

ABSTRACT
Background: We investigated a family that presented with an infantile‐onset chorea‐predominant movement disorder, negative for NKX2‐1, ADCY5, and PDE10A mutations. Methods: Phenotypic characterization and trio whole‐exome sequencing was carried out in the family. Results: We identified a homozygous mutation affecting the GAF‐B domain of the 3’,5’‐cyclic nucleotide phosphodiesterase PDE2A gene (c.1439A>G; p.Asp480Gly) as the candidate novel genetic cause of chorea in the proband. PDE2A hydrolyzes cyclic adenosine/guanosine monophosphate and is highly expressed in striatal medium spiny neurons. We functionally characterized the p.Asp480Gly mutation and found that it severely decreases the enzymatic activity of PDE2A. In addition, we showed equivalent expression in human and mouse striatum of PDE2A and its homolog gene, PDE10A. Conclusions: We identified a loss‐of‐function homozygous mutation in PDE2A associated to early‐onset chorea. Our findings possibly strengthen the role of cyclic adenosine monophosphate and cyclic guanosine monophosphate metabolism in striatal medium spiny neurons as a crucial pathophysiological mechanism in hyperkinetic movement disorders. © 2018 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: phosphodiesterase, striatum, chorea, movement disorders, PDE2A
Chorea is a hyperkinetic movement disorder characterized by an excess of brief, continuous, unpatterned involuntary movements.1 Focal lesions of the striatum, degeneration, or functional dysregulation of medium spiny neurons (MSNs) that constitute ∼95% of the striatal cells are considered to be crucially implicated in the pathophysiology of choreic movements.2, 3, 4, 5 A variety of acquired causes may underlie chorea in the pediatric age group (e.g., Sydenham chorea, cerebral palsy), but genetic etiologies also play a role in different early‐onset choreic syndromes. Among the possible genetic causes, dominantly inherited (or de novo) mutations in NKX2‐1 (MIM #600635), encoding a transcription factor essential for striatum development, cause a variable spectrum of childhood‐onset disorders ranging from choreoathetosis to myoclonus, congenital hypothyroidism, and respiratory distress.6 In addition, de novo or dominantly inherited mutations in ADCY5 (MIM #600293), encoding an enzyme crucial to the synthesis of cyclic adenosine monophosphate (cAMP) in MSNs, have been linked to different infantile/childhood‐onset phenotypes defined as “ADCY5‐related dyskinesias” that include nonprogressive choreiform movement disorders as well as pleiotropic fluctuating/paroxysmal dyskinesias.7, 8 Furthermore, both de novo dominant and biallelic mutations in the PDE10A gene (MIM #610652), encoding an enzyme involved in the hydrolysis/degradation of cAMP and cyclic guanosine monophosphate (cGMP) in MSNs, have been reported in patients with infantile/childhood‐onset chorea usually exhibiting a generalized distribution.9, 10 In this study, we performed trio‐based whole‐exome sequencing (WES) in a 12‐year‐old patient presenting a childhood‐onset chorea‐predominant movement disorder and identified a homozygous missense mutation (c.1439A>G; p.Asp480Gly) in PDE2A (MIM #602658), a gene mainly involved in brain cAMP and cGMP metabolism and not previously associated with human phenotypes, as the possible novel genetic cause of chorea in the patient. Screening of genomic data from a number (n = 62) of similarly affected individuals did not identify additional PDE2A likely pathogenic variants.
Materials and Methods
Genetic Analysis
The 12‐year‐old male patient is the second child of unrelated healthy parents, both originally from the Canary Island of Tenerife, and he was seen at the Hospital Sant Joan de Déu in Barcelona (Spain) by movement disorder specialists because of a history of early‐onset fluctuating dyskinesia associated with chronic chorea. To molecularly investigate the cause of the disease in this child, the 4 family members (Fig. 1A) donated their blood samples after informed consent and DNA was extracted using standard procedures. A trio WES study of the family (Fig. 1A, I‐1, I‐2, II‐2) was then performed. Genomic pipeline and variants annotation were carried out as previously reported11, 12 and described in the Supplementary Information. In accord with the pedigree and phenotype, our filtering strategy prioritized rare (<1% in public databases, including 1000 Genomes project and Exome Aggregation Consortium [ExAC v0.2]) variants that were fitting a de novo or a recessive model (Supplementary Information). We also performed homozygosity mapping in the family (Supplementary Information).
Figure 1.
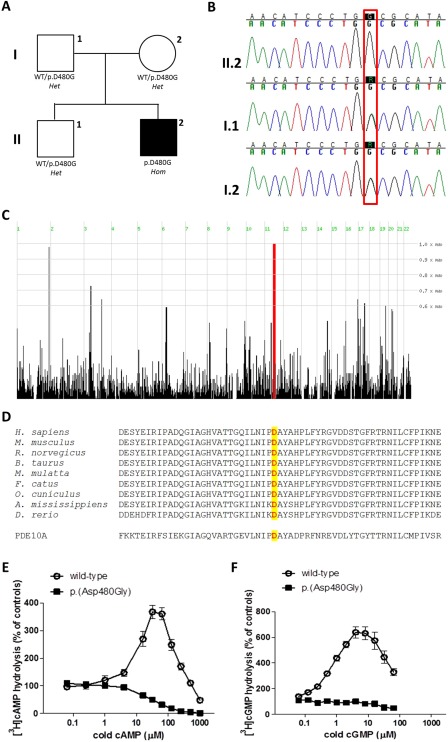
Family tree and genetic and functional studies. (A) Family tree. (B) Chromatograms from Sanger sequencing from individuals I‐1, I‐2, and II‐2. (C) Genetic analysis showing the homozygous block of the proband on chromosome 11 (chr11: 63138482‐76853783). (D) Multiple‐sequence alignment showing conservation of protein sequence across species and PDE homolog (PDE10A) in the GAF‐B domain, in which the p.Asp480Gly homozygous mutation (underlined) was found. (E) Catalytic activity of wild‐type and mutant PDE2A in the presence of 70 nM of [3H]cAMP. (F) Catalytic activity of wild‐type and mutant PDE2A in the presence of 70 nM of [3H]cGMP. [Color figure can be viewed at wileyonlinelibrary.com]
Functional Characterization of the p.Asp480Gly PDE2A Mutation
Materials
cAMP and cGMP were purchased from Sigma‐Aldrich (St. Louis, MO). The [3H]‐labeled nucleotides, [3H]cAMP (31.3 Ci/mmol) and [3H]cGMP (14.3 Ci/mmol), were purchased from PerkinElmer (Waltham, MA).
Cloning and Expression of Constructs
Complementary DNA for human PDE2A3 (GenBank: U67733) was used as a template and the mutant, c.1439A>G; p.(Asp480Gly), was constructed by site‐directed point mutation. All constructs were cloned into the pcDNA3.1(+)neo vector (Thermo Fisher Scientific, Inc., Waltham, MA) and transfected into COS‐7 cells (ECACC, Salisbury, UK). The membrane fractions were used for the enzyme assay.
In Vitro Phosphodiesterase Enzyme Assay
Phosphodiesterase (PDE) activities were measured using a scintillation proximity assay (SPA)‐based method.13 In this assay, the product of the PDE reaction, either [3H]AMP or [3H]GMP, can bind directly to yttrium silicate PDE SPA beads (GE Healthcare Ltd., Little Chalfont, UK), leading to light emission from the scintillant in the beads. The enzyme assays were conducted in a buffer (50 mM of HEPES‐NaOH, 8.3 mM of MgCl2, 1.7 mM of ethylene glycol tetraacetic acid, and 0.1% bovine serum albumin [pH 7.4]) in 96‐well half‐area plates (Corning Inc., Corning, NY). For enzyme studies, reaction was conducted at the presence of the indicated concentrations of substrate using a mixture (20 µL) of [3H]‐labeled and unlabeled cAMP or [3H]‐labeled and unlabeled cGMP with the 20 µL of membrane fractions of PDE2A‐expressing COS‐7 cells at 37oC, followed by reaction termination by SPA beads addition (20 µL of 20 mg/mL). Degradation of each radiolabeled substrate ([3H]cAMP or [3H]cGMP) at the presence of various concentrations of cold cAMP or cGMP, respectively, was measured using the SPA‐based assay method.
PDE2A Messenger RNA Expression Studies
Overall brain expression and cell‐specific expression data were initially obtained using BRAINEAC and BacTRAP mice data, respectively.14, 15 Then, we analyzed in vivo PDE2A and PDE10A messenger RNA (mRNA) expression patterns using mice and human brains, as described in detail in the Supplementary Information.
Results
Disease onset in our patient mainly consisted in fluctuating attacks of sudden falls, followed by dystonic postures and generalized choreic movements. Subsequently, he developed since the age of 9 years a slowly progressive choreic movement disorder associated with dystonic features. In addition to his movement disorder, he also had language difficulties, cognitive impairment (total score 44 with Kauffman Brief Intelligence Test), and a history of interictal epileptic features (Supplementary Information). We performed a trio‐based WES, and, after applying our filtering criteria, we identified a number of possibly pathogenic variants according to guidelines for variants interpretation (Supplementary Information; Supplementary Table 1).16 The proband of our family carried a single de novo variant in the gene, STRADA (NM_153335.5; c.1042G>T: p.Gly348Trp); homozygous intragenic deletions (or biallelic truncating mutations) in this gene were previously associated with polyhydramnios, megalencephaly, distinctive facial features, and symptomatic epilepsy (#MIM 611087),17, 18 a phenotype not consistent with the clinical presentation of our patient. In addition, we identified a homozygous variant (NM_002599.4; c.1439A>G: p.Asp480Gly) in PDE2A (#MIM 602658) as the only biallelic variant segregating with the disease within the family. Also, the PDE2A homozygous mutation was inherited within the only significant homozygous block (chr11: 63138482‐76853783) identified in the proband by homozygosity mapping analysis (Fig. 1B–D; Supplementary Information). Additionally, our in vitro functional studies showed that both cAMP and cGMP hydrolysis were markedly increased for wild‐type PDE2A compared to the p.Asp480Gly mutant (approximately 3.7‐fold and 6.4‐fold, respectively). On the contrary, neither cAMP nor cGMP increased enzyme activities of the mutant enzyme (Fig. 1E,F; Supplementary Tables 2‐5), indicating a severe disruption of catalytic activity of mutated PDE2A.
Discussion
The genetic analysis of this family using WES and homozygosity mapping indicated a homozygous missense mutation in PDE2A as the most likely explanation for the patient's disease. This is supported by: (1) in silico pathogenic predictors, conservation, and cosegregation analysis (Supplementary Table 1; Fig. 1D); (2) previous9, 10, 19 and current (Fig. 2A–C) studies, which indicates a high (predominant) expression of PDE2A in the striatum (qualitatively equivalent to PDE10A striatal expression); (3) biological importance of the PDE2A GAF‐B domain (where the p.Asp480Gly mutation is located) in the metabolism of cAMP and cGMP (and implication of these cyclic nucleotides in the pathogenesis of similar phenotypes including ADCY5‐ and PDE10A‐related movement disorders)5, 6, 7, 8, 9, 10, 20, 21, 22, 23; and (4) in vitro functional studies that showed a severe disruption of the mutant PDE2A enzymatic activity (Fig. 2E; Supplementary Tables 2‐5).
Figure 2.
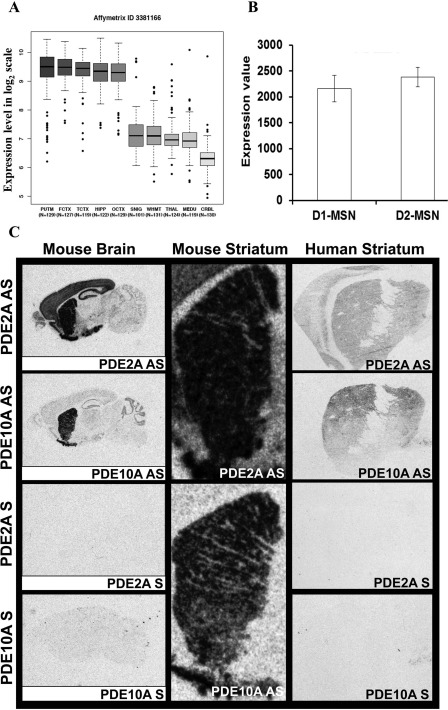
PDE2A expression studies. (A) Brain expression values of PDE2A show higher expression levels in the striatum and also other CNS regions, especially FCTX (frontal cortex) and TCTX (temporal cortex), and less in HIPP (hippocampus), OCTX (occipital cortex), WHMT (white matter), SNIG (substantia nigra), MEDU (medulla), THAL (thalamus), and CRBL (cerebellar cortex). (B) Expression values of PDE2A mRNA (1447707_s_at) in MSNs of direct and indirect pathways (D1 + and D2 + MSNs). (C) mRNA expression analysis on mice and human brains of PDE2A and PDE10A show similar expression patterns in striatum, but different expression patterns elsewhere in mouse and human brain.
Importantly, the amino acid involved by the mutation we identified (aspartic acid at position 480) is located in the presumed cyclic nucleotide binding pocket of GAF‐b domain.24 To assess enzyme activity of PDE2A, we measured degradation of radiolabeled substrate ([3H]cAMP or [3H]cGMP) and showed a significant decrease in the hydrolytic activity of the mutant PDE2A enzyme. Of note, we screened genomic data from 17 individuals with childhood‐onset chorea and from 45 patients presenting fluctuating or paroxysmal dyskinesia and failed to identify further mutations in PDE2A, suggesting that this might represent a very rare genetic cause of early‐onset choreic/hyperkinetic movement disorders. The PDE2A p.Asp480Gly mutation was inherited within the only homozygous block on a shared haplotype (Supplementary Fig. 1), indicating that this could possibly represent a founder mutation from the genetic isolate of Canary Islands. Supporting this Canary founder effect, in the ExAC database (http://exac.broadinstitute.org, last accessed October 2017) containing 60,706 individuals, the PDE2A gene was found to be highly constrained for missense variation (z = 4.78), with only 27 individuals carrying nonsynonymous (heterozygous) variants affecting the GAF‐B domain.
Of interest, monogenic etiologies underlying infantile‐ and childhood‐onset choreic movement disorders are being increasingly recognized.7, 8, 9, 10 In this regard, the identification and characterization of the ADCY5‐ and PDE10A‐related spectrum of disorders shed new light on the key role of cAMP and cGMP signaling in basal ganglia circuit and the control of movements.5, 6, 7 The adenyl cyclase 5 enzyme catalyzes cAMP formation and pathogenic ADCY5 mutations might thus increase the synthesis of cAMP because of a possible enhancement of Ac5 enzymatic activity.5, 7, 8 The PDE10A enzyme is involved in cAMP hydrolysis in MSNs, and in vitro assessment of PDE10A mutations showed that both dominant and recessive variants lead to a loss‐of‐function effect with consequent impairment of cAMP and cGMP hydrolysis/degradation.9, 10 Thus, elevated cAMP (and possibly cGMP) intracellular levels in MSNs (either attributed to increased synthesis or reduced hydrolysis) could represent a central mechanism for molecular pathogenesis of different hyperkinetic movement disorders.5 Notably, the cyclic nucleotides cAMP and cGMP are ubiquitous intracellular second messengers regulating a variety of biological processes.25 The intracellular concentration of these molecules is modulated by the activity of PDEs, a class of several different enzymes currently grouped overall in 11 families, and, among the PDEs, PDE2A and PDE10A are among the most highly enriched in the striatum.26, 27 PDE2A expression is highest in the striatum, cortex, and hippocampus compared to other central nervous system (CNS) regions (Fig. 2A), with similar expression in direct and indirect MSN pathways (Fig. 2B). Furthermore, our comparative expression analysis on human and mice brain found that striatal mRNA levels of PDE2A and its paralogue gene, PDE10A, are qualitatively equivalent, with a more generalized PDE2A expression in some additional CNS regions (Fig. 2C). These PDE2A more generalized brain expression patterns may explain the broad neurological features (including interictal epileptic discharges and cognitive impairment) we observed in our patient in addition to his movement disorder. Notably, the PDE2A enzyme is a dual‐substrate PDE that hydrolyzes both cAMP and cGMP and has two N‐terminal tandem noncatalytic domains, named GAF‐A and GAFB.18, 19, 28 PDE2A exhibits a low level of basal hydrolytic activity that is further stimulated when its GAF‐B domain binds cAMP or cGMP.20, 21, 29 However, it is most likely that only cGMP binds the PDE2A GAF‐B domain in vivo, thereby selectively stimulating its cAMP hydrolytic activity.20, 30
In conclusion, results from our genetic and functional studies indicate a PDE2A p.Asp480Gly homozygous loss‐of‐function mutation as the likely genetic cause of early‐onset hereditary chorea in our family, thus possibly expanding the genetic aetiology of early‐onset choreic/hyperkinetic movement disorders associated to abnormal c‐AMP and c‐GMP metabolism in striatal MSNs.
Legend to the Video
The episodes of dyskinesia initiated with neck extension, backward falling, and dystonic posturing of the four limbs, followed by choreic movements, facial grimacing, blinking, and orolingual movements. During execution of motor task (writing), the child showed a complex hyperkinetic movement consisting in baseline chorea associated with dystonic posturing (predominantly in the left foot).
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
V.S.: 3A, 3B
B.P.‐D.: 1A, 1B, 1C, 3A
K.N.: 1A, 1B, 1C, 3A
V.S.A.‐A.: 1A, 1B, 1C, 3A
A. Manole: 3B
S.E.: 1A, 1B, 1C, 3A
J.V.: 1B, 1C, 3A
C.B.: 1A, 1B, 1C, 3A
N.E.M.: 1A, 1B, 1C, 3A
C.K.: 3B
M.P.K.: 1A, 1B, 1C, 3A
C.H.D.: 3B
H.K.: 3B
A. Macaya: 3B
H.H.: 3B
Financial Disclosures
Nothing to report.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website
Supplementary Information
Supplementary FIG. 1. Haplotype analysis for the region on chromosome 11 surrounding PDE2A c.1439A>G (indicated in red), with markers and their positions (Bp) displayed on the left.
Supplementary Information Tables
Supplementary Information Movie
Acknowledgments
This study was supported by the Wellcome Trust (WT093205MA, WT104033AIA), Medical Research Council (to V.S. and H.H.), European Community's Seventh Framework Programme (FP7/2007‐2013, under grant agreement No. 2012‐305121; to H.H.), and National Institute for Health Research University College London Hospitals Biomedical Research Centre (to H.H.). M.P.K. is a recipient of an award from NIMH 1R01MH101130. C.K. is the recipient of a career development award from the Hermann and Lilly Schilling Foundation. The authors acknowledge Masaaki Nishimura for constructing the plasmids carrying genes of wild‐type PDE2A and a mutant, c.1439A>G; p.(Asp480Gly). The authors acknowledge the individuals (and their family) who donated their brain to the Stanley Foundation and the Stanley Foundation for sharing this donation with our laboratory.
Relevant conflicts of interest/financial disclosures: Nothing to report.
Full financial disclosures and author roles may be found in the online version of this article.
References
- 1. Gittis AH, Kreitzer AC. Striatal microcircuitry and movement disorders. Trends Neurosci 2012;35:557‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci 1990;13:244‐254. [DOI] [PubMed] [Google Scholar]
- 3. Graybiel AM. The basal ganglia. Curr Biol 2000;10:R509‐R511. [DOI] [PubMed] [Google Scholar]
- 4. Obeso JA, Rodríguez‐Oroz MC, Rodríguez M, et al., The basal ganglia and disorders of movement: pathophysiological mechanisms. News Physiol Sci 2002;17:51‐55. [DOI] [PubMed] [Google Scholar]
- 5. Mencacci NE, Carecchio M. Recent advances in genetics of chorea. Curr Opin Neurol 2016;29:486‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kleiner‐Fisman G, Rogaeva E, Halliday W, et al. Benign hereditary chorea: clinical, genetic, and pathological findings. Ann Neurol 2003;54:244‐247. [DOI] [PubMed] [Google Scholar]
- 7. Chang FC, Westenberger A, Dale RC, et al. Phenotypic insights into ADCY5‐associated disease. Mov Disord 2016;31:1033‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friedman JR, Méneret A, Chen DH, et al. ADCY5 mutation carriers display pleiotropic paroxysmal day and nighttime dyskinesias. Mov Disord 2016;31:147‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diggle CP, Sukoff Rizzo SJ, Popiolek M, et al. Biallelic Mutations in PDE10A Lead to Loss of Striatal PDE10A and a Hyperkinetic Movement Disorder with Onset in Infancy. Am J Hum Genet 2016;98:735‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mencacci NE, Kamsteeg EJ, Nakashima K, et al. De Novo Mutations in PDE10A Cause Childhood‐Onset Chorea with Bilateral Striatal Lesions. Am J Hum Genet 2016; 98:763‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zollo M, Ahmed M, Ferrucci V, et al. PRUNE is crucial for normal brain development and mutated in microcephaly with neurodevelopmental impairment. Brain 2017;140:940‐952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salpietro V, Lin W, Delle Vedove A, et al. Homozygous mutations in VAMP1 cause a presynaptic congenital myasthenic syndrome. Ann Neurol 2017;81:597‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matthiesen K, Nielsen J. Binding of cyclic nucleotides to phosphodiesterase 10A and 11A GAF domains does not stimulate catalytic activity. Biochem J 2009;423:401‐409. [DOI] [PubMed] [Google Scholar]
- 14. Ramasamy A, Trabzuni D, Guelfi S, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci 2014;17:1418‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doyle, J.P. , Dougherty JD, Heiman M, et al., Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 2008;135:749‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the nterpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Puffenberger EG, Strauss KA, Ramsey KE, et al. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7‐kilobase deletion in LYK5. Brain 2007;130:1929‐1941. [DOI] [PubMed] [Google Scholar]
- 18. Bi W, Glass IA, Muzny DM, Gibbs RA, Eng CM, Yang Y, Sun A. Whole exome sequencing identifies the first STRADA point mutation in a patient with polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome (PMSE). Am J Med Genet A 2016;170:2181‐2185. [DOI] [PubMed] [Google Scholar]
- 19. Stephenson DT, Coskran TM, Kelly MP, et al. The distribution of phosphodiesterase 2A in the rat brain. Neuroscience 2012;226:145‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beavo JA, Hardman JG, Sutherland EW. Stimulation of adenosine 3’,5’ monophosphate hydrolysis by guanosine 3’,5’‐monophosphate. J Biol Chem 1971;246:3841‐3846. [PubMed] [Google Scholar]
- 21. Erneux C, Couchie D, Dumont JE, et al. Specificity of cyclic GMP activation of a multi‐substrate cyclic nucleotide phosphodiesterase from rat liver. Eur J Biochem 1981;115:503‐510. [DOI] [PubMed] [Google Scholar]
- 22. Martins TJ, Mumby MC, Beavo JA. Purification and characterization of a cyclic GMP‐stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J Biol Chem 1982;257:1973‐1979. [PubMed] [Google Scholar]
- 23. Rosman GJ, Damico CA, Cross AH, et al. Isolation and characterization of human cDNAs encoding a cGMP‐stimulated 3’,5’‐cyclic nucleotide phosphodiesterase. Gene 1997;191:89‐95. [DOI] [PubMed] [Google Scholar]
- 24. Wu AY, Tang XB, Martinez Se, et al. Molecular determinants for cyclic nucleotide binding to the regulatory domains of phosphodiesterase 2A. J Biol Chem 2004;279:37928‐37938. [DOI] [PubMed] [Google Scholar]
- 25. Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 2006;58:488‐520. [DOI] [PubMed] [Google Scholar]
- 26. Kelly MP, Adamowicz W, Bove S, et al. Select 3’,5’‐cyclic nucleotide phosphodiesterases exhibit altered expression in the aged rodent brain. Cell Signal 2014;26:383‐397. [DOI] [PubMed] [Google Scholar]
- 27. Lakics V, Karran EH, Boess FG. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010;59:367‐374. [DOI] [PubMed] [Google Scholar]
- 28. Pandit J, Forman MD, Fennell KF, et al. Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X‐ray structure of a near full‐length construct. Proc Natl Acad Sci U S A 2009;106:18225‐18230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev 2011;91:651‐690. [DOI] [PubMed] [Google Scholar]
- 30. Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 2006;58:488‐520. [DOI] [PubMed] [Google Scholar]
- 31. Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res 2012;22:1525‐1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Backenroth D, Homsy J, Murillo LR, et al. CANOES: detecting rare copy number variants from whole exome sequencing data. Nucleic Acids Res 2014;42:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website
Supplementary Information
Supplementary FIG. 1. Haplotype analysis for the region on chromosome 11 surrounding PDE2A c.1439A>G (indicated in red), with markers and their positions (Bp) displayed on the left.
Supplementary Information Tables
Supplementary Information Movie