocked by targeting soluble TNFα with neutralizing antibodies. We also found that mutant astrocytes trigger changes to motor neuron AMPA receptors (AMPAR) that render them susceptible to excitotoxicity and AMPAR‐mediated cell death. Our data provide the first evidence of astrocytic involvement in FUS‐ALS, identify TNFα as a mediator of this toxicity, and provide several potential therapeutic targets to protect motor neurons in FUS‐linked ALS.
Keywords: AMPA receptor, cytokine, excitotoxicity, neurodegeneration
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is an incurable progressive neurodegenerative disease characterized by loss of motor function leading to respiratory failure and death within 3 to 5 years after onset. While the majority of cases occur with no family history and are sporadic (sALS), ∼10% of patients have an inherited familial form (fALS). fALS cases can be caused by genetic mutations in several genes, including C9orf72, Cu/Zn superoxide dismutase (SOD1), TAR‐DNA binding protein 43 (TDP43), fused in sarcoma/translocated in liposarcoma (FUS/TLS) and others. Over 35 mutations have been described in FUS, which has been identified as the primary cause of ALS type 6 (Vance et al., 2009). In healthy controls this multifunctional protein, with roles ranging from DNA repair, transcriptional regulation and mRNA transport (Lagier‐Tourenne, Polymenidou, & Cleveland, 2010), is located primarily in the nucleus (Dormann et al., 2010); however, post mortem tissues of FUS‐ALS patients reveal cytoplasmic inclusions in affected neurons and glia (Kwiatkowski et al., 2009). Specifically, carriers of the FUS R521C, R521G, R521H, R524W, or G507N mutations show wide‐spread FUS pathology (Blair et al., 2009; Hewitt et al., 2010; Rademakers et al., 2010), including glial and neuronal cell loss, with increasing distribution of FUS‐immunoreactive inclusions in patients with longer disease durations (Suzuki et al., 2012). Carriers of FUS R521C mutation also show numerous oligodendroglial cytoplasmic inclusions (Mackenzie et al., 2011). Abundant FUS‐immunoreactive pathology is also observed in some sALS cases (Oketa, Higashida, Fukasawa, Tsukie, & Ono, 2013).
Several lines of evidence suggest that non‐neuronal cells play an active role in driving neuronal dysfunction and neurodegeneration in ALS [reviewed (Lasiene and Yamanaka, 2011)]. Among these non‐neuronal cells, astrocytes are considered key mediators of disease progression in vivo and of motor neuron death in vitro. Chimeric mice in which mutant SOD1 was selectively deleted in astrocytes showed substantially slowed disease progression (Yamanaka et al., 2008) while both rodent and human patient‐derived astrocytes have been shown to play a toxic role through the release of, yet identified, soluble factors [reviewed (Haidet‐Phillips et al., 2011; Lasiene and Yamanaka, 2011). Furthermore, we recently showed that ALS astrocytes, including mutFUS expressing patient astrocytes, drive up‐regulation of the multidrug resistance transporter P‐Glycoprotein (P‐gp) in endothelial cells of the blood‐brain barrier (Qosa et al., 2016). Astrocytes participate in disease also through a “loss of function” [reviewed (Maragakis and Rothstein, 2006)] as demonstrated by several human and rodent studies that have observed defects and/or loss of astrocytic glutamate transporter EAAT2/GLT1 for several ALS subtypes including SOD1‐ALS (Benkler, Ben‐Zur, Barhum, & Offen, 2013; Howland et al., 2002) sALS (Lin et al., 1998), and C9orf72‐ALS (Kwon et al., 2014). In contrast, studies of astrocytic contributions in TDP‐43‐mediated ALS have shown conflicting results for both gain and loss‐of‐function toxicity (Haidet‐Phillips et al., 2013; Serio et al., 2013; Tong et al., 2013). Important questions still remain about astrocytic contributions to disease across different ALS‐subtypes, and in this work, we focused on the role played by astrocytes in FUS‐linked ALS.
One of the striking hallmarks of ALS is neuroinflammation. This is characterized by the appearance of reactive astrocytes and microglia, as well as macrophage and T‐lymphocyte infiltration, further strengthening the evidence that ALS is a noncell autonomous disease where non‐neuronal cells actively participate in neurodegeneration (Keller, Gravel, & Kriz, 2009; Levine, Kong, Nadler, & Xu, 1999; Rizzo et al., 2013). Proinflammatory cytokines such as tumor necrosis factor alpha (TNFα) are upregulated in the spinal cord of ALS patients, and presymptomatically in SOD1 G93A mice (Philips and Robberecht, 2011). Inflammation may also have a causal link to ALS as haploinsufficiency of the TBK1 gene, a regulator of inflammatory signaling, has been recently shown to cause ALS and frontotemporal lobe dementia (FTD) (Freischmidt et al., 2015). Though inflammation and astrocyte‐mediated toxicity have been identified as part of the pathogenic process of ALS [reviewed (Philips and Robberecht, 2011)], the role of TNFα has been widely debated, at least in the SOD1 mouse model. Approaches to globally eliminate single inflammatory genes in SOD1‐ALS mouse models have largely failed to extend survival (Gowing, Dequen, Soucy, & Julien, 2006) and treatment with the TNFα inhibitor, thalidomide, showed some promising results in rodents but did not appear to effectively modulate disease progression in patients and was additionally accompanied by significant side‐effects which prevented patients from reaching the target dosage (Stommel et al., 2009). However, whether inflammation in general and TNFα in particular play different pathogenic roles in other forms (non‐SOD1) of ALS remains to be solved.
Nuclear factor kappa B (NF‐κB), an inducible cellular transcription factor found in almost all cell types, plays a key role in regulating the inflammatory response and cellular stress response. Dysregulation of NF‐κB has been linked to cancer and neurodegenerative diseases such as Huntington's (Ghose, Sinha, Das, Jana, & Bhattacharyya, 2011) and ALS (Frakes et al., 2014; Prell et al., 2014). Interestingly, FUS augments NF‐ĸB‐dependent gene expression induced by physiological stimuli such as TNFα (Uranishi, 2001). TNFα and NF‐ĸB have been shown to participate in oscillatory positive feedback loops wherein NF‐ĸB can regulate TNFα transcription while autocrine and paracrine TNFα signals can in turn trigger patterns of NF‐ĸB activation (Pękalski et al., 2013). This process plays a key role in regulating inflammatory responses across a variety of cell types.
TNFα is a pleiotropic molecule and well‐studied trigger of apoptosis, however, there has recently been an increased interest in its role in CNS physiology and synaptic functions [reviewed (Santello and Volterra, 2012)]. In addition to classical caspase‐8 activation, one mechanism proposed for TNFα‐induced neuron death is through the rapid TNFα‐induced surface expression changes of AMPA‐type glutamate receptors (AMPARs) (Ferguson et al., 2008). Dysregulation of the precise trafficking can alter neuronal calcium permeability and contribute to excitotoxic vulnerability (Ferguson et al., 2008; Leonoudakis, Zhao, & Beattie, 2008; Olmos and Lladó, 2014).
Here, using primary astrocytes expressing mutFUS cocultured with primary motor neurons, we studied whether mutFUS‐expressing astrocytes mediate motor neuron dysfunction and began identification of the mechanisms by which mutFUS astrocytes damage the motor neurons. We show that mutFUS‐expressing astrocytes damage motor neurons through release of toxic factor(s) in the condition medium (ACM). Using microarray analysis of mutFUS astrocytes and ELISA and Western Blot analysis of mutFUS ACM and cell lysates, we identified secreted TNFα as the predominant factor, whose toxicity is blocked by anti‐TNFα neutralizing antibodies. Preventing astrocytic NF‐kB activation inhibits TNFα accumulation in the ACM and prevents mutFUS ACM induced motor neuron degeneration. Further, mutFUS ACM alters expression levels of AMPA receptor subunits GluA1 and GluA2 in motor neurons. We show that these changes sensitize motor neurons to AMPA receptor stimulation resulting in increased calcium influx leading to excitotoxic damage and cell death. We also show that these AMPAR changes are a result of TNFα acting on motor neurons. Our data provide the first evidence of astrocytic involvement in FUS‐ALS and identify TNFα and components of the AMPA‐mediated excitotoxic pathway as potential therapeutic targets for mutFUS‐ALS.
2. MATERIALS AND METHODS
2.1. Primary astrocytic culture
Primary astrocytes were prepared from spinal cord of nontransgenic mice (P2–P4). Briefly, flushed spinal cords were cut into small pieces followed by trypsin‐DNase treatment and mechanical resuspension. After plating for 2 weeks, cells were then shaken overnight for 4 days to remove nonspecific glia. Cell monolayers were >95% astrocyte‐pure as determined by glial fibrillary acidic protein (GLAST) immunostaining. Cells were maintained in culture in DMEM F12 50/50 (Cellgro, 10–092CV), 20% heat‐inactivated FBS (Cellgro, 35‐016‐CV), 1% Penn‐Strep (Cellgro, 30‐002‐CI), 1X Fungin (InVivoGen, ant‐fn‐2), 1X Primocin (InVivoGen, ant‐pm‐2), 0.25% Gentamicin (Invitrogen, 15710‐064).
2.2. Primary rat motor neuron culture
Motor neurons were prepared from e14.5 rat spinal cords. Briefly, 10–20 spinal cords were dissected and mechanically broken into small fragments before being incubated in 0.025% trypsin, followed by DNase. After a 4% w/v BSA cushion, cells were centrifuged for 55 min without brake through a 10.4% (v/v) Optiprep (Nycomed Pharma) cushion. Collected bands were then spun through a 4% w/v BSA cushion and treated with rat anti‐mouse p75NTR antibody for 30 min. Cells were then washed, spun through a 4% w/v BSA cushion and resuspended in goat anti‐mouse IgG microbeads for 15 min at +4°C. After 4% w/v BSA cushion magnetically labeled cells were loaded into a column attached to a magnetic base and allowed to pass through, collecting the effluent as a negative fraction on a centrifuge tube. After three rinses, the column was removed from the magnet and a flow resister was used to collect the positive fraction. A final 4% w/v BSA cushion was used to collect motor neurons in complete neurobasal medium (Nb‐C) (Neurobasal [Gibco, 21103], B27 supplement (2%), glutamine (0.25%), 2‐mercaptoethanol (0.1%), horse serum (2%). Cells were plated on polyornithine‐laminin coated coverslips at a density of 5000/18 mm coverslip. Motor neurons were maintained in Nb‐C for up to 2.5 weeks.
2.3. Coculture system
A confluent monolayer of astrocytes was plated a minimum of 48 hr before viral treatment in two‐chamber slides (Lab Tek, 177380) at a density of 5× 104 cells/cm2. Media was washed off 24 hr after viral induction and motor neurons were seeded on top at a density of 2 × 104 cells/cm2. Cocultures were maintained for additional 72 hr in L15‐complete (L15‐C) (L15 media [Cellgro, 10–045‐CV], 0.63 mg mL−1 NaHCO3 [Fisher, S233500], 5 mg mL−1 insulin [Sigma, I6634], 0.1 mg mL−1 conalbumin [Sigma, C7786], 0.1 mM putrescine [Sigma, P5780], 30 nM sodium selenite [Sigma, S5261], 20 nM progesterone [Sigma, P8783], 20 mM glucose, 0.1 mg mL−1 Primocin (InVivoGen, ant‐pm‐2), and 2% horse serum [Cellgro, 35‐015‐CV].)
2.4. Constructs
N‐terminally eGFP tagged human FUS in a pcDNA3.1/nV5‐DEST backbone was used to create R521G (CGC to GGC) mutant using the QuikChange Lightning Site‐Directed Mutagenesis Kit (Agilent 210518). Samples were sequenced using BGH pA forward and reverse primers. To allow for Gateway recombination into adenoviral expression plasmids, pENTR4 (Invitrogen) was opened at EcoR1 and BamH1 sites and the expression cassettes for wild‐type and mutant human FUS were released from pcDNA3.1/nV5‐DEST plasmids at BglII and NaeI sites and ligated to pENTR4 with EcoRV and BamHI. Finally, both expression cassettes were transferred by Gateway recombinase into adenoviral expression constructs, which were produced and amplified in HEK 293A cells (ViraPower Adenoviral Expression System; Invitrogen).
2.5. Viral induction
After astrocytes were allowed to adhere following trypsinization, an adenoviral N‐terminal GFP tagged FUS or C‐terminally eGFP tagged SOD1 construct was incubated with cells for 24 hr before being washed with HBSS and cell media. Multiplicity of infection (MOI) ranged from 3 to 5. Cells were then incubated for 3–4 days to allow maximal protein expression and used for experiments. Protein expression stabilized after 4 days and remained constant for 2 weeks or more.
2.6. Protein analysis
To access protein levels, western blotting was done per standard protocols and probed against GFP (Clontech, 632459) and Actin (Abcam, 8226), GAPDH (Fitzgerald, 10R‐G109A), GFAP (Cell Signaling, 3670), TNFa (Abcam, ab6671) with respective HRP‐conjugated secondary. Total protein levels were assessed by imaging the Mini‐PROTEAN TGX stain‐free gel prior to transfer. To visualize cells, pictures were taken while in culture of astrocytic GFP‐virus expression, as well as bright‐field motor neuron morphology during development. For immunofluorescence, cells were covered to a depth of 2–3 mm with 4% formaldehyde in PBS. After 15 min of fixation, cells were rinsed in PBS and blocked (PBS, 10% horse serum, 0.3% triton X‐100) for 60 min. Cells were then incubated overnight in primary antibody at 4°C. After PBS rinses and 1‐hr incubation in secondary antibody cells were then washed, aspirated, wells removed and coversliped with Prolong Gold Antifade DAPI Reagent (Invitrogen, P36935). The following primary antibodies were used for characterization and neuronal morphology: p75NTR 1:200 (Millipore, 07–476), GLAST 1:200 (Abcam, ab416 or A522 custom polyclonal where notated), ChAT 1:100 (Millipore, AB144P). Slides were imaged using either Zeiss LSM 510 Meta Confocal Laser Scanning Microscope (Bioimaging Shared Resource of the Kimmel Cancer Center [NCI 5 P30 CA‐56036]) or using an Olympus BX51with MBF bioscience Stereo Investigator
For AMPAR visualization, DIV 9 motor neurons were treated with ACM for 72 hr following which they were probed for GluA1 and GluA2 by immunocytochemistry. The following primary antibodies were used: GluA1 (1:200, Millipore AB1504) GluA2 (1:200, Millipore MAB397), MAP‐2 (1:500, Millipore AB5622), and ChAT (1:100, Millipore AB15468). Coverslips were imaged using a ×60 oil immersion objective of a confocal microscope (Olympus FV1000). Laser and image acquisition settings were kept constant for all the conditions compared. ROIs along the dendrites of motor neurons were selected on the basis of MAP‐2 labeling. Fluorescence intensity of the ROIs was measured with Fluoview Viewer software (Olympus) on a scale of 0–4095 pixels
2.7. Assessment of neuronal length
Each slide was scanned systematically to cover every field of view and each motor neuron was imaged and saved. Motor neurons were identified by their lack of GFP‐virus expression, clear morphology, and positive staining of markers. Images were then loaded into Image J and subsequently analyzed using the Neuron J plug‐in. Each neurite was individually traced and the sum of neurites for each individual neuron was then used for analysis.
2.8. Enzyme‐linked immunosorbent assay
Levels of TNFα in astrocyte culture medium were determined by ELISA (DuoSet ELISA Development System, R&D Systems, DY410). Briefly, 96‐well microplates were incubated with the capture antibody overnight, at room temperature. Following coating, plates were blocked with 1% BSA in PBS and then washed with 0.05% Tween 20 in PBS. Samples and standards were then added and allowed to incubate for 2 hr while shaking at room temperature. Plates were then washed and allowed to incubate with the biotinylated goat anti‐mouse TNFα detection antibody for 2 hr while shaking at room temperature, then washed again. Finally, we incubated the plates with Streptavidin‐HRP and a substrate solution for 30‐min each. A stop solution of 1N H2SO4 was added just prior to plate reading. The optical‐density of each well was determined using a microplate reader, set to 450 nm. For inflammatory array, Mouse Inflammatory Cytokines Multi‐Analyte ELISArray was run per manufacture instructions (Qiagen, MEM‐004A).
2.9. Astrocyte‐conditioned medium experiments
Medium was collected from fully confluent cultures of virally transduced astrocytes beginning 48 hr after transduction and at two subsequent time points of 72 and 96 hr. At these three time points, a mixture of 25% astrocyte‐conditioned medium and 75% neuronal medium was added onto cultures of primary motor neurons at DIV 4–5. For TNFα neutralization, the medium was incubated with a TNFα neutralizing antibody (abcam ab6671) at a concentration of 5 ng μL−1 for 30 min prior to addition onto motor neurons. SN50 experiments were carried out by pretreating the astrocyte cultures from which the conditioned medium was collected with 18 μM SN50 (Calbiochem 481480). For the CNQX experiments, CNQX (Tocris Bioscience 0190) was added to the conditioned medium just prior to motor neuron treatment, at a final concentration of 10 μM. Individual motor neurons were tracked an imaged each day at 37°C and 5% CO2 using a semiautomated inverted Nikon Eclipse Ti microscope equipped with a Tokai Hit stage top incubator with gas and temperature controller. Neurons were marked as dead by their disappearance or obvious morphological indicators such as fragmentation of the soma and neurites.
2.10. Neuronal calcium imaging
DIV 7 motor neurons were transfected with GCaMP6m (pGP‐CMV‐GCaMP6m was a gift from Douglas Kim (Addgene plasmid # 40754)) using lipofectamine‐based DNA transfection. After 24 hr, motor neurons were treated with ACM similarly to what we have described for other experiments. Following 48h after ACM addition, coverslips were mounted on a closed‐bath RC‐20 perfusion chamber (Warner Instruments 64–0222) and kept in freshly made artificial CSF (aCSF). Green fluorescence signal was captured using an inverted Nikon Eclipse Ti epifluoresence microscope at 5‐s intervals. The plane of focus was stabilized using the Nikon Perfect Focus System throughout the duration of imaging. After ∼3 min of baseline images, 10 μm AMPA + 10 μm cyclothiazide in aCSF was perfused onto the cells, and imaging continued for 20 min. Cyclothiazide is a positive allosteric modulator of AMPARs and a blocker of rapid AMPAR desensitization and was included to allow for maximal AMPAR stimulation. GCaMP6 fluorescence changes were analyzed using the ROI intensity evolution plugin‐in for Icy, and open bioimage informatics platform. After background subtraction, ROIs of the cell soma were taken and the change in fluorescence (ΔF) normalized to baseline fluorescence (F 0) or ΔF/F 0 was calculated for responding neurons. The mean intensity of the 3‐min baseline period was used for normalization. A total of >50 cells per condition were pooled from multiple independent experiments and analyzed.
2.11. Microarray expression profiling for astrocytes
RNA purification was performed per manufacturer instructions (RNeasy Plus Micro, Qiagen). Amplification of cDNA was performed using the Ovation Pico WTA‐system V2 RNA amplification system (NuGen Technologies). Briefly, 50 ng of total RNA was reverse transcribed using a chimeric cDNA/mRNA primer, and a second complementary cDNA strand was synthesized. Purified cDNA was then amplified with ribo‐SPIA enzyme and SPIA DNA/RNA primers (NuGEN Technologies, Inc.). Amplified ST‐cDNA was purified with Qiagen MinElute reaction cleanup kit. The concentration of Purified ST‐cDNA was measured using the Nanodrop. 2.5 µg ST‐cDNAs were fragmented and chemically labeled with biotin to generate biotinylated ST‐cDNA using FL‐Ovation cDNA biotin module V2 (NuGen Technologies).
Affymetrix gene chips, Mouse gene 1.0 ST array (Affymetrix, Santa Clara, CA), were hybridized with fragmented and biotin‐labeled target (2.5 µg) in 110 μL of hybridization cocktail. Target denaturation was performed at 99°C for 2 min and then 45°C for 5 min, followed by hybridization for 18 h. Arrays were then washed and stained using Gene chip Fluidic Station 450, and hybridization signals were amplified using antibody amplification with goat IgG and anti‐streptavidin biotinylated antibody. Chips were scanned on an Affymetrix Gene Chip Scanner 3000, using Command Console Software. Background correction and normalization were done using Iterative Plier 16 with GeneSpring V12.0 software (Agilent, Palo Alto, CA). The 1.3‐fold (p value < 0.05) differentially expressed gene list was generated. The differentially expressed gene list was loaded into Affymetrix Transcriptome Analysis Console software to generate the volcano plot and run the WikiPathways pathway analysis.
2.12. Quantitative real‐time PCR analysis
Measurement of motor neuron GluA1 and GluA2 mRNA levels was made by qPCR. Briefly, DIV 4–5 primary motor neuron cultures were treated with astrocyte conditioned medium and left for 72 hr. Total RNA was isolated by lysing the motor neuron cultures with TRIZOL. Next, phase separation of the sample was done using chloroform, and the upper aqueous phase was retrieved. RNA was then precipitated by isopropanol and washed with ethanol. The subsequent pellet was dissolved in DEPC‐treated water and those samples were used in a standard reverse transcription protocol using the reverse transcriptase SuperScript IV (Invitrogen). The cDNA was added with TaqMan qPCR primers for rat GAPDH (Rn01775763_g1), and Gria1 (Rn00709588_m1) or Gria2 (Rn00568514_m1). The multi‐plex qPCR assay was then performed and analyzed using the QuantStudio 5 Real‐Time PCR system (ThermoScientific).
2.13. Statistical analysis
Relative fluorescence levels, neurite lengths, % cells remaining, and time to peak data are presented as mean ± SEM. Statistical significance was evaluated using Student's t test or one‐way analysis of variance with post‐hoc Tukey's HSD. p < 0.05 or 0.01 was used to determine significance where indicated. A data transformation was used prior to statistical testing in one case for determining differences between neurite lengths as indicated. A logarithmic transformation was required to address a skew in the distribution.
3. RESULTS
3.1. MutFUS mislocalizes to the cytoplasm and increases GFAP expression in primary astrocytes
Carriers of FUS mutations, including R521G, show widespread FUS pathology, including neuronal and glial cytoplasmic inclusions in the absence of basophilic inclusions (Blair et al., 2009; Hewitt et al., 2010). Because the redistribution of nuclear FUS has been observed in a variety of cell culture models (Bosco et al., 2010; Dormann et al., 2010; Farg et al., 2012), we examined it in our in vitro system of cultured astrocytes transduced with mutFUS. Isolated primary astrocytes were firstly tested for purity using the glutamate transporter GLAST as a marker of mature astrocytes (Supporting Information Figure S1a,c). Immunofluorescence analysis showed negligible staining for cellular markers of other forms of glia or neurons (Supporting Information Figure S1B). Viral transduction was used to introduce N‐terminally GFP‐tagged wild‐type (WTFUS) and R521G‐FUS mutFUS into primary astrocytes with efficiency around 80% (Supporting Information Figure S1e). Expression levels of both WT FUS and mutFUS in astrocytes were comparable as shown in western blot analysis (Figure 1c). Expression of mutFUS led to cytoplasmic mislocalization in ∼90% of the transduced astrocytes (Figure 1a,b) thus recapitulating the mislocalization pattern seen in ALS patients. We next asked if mutFUS expression would be sufficient to cause toxicity in astrocytes. Toxicity was measured using CellTiter‐Glo Luminescent Cell Viability Assay (CTG), which detects changes in metabolic activity as a measure of overall cell viability (Pedrini et al., 2010). This assay showed that, while 0.5M thapsigargin used as positive control significantly reduced viability, null virus treatment was not significantly different than either WTFUS or mutFUS (Supporting Information Figure S1f). These data suggest that expression of mutFUS alone does not result in astrocytic cell death. However, immunofluorescence for GFAP, an indicator of astrogliosis was enhanced in mutFUS astrocytes (Figure 1d). Interestingly the levels of GFAP expression in mutFUS astrocytes were significantly higher compared to WTFUS and null virus (NT) astrocytes (Figure 1e,f). WTFUS‐expressing astrocytes, on the other hand, only showed a modest increase in GFAP expression compared to NT astrocytes. These results suggest that although mutFUS expression is non‐toxic to astrocytes, mutFUS induces enhanced astrogliosis.
Figure 1.
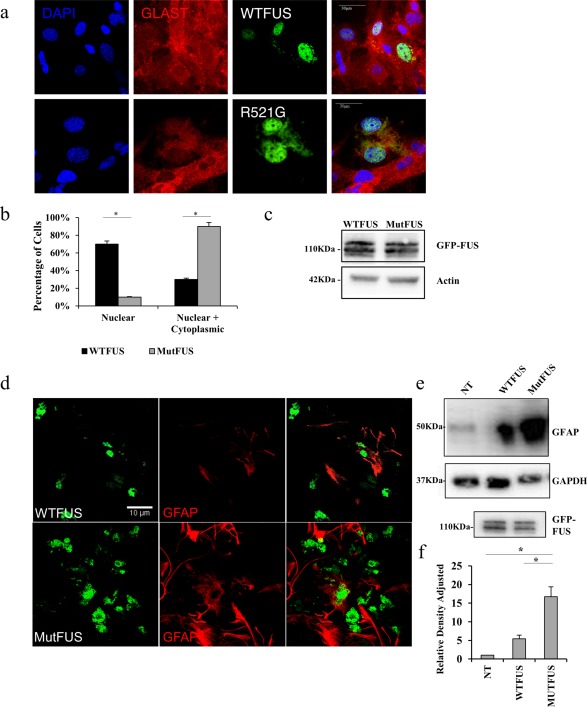
MutFUS mislocalizes to the cytoplasm and increases GFAP expression in primary astrocytes: (a) Immunofluorescence staining of astrocytes infected with GFP‐FUS, 5 days after infection, for GLAST and DAPI. Scale bar 30 μm. (b) Quantification of percentage of astrocytes displaying cytoplasmic GFP‐FUS. Statistical significance was evaluated using Student's t test, n = 3, *p < 0.05 (c) Western blots of astrocytic total lysate confirming equal levels of expression: top GFP, middle actin. (d) Immunofluorescence staining of astrocytes infected with GFP‐FUS, 5 days after infection, for GFAP. (e) Western blot of total astrocytic lysate: top GFAP, bottom GAPDH; lower panel, GFP‐FUS. (f) Quantification of GFAP levels displayed in Panel (e). Densitometry data are presented as mean ± SEM. GFAP levels were normalized to the GAPDH loading control and presented relative to nontransduced control. Statistical significance was evaluated using one‐way ANOVA, n = 3, *p < 0.01
3.2. MutFUS‐expressing astrocytes are toxic to cocultured motor neurons
We next tested whether mutFUS‐expressing astrocytes are toxic to co‐cultured motor neurons. Immunopanned E14.5 primary rat motor neurons were plated on top of a bed of astrocytes previously transduced with FUS (WT or R521G) and allowed to grow in co‐culture for 3 days. Cultures were then fixed and stained with antibodies for motor neuron markers choline acetyltransferase (ChAT), and the 75‐kDa low‐affinity neurotrophin receptor (p75‐NTR). We then assessed neurite lengths in both groups to gauge toxic phenotype as previously done by others (Han et al., 2013; Lasiene and Yamanaka, 2011). Motor neurons were selected based on ChAT‐positive immunostaining (Supporting Information Figure S1d), and neurite length measured using p75‐NTR (Anderson et al., 2004). Contrary to motor neurons co‐cultured with WTFUS‐astrocytes, motor neurons plated on mutFUS containing astrocytes had less extensive branching and shorter neurites (Figure 2a). To quantify reductions in neurite length, tracings of individual neurons were used (Figure 2b) to create cumulative frequency plots (Figure 2c). Motor neurons cocultured with mutFUS astrocytes, exhibited a 34% ± 6% reduction in neurite length (Figure 2d), compared to those cocultured with WTFUS‐expressing astrocytes. As additional controls, we demonstrated that damage to neurite morphology is also evident in motor neurons co‐cultured with mutant, but not WT, SOD1 astrocytes (Figure 2c). These data suggest that mutFUS expression in astrocytes can damage neurite morphology of wild‐type neurons.
Figure 2.
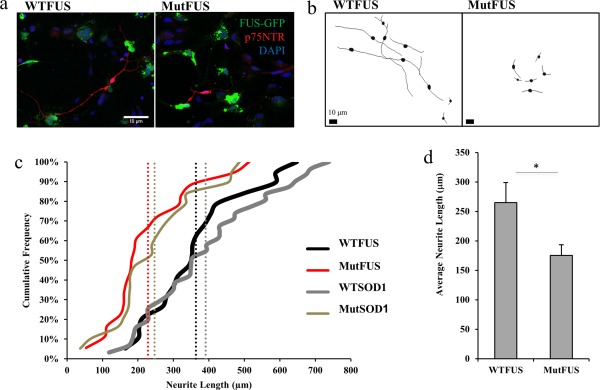
MutFUS‐expressing astrocytes are toxic to motor neurons: (a) Motor neurons plated on a bed of mutFUS‐infected astrocytes displayed shorter neurites. Immunostained for p75NTR and DAPI. (b) Tracings of co‐cultured motor neurons on a bed of virally infected FUS astrocytes. Scale bar 10 μm. (c) Representative cumulative frequency graph show motor neurons co‐cultured with MutFUS astrocytes exhibit a reduction in average neurite length. WTSOD1 and MutSOD1 used for comparison due to their known effects on motor neurons. Average neurite length of each condition shown as vertical dotted line. >100 neurons traced per condition. (d) Neurite length data are presented as mean ± SEM. Data were assessed after a logarithmic transformation to normalize the distribution and statistical significance was evaluated using Student's t test, >100 neurons traced per condition, n = 4, *p < 0.05. The data shown are the raw untransformed data in the original units
3.3. MutFUS astrocytic conditioned medium is toxic to motor neurons
We next asked if the mutFUS‐mediated defect in neurite morphology was due to a contact‐dependent mechanism or the release of soluble factor(s). Motor neurons were cultured alone and treated with ACM from transduced astrocytes (Figure 3a). Motor neurons were then analyzed for cumulative neurite lengths as they were in Figure 2. We observed reduced neurite length in motor neurons treated with mutFUS but not WTFUS ACM (Figure 3b). While neurite length has been previously used reliably as a measure of neuronal viability, nevertheless, it is not a measure of neuronal degeneration. Thus, to measure motor neuron degeneration over time after exposure of ACM, we tracked individual neurons in 24‐hr intervals by time lapse‐imaging (Figure 3c). Neurons were marked as dead by their disappearance from the field of imaging or obvious morphological indicators such as fragmentation as shown in Figure 3c. At a time point of 96 hr after ACM treatment, we measured a significant motor neuron loss (43% ± 3.7%) in cultures treated with mutFUS ACM compared to WT FUS ACM (11% ± 1.9%) (Figure 3d). As a comparison, neuronal death caused by mutant SOD1 ACM is shown (Figure 3d). These results indicate that motor neuron death triggered by mutFUS astrocytic expression involves a soluble factor(s).
Figure 3.
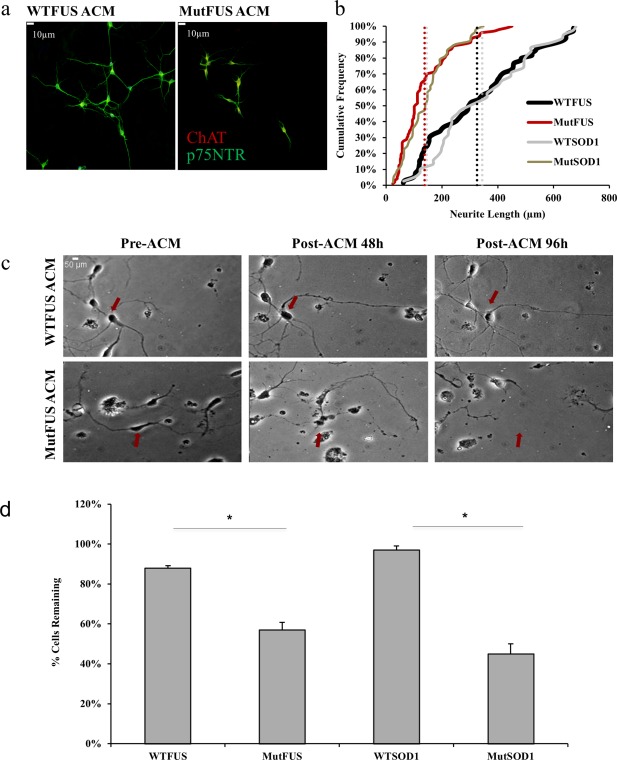
MutFUS ACM is toxic to motor neurons: (a) Conditioned media from FUS infected astrocytes used to treat motor neurons plated in isolation. Immunostained for p75NTR and ChAT. Scale bar 10 μm. (b) Representative cumulative frequency data demonstrating a decrease in neurite length of isolated motor neurons treated with mutFUS ACM. Vertical dotted lines represent average neurite length. >100 neurons traced per condition, n = 3, *p < 0.05. (c) Individual motor neurons were tracked over the course of 4 days and assessed for fragmented morphology and distorted cell bodies or complete disappearance every 24 hr. The red arrows indicate examples of the same motor neurons starting from pretreatment and continuing to mark their presence/absence through to endpoint. (d) Quantification of the percentage of motor neurons remaining after 96 hr of ACM addition. Survival data are presented as mean ± SEM and statistical significance was evaluated using Student's t test, 50 motor neurons counted per condition, n = 3, *p < 0.01
3.4. MutFUS astrocytes secrete TNFα
Because mutFUS ACM is sufficient to damage the motor neurons, we used a combination of gene expression profiling and protein analysis to begin to identify astrocytic changes that might contribute to toxic factor(s) being released by mutFUS‐astrocytes. Microarray profiling was first used to compare gene expression changes between WTFUS and mutFUS‐expressing astrocytes. A total of ∼1500 significant gene changes of 1.3 fold change were found in mutFUS astrocytes versus WTFUS (Figure 4a). We subsequently analyzed these changes at the level of gene networks using the Affymetrix Transcriptome Analysis software and WikiPathways analysis. From the list of the most affected gene networks (as ranked by the total number of significantly changed genes found in each pathway), we focused on the secretome pathway, as we have seen ACM toxicity. The top pathway hit that we predicted would influence broad secretomic changes was the TNF‐alpha and NF‐KB signaling pathway (Table 1), which has been studied extensively for its role in mediating pro‐inflammatory cell‐to‐cell signaling. On the basis of these results, we then measured common pro‐inflammatory cytokine levels in the WTFUS and mutFUS ACM by ELISA (Figure 4b). Among those, we identified TNFα as the most highly altered secreted factor in mutFUS ACM (3.8‐fold increase over WTFUS ACM). Further, we validated this result using a second ELISA containing a new set of sandwich antibodies, and found mutFUS ACM contains significantly more TNFα compared to WTFUS or Non‐transduced (NT) ACM (Figure 4c). Additionally, we probed astrocyte cell‐lysates for total TNFα via western blot, and found that astrocytes expressing mutFUS have increased expression levels compared to WTFUS and NT astrocytes (Figure 4d). Together, these data clearly show that TNFα is aberrantly upregulated and secreted by mutFUS astrocytes.
Figure 4.
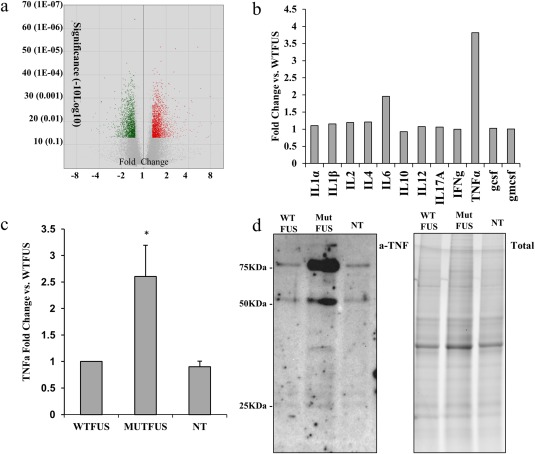
MutFUS astrocytes release TNFα: (a) Volcano plot of microarray data displaying significance against fold change. Significantly altered genes above 1.3 fold are shown as increased (red) or decreased (green) over the total set of genes assayed (gray). (b) ELISA assay of inflammatory factors in astrocyte conditioned media. Values expressed as fold change of MutFUS ACM over WTFUS ACM. (c) Anti‐TNFa ELISA of ACM samples. Values expressed as fold change versus WTFUS. (d) Left: Anti‐TNFα Western Blot of astrocyte cell lysates. WTFUS, mutFUS, and nontransduced astrocytes are compared. Positive TNFα bands can be observed at ∼26, 52, and 78 kDa, which correspond to TNFα monomers, dimers, and trimers respectively. Right: Loading control showing total protein. Total protein levels were assessed by imaging the Mini‐PROTEAN TGX stain‐free gel prior to transfer
Table 1.
TNFa & NFkB related transcripts are highly altered Pathway analysis results of microarray data processed through WikiPathways analysis.
| Pathway | Total number of changes | Up in MUTFUS | Down in MUTFUS | Significance (z score) |
|---|---|---|---|---|
| Protein‐protein interactions in the podocyte (XPodNet) | 209 | 92 | 117 | 7.77 |
| Pluripotency associated networks (PluriNetWork) | 80 | 43 | 37 | 4.79 |
| Focal Adhesion Kinase‐PI3K‐AKT‐mTOR signaling | 68 | 27 | 41 | 1.04 |
| mRNA processing | 60 | 32 | 28 | 2.31 |
| TNF‐alpha and NF‐KB signaling | 48 | 23 | 25 | 2.5 |
| MAPK | 43 | 24 | 19 | 2.12 |
| EGFR1 | 42 | 21 | 21 | 1.56 |
| Adipogenesis | 37 | 22 | 15 | 2.59 |
| Myometrial relaxation and contraction | 37 | 19 | 18 | 1.36 |
| Purine metabolism | 37 | 23 | 14 | 0.9 |
| Insulin signaling | 36 | 16 | 20 | 1.03 |
| TGF‐beta receptor signaling | 36 | 18 | 18 | 1.41 |
| Chemokine signaling | 35 | 19 | 16 | 0.2 |
| Androgen receptor signaling | 30 | 13 | 17 | 1.93 |
| Wnt signaling | 29 | 14 | 15 | 2.57 |
| Regulation of actin cytoskeleton | 28 | 12 | 16 | 0.18 |
| T cell receptor signaling | 28 | 16 | 12 | 0.6 |
| B cell receptor signaling | 26 | 13 | 13 | 0.04 |
| Non‐odorant GPCRs | 26 | 13 | 13 | 3.22 |
| Il‐3 signaling | 25 | 11 | 14 | 1.33 |
Shown are the top 20 most altered pathways as determined by the combined number of significantly altered genes within each pathway along with the number of changes and the significance score ranking. In bold is the TNF‐alpha and NF‐KB signaling pathway which was chosen as a candidate pathway for further analysis based on previous evidence of role of the TNF‐alpha and NF‐KB pathways in regulating cytokines and inflammatory cell‐to‐cell signals.
3.5. TNFα neutralization in mutFUS ACM rescues motor neuron degeneration and death
To test the role of TNFα in mutFUS‐mediated toxicity, we plated motor neurons in isolation and imaged individual cells before and after ACM treatment ± 5 ng µL−1 anti‐TNFα neutralizing antibody (Abcam, ab6671) and tracked cell morphological changes and viability over 96 hr (Figure 5a and Supporting Information Figure S2a). Remarkably, when anti‐TNFα antibody is added to mutFUS ACM, motor neurons display a 2.3‐fold greater average neurite length compared to motor neurons treated with mutFUS ACM alone (Figure 5b,c). Further, addition of anti‐TNFα antibody to mutFUS ACM, significantly prevented mutFUS induced death of motor neurons [cell loss of 12.2% in mutFUS ACM+ anti‐TNFα antibody compared to 45.6% with mutFUS ACM alone (Figure 5d)]. While TNFα neutralization inhibits mutFUS‐mediated toxicity, it does not block mutSOD1 astrocytic toxicity, suggesting that release of TNFα is specific to mutFUS astroctyes (data not shown). These data demonstrate that in mutFUS, astrocytic TNFα is a mediator of motor neuron viability. In our previous work, we have also demonstrated that TNFα is specifically released from mutFUS iPSC‐astrocytes and drives pathological blood–brain‐barrier changes that lead to pharmacoresistance (Qosa et al., 2016). Thus, reducing astrocytic TNFα production in patients may have therapeutic potential for multiple features of disease.
Figure 5.
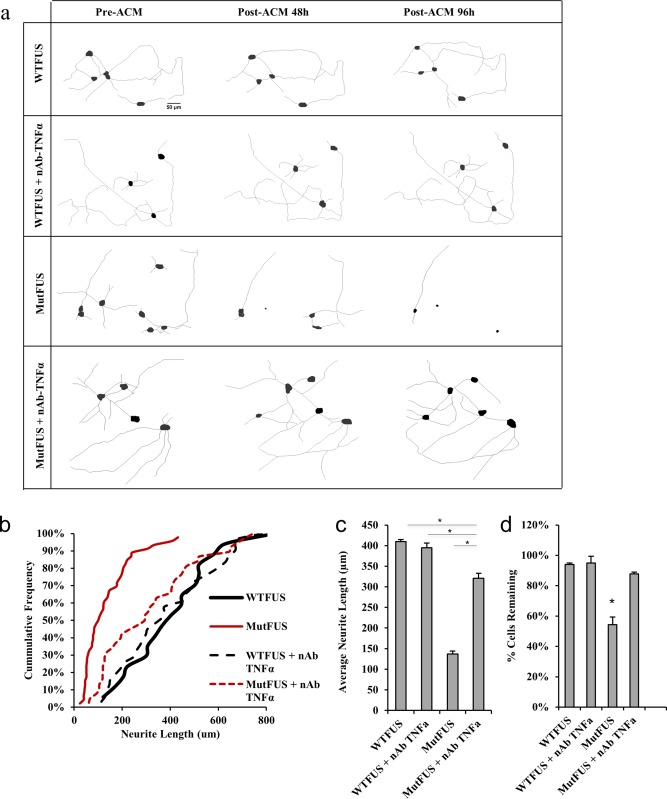
TNFα neutralization in MutFUS ACM rescues neurite loss and motor neuron death: (a) Tracings made of individually tracked live motor neurons, cultured in isolation, and treated with ACM. TNFα neutralization antibody (nAb) diluted directly into ACM. (b) Representative cumulative frequency of neurite lengths of motor neurons treated with ACM and TNFα nAb. >100 neurons traced per condition. (c) Neurite length data are presented as mean ± SEM. Statistical significance was evaluated using one‐way ANOVA, >100 neurons traced per condition, n = 4, *p < 0.05. (d) Quantification of the percentage of motor neurons remaining after 96 hr of ACM addition. Survival data are presented as mean ± SEM and statistical significance was evaluated by one‐way ANOVA, >50 neurons per condition, n = 3, *p < 0.01
3.6. Pharmacological inhibition of NF‐ĸB in astrocytes rescues motor neuron neurite loss and cell death
Astrocytes can respond to inflammatory cytokines such as TNFα by inducing classical NF‐kappaB (NF‐ĸB) signaling to regulate inflammation and drive the expression of many chemokines and molecules upregulated in ALS. NF‐ĸB is activated in glia in fALS and sALS patients, and astrocytes derived from human postmortem ALS patients showed NF‐ĸB as the highest‐ranked regulator of inflammation in gene array data (Lasiene and Yamanaka, 2011; Swarup et al., 2011). NF‐ĸB can also drive the transcription of TNFα and many other inflammatory molecules (Pękalski et al., 2013). Further, FUS has been shown to act as a co‐activator of NF‐ĸB by enhancing the NF‐ĸB‐mediated transactivation induced by TNFα (Uranishi, 2001). We next asked if selectively inhibiting NF‐ĸB in mutFUS astrocytes could block TNFα–induced motor neuron loss. To test this, we used SN50, a cell‐permeable inhibitor peptide that inhibits translocation of the NF‐ĸB active complex into the nucleus, and subsequent NF‐kB activation. Treating astrocytes with this peptide reduced TNFα secretion by 67% (data not shown) and, consequently, alleviated mutFUS‐induced, reduction of neurite length and reduced motor neuron cell loss by 23% (Figure 6 and Supporting Information Figure S2b).
Figure 6.
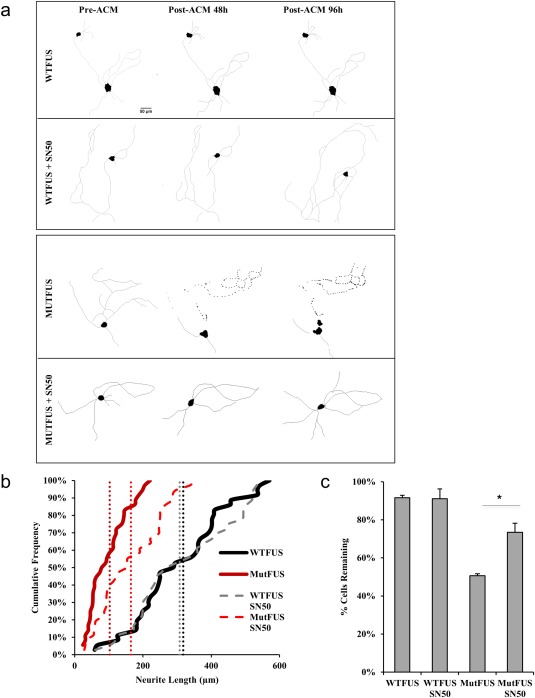
Pharmacological inhibition of NF‐κB in astrocytes rescues motor neuron neurite loss and cell death: (a) Representative tracings of motor neurons treated with ACM. Astrocyte cultures for ACM samples were pretreated with the NF‐kB inhibitor SN50. (b) Representative cumulative frequency of neurite lengths of motor neurons treated with ACM similarly to 6A. >100 neurons traced per condition, n = 3, *p < 0.05. (c) Percent survival of motor neurons before and after ACM. Survival data are presented as mean ± SEM and statistical significance was evaluated by one‐way ANOVA, >50 neurons per condition, n = 3, *p < 0.05
3.7. MutFUS ACM triggers AMPA receptor changes and AMPA receptor‐mediated cell death in motor neurons
Because TNFα has been shown to induce surface expression changes of AMPA‐type glutamate receptors (AMPARs), leading to excitotoxicity (Ferguson et al., 2008; Leonoudakis et al., 2008; Santello and Volterra, 2012), we hypothesized that the aberrant release of TNFα by mutFUS astrocytes may also lead to AMPA receptor dysregulation in motor neurons. To test this, we treated motor neurons with ACM from WTFUS or mutFUS‐expressing astrocytes and stained them for glutamate receptor 1 (GluA1), glutamate receptor 2 (GluA2) and microtubule‐associated protein 2 (MAP2) as dendritic marker. Measurement of dendritic immunofluorescence intensity in at least 250 regions of interest (ROIs) in motor neurons revealed that motor neurons treated with mutFUS ACM had increased GluA1 and decreased GluA2 expression compared to motor neurons treated with WTFUS ACM (Figure 7a–c). We then determined whether AMPAR changes occur also at the mRNA level. We therefore probed GluA1 and GluA2 mRNA in ACM‐treated motor neurons and found GluA1 mRNA to be significantly increased at a time point of 72 hr in the mutFUS ACM condition (Figure 7d). Although GluA2 mRNA remain unchanged at this time point, with the significant increase in GluA1 mRNA levels, the GluA1/GluA2 ratio significantly shifts, similar to the immunofluorescence data shown in Figure 7a–c. GluA2 subunits confer calcium impermeability on AMPARs and their decrease relative to GluA1 is associated with the addition of more calcium‐permeable AMPARs (Mansour, Nagarajan, Nehring, Clements, & Rosenmund, 2001). Given that AMPA receptor subunit composition was altered in motor neurons exposed to mutFUS ACM, we hypothesized that AMPA receptor stimulation in these neurons may result in a higher calcium entry. To test this idea, we measured intracellular calcium levels following AMPA stimulation in motor neurons treated with ACM, using the genetically encoded calcium sensor GCaMP6m (Figure 7e). Motor neurons treated with mutFUS ACM for 48 hr showed significantly larger intracellular calcium increases following stimulation with 10 μm AMPA + 10 μm cyclothiazide compared to WT ACM. When comparing peak GCaMP6m ΔF/F 0 we found that mutFUS ACM treated motor neurons displayed a ∼1.8‐fold increase over WTFUS ACM‐treated neurons on average (Figure 7f). To test the idea that excitotoxic damage might be contributing to neuronal death in our conditioned medium experiments we sought determine whether pharmacological inhibition of AMPA receptors could offer neuroprotection from mutFUS ACM. We treated motor neurons with the AMPA/Kainate receptor antagonist 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX) alongside treatments of mutFUS astrocytic media. Addition of CNQX significantly increased survival (79% ± 1.3% compared to 59% ± 2.5%) in neurons treated with mutFUS ACM (Figure 7g). Taken together, these results suggest that mutFUS astrocytes sensitize motor neurons to excitotoxic damage.
Figure 7.
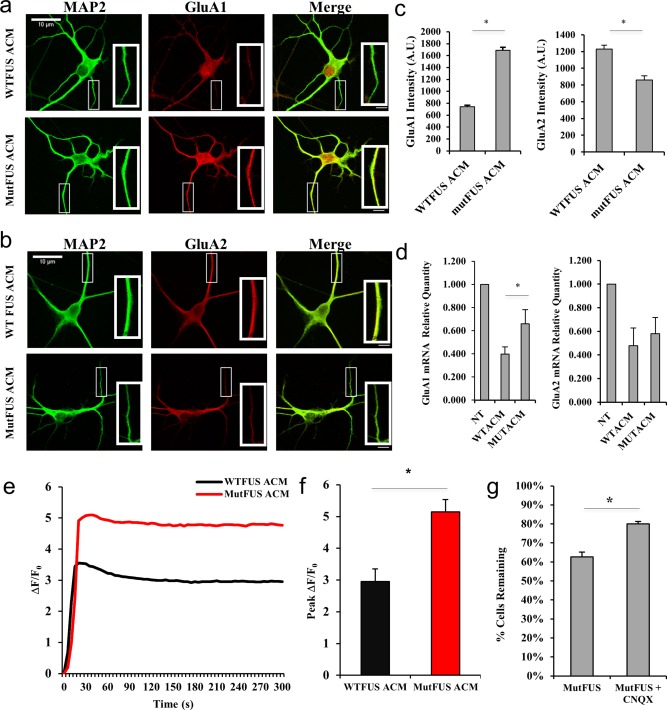
MutFUS ACM triggers AMPA receptor changes and AMPA receptor‐mediated cell death in motor neurons: (a) 9 DIV motor neurons were treated with ACM for 72 hr then probed for GluA1 by immunocytochemistry [MAP‐2 (green) and GluA1 (red)]. (b) 9 DIV motor neurons were treated and analyzed for GluA2 similarly to panel A. (c) Quantification of dendritic GluA1 and GluA2 signal. Fluorescence levels are presented as mean ± SEM and statistical significance was evaluated using Student's t test. A total of >50 dendrites from >15 cells were analyzed per condition. (d) Relative quantity of ACM‐treated motor neuron AMPAR RNA at 72 hr post‐ACM, determined by qPCR ΔCT. GluA1 or GluA2 levels were normalized to GAPDH and then displayed as relative to a nontreated control. Statistical significance was evaluated using Student's t test, n = 4, *p < 0.05. (e) Representative single‐traces of the change in background‐subtracted GCaMP6m fluorescence normalized to baseline (ΔF/F 0) after stimulation with 10 μM AMPA + 10 μM cyclothiazide for motor neurons pretreated (48 hr) with WT or mutFUS‐ACM. (f) Quantification of peak ΔF/F 0 values for calcium imaging experiments represented in (e). Peak values are presented as mean ± SEM and statistical significance was evaluated using Student's t test. n ≥ 50, *p < 0.01. (g) Quantification of motor neurons remaining after 96 hr treatment with ACM ± 10 μM CNQX (6‐cyano‐7‐nitroquinoxaline‐2,3‐dione). Survival data are presented as mean ± SEM and statistical significance was evaluated using Student's t test, > 60 neurons counted per condition, n ≥ 3, *p < 0.01
3.8. TNFα neutralization in mutFUS‐ACM prevents AMPA receptor alterations
Next, we sought to determine whether TNFα released by mutFUS astrocytes contributes to motor neuron AMPAR changes as hypothesized. To test this, we measured dendritic GluA1 levels in ACM‐treated motor neurons by immunofluorescence (as done in Figure 7a), under conditions in which ACM‐TNFα has been ablated. ACM collected from WT or mutFUS expressing astrocytes was incubated with or without a TNFα neutralizing antibody prior treatment of motor neurons. Here, we found that the increase in GluA1 levels observed after treatment with mutFUS ACM was blocked by preincubating the ACM with a TNFα antibody (Figure 8a,b). The results indicate that TNFα signaling is necessary for motor neuron AMPAR changes driven by mutFUS astrocytes.
Figure 8.
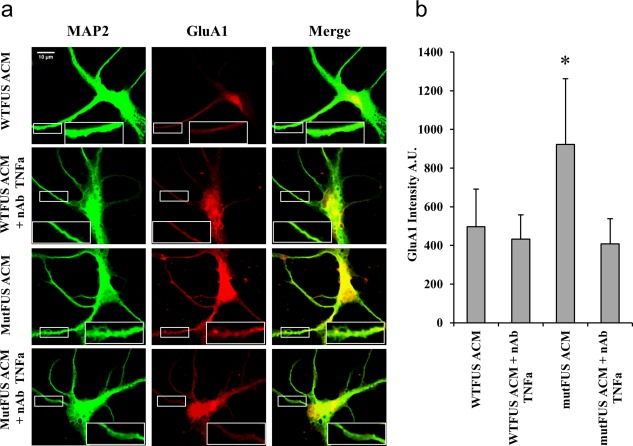
TNFα neutralization in mutFUS‐ACM prevents AMPA receptor alterations: (a) 9 DIV motor neurons were treated with ACM for 72 hr following which they were probed for GluA1 and MAP‐2 by immunocytochemistry. A representative image is shown along with cropped images of the highlighted region of a single neuron for MAP‐2 (green) and GluA1 (red). For TNFα neutralization, ACM was incubated with a TNFα neutralizing antibody (nAb) prior to treatment of motor neurons. (b) Quantification of dendritic GluA1 signal. ROIs along the dendrites of motor neurons were selected on the basis of MAP‐2 labeling and fluorescence intensity was measured. A total of >50 dendrites from >15 cells were analyzed per condition. Fluorescence levels are presented as mean ± SEM. Statistical significance was evaluated using one‐way ANOVA, n = 5, *p < 0.05
4. DISCUSSION
Here, we show for the first time that ALS‐causative mutations in FUS can cause noncell autonomous motor neuron degeneration. While expression of mutFUS‐R521G is not toxic to astrocytes, these cells adopt a reactive phenotype as evidenced by an increase in GFAP expression and a selective increase in some pro‐inflammatory cytokines. Importantly, these astrocytes become toxic to motor neurons via the release of harmful factor(s). Interestingly, mutFUS astrocyte toxicity is driven by a cascade of events that include astrocytic NF‐kB activation, release of TNFα and AMPA receptor alterations in motor neurons that allow for dysregulated calcium influx and cell death.
Perhaps with the exclusion of TDP‐43 astrocytes, it is widely recognized that ALS astrocytes actively kill motor neurons through the release of toxic factor(s) (Haidet‐Phillips et al., 2013). While the identification of factors responsible for transmitting toxicity of mutant SOD1 and sporadic ALS astrocytes is still under intense investigation, our work identifies a key toxic factor in mutFUS astrocytes. We show that mutFUS expressing astrocytes have increased expression of TNFα and increased production of soluble TNFα. This is unique to mutFUS astrocytes as mutant SOD1 astrocytes do not release TNFα, as we have shown using patients‐derived iPSc‐astrocytes cocultured with iPSc‐endothelial cells (Qosa et al., 2016). Elevated TNFα production has been observed in a number of neurodegenerative diseases including ALS [reviewed (Philips and Robberecht, 2011)]. TNFα is a pleiotropic molecule that can be produced by a number of different cell‐types, including astrocytes (Chung and Benveniste, 1990; Santello and Volterra, 2012). In the CNS in particular, TNFα signaling is complex, and its role in regulating synaptic transmission both physiologically and pathologically is increasingly being appreciated (Ferguson et al., 2008; Leonoudakis et al., 2008; Olmos and Lladó, 2014; Santello and Volterra, 2012). Importantly, soluble recombinant TNFα has been shown to be directly toxic to human fetal tissue derived neurons and motor neuron‐like NSC‐34 in vitro and knockout of TNFα receptors prevents motor neuron cell death in an axonal injury mouse model, indicating that TNFα can damage motor neurons (D'Souza, Alinauskas, McCrea, Goodyer, & Antel, 1995; He, Wen, & Strong, 2002; Raivich et al., 2002). Accordingly, in our model, the TNFα produced by mutFUS astrocytes is toxic to motor neurons and motor neuron death can be rescued by neutralizing anti‐TNFα antibodies. In contrast to TNFα, we did not see wholesale changes to a larger set of inflammatory molecules with the exception of IL‐6. Although our in vitro results are unique for TNFα, we cannot exclude that in vivo, over a progressing disease, perturbing this key inflammatory mediator may trigger and/or mediate a more robust proinflammatory response involving several cell‐types to sustain gliosis, driving disease progression in later stages. Similarly, although in vitro blocking of TNFα with neutralizing antibodies completely rescues motor neuron death here, we cannot exclude the role of other toxic factors in the complex in vivo context.
In addition to TNFα, our study also points to astrocytic NF‐kB as a driver of neurodegeneration. It is well‐known that NF‐kB plays a role in both driving the expression of TNFα and mediating TNFα signaling pathways (Pękalski et al., 2013). In our mutFUS astrocytes, inhibiting the activation of NF‐kB reduces the production of TNFα and protects motor neurons, strongly suggesting that NF‐kB and TNFα pathways are inter‐related. In ALS, NF‐kB has been shown to be activated in glial cells in vitro and in postmortem tissues of fALS and sALS and its dysregulation has been shown to have a significant impact on neuroinflammation (Frakes et al., 2014; Haidet‐Phillips et al., 2011; Prell et al., 2014; Swarup et al., 2011). Interestingly, studies by Frakes et al. (2014) have found that microglia containing mutant SOD1 also increase their production of TNFα and acquire a neurotoxic phenotype that can be prevented through NF‐kB inhibition. Although the sources of TNFα production are different between their study and ours, they may point to some shared mechanisms of noncell autonomous toxicity between different forms of ALS. Moreover, other ALS causing genes may also implicate this pathway. Mutations in optineurin and more recently, tank binding kinase 1, have been discovered to cause ALS and are known regulators of TNFα signaling and NF‐kB. Interestingly, both TDP‐43 and FUS have been previously been shown to directly bind to NF‐ĸB (Swarup et al., 2011; Uranishi, 2001) and augment its function. The study of FUS and NF‐kB interactions however is limited to just a single study that was carried out prior to discovery of FUS as an ALS gene. Future studies will be required to uncover more about interactions between NF‐kB and mutFUS variants and the role of these interactions in ALS.
In line with a number of other studies we also found AMPARs as an important component of neuronal toxicity in our model. AMPAR changes have been demonstrated as a contributor in both in vitro and in vivo models of SOD1‐ALS (Van Damme et al., 2007), in vitro studies of C9orf72‐ALS (Donnelly et al., 2013), postmortem studies of sporadic ALS (Kwak, Hideyama, Yamashita, & Aizawa, 2010), and cell‐autonomous studies of FUS‐ALS (Udagawa et al., 2015). Motor neurons are known to be especially vulnerable to AMPA‐mediated excitotoxicity due to an unusually high density of Ca2+‐permeable AMPA receptors and low calcium buffering ability (Palecek, Lips, & Keller, 1999; Vandenberghe et al. 2000). Furthermore, neighboring astrocytes are known to be key regulators of these AMPA receptors (Beattie, 2002; Van Damme et al., 2007). Here, we show that the AMPAR GluA2 subunits are decreased, while GluA1 subunits are increased in the dendrites of motor neurons exposed to mutFUS ACM but not WTFUS ACM. Edited GluA2 subunits are known to confer calcium impermeability to AMPARs and, therefore, we hypothesized that motor neuron cell death following mutFUS ACM treatment might be attributable to an excitotoxic mechanism involving this AMPAR change. Corroborating with this hypothesis, we found intracellular calcium levels following AMPA stimulation are elevated in motor neurons exposed to mutFUS ACM. Using an AMPA receptor antagonist we then rescued the motor neuron death resulting from mutFUS ACM, indicating the involvement of excitoxicity in our model. Our results extend current evidence for AMPAR involvement in ALS and could prompt further studies of noncell autonomous AMPA regulation in ALS and FUS‐ALS. Interestingly, several groups have reported a role for TNFα as a regulator of surface AMPAR expression in spinal cord motor neurons. These studies have shown that TNFα triggers a change in AMPARs which can promote a shift towards surface expression of GluA2‐lacking receptors (Ferguson et al., 2008; Ogoshi et al., 2005; Stellwagen, 2005). Although these studies focused on the rapid effects of short‐term TNFα exposure, we have demonstrated here that longer incubations with TNFα–containing mutFUS‐ACM, not only alter GluA1 & GluA2 protein levels but also trigger an increase mRNA levels for GluA1. Furthermore, our results indicate that AMPAR changes driven by mutFUS‐ACM can be attributed to TNFα signaling, as ablation of soluble TNFα in the ACM prevents an increase in dendritic GluA1 protein levels.
In addition to paracrine effects on motor neurons, astrocytes also express both TNFα receptors (TNFR1 and TNFR2) and TNFα can act on astrocytes to cause a diverse set of changes (Fischer, Wajant, Kontermann, Pfizenmaier, & Maier, 2014; Tezel, Li, Patil, & Wax, 2001) Thus, TNFα released from mutFUS astrocytes may contribute to astrocytic changes through autocrine mechanisms. Two recent studies have demonstrated that TNFα contributes to the induction of neurotoxic astrocytes. Liddelow et al. (2017) have reported that TNFα, in combination with cytokines Il‐1α and C1q, produces a reactive and neurotoxic phenotype characterized by loss of supportive functions and the release of soluble toxic factor(s). Additionally, Almad et al. (2016) have recently shown that soluble TNFα acts on astrocytes to enhance Cx43 hemichannel function. Astrocytes derived from ALS mice are primed for a larger magnitude of this effect, and these increases reduce neuronal viability in coculture studies. Furthermore, as we have discussed here, TNFα can induce activation of NF‐kB transcription factors. Previous studies suggest that the panel of cytokines and chemokines produced by human astrocytes is made up largely of NF‐kB targets in both resting and activated conditions (Choi, Lee, Lim, Satoh, & Kim, 2014). While these effects have not been directly addressed here, further study of the effects of TNFα signaling on astrocytes in FUS‐ALS is warranted.
Despite numerous studies showing both fALS and sALS astrocytes being toxic to motor neurons, the identification of a toxic factor remained elusive. Here we demonstrate that mutant FUS astrocytes contribute to motor neuron death and the principal factor of this toxicity is TNFα secreted from these astrocytes. Although our studies clearly show mutFUS astrocytes are primed to secrete toxic levels of TNFα, the underlying mechanism for such an aberrant secretion remains to be studied. In future studies, it will also be interesting to understand whether changes to the RNA/DNA binding properties of mutant FUS or other cellular stress induced by the mislocalization of mutant FUS mediates the aberrant NF‐kB activation and production of TNFα.
In summary, we have extended a significant body of work pointing to a key role for astrocytes in ALS by demonstrating for the first time the neurotoxic properties of astrocytes expressing mutFUS, an ALS causative gene. In our model, we have found key proinflammatory changes to astrocytes, which trigger excitotoxic vulnerability in motor neurons. Although the underlying connection between different genetic forms of ALS is complex and at this time unclear, this work indicates that strategies targeting astrocyte contributions to disease may be useful for a variety of ALS cases. Although future work must be done to confirm these findings in vivo and, eventually in patients, we recently confirmed the clinical relevance of our findings showing that in a patient‐derived in vitro model of the blood–brain‐barrier, iPSC‐derived astrocytes from a mutFUS patient, drive drug‐efflux transporter mediated pharmacoresistance through TNFα (Qosa et al., 2016). Thus, underscoring the role of TNFα in driving mutFUS‐ALS through astrocytes and identifying TNFα as an important therapeutic target in this form of the disease. Understanding how astrocyte contributions may differ between disease subtypes could aid in the development of more effective, individually tailored therapies, for what is a highly heterogeneous disease.
AUTHOR CONTRIBUTIONS
A.K; K.M; K.K; D.T. and P.P conceived and designed the experiments. A.K; K.M; and K.K performed the experiments. A.K; K.M and K.K analyzed the data. A.K; K.M; K.K; D.T. and P.P wrote the article. D.T and P.P coordinated and supervised.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Figure S1
Figure S2
ACKNOWLEDGMENT
We thank Drs. Alexey Bogush and Wenzhi Tan for the production of adenoviral constructs. N‐terminally EGFP tagged human FUS in a pcDNA3.1/nV5‐DEST backbone was received as a kind gift from Dr. Aaron Gitler. This work was supported by the National Institute of Neurological Disorders and Stroke ‐RO1 NS051488. The Jefferson Weinberg ALS Center and the Frances and Joseph Weinberg Research Unit are also supported by the Farber Family Foundation.
Kia A, McAvoy K, Krishnamurthy K, Trotti D, Pasinelli P. Astrocytes expressing ALS‐linked mutant FUS induce motor neuron death through release of tumor necrosis factor‐alpha. Glia. 2018;66:1016–1033. https://doi.org/10.1002/glia.23298
Funding information National Institute of Neurological Disorders and Stroke, Grant Number: RO1 NS051488, R56 NS092572‐01; Farber Family Foundation
Azadeh Kia and Kevin McAvoy contributed equally to this work.
REFERENCES
- Almad, A. A. , Doreswamy, A. , Gross, S. K. , Richard, J. P. , Huo, Y. , Haughey, N. , & Maragakis, N. J. (2016). Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia, 64(7), 1154–1169. https://doi.org/10.1002/glia.22989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, K. N. , Potter, A. C. , Piccenna, L. G. , Quah, A. K. J. , Davies, K. E. , & Cheema, S. S. (2004). Isolation and culture of motor neurons from the newborn mouse spinal cord. Brain Research Protocols, 12(3), 132–136. https://doi.org/10.1016/j.brainresprot.2003.10.001 [DOI] [PubMed] [Google Scholar]
- Beattie, E. C. (2002). Control of synaptic strength by glial TNFalpha. Science, 295(5563), 2282–2285. https://doi.org/10.1126/science.1067859 [DOI] [PubMed] [Google Scholar]
- Benkler, C. , Ben‐Zur, T. , Barhum, Y. , & Offen, D. (2013). Altered astrocytic response to activation in SOD1(G93A) mice and its implications on amyotrophic lateral sclerosis pathogenesis. Glia, 61(3), 312–326. https://doi.org/10.1002/glia.22428 [DOI] [PubMed] [Google Scholar]
- Blair, I. P. , Williams, K. L. , Warraich, S. T. , Durnall, J. C. , Thoeng, A. D. , Manavis, J. , … Nicholson, G. A. (2009). FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. Journal of Neurology, Neurosurgery & Psychiatry, 81(6), 639–645. https://doi.org/10.1136/jnnp.2009.194399 [DOI] [PubMed] [Google Scholar]
- Bosco, D. A. , Lemay, N. , Ko, H. K. , Zhou, H. , Burke, C. , Kwiatkowski, T. J. , … Hayward, L. J. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Human Molecular Genetics, 19(21), 4160–4175. https://doi.org/10.1093/hmg/ddq335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. S. , Lee, H. J. , Lim, I. , Satoh, J. , & Kim, S. U. (2014). Human astrocytes: secretome profiles of cytokines and chemokines. PLoS One, 9(4), e92325 https://doi.org/10.1371/journal.pone.0092325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, I. Y. , & Benveniste, E. N. (1990). Tumor necrosis factor‐alpha production by astrocytes. Induction by lipopolysaccharide, IFN‐gamma, and IL‐1 beta. Journal of Immunology, 144(8), 2999–3007. [PubMed] [Google Scholar]
- Donnelly, C. J. , Zhang, P.‐W. , Pham, J. T. , Haeusler, A. R. , Mistry, N. A. , Vidensky, S. , … Rothstein, J. D. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron, 80(2), 415–428. https://doi.org/10.1016/j.neuron.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann, D. , Rodde, R. , Edbauer, D. , Bentmann, E. , Fischer, I. , Hruscha, A. , … Haass, C. (2010). ALS‐associated fused in sarcoma (FUS) mutations disrupt Transportin‐mediated nuclear import. EMBO Journal, 29(16), 2841–2857. https://doi.org/10.1038/emboj.2010.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza, S. , Alinauskas, K. , McCrea, E. , Goodyer, C. , & Antel, J. P. (1995). Differential susceptibility of human CNS‐derived cell populations to TNF‐dependent and independent immune‐mediated injury. Journal of Neuroscience, 15(11), 7293–7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farg, M. A. , Soo, K. Y. , Walker, A. K. , Pham, H. , Orian, J. , Horne, M. K. , … Atkin, J. D. (2012). Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide‐isomerase. Neurobiology of Aging, 33(12), 2855–2868. https://doi.org/10.1016/j.neurobiolaging.2012.02.009 [DOI] [PubMed] [Google Scholar]
- Ferguson, A. R. , Christensen, R. N. , Gensel, J. C. , Miller, B. A. , Sun, F. , Beattie, E. C. , … Beattie, M. S. (2008). Cell death after spinal cord injury is exacerbated by rapid TNF‐induced trafficking of GluR2‐lacking AMPARs to the plasma membrane. Journal of Neuroscience, 28(44), 11391–11400. https://doi.org/10.1523/jneurosci.3708-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, R. , Wajant, H. , Kontermann, R. , Pfizenmaier, K. , & Maier, O. (2014). Astrocyte‐specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia, 62(2), 272–283. https://doi.org/10.1002/glia.22605 [DOI] [PubMed] [Google Scholar]
- Frakes, A. E. , Ferraiuolo, L. , Haidet, P. , Amanda, M. , Schmelzer, L. , Braun, L. , … Kaspar, B. K. (2014). Microglia induce motor neuron death via the classical NF‐κB pathway in amyotrophic lateral sclerosis. Neuron, 81(5), 1009–1023. https://doi.org/10.1016/j.neuron.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt, A. , Wieland, T. , Richter, B. , Ruf, W. , Schaeffer, V. , Müller, K. , … Weishaupt, J. H. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto‐temporal dementia. Nature Neuroscience, 18(5), 631–636. https://doi.org/10.1038/nn.4000 [DOI] [PubMed] [Google Scholar]
- Ghose, J. , Sinha, M. , Das, E. , Jana, N. R. , & Bhattacharyya, N. P. (2011). Regulation of miR‐146a by RelA/NFkB and p53 in STHdhQ111/HdhQ111 cells, a cell model of Huntington's disease. PLoS ONE, 6(8), e23837 https://doi.org/10.1371/journal.pone.0023837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowing, G. , Dequen, F. , Soucy, G. , & Julien, J. P. (2006). Absence of tumor necrosis factor‐alpha does not affect motor neuron disease caused by superoxide dismutase 1 mutations. Journal of Neuroscience, 26(44), 11397–11402. https://doi.org/10.1523/JNEUROSCI.0602-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidet‐Phillips, A. M. , Gross, S. K. , Williams, T. , Tuteja, A. , Sherman, A. , Ko, M. , … Maragakis, N. J. (2013). Altered astrocytic expression of TDP‐43 does not influence motor neuron survival. Experimental Neurology, 250, 250–259. doi:https://doi.org/10.1016/j.expneurol.2013.10.004 [DOI] [PubMed] [Google Scholar]
- Haidet‐Phillips, A. M. , Hester, M. E. , Miranda, C. J. , Meyer, K. , Braun, L. , Frakes, A. , … Kaspar, B. K. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nature of Biotechnology, 29(9), 824–828. https://doi.org/10.1038/nbt.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J.‐H. , Yu, T.‐H. , Ryu, H.‐H. , Jun, M.‐H. , Ban, B.‐K. , Jang, D.‐J. , & Lee, J.‐A. (2013). ALS/FTLD‐linked TDP‐43 regulates neurite morphology and cell survival in differentiated neurons. Experimental Cell Research, 319(13), 1998–2005. https://doi.org/10.1016/j.yexcr.2013.05.025 [DOI] [PubMed] [Google Scholar]
- He, B. P. , Wen, W. , & Strong, M. J. (2002). Activated microglia (BV‐2) facilitation of TNF‐alpha‐mediated motor neuron death in vitro. Journal of Neuroimmunology, 128(1/2), 31–38. [DOI] [PubMed] [Google Scholar]
- Hewitt, C. , Kirby, J. , Highley, J. R. , Hartley, J. A. , Hibberd, R. , Hollinger, H. C. , … Shaw, P. J. (2010). Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Archives in Neurology, 67(4), 455–461. doi:10.1001/archneurol.2010.52 [DOI] [PubMed] [Google Scholar]
- Howland, D. S. , Liu, J. , She, Y. , Goad, B. , Maragakis, N. J. , Kim, B. , … Rothstein, J. D. (2002). Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant‐mediated amyotrophic lateral sclerosis (ALS). Proceedings of the National Academy of Sciences of the United States of America, 99(3), 1604–1609. https://doi.org/10.1073/pnas.032539299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, A. F. , Gravel, M. , & Kriz, J. (2009). Live imaging of amyotrophic lateral sclerosis pathogenesis: Disease onset is characterized by marked induction of GFAP in Schwann cells. Glia, 57(10), 1130–1142. https://doi.org/10.1002/glia.20836 [DOI] [PubMed] [Google Scholar]
- Kwak, S. , Hideyama, T. , Yamashita, T. , & Aizawa, H. (2010). AMPA receptor‐mediated neuronal death in sporadic ALS. Neuropathology, 30(2), 182–188. https://doi.org/10.1111/j.1440-1789.2009.01090.x [DOI] [PubMed] [Google Scholar]
- Kwiatkowski, T. J. , Bosco, D. A. , LeClerc, A. L. , Tamrazian, E. , Vanderburg, C. R. , Russ, C. , … Brown, R. H. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science, 323(5918), 1205–1208. https://doi.org/10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- Kwon, I. , Xiang, S. , Kato, M. , Wu, L. , Theodoropoulos, P. , Wang, T. , … McKnight, S. L. (2014). Poly‐dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science, 345(6201), 1139–1145. https://doi.org/10.1126/science.1254917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier‐Tourenne, C. , Polymenidou, M. , & Cleveland, D. W. (2010). TDP‐43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Human Molecular Genetics, 19(R1), R46–R64. https://doi.org/10.1093/hmg/ddq137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasiene, J. , & Yamanaka, K. (2011). Glial cells in amyotrophic lateral sclerosis. Neurology Research International, 2011, 1–7. https://doi.org/10.1155/2011/718987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis, D. , Zhao, P. , & Beattie, E. C. (2008). Rapid tumor necrosis factor‐induced exocytosis of glutamate receptor 2‐lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. Journal of Neuroscience, 28(9), 2119–2130. https://doi.org/10.1523/jneurosci.5159-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, J. B. , Kong, J. , Nadler, M. , & Xu, Z. (1999). Astrocytes interact intimately with degenerating motor neurons in mouse amyotrophic lateral sclerosis (ALS). Glia, 28(3), 215–224. [PubMed] [Google Scholar]
- Liddelow, S. A. , Guttenplan, K. A. , Clarke, L. E. , Bennett, F. C. , Bohlen, C. J. , Schirmer, L. , … Barres, B. A. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541(7638), 481–487. https://doi.org/10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. L. , Bristol, L. A. , Jin, L. , Dykes‐Hoberg, M. , Crawford, T. , Clawson, L. , & Rothstein, J. D. (1998). Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron, 20(3), 589–602. [DOI] [PubMed] [Google Scholar]
- Mackenzie, I. R. A. , Ansorge, O. , Strong, M. , Bilbao, J. , Zinman, L. , Ang, L.‐C. , … Neumann, M. (2011). Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: Two distinct patterns correlating with disease severity and mutation. Acta Neuropathologica, 122(1), 87–98. https://doi.org/10.1007/s00401-011-0838-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour, M. , Nagarajan, N. , Nehring, R. B. , Clements, J. D. , & Rosenmund, C. (2001). Heteromeric AMPA receptors assemble with a preferred subunit stoichiometry and spatial arrangement. Neuron, 32(5), 841–853. https://doi.org/10.1016/s0896-6273(01)00520-7 [DOI] [PubMed] [Google Scholar]
- Maragakis, N. J. , & Rothstein, J. D. (2006). Mechanisms of disease: Astrocytes in neurodegenerative disease. Nature Clinical Practice Neurology, 2(12), 679–689. https://doi.org/10.1038/ncpneuro0355 [DOI] [PubMed] [Google Scholar]
- Ogoshi, F. , Yin, H. Z. , Kuppumbatti, Y. , Song, B. , Amindari, S. , & Weiss, J. H. (2005). Tumor necrosis‐factor‐alpha (TNF‐α) induces rapid insertion of Ca2+‐permeable α‐amino‐3‐hydroxyl‐5‐methyl‐4‐isoxazole‐propionate (AMPA)/kainate (Ca‐A/K) channels in a subset of hippocampal pyramidal neurons. Experimental Neurology, 193(2), 384–393. https://doi.org/10.1016/j.expneurol.2004.12.026 [DOI] [PubMed] [Google Scholar]
- Oketa, Y. , Higashida, K. , Fukasawa, H. , Tsukie, T. , & Ono, S. (2013). Abundant FUS‐immunoreactive pathology in the skin of sporadic amyotrophic lateral sclerosis. Acta Neurologica Scandinavica, 128(4), 257–264. https://doi.org/10.1111/ane.12114 [DOI] [PubMed] [Google Scholar]
- Olmos, G. , & Lladó, J. (2014). Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediators of Inflammation, 2014, 1–12. https://doi.org/10.1155/2014/861231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek, J. , Lips, M. B. , & Keller, B. U. (1999). Calcium dynamics and buffering in motoneurones of the mouse spinal cord. The Journal of Physiology, 520(2), 485–502. https://doi.org/10.1111/j.1469-7793.1999.00485.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrini, S. , Sau, D. , Guareschi, S. , Bogush, M. , Brown, R. H. , Naniche, N. , … Pasinelli, P. (2010). ALS‐linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl‐2. Human Molecular Genetics, 19(15), 2974–2986. https://doi.org/10.1093/hmg/ddq202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pękalski, J. , Zuk, P. J. , Kochańczyk, M. , Junkin, M. , Kellogg, R. , Tay, S. , & Lipniacki, T. (2013). Spontaneous NF‐κB activation by autocrine TNFα signaling: A computational analysis. PLoS ONE, 8(11), e78887 https://doi.org/10.1371/journal.pone.0078887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips, T. , & Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. The Lancet Neurology, 10(3), 253–263. https://doi.org/10.1016/s1474-4422(11)70015-1 [DOI] [PubMed] [Google Scholar]
- Prell, T. , Lautenschläger, J. , Weidemann, L. , Ruhmer, J. , Witte, O. W. , & Grosskreutz, J. (2014). Endoplasmic reticulum stress is accompanied by activation of NF‐κB in amyotrophic lateral sclerosis. Journal of Neuroimmunology, 270(1/2), 29–36. https://doi.org/10.1016/j.jneuroim.2014.03.005 [DOI] [PubMed] [Google Scholar]
- Qosa, H. , Lichter, J. , Sarlo, M. , Markandaiah, S. S. , McAvoy, K. , Richard, J. P. , … Trotti, D. (2016). Astrocytes drive upregulation of the multidrug resistance transporter ABCB1 (P‐Glycoprotein) in endothelial cells of the blood‐brain barrier in mutant superoxide dismutase 1‐linked amyotrophic lateral sclerosis. Glia, 64(8), 1298–1313. https://doi.org/10.1002/glia.23003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers, R. , Stewart, H. , Dejesus‐Hernandez, M. , Krieger, C. , Graff‐Radford, N. , Fabros, M. , … Mackenzie, I. R. A. (2010). Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle & Nerve, 42(2), 170–176. https://doi.org/10.1002/mus.21665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich, G. , Liu, Z. Q. , Kloss, C. U. , Labow, M. , Bluethmann, H. , & Bohatschek, M. (2002). Cytotoxic potential of proinflammatory cytokines: Combined deletion of TNF receptors TNFR1 and TNFR2 prevents motoneuron cell death after facial axotomy in adult mouse. Experimental Neurology, 178(2), 186–193. [DOI] [PubMed] [Google Scholar]
- Rizzo, F. , Riboldi, G. , Salani, S. , Nizzardo, M. , Simone, C. , Corti, S. , & Hedlund, E. (2013). Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cellular & Molecular Life Sciences, 71(6), 999–1015. https://doi.org/10.1007/s00018-013-1480-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello, M. , & Volterra, A. (2012). TNFα in synaptic function: Switching gears. Trends in Neurosciences, 35(10), 638–647. https://doi.org/10.1016/j.tins.2012.06.001 [DOI] [PubMed] [Google Scholar]
- Serio, A. , Bilican, B. , Barmada, S. J. , Ando, D. M. , Zhao, C. , Siller, R. , … Chandran, S. (2013). Astrocyte pathology and the absence of non‐cell autonomy in an induced pluripotent stem cell model of TDP‐43 proteinopathy. Proceedings of the National Academy of Sciences of the United States of America, 110(12), 4697–4702. https://doi.org/10.1073/pnas.1300398110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen, D. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor. Journal of Neuroscience, 25(12), 3219–3228. https://doi.org/10.1523/jneurosci.4486-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel, E. W. , Cohen, J. A. , Fadul, C. E. , Cogbill, C. H. , Graber, D. J. , Kingman, L. , … Harris, B. T. (2009). Efficacy of thalidomide for the treatment of amyotrophic lateral sclerosis: A phase II open label clinical trial. Amyotrophic Lateral Sclerosis, 10(5/6), 393–404. https://doi.org/10.3109/17482960802709416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, N. , Kato, S. , Kato, M. , Warita, H. , Mizuno, H. , Kato, M. , … Aoki, M. (2012). FUS/TLS‐immunoreactive neuronal and glial cell inclusions increase with disease duration in familial amyotrophic lateral sclerosis with an R521CFUS/TLSMutation. Journal of Neuropathology & Experimental Neurology, 71(9), 779–788. https://doi.org/10.1097/nen.0b013e318264f164 [DOI] [PubMed] [Google Scholar]
- Swarup, V. , Phaneuf, D. , Dupré, N. , Petri, S. , Strong, M. , Kriz, J. , & Julien, J.‐P. (2011). Deregulation of TDP‐43 in amyotrophic lateral sclerosis triggers nuclear factor κB‐mediated pathogenic pathways. The Journal of Experimental Medicine, 208(12), 2429–2447. https://doi.org/10.1084/jem.20111313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel, G. , Li, L. Y. , Patil, R. V. , & Wax, M. B. (2001). TNF‐alpha and TNF‐alpha receptor‐1 in the retina of normal and glaucomatous eyes. Invest Ophthalmology & Visual Sciences, 42(8), 1787–1794. [PubMed] [Google Scholar]
- Tong, J. , Huang, C. , Bi, F. , Wu, Q. , Huang, B. , Liu, X. , … Xia, X.‐G. (2013). Expression of ALS‐linked TDP‐43 mutant in astrocytes causes non‐cell‐autonomous motor neuron death in rats. EMBO J, 32(13), 1917–1926. https://doi.org/10.1038/emboj.2013.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udagawa, T. , Fujioka, Y. , Tanaka, M. , Honda, D. , Yokoi, S. , Riku, Y. , … Sobue, G. (2015). FUS regulates AMPA receptor function and FTLD/ALS‐associated behaviour via GluA1 mRNA stabilization. Nature Communications, 6, 7098 https://doi.org/10.1038/ncomms8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranishi, H. (2001). Involvement of the Pro‐oncoprotein TLS (Translocated in Liposarcoma) in nuclear factor‐kappa B p65‐mediated transcription as a coactivator. Journal of Biological Chemistry, 276(16), 13395–13401. https://doi.org/10.1074/jbc.m011176200 [DOI] [PubMed] [Google Scholar]
- Van Damme, P. , Bogaert, E. , Dewil, M. , Hersmus, N. , Kiraly, D. , Scheveneels, W. , … Robberecht, W. (2007). Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proceedings of the National Academy of Sciences of the United States of America, 104(37), 14825–14830. https://doi.org/10.1073/pnas.0705046104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance, C. , Rogelj, B. , Hortobagyi, T. , De Vos, K. J. , Nishimura, A. L. , Sreedharan, J. , … Shaw, C. E. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science, 323(5918), 1208–1211. https://doi.org/10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe, W. , Ihle, E. C. , Patneau, D. K. , Robberecht, W. , Robberecht, W. , & Brorson, J. R. (2000). AMPA receptor current density, not desensitization, predicts selective motoneuron vulnerability. Journal of Neuroscience, 20(19), 7158–7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka, K. , Chun, S. J. , Boillee, S. , Fujimori‐Tonou, N. , Yamashita, H. , Gutmann, D. H. , … Cleveland, D. W. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nature Neuroscience, 11(3), 251–253. https://doi.org/10.1038/nn2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Figure S1
Figure S2