

Abstract
Aims
To investigate whether the pharmacokinetic characteristics of semaglutide were altered in people with hepatic impairment, assessed using Child–Pugh criteria, vs those with normal hepatic function.
Methods
In this multicentre, open‐label, parallel‐group trial (sponsor Novo Nordisk, ClinicalTrials.gov ID NCT02210871), four groups of participants with normal hepatic function (n = 19) or mild (n = 8), moderate (n = 10) or severe (n = 7) hepatic impairment received a single, subcutaneous dose of 0.5 mg semaglutide. Semaglutide plasma concentrations were assessed frequently for 35 days after dosing. The primary endpoint was area under the semaglutide plasma concentration–time curve from time zero to infinity (AUC0‐∞). No effect of hepatic impairment was declared if the 90% confidence interval (CI) for the between‐group ratio (hepatic impairment/normal function) was within the interval 0.70 to 1.43.
Results
Semaglutide exposure was similar across all groups, with AUC0‐∞ treatment ratios for mild impairment/normal function of 0.95 (90% CI 0.77, 1.16), moderate impairment/normal function 1.02 (90% CI 0.93, 1.12), and severe impairment/normal function 0.97 (90% CI 0.84, 1.12). The maximum plasma semaglutide concentration (Cmax) did not appear to be influenced by hepatic function, with mild impairment/normal function treatment ratios of 0.99 (90% CI 0.80, 1.23), moderate impairment/normal function 1.02 (90% CI 0.88, 1.18) and severe impairment/normal function 1.15 (90% CI 0.89, 1.48; sensitivity analysis excluding one extreme semaglutide concentration: 1.05 [90% CI 0.88, 1.25]). In all, 10 participants reported 12 mild or moderate non‐serious adverse events. No unexpected safety or tolerability issues were observed.
Conclusions
Semaglutide exposure did not appear to be affected by hepatic impairment, suggesting that dose adjustment may not be necessary in patients with hepatic impairment. Semaglutide was well tolerated and there were no unexpected safety issues.
Keywords: GLP‐1, GLP‐1 analogue, liver, pharmacokinetics, type 2 diabetes
1. INTRODUCTION
Type 2 diabetes is a complex disease that remains one of the greatest challenges in terms of both health and economic costs.1, 2 Increased insulin resistance, coupled with progressive β‐cell failure, are essential components of the pathogenesis of the disease, which is associated with multiple morbidities.3, 4 Glucagon‐like peptide‐1 (GLP‐1) receptor agonists (RAs) have gained recognition in recent years for the treatment of type 2 diabetes. In clinical trials, GLP‐1RAs were associated with reductions in glycated haemoglobin (HbA1c), fasting plasma glucose and body weight, with the most frequently observed side effects being nausea, vomiting and diarrhoea.5, 6
Semaglutide is a GLP‐1 analogue under development for the treatment of type 2 diabetes. Semaglutide has 94% homology to native GLP‐1, with structural modifications that make semaglutide less susceptible to degradation by dipeptidyl peptidase‐4 (DPP‐4) enzymes.7 Moreover, the modifications improve the specific high‐affinity binding to albumin,7 which slows down the degradation of semaglutide in plasma and results in decreased renal clearance.8 The structural modifications prolong the half‐life of semaglutide to ~1 week, making it appropriate for once‐weekly administration.7, 9 In phase III trials, semaglutide demonstrated superior reductions in HbA1c and body weight compared with placebo and active comparators,10, 11, 12 as well as a decrease in cardiovascular risk.13
Native GLP‐1 is rapidly metabolized by enzymes such as DPP‐4, which is found in many tissues and cell types.14 Clearance of native GLP‐1 and its metabolites is largely mediated via the kidneys14 and, in general, GLP‐1RAs require no dose adjustment for hepatic impairment.15 In humans, semaglutide is metabolized via proteolytic cleavage of the peptide backbone and sequential β‐oxidation of the fatty acid chain, with no single organ acting as the major route of elimination.8 Semaglutide degradation products are excreted via urine and faeces,8 implying at least partial involvement of the liver in semaglutide elimination; therefore, impaired hepatic function may affect the pharmacokinetics of semaglutide. Moreover, semaglutide binds to albumin and the concentration of albumin may be lower in people with hepatic impairment than in those with normal hepatic function.
The rationale for the present study was to provide information on whether the pharmacokinetics of a single subcutaneous dose of 0.5 mg semaglutide was altered to such an extent that people with impaired hepatic function should be dosed differently from those with normal hepatic function. The safety and tolerability of semaglutide were also assessed.
2. METHODS
2.1. Trial population
The trial was conducted in men or women aged ≥18 years with a body mass index (BMI) of 18.5 to 40.0 kg/m2, who provided written informed consent before starting any trial‐related activities. At screening, participants were allocated into four groups: normal hepatic function; or mild; moderate; or severe hepatic impairment. Participants with hepatic impairment were those with a diagnosis of cirrhosis attributable to parenchymal liver disease, classified according to the Child–Pugh criteria,16 as indicated in Table 1. The diagnosis of liver cirrhosis attributable to parenchymal liver disease was confirmed and documented according to the participant's medical history, physical examination and at least one of the following: hepatic ultrasonography, computed axial tomography scan, magnetic resonance imaging, and/or liver biopsy. The hepatic impairment was to be stable, defined as no clinically significant change in disease status in the 30 days before screening, according to recent medical history.
Table 1.
Child–Pugh criteria for assessment of impaired liver function
| Points scored for observed findings | |||
|---|---|---|---|
| 1 | 2 | 3 | |
| Encephalopathy gradea | 0 | 1 or 2 | 3 or 4b |
| Ascitesc | Absent | Slight | Moderate |
| Serum bilirubin, μmol/L | < 34.2 | 34.2‐51.3 | > 51.3 |
| Serum albumin, g/L | > 35 | 28‐35 | < 28 |
| Prothrombin time (seconds prolonged) | < 4 | 4‐6 | >6 |
Grade 0: normal consciousness, personality, neurological examination, electroencephalogram; Grade 1: restless, sleep disturbed, irritable/agitated tremor, impaired handwriting, 5 cycles per second (cps) waves; Grade 2: lethargic, time‐disorientated, inappropriate, asterixis, ataxia, slow triphasic waves; Grade 3: somnolent, stuporous, place‐disorientated, hyperactive reflexes, rigidity, slower waves; Grade 4: unrousable coma, no personality/behaviour, decerebrate, slow 2‐3 cps delta activity.
People with encephalopathy grades 3 or 4 were excluded from the study.
People with advanced ascites were excluded from the study.
Mild hepatic impairment = 5‐6 points. Moderate hepatic impairment = 7‐9 points. Severe hepatic impairment = 10‐15 points.
Participants were matched to the extent possible across groups with respect to age, sex and body weight. The median body weight and age ranges in the three hepatic impairment groups were calculated after ≥5 participants in each group had been dosed. Participants with normal hepatic function were subsequently included within these ranges and with ~50% of participants on each side of the median.
Exclusion criteria included: history of acute or chronic pancreatitis; human immune‐deficiency virus (HIV) positive; uncontrolled hypertension (systolic blood pressure ≥ 180 mmHg and/or diastolic blood pressure ≥ 100 mmHg); and any disorder, except for conditions associated with hepatic impairment in participants with impaired hepatic function, which might jeopardize the participant's safety or compliance with the protocol and/or ability to complete the trial.
Specific exclusion criteria for participants with hepatic impairment included: clinically significant renal disease (estimated glomerular filtration rate < 60 mL/min/1.73 m2 using the Modification of Diet in Renal Disease formula17); liver transplantation; biliary obstruction and/or other causes of hepatic impairment not related to parenchymal disorders and/or diseases; history or presence of severe hepatic encephalopathy (≥ grade 3); advanced ascites, and ascites which required emptying and albumin supplementation; and oesophageal variceal bleeding <3 months prior to screening.
Participants who used any prescription or non‐prescription medication which could interfere with the trial pharmacokinetic results, as judged by the investigator, were to be withdrawn.
2.2. Trial design and treatment
This was a multicentre, single‐dose, parallel‐group, open‐label trial. Prior to trial initiation, the protocol was approved by the appropriate health authorities and independent ethics committees. The trial was registered at Clinicaltrials.gov (identifier NCT02210871) and conducted in accordance with guidelines for trials in people with impaired hepatic function,18, 19 as well as the Declaration of Helsinki20 and Good Clinical Practice guidelines.21 Four clinical research sites in two countries were involved in the trial: two sites in Poland and two in Slovakia.
Participants attended a screening visit to assess their eligibility and were scheduled for an in‐house visit within the next 28 days. The in‐house visit began on day −1 (pre‐dose). On the dosing day (day 1), participants received a single injection of 0.5 mg semaglutide (Novo Nordisk A/S, Bagsværd, Denmark), administered subcutaneously in the thigh using a pre‐filled PDS290 pen‐injector (Novo Nordisk). Participants remained in‐house for a minimum of 5 days (120 hours) after dosing. Ambulant visits took place on days 7, 8, 15, 22 and 29, with a follow‐up visit on day 36. Blood samples were taken to determine the plasma concentration profiles of semaglutide 15 minutes prior to dosing, at time zero, and at 6, 12, 18, 24, 30, 36, 42, 48, 54, 60, 72, 96, 120, 144, 168, 336, 504, 672 and 840 hours (up to 35 days) after administration of semaglutide. Furthermore, a blood sample was taken on day 1 prior to dosing to assess protein binding. Also on day 1, a baseline urine sample was collected before dosing. From 24 to 72 hours after dosing, which was the time period expected to include the maximum semaglutide concentration, fractionated urine collections were made in predefined intervals: 24 to 36 hours (nominal time 36 hours), 36 to 48 hours (nominal time 48 hours), 48 to 60 hours (nominal time 60 hours) and 60 to 72 hours (nominal time 72 hours). Participants were to empty their bladders immediately before the end of a collection period.
2.3. Pharmacokinetic and safety endpoints
The primary endpoint was the area under the semaglutide plasma concentration–time curve (AUC) from time zero to infinity after a single 0.5 mg dose of semaglutide (AUC0‐∞). Supportive secondary pharmacokinetic endpoints included the AUC from time zero to the last quantifiable measurement (AUC0‐last), the maximum observed semaglutide plasma concentration (Cmax), the time to Cmax (tmax), the terminal elimination half‐life for semaglutide (t½), the total apparent clearance of semaglutide (CL/F), the renal clearance and the fraction of unbound semaglutide.
Supportive safety and tolerability endpoints included adverse events, hypoglycaemic episodes, haematology, biochemistry (including amylase and lipase), coagulation measures, calcitonin, urine analysis, vital signs, physical examination, ECG and fasting plasma glucose.
2.4. Laboratory assessments
Semaglutide concentrations in plasma were analysed using liquid chromatography with tandem mass spectrometry (LC‐MS/MS; Celerion Inc., Fehraltorf, Switzerland), as previously reported.9 Blood samples were drawn and stored at −20°C until analysed. Prior to analysis, plasma proteins were precipitated. The LC‐MS/MS assay was validated according to current guidelines for analysing plasma samples in the concentration range 0.729 to 60.8 nmol/L. A 5‐fold dilution of each sample was validated to extend the assay range above 60.8 nM and a semaglutide analogue was used as an internal standard. Protein binding of semaglutide was determined using surface plasmon resonance technology, as described elsewhere.22 The assay measured the binding of semaglutide to protein, and the fraction of unbound semaglutide was calculated. Semaglutide in urine was measured after adding 1% Triton X‐100 (to prevent non‐specific binding of semaglutide to the container); the same assay as described above for semaglutide in plasma was subsequently used, with a lower limit of quantification of 0.810 nmol/L for semaglutide.
2.5. Statistical analyses
The trial was designed with 78% power to establish no effect on overall semaglutide exposure as measured by the primary endpoint (AUC0‐∞) across the three hepatic impairment groups, all compared to the normal hepatic function group. The sample size was based on a 2:1:1:1 allocation of participants with normal hepatic function and each of the three hepatic impairment groups, a true between‐group ratio of 1.0 (no exposure difference) and a standard deviation on the log‐scale for AUC0‐∞ of 0.25.22 No effect of hepatic impairment was declared if the 90% confidence interval (CI) for the between‐group ratio was within the interval 0.70 to 1.43. An overall power of 78% required a total of 45 completing participants (18:9:9:9 allocated across groups).
The primary endpoint, derived from the semaglutide plasma profiles, was the sum of the AUC from time point zero to the last quantifiable measurement and the AUC from the last quantifiable measurement to infinity, estimated using standard non‐compartmental methods. The primary endpoint was compared among the four groups of participants using an analysis of covariance (ANCOVA) with log‐transformed AUC0‐∞ as the dependent variable, age and log‐transformed weight as continuous covariates, and sex and hepatic function group as categorical factors. A hierarchical testing scheme was prespecified to adjust the three confirmatory comparisons for multiplicity. The normal hepatic function group was compared to mild, moderate and severe hepatic impairment in order.
The variables AUC0‐last and Cmax were analysed using the same model as used for the primary endpoint. An exploratory linear regression analysis was performed to examine the influence of the individual Child–Pugh variables albumin (serum), total bilirubin (serum) and prothrombin time prolongation (plasma) on the primary endpoint, AUC0‐∞, as well as on Cmax. In each analysis, the log‐transformed parameter in question was included as an independent variable in the model. In addition, a sensitivity analysis was performed for Cmax, using the same model as for the primary endpoint, whereby a single sample with an unphysiologically high value was excluded from the pharmacokinetic profile.
The value of t½ was determined using t½ = log(2)/λz, where λz was estimated by log‐linear regression on the terminal part of the concentration‐time curve. CL/F was calculated as the dose of semaglutide divided by AUC0‐∞. Renal clearance was calculated as the amount excreted from 24 to 72 hours, divided by the total AUC of the compound during that period of sampling. Other secondary pharmacokinetic analyses and safety endpoints were descriptively described. SAS software, version 9.4 (SAS Institute, Cary, North Carolina) was used to perform the statistical analyses.
3. RESULTS
3.1. Participant characteristics
Of the 65 participants screened, 44 were exposed to subcutaneous semaglutide. Of those exposed participants, 1 in the normal hepatic function group withdrew their consent to remain in the trial on day 1 after dosing and 1 in the severe hepatic impairment group was withdrawn for “other” reasons on day 22 after dosing. Hence, 42 participants completed the trial, which was conducted between August 7, 2014, and June 3, 2015. Both the safety and the full analysis sets included all 44 exposed participants.
Instead of 9 participants in each of the hepatic impairment groups, there were 8 in the mild, 10 in the moderate and 7 in the severe group, as a result of a participant with moderate hepatic impairment being miscategorized as having mild impairment (discovered during data review), and also difficulty in recruiting participants with severe impairment.
Baseline characteristics of the trial population are included in Table 2. The groups were generally comparable with respect to age, body weight and BMI, although the proportions of men and women varied across groups. All participants were white and not of Hispanic or Latino origin.
Table 2.
Baseline demographics and participant characteristics
| Hepatic impairment group | ||||
|---|---|---|---|---|
| No impairment n = 19 | Mild n = 8 | Moderate n = 10 | Severe n = 7 | |
| Age, years | 52 (34‐67) | 52 (34‐64) | 56 (35‐67) | 55 (45‐61) |
| Sex, number | ||||
| Women | 10 | 3 | 8 | 2 |
| Men | 9 | 5 | 2 | 5 |
| Body weight, kg | 80.5 (52.4‐111.4) | 80.6 (51.9‐101.0) | 75.6 (52.2‐103.7) | 82.8 (61.1‐114.0) |
| BMI, kg/m2 | 27.8 (21.1‐39.6) | 28.0 (22.5‐37.8) | 28.2 (18.7‐38.9) | 27.0 (19.7‐34.2) |
| Type 2 diabetes, number | 0 | 0 | 1 | 1 |
| Child–Pugh score | NA | 5.5 (5‐6) | 7.4 (7‐9) | 10.4 (10‐12) |
| Bilirubin, μmol/L | 11.5 (4.4‐24.4) | 15.5 (6.7‐29.2) | 18.9 (7.1‐36.9) | 53.4 (32.1‐94.9) |
| Albumin, g/L | 42.4 (38.4‐45.5) | 42.5 (39.1‐45.3) | 39.6 (35.0‐46.4) | 32.7 (28.9‐39.0) |
| Prothrombin time (seconds prolonged) | 0.5 (−0.5‐2.0) | 1.3 (0.3‐2.6) | 1.0 (−0.4‐3.5) | 3.7 (1.5‐6.0) |
Abbreviations: BMI, body mass index; NA, not applicable.
Data are presented as means (range) unless otherwise stated. Albumin and bilirubin were measured in serum, prothrombin time in plasma.
3.2. Pharmacokinetics
Semaglutide concentration‐over‐time profiles are shown in Figure 1. The mean profiles (Figure 1A) showed a similar increase in plasma semaglutide concentration in all hepatic function groups after administration of a single 0.5 mg dose of semaglutide. Individual participant profiles for each hepatic function group are shown in Figure 1B–E.
Figure 1.
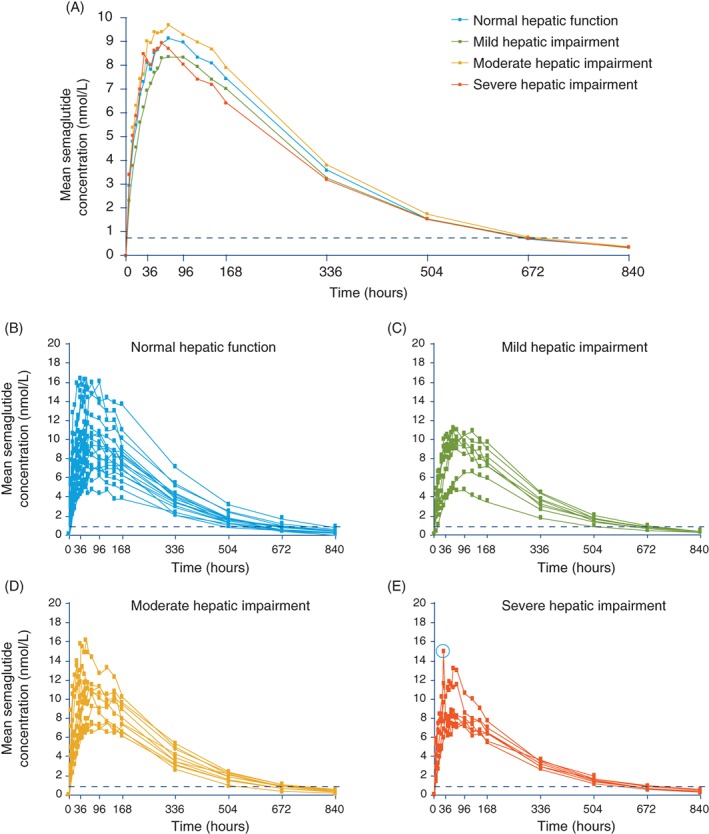
Plasma semaglutide concentration–time profiles in participants with normal hepatic function and those with hepatic impairment after a single dose of subcutaneous semaglutide 0.5 mg. A, Geometric mean profiles. B–E, Individual participant profiles. The circled data point in E represents the outlier excluded from the sensitivity analysis for Cmax. Data are geometric means. Values below the lower limit of quantification (represented by the dotted line) were imputed
The estimated mean values for AUC0‐∞ are shown in Table 3. The 90% CI for the estimated ratios of the mean total exposure of semaglutide between each group tested were all within the prespecified interval of 0.70 to 1.43, indicating that semaglutide exposure was independent of hepatic impairment.
Table 3.
Pharmacokinetic characteristics of semaglutide in hepatic impairment groups
| Hepatic impairment group | ||||
|---|---|---|---|---|
| No impairment n = 18a | Mild n = 8 | Moderate n = 10 | Severe n = 7 | |
| Primary endpoint | ||||
| AUC0‐∞, nmol×h/L | ||||
| Estimated mean | 3026 | 2872 | 3080 | 2937 |
| (95% CI) | (2735, 3349) | (2252, 3663) | (2895, 3277) | (2505, 3444) |
| ER (90% CI) vs no impairment | – | 0.95 (0.77, 1.16) | 1.02 (0.93, 1.12) | 0.97 (0.84, 1.12) |
| Secondary endpoints | ||||
|---|---|---|---|---|
| AUC0‐last, nmol×h/L | ||||
| Estimated mean | 2731 | 2621 | 2807 | 2539 |
| (95% CI) | (2442, 3055) | (2000, 3433) | (2597, 3035) | (2013, 3203) |
| ER (90% CI) vs no impairment | – | 0.96 (0.76, 1.20) | 1.03 (0.92, 1.15) | 0.93 (0.77, 1.13) |
| Cmax, nmol/L | ||||
| Estimated mean | 9.5 | 9.3 | 9.7 | 10.9 |
| (95% CI) | (8.4, 10.6) | (7.2, 12.1) | (8.3, 11.2) | (7.9, 14.9) |
| ER (90% CI) vs no impairmentb | – | 0.99 (0.80, 1.23) | 1.02 (0.88, 1.18) | 1.15 (0.89, 1.48) |
| Tmax (h) | ||||
| Median | 65.8 | 65.9 | 77.8 | 53.6 |
| Range | 30.0‐167.5 | 54.4‐119.8 | 23.8‐144.1 | 29.9‐144.9 |
| T½ (h) | ||||
| Geometric mean (CV) | 150 (8.7) | 155 (6.0) | 151 (11.2) | 163 (12.3) |
| Range | 124‐169 | 144‐171 | 118‐171 | 142‐191 |
| CL/F (L/h) | ||||
| Geometric mean (CV) | 0.040 (32.8) | 0.043 (29.0) | 0.037 (19.0) | 0.043 (10.0) |
| Range | 0.021‐0.072 | 0.034‐0.079 | 0.028‐0.048 | 0.037‐0.050 |
Abbreviations: AUC0‐∞, area under the semaglutide concentration‐time curve from time 0 to infinity; AUC0‐last, area under the semaglutide concentration‐time curve from time 0 to last quantifiable observation; CI, confidence interval; CL/F, total apparent clearance of semaglutide, calculated as the semaglutide dose divided by AUC0‐∞; Cmax, maximum semaglutide concentration; CV, coefficient of variation in %; ER, estimated ratio; n, number of participants with available data; tmax, time to maximum semaglutide concentration; t½, elimination half‐life, determined using t½ = log (2)/λz, where λz was estimated by log‐linear regression on the terminal part of the concentration‐time curve.
In the no impairment group, 1 participant withdrew consent to remain in the trial on the dosing day (after dosing) and was excluded from the analyses. In addition, 1 participant was excluded from the analysis of the AUC0‐∞, t½ and CL/F due to lack of data points for the calculation of t½.
A sensitivity analysis for Cmax provided a mean ER (90% CI) of 1.05 (0.88, 1.25) for the severe vs no hepatic impairment comparison.
Secondary pharmacokinetic endpoints are shown in Table 3. For AUC0‐last and Cmax, the 90% CI for the estimated ratios between each group indicated similar effects across hepatic function groups and supported the results for the primary endpoint. A sensitivity analysis for Cmax provided a ratio closer to 1 for the severe/no hepatic impairment comparison, with an estimated mean ratio (90% CI) of 1.05 (0.88, 1.25; see Figure 1E for the excluded data point). tmax, t½ and CL/F also appeared to be similar across the hepatic function groups (Table 3); no formal statistical testing was carried out.
Results of the linear regression exploratory analysis of the influence of the individual Child–Pugh variables on the primary endpoint AUC0‐∞ as well as Cmax are given in Table 4. Non‐significant P values for each analysis suggested that the individual variables did not influence the AUC0‐∞ or Cmax of semaglutide, supporting the earlier main results.
Table 4.
Exploratory linear regression statistical analysis of the influence of the individual Child–Pugh parameters on the primary endpoint AUC0‐∞ and Cmax
| Estimate of coefficient | (95% CI) | P | |
|---|---|---|---|
| AUC0‐∞, nmol×h/L (N = 42a) | |||
| Albumin (serum) | −0.012 | (−0.714, 0.690) | .97 |
| Total bilirubin (serum) | 0.080 | (−0.085, 0.245) | .33 |
| Prothrombin time prolongation (plasma) + 1b | −0.099 | (−0.270, 0.072) | .25 |
| Cmax, nmol/L (N = 43a) | |||
| Albumin (serum) | −0.396 | (−1.302, 0.509) | .38 |
| Total bilirubin (serum) | 0.077 | (−0.121, 0.274) | .44 |
| Prothrombin time prolongation (plasma) + 1b | −0.087 | (−0.293, 0.118) | .39 |
Abbreviations: AUC0‐∞, area under the semaglutide concentration‐time curve from time 0 to infinity; CI, confidence interval; Cmax, maximum semaglutide concentration; N, number of participants contributing to the analysis.
The endpoint was logarithmically transformed and analysed using a linear regression model with log(albumin), log(total bilirubin), log(prothrombin time prolongation + 1), log(weight) and age assessed at baseline as continuous independent variables. Sex was included as a categorical factor.
In the no impairment group, 1 participant withdrew consent to remain in the trial on the dosing day (after dosing) and was excluded from the Cmax analysis. In addition, 1 participant was excluded from the analysis of the AUC0‐∞ because of lack of data points for the calculation of t½.
For prothrombin time prolongation (plasma), 1 was added before log transformation, as this parameter could become slightly negative.
The median fraction of unbound semaglutide was <1% across the groups (ranging from 0.13% to 0.36%), corresponding to a plasma protein binding of >99% in all participants.
Semaglutide was not quantifiable in any urine sample (all results were below the lower limit of quantification) and renal clearance was therefore not estimated.
3.3. Safety and tolerability
No serious adverse events or events leading to participant withdrawal were reported in this trial. Ten participants across all groups reported 12 adverse events. These comprised: 2 events of headache and single events of asthenia, dyspnoea, oropharyngeal pain and hypertension in the normal hepatic function group; an event of back pain in a participant with mild hepatic impairment; events of viral infection, vomiting, eye injury and allergic dermatitis in the moderate hepatic impairment group; and an event of gastroenteritis in a participant with severe impairment. All adverse events were of mild or moderate severity (moderate events included headache [1 case] and back pain, vomiting, eye injury, allergic dermatitis and gastroenteritis events). All participants recovered from the events, which were judged unlikely to be related to treatment, except for the vomiting, hypertension, asthenia and 1 headache event, which were considered possibly related to treatment.
No severe hypoglycaemic episodes were reported in the trial and there were no blood glucose‐confirmed symptomatic episodes. Three asymptomatic hypoglycaemic episodes were reported by 3 participants: 2 in the normal hepatic function group and 1 in the moderate hepatic impairment group.
No clinically relevant changes in vital signs (systolic and diastolic blood pressure and pulse), clinical laboratory assessments, physical examination or ECG were observed in participants with normal or impaired hepatic function.
4. DISCUSSION
The present study has provided important information with respect to the use of semaglutide in people with hepatic impairment. Based on the exposure results for the primary endpoint AUC0‐∞ and supported by the other pharmacokinetic measures, there appears to be no requirement for dose adjustment of semaglutide in these people. In the present trial, semaglutide was well tolerated overall and no unexpected safety issues related to treatment with a single 0.5 mg dose of semaglutide were identified.
Administration of treatment in patients with hepatic impairment could cause problems if the drug is metabolized in the liver, leading to increased exposure and the potential to increase side effects. Semaglutide appears to be at least partly excreted by the liver;8 therefore, the pharmacokinetics of semaglutide might be affected with impaired hepatic function. For GLP‐1RAs other than semaglutide, such as dulaglutide and albiglutide, limited experience is available in people with hepatic impairment, although no evidence suggests that dose adjustment is required.15 No pharmacokinetic studies in participants with hepatic impairment have been conducted with exenatide; however, such impairment is not expected to have an effect, as exenatide is cleared mainly by the kidney.23 With liraglutide, which has a similar structure to semaglutide, but a different pharmacokinetic profile, the exposure was not increased by impaired liver function; rather, the results suggested a decreased exposure with increasing degree of hepatic impairment;24 however, the data were not conclusive to suggest a dose increase of liraglutide and the results thus indicated that patients with type 2 diabetes mellitus and hepatic impairment can use standard treatment regimens of liraglutide. Results of another trial with semaglutide concluded that dose adjustment may likewise not be needed for people with renal impairment, as exposure was comparable to that in people with normal renal function after adjusting for specific baseline characteristics.22
Hepatic impairment is associated with decreased concentrations of albumin.25 As semaglutide is bound to albumin, an increase in the free fraction might decrease semaglutide exposure because albumin binding slows the degradation of semaglutide in plasma and results in decreased renal clearance.9 In the present trial, the fraction of protein‐bound semaglutide was >99% in all participants across the hepatic function groups, indicating that the large majority of semaglutide molecules remained bound to albumin. The reported amount of freely available semaglutide (<1% across groups) should be interpreted with care as the protein‐binding of semaglutide was carried out in vitro on blood samples taken prior to dosing. Furthermore, in order to be able to measure the protein binding in vitro, the concentration of semaglutide relative to albumin was much higher in vitro than in the participants dosed with semaglutide; in vivo, the concentration of semaglutide9, 26 is very low compared with physiological albumin concentrations of 0.5 to 0.7 mM (> 10 000‐fold lower).
Limitations of the present study include the fact that there was only a single dose of treatment and also that there were not the same number of participants in each treatment group, with fewest in the severely impaired hepatic function group. The latter was the result of difficulties in recruiting participants with severe impairment. Furthermore, for safety and ethical reasons, and also as per Good Clinical Practice guidelines,21 those participants with severe hepatic encephalopathy were excluded. In addition, people with advanced ascites and those for whom ascites required emptying were excluded because uncertainty in the volume of distribution in these cases could have affected the trial results. Nevertheless, the trial was conducted in accordance with the regulatory guidelines for these types of trial, and a single‐dose study is sufficient when the drug exhibits linear pharmacokinetics.18, 19 The 0.5‐mg dose was the lower of the two maintenance doses investigated in the semaglutide phase III trials, and was chosen to ensure optimal pharmacokinetic profiles from single dosing and also because a higher dose would not be tolerated without dose escalation. It is recognized that it might not be feasible to conduct the study in people with the condition for which the drug is under development.19 Volunteers with hepatic disease are an acceptable alternative and therefore results may not be directly applied to patients with type 2 diabetes and hepatic impairment. Nevertheless, the pharmacokinetics of semaglutide were similar in healthy individuals and those with type 2 diabetes in previous reports.9, 26, 27 Finally, semaglutide could not be quantified in urine in the present study using an LC‐MS/MS assay. A study investigating the absorption, metabolism and excretion of semaglutide using radioactive‐labelled compound showed that only 3.1% of the administered dose was excreted in urine as intact semaglutide8 and therefore the methodology used in the present study would most likely not be sensitive enough to detect these small amounts.
In terms of safety, no new issues related to treatment with a single 0.5 mg dose of semaglutide were identified in this trial. Few adverse events were reported and no pattern in reported events was seen across hepatic function groups. Of the 3 asymptomatic hypoglycaemic episodes reported, 2 occurred in 2 participants in the normal hepatic function group, and 1 in a participant in the moderate impairment group, indicating no increased risk of hypoglycaemia with hepatic impairment in this trial.
In summary, exposure of semaglutide did not appear to be affected by hepatic impairment and the pharmacokinetic properties of semaglutide in participants with hepatic impairment were similar to those of participants with normal hepatic function. Semaglutide was well tolerated and there were no unexpected safety concerns for the use of semaglutide in participants with hepatic impairment. Taken together, the results suggest that no semaglutide dose adjustment may be necessary in people with hepatic impairment.
CONFLICT OF INTEREST
L.J., J.P. and J.B.H. are employed by and hold stock in Novo Nordisk. G.A. is employed at PRA Healthsciences. V.K. has nothing to disclose.
AUTHOR CONTRIBUTIONS
J.B.H. planned and designed the study with contribution from G.A. All authors participated in the collection and interpretation of the data. All authors were involved in the writing, reviewing and editing of the manuscript, gave final approval and agreed to be accountable for all aspects of the work.
ACKNOWLEDGMENTS
Novo Nordisk A/S, Bagsværd, Denmark provided the trial products. The contribution of the trial participants and trial site personnel is gratefully acknowledged. We would also like to thank Gurudutt Nayak, MBBS, MD (Novo Nordisk) for providing input to the manuscript reviews, as well as Angela Stocks, PhD (Larix A/S, Copenhagen, Denmark) for editorial and medical writing services, which were funded by Novo Nordisk.
Jensen L, Kupcova V, Arold G, Pettersson J, Hjerpsted JB. Pharmacokinetics and tolerability of semaglutide in people with hepatic impairment. Diabetes Obes Metab. 2018;20:998–1005. https://doi.org/10.1111/dom.13186
Funding information Funding for this trial and the trial products was provided by Novo Nordisk A/S, Bagsværd, Denmark.
REFERENCES
- 1. Tahrani AA, Bailey CJ, Del Prato S, Barnett AH. Management of type 2 diabetes: new and future developments in treatment. Lancet. 2011;378:182‐197. [DOI] [PubMed] [Google Scholar]
- 2. Hex N, Bartlett C, Wright D, Taylor M, Varley D. Estimating the current and future costs of type 1 and type 2 diabetes in the UK, including direct health costs and indirect societal and productivity costs. Diabet Med. 2012;29:855‐862. [DOI] [PubMed] [Google Scholar]
- 3. International Diabetes Federation . IDF Diabetes Atlas. 7th ed. ; 2015. https://www.idf.org/e-library/epidemiology-research/diabetes-atlas/13-diabetes-atlas-seventh-edition.html. Accessed June 8, 2017. [Google Scholar]
- 4. Halban PA, Polonsky KS, Bowden DW, et al. ß‐cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care. 2014;37:1751‐1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Htike ZZ, Zaccardi F, Papamargaritis D, Webb DR, Khunti K, Davies MJ. Efficacy and safety of glucagon‐like peptide‐1 receptor agonists in type 2 diabetes: a systematic review and mixed‐treatment comparison analysis. Diabetes Obes Metab. 2017;19:524‐536. [DOI] [PubMed] [Google Scholar]
- 6. Madsbad S. Review of head‐to‐head comparisons of glucagon‐like peptide‐1receptor agonists. Diabetes Obes Metab. 2016;18:317‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lau J, Bloch P, Schaffer L, et al. Discovery of the once‐weekly glucagon‐like peptide‐1 (GLP‐1) analogue semaglutide. J Med Chem. 2015;58:7370‐7380. [DOI] [PubMed] [Google Scholar]
- 8. Jensen L, Helleberg H, Roffel A, et al. Absorption, metabolism and excretion of the GLP‐1 analogue semaglutide in humans and nonclinical species. Eur J Pharm Sci. 2017;104:31‐41. [DOI] [PubMed] [Google Scholar]
- 9. Kapitza C, Nosek L, Jensen L, Hartvig H, Jensen CB, Flint A. Semaglutide, a once‐weekly human GLP‐1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J Clin Pharmacol. 2015;55:497‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sorli C, Harashima S, Tsoukas GM, et al. Efficacy and safety of once‐weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double‐blind, randomised, placebo‐controlled, parallel‐group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017;5:251‐260. [DOI] [PubMed] [Google Scholar]
- 11. Ahren B, Masmiquel L, Kumar H, et al. Efficacy and safety of once weekly semaglutide versus sitagliptin as add‐on to metformin and/or thiazolidinediones in subjects with type 2 diabetes (SUSTAIN 2): a 56‐week randomised, controlled clinical trial. Lancet Diabetes Endocrinol. 2017;5:341‐354. [DOI] [PubMed] [Google Scholar]
- 12. Aroda VR, Bain SC, Cariou B, et al. Efficacy and safety of once‐weekly semaglutide versus once‐daily insulin glargine in insulin‐naïve subjects with type 2 diabetes (SUSTAIN 4): a randomised open‐label clinical trial. Lancet Diabetes Endocrinol. 2017;5:355‐366. [DOI] [PubMed] [Google Scholar]
- 13. Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834‐1844. [DOI] [PubMed] [Google Scholar]
- 14. Baggio LL, Drucker DJ. Biology of incretins: GLP‐1 and GIP. Gastroenterology. 2007;132:2131‐2157. [DOI] [PubMed] [Google Scholar]
- 15. Prasad‐Reddy L, Isaacs D. A clinical review of GLP‐1 receptor agonists: efficacy and safety in diabetes and beyond. Drugs Context. 2015;4:212283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pugh RN, Murray‐Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646‐649. [DOI] [PubMed] [Google Scholar]
- 17. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. FDA Guidance for Industry . Pharmacokinetics in patients with impaired hepatic function ‐ trial design, data analysis, and impact on dosing and labelling. 2003. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072123.pdf. Accessed June 8, 2017.
- 19. EMA, Committee for Medicinal Products for Human Use CPMP . Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function (CHMP/EWP/2339/02). 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003122.pdf. Accessed June 8, 2017.
- 20. World Medical Association Declaration of Helsinki . Ethical principles for medical research involving human subjects. JAMA. 2000;284:3043‐3045. [PubMed] [Google Scholar]
- 21. International Conference on Harmonisation . ICH Harmonised Tripartite Guideline. Good Clinical Practice. 1996. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed June 8, 2017.
- 22. Marbury TC, Flint A, Jacobsen JB, Karsbøl JD, Lasseter K. Pharmacokinetics and tolerability of a single dose of semaglutide, a human GLP‐1 analog, in subjects with and without renal impairment. Clin Pharmacokinet. 2017;56(11):1381‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giorda CB, Nada E, Tartaglino B. Pharmacokinetics, safety, and efficacy of DPP‐4 inhibitors and GLP‐1 receptor agonists in patients with type 2 diabetes mellitus and renal or hepatic impairment. A systematic review of the literature. Endocrine. 2014;46:406‐419. [DOI] [PubMed] [Google Scholar]
- 24. Flint A, Nazzal K, Jagielski P, Hindsberger C, Zdravkovic M. Influence of hepatic impairment on pharmacokinetics of the human GLP‐1 analogue, liraglutide. Br J Clin Pharmacol. 2010;70:807‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Quinlan GJ, Martin GS, Evans TW. Albumin: biochemical properties and therapeutic potential. Hepatology. 2005;41:1211‐1219. [DOI] [PubMed] [Google Scholar]
- 26. Kapitza C, Dahl K, Bonde Jacobsen J, Axelsen MB, Flint, A . The effects of once‐weekly semaglutide on beta‐cell function in subjects with type 2 diabetes. EASD abstract 754. 2016.
- 27. Hausner H, Karsbøl JD, Holst AG, et al. Effect of semaglutide on the pharmacokinetics of metformin, warfarin, atorvastatin and digoxin in healthy sbjects. Clin Pharmacokinet. 2017;56:1391‐1401. https://doi.org/10.1007/s40262-017-0532-6. [DOI] [PMC free article] [PubMed] [Google Scholar]