

Abstract
A large body of evidence that has accumulated over the past decade strongly supports the role of inflammation in the pathophysiology of human epilepsy. Specific inflammatory molecules and pathways have been identified that influence various pathologic outcomes in different experimental models of epilepsy. Most importantly, the same inflammatory pathways have also been found in surgically resected brain tissue from patients with treatment-resistant epilepsy. New anti-seizure therapies may be derived from these novel potential targets. An essential and crucial question is whether targeting these molecules and pathways may result in anti-ictogenesis, anti-epileptogenesis and/or disease-modification effects. Therefore, preclinical testing in models mimicking relevant aspects of epileptogenesis is needed to guide integrated experimental and clinical trial designs. We discuss the most recent preclinical proof-of-concept studies validating a number of therapeutic approaches against inflammatory mechanisms in animal models that could represent novel avenues for drug development in epilepsy. Finally, we suggest future directions to accelerate preclinical to clinical translation of these recent discoveries.
Keywords: Inflammation, Immune response, Drug development, Anti-ictogenesis, Anti-epileptogenesis, Disease-modification, Epilepsy
Introduction
There is an urgent need for the development of new drugs for patients with drug-resistant epilepsy. In particular, anti-epileptogenesis or disease-modifying therapies for preventing or delaying the onset of the disease are still missing. In the last decade an increasing body of clinical and experimental evidence supports the role of inflammation in the pathophysiology of epilepsy.1 In particular, different inflammatory molecules and pathways have been shown to significantly contribute to the mechanisms of seizure generation and progression in different experimental models.2-6
Here, we summarize presentations concerning targets and treatments validated in experimental models [i.e., Cyclooxygenase-2 (COX-2), Prostaglandin (PG) EP2 receptor, Monoacylglycerol lipase (MAGL), Interleukin-(IL)1β, High Mobility Group Box 1 (HMGB1)/Toll-like receptor (TLR) signaling, P2X7 receptor, Immunoproteasome, Mammalian target of rapamycin (mTOR), Transforming Growth Factor-β, Metalloproteinases, Chemokines; Fig. 1]. We will discuss strategies related to target validation, controversies and open questions that may hopefully help to improve research efforts for novel drug development and foster more coordinated experimental and clinical trial designs.
Figure 1. Schematic representation of the cascade of inflammatory processes in which neurons, astrocytes, microglia and endothelial cells are involved.
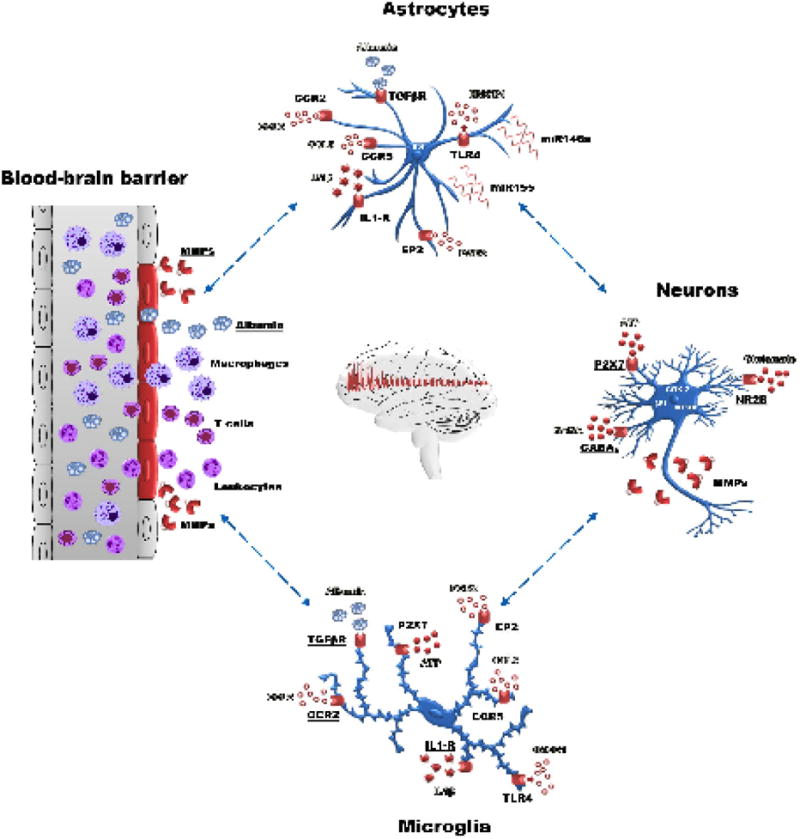
Several inflammatory targets have been validated in experimental models (i.e. cyclooxygenase-2, prostaglandin EP2 receptor, Interleukin‐1β, HMGB1/Toll‐like receptor signaling, P2X7 receptor, immunoproteasome, mTOR, TGF-beta, metalloproteinases (MMP), chemokines). The complement activation (not shown) could also contribute to a sustained inflammatory response98,99, deserving further investigation as potential target of therapy.
COX-2, PGE2 and MAGL
(W. Löscher, R. Dingledine, G. Terrone)
The enzyme COX-2, which catalyzes the production of prostaglandins, is induced rapidly and strongly in the brain after various insults, thus contributing to brain inflammatory processes involved in the long-term consequences of such insults. The use of selective COX-2 inhibitors has been considered as a novel approach for anti-epileptogenesis or disease-modification after brain injuries such as head trauma, cerebral ischemia or status epilepticus (SE).7 Accordingly, conditional ablation of COX-2 limited to forebrain neurons is neuroprotective and reduces brain inflammation after SE.8 Löscher discussed previously reported conflicting results related to prophylactic administration of different COX-2 inhibitors after SE in rodents. Jung et al. 9 showed that prolonged administration of the COX-2 inhibitor celecoxib after pilocarpine-induced SE in rats prevents neuronal damage in the hippocampus and reduces incidence, frequency and duration of spontaneous recurrent seizures, indicating an anti-epileptogenic or disease-modifying effect in this post-SE model of temporal lobe epilepsy. In apparent contrast to the study of Jung et al.9, Holtman et al.10 did not find any evidence of anti-epileptogenic, disease-modifying or neuroprotective effects of prolonged treatment with another COX-2 inhibitor, SC58236, after electrically-induced SE. Similar experiments were performed by Polascheck et al.11, using parecoxib, a prodrug of the highly potent and selective COX-2 inhibitor valdecoxib, because celecoxib has been reported to exert also COX-2 independent actions. Using an electrical model of SE, Löscher and colleagues partially confirmed the findings of Jung et al.9 in the pilocarpine model, thus allowing to evaluate which experimental factors may affect the outcome of such studies. Prophylactic treatment with parecoxib prevented the SE-induced increase in PGE2 and reduced neuronal damage in the hippocampus and piriform cortex. However, the incidence, frequency or duration of spontaneous seizures developing after SE or the behavioral and cognitive alterations associated with epilepsy was not affected by parecoxib. Only the severity of spontaneous seizures was reduced, suggesting a disease-modifying effect. These results substantiated that COX-2 contributes to neuronal injury developing after SE, but inhibition of COX-2 does not appear to provide an effective strategy to modify epileptogenesis. This implies that neuroprotective strategies alone after SE are unlikely to affect epileptogenesis. In view of the complexity of the inflammatory alterations after brain insults such as SE, it is more likely that a multi-targeted combination of different anti-inflammatory drugs is needed for efficient control of the pathologic inflammatory cascade. Indeed, Kwon et al. 12 studied combinations of anti-inflammatory drugs in the pilocarpine model in developing rats (2-3 weeks of age) and found that none of these drugs affected epilepsy when administered alone, whereas the blockade of IL-1 receptor type 1 (IL-1R1) using anakinra and the concomitant COX-2 inhibition greatly reduced the development of spontaneous recurrent seizures and limited the extent of CA1 injury and mossy fiber sprouting. However, treatment started shortly before SE induction, so that it is not clear whether the effects of treatment were due to initial insult modification or to genuine antiepileptogenesis. More recently, Citraro et al. 13 reported that early long-term treatment with the COX-2 selective inhibitor etericoxib reduced by ~40% the development of spontaneous absence seizures in WAG/Rij rats, a recognized animal model of absence epilepsy. The latter finding could either indicate that COX-2 is more critical in epileptogenesis underlying absence epilepsy than in temporal lobe epilepsy or, more likely, post-SE models of epilepsy are too severe to disclose anti-epileptogenic effects of COX-2 inhibitors.
Notably, several COX-2 inhibitors have been withdrawn from the market due to cardiac events.14 Therefore, an alternative strategy to increase target selectivity with milder adverse event profile, might be achieved by a drug acting on a downstream prostanoid receptor. Because PGE2 is a major product of COX-2 in the brain, and two of its receptors, EP1 and EP2, have been implicated in neuronal injury and inflammation in other disorders15, Dingledine and colleagues focused on these two prostanoid receptors. A higher dose of systemic kainate was required to produce SE in EP1 knockout mice (KO) compared to wild-type (WT) controls, but EP1 KO mice that did experience typical SE had reduced hippocampal neurodegeneration and a blunted inflammatory response 16 Because this is a global KO, the location of the responsible EP1 receptors is, of course, unspecified by this experiment. Co-expression of recombinant EP1 and kainate receptors in cell cultures demonstrated that EP1 receptor activation potentiates heteromeric but not homomeric kainate receptors via a second messenger cascade involving phospholipase C, calcium and protein kinase C. Turning to EP2, Dingledine and colleagues used high throughput screening and medicinal chemistry to develop a small molecule acting as EP2 competitive antagonist that is potent (Schild Kb=18 nM for human EP2), selective (10 to >500-fold over other prostanoid receptors as well as 38 other receptors, enzymes and channels), brain-permeant (brain/plasma = 0.8-1.6), orally available and with sufficient plasma half-life (1.6-2.5 hr) in mouse and rat to be useful for in vivo studies.17-19 Brief (2-3 days) treatment with the EP2 antagonist beginning 2-4 h after SE onset, during the time in which neuronal COX-2 is induced, appeared to fully replicate the effects of neuron-selective conditional COX-2 ablation. Thus, mice or rats treated with the EP2 antagonist showed neuroprotection, less neuroinflammation and blood-brain barrier (BBB) permeability function was preserved. Moreover, the typical development of a deficit in the novel object recognition test was prevented in rats treated briefly with the EP2 antagonist after SE onset.20 This suggests that much of the morbidity associated with COX-2 induction after SE is mediated by the activation of EP2, and possibly EP1, by COX-2 derived PGE2. Delayed inhibition of EP2 could therefore represent a viable adjunctive treatment for alleviating the deleterious consequences of SE. In studies of cultured microglia, EP2 has been shown to act as an immunomodulator of microglial activation, with much of its effects being mediated through ePAC (cAMP-regulated guanine nucleotide exchange factors) rather than protein kinase A.21; 22 Excessive EP2 activation can eventually kill activated microglia in vitro and thus might participate in resolution as well as generation of inflammation.
Terrone and colleagues provided evidence supporting that MAGL is a potential target for drug development in epilepsy, in particular for the treatment of drug-refractory SE.23 MAGL is a key enzyme in the hydrolysis of the endocannabinoid 2-arachidonoylglycerol (2-AG) and represents the major brain source of arachidonic acid (AA) and eicosanoids.24 They showed that the severity and duration of benzodiazepine-refractory SE, and the consequent cell loss and cognitive deficits, were significantly reduced in mice treated with a potent and selective irreversible MAGL inhibitor, and these therapeutic effects were potentiated by the ketogenic diet (KD). Notably, when MAGL was inhibited in KD-fed mice, SE was virtually abolished with immediate effect. Moreover, KD itself, although not affecting SE, prevented the hippocampal cell loss. Based on these positive results, they are further evaluating the mechanisms of therapeutic action of MAGL inhibition to determine if it is mainly mediated by 2-AG accumulation, thereby activating CB1 receptors, or by reduction of arachidonic acid availability to COX-2, thus providing anti-inflammatory effects (unpublished observations).
IL-1R1//TLR4 signaling, microRNAs, P2X7 receptors and immunoproteasome
(A. Vezzani, S. Bauer, B. A. Norwood, E. Aronica, D. Henshall, M. Mishto)
The activation of the IL-1R1 and TLR4 signaling pathways by their endogenous ligands IL-1β and HMGB1, respectively, is pivotal for the generation of neuroinflammatory responses during seizures or after epileptogenic injuries, and their pharmacological blockade or genetic inactivation results in powerful anticonvulsive effects.5 These pathways are concomitantly activated during epileptogenesis, thus likely requiring a rational drug combination for their effective blockade. Bauer and colleagues performed longitudinal cerebral microdialysis monitoring of hippocampal cytokine release in experimental epileptogenesis, detecting consistent elevations of IL-1β and Norwood provided evidence of endosomal Toll-like receptor dysregulation in experimental epilepsy (unpublished observations). Vezzani and colleagues studied whether a combination of anti-inflammatory drugs targeting the ictogenic IL-1β-IL-1R1 and HMGB1-TLR4 pro-inflammatory signals affects spontaneous seizure onset and disease progression in two rodent models of acquired epilepsy. They used two different schedules of intervention: male adult Sprague-Dawley rats exposed to electrical SE were given the drugs during the prodromal phase of epilepsy, while C57BL6 adult male mice exposed to intraamygdala kainate-induced SE were treated after the onset of the disease (i.e., after the first two unprovoked seizures). Rats were injected daily for 7 days with a combination of anakinra (IL-1 receptor antagonist), BoxA (HMGB1 antagonist) and ifenprodil (NR2B-NMDA receptor antagonist) starting 1 h post-SE. Mice were injected daily for 7 days with a combination of VX-765 (IL-1β biosynthesis inhibitor) and Cyanobacterial LPS (TLR4 antagonist) starting after the onset of epilepsy in each mouse. EEG recording (24/7) was performed from SE induction until the onset of spontaneous seizures, and for an additional 4 weeks in the chronic epilepsy phase. The combined treatments blocked seizure progression occurring in this model over 3 months post-SE leading to a reduction in the frequency of spontaneous seizures by 70-90% in the chronic epilepsy phase both in rats and mice as compared to corresponding vehicle-treated SE-exposed animals 25. When drugs were given before the onset of epilepsy, they additionally reduced neurodegeneration in forebrain and improved memory deficits in epileptic animals. Similar therapeutic effects were attained when treating electrical SE-exposed rats with a combination of drugs targeting oxidative stress mechanisms (unpublished observations).
The targeting of IL-1R/TLR4 signaling has been attempted also by epigenetic intervention with microRNAs (miRNAs). Molecular biology has been revolutionized by the discovery of these small regulatory molecules that regulate gene expression. Several miRNAs have been found in human brain to have crucial roles in a wide range of normal biological processes, including inflammation.26; 27. In particular, Aronica and colleagues showed an upregulation of miRNAs involved in the modulation of the IL-1R/TLR pathway, including miR146a, miR21 and miR155, during epileptogenesis.28 These miRNAs were also shown to be deregulated in different human epilepsy-associated pathologies including hippocampal sclerosis, malformations of cortical development and glioneuronal tumors.29-31 Interestingly, tumor and peritumoral miR146a expression was negatively correlated with frequency of seizures.30 The expression of miR21, miR146a and miR155 in astrocytes within epileptogenic brain lesions points to the role of these cell types as both source and targets of these miRNAs. IL-1β stimulation strongly induced miR146a extracellular release in human astrocytes in culture and IL-1β signaling was differentially modulated by overexpression of miR155 or miR146a, resulting in pro- or anti-inflammatory effects, respectively. 31 It is likely that expression of miR146a in astrocytes may represent a homeostatic mechanism aimed at counteracting the inflammatory response triggered by either IL-1β or other pro-inflammatory miRNAs.
To provide a preclinical proof-of-concept evidence of a therapeutic treatment targeting this miRNA, Vezzani, Aronica and colleagues tested the effect of a synthetic oligonucleotide analog of miR-146a in murine models of acute seizures and chronic epilepsy. Intracerebroventricular injection of a synthetic miR-146a mimic significantly reduced neuronal excitability and acute seizure susceptibility. Moreover, the mimic stopped the disease development when injected in mice after the epilepsy onset, and reduced cell loss and improved memory deficit when given shortly after the epileptogenic event. These therapeutic effects were similar to those attained with anti-inflammatory drug intervention against the IL-1R1 and TLR4 receptors, and were associated with broad modifications in immune/inflammatory pathways in the brain. 25
Another key driver of neuroinflammation is the P2X7 receptor, an ATP-gated ionotropic purinergic receptor predominantly expressed in microglia, and also in presynaptic nerve terminals. Henshall reported that activation of these receptors results in cell depolarization and activation of signaling pathways that promote microglial activation and inflammasome-mediated IL-1β release leading to acute and chronic neuroinflammation 32; 33. Previous studies found increased expression of the P2X7 receptor in experimental and human epilepsy and antagonists of this receptor have been reported to reduce seizure severity in certain models of SE.34-36 In recent studies, a reporter mouse expressing enhanced green fluorescent protein driven by the P2rx7 gene promoter has been used to identify the cell types involved. These studies show that the receptor is expressed by neurons as well as microglia in mice that develop epilepsy after intraamygdala kainic acid-induced SE.37 Intracellular recordings and analysis of synaptoneurosomes indicated enhanced agonist-evoked and altered calcium responses in hippocampal neurons from epileptic mice. Increased P2X7 receptor levels were observed in hippocampus resected from patients with pharmacoresistant temporal lobe epilepsy, as well as in patients with focal cortical dysplasia.37; 38 Moreover, twice daily systemic injections of the centrally-available, potent and specific P2X7 receptor antagonist, JNJ-47965567 (30 mg/kg), significantly reduced spontaneous seizures during continuous video-EEG monitoring. Interestingly, the spontaneous seizure rate did not return to baseline during a relatively short monitoring after drug washout. The hippocampus of epileptic mice treated with the P2X7 receptor antagonist showed markedly reduced microgliosis and astrogliosis. These latest studies suggest that targeting the P2X7 receptor has anticonvulsant and possibly disease-modifying effects in experimental epilepsy.37
A potential novel target that was discussed by Mishto is the proteasome, the core of the ubiquitin proteasome system, which degrades the large majority of the cytoplasm proteins and thus it is involved in several metabolic pathways including synaptic protein metabolism.39 The incorporation of the three inducible subunits β1i, β2i, and β5i into newly formed proteasomes results in the generation of the so-called immunoproteasome. This proteasome isoform is normally induced during inflammation, has altered polypeptide degradation dynamics compared to the standard proteasome isoform 40-42, and is endowed of pathophysiological functions related to immunity and inflammation responses.43 In healthy young human brain, the expression of the immunoproteasome is almost absent. 43 On the contrary, it is expressed in the brain of elderlies and patients with different neurological diseases with an inflammatory component such as Huntington disease, Alzheimer disease, multiple sclerosis 44-47, as well as with temporal lobe epilepsy and malformations of cortical development.48; 49 In agreement with the latter observations, the β5i immuno-subunit expression is induced also in experimental epilepsy, and its selective pharmacological inhibition reduced the incidence and delayed the occurrence of 4-aminopyridine-induced seizure-like events evoked in acute rat hippocampal/entorhinal cortex slices.50 These effects were stronger in slices obtained from epileptic vs normal rat brain, likely due to the prominent β5i subunit induction in neurons and glia in diseased tissue. TLR4 signaling activation is likely involved in the transcriptional induction of β5i, an effect which was independent on promoter methylation.50 The proteasome subunit expression is also induced by IL-1β in human astrocytes in vitro and is negatively regulated by treatment with the immunomodulatory drug rapamycin, an inhibitor of the mTOR pathway.49 How the immunoproteasome could have a pro-ictogenic effect is unknown. It could have either a specific role in synaptic density and as enhancer of inflammatory milieu as demonstrated for other neurological diseases.47; 51 Of note, the immunoproteasome could regulate (neuro)inflammation not only as intracellular protease but also in the extracellular space. There, it could regulate pro-inflammatory functions of cytokines, as recently demonstrated for osteopontin in the multiple sclerosis context. 52 Therefore, the immunoproteasome might be considered as therapeutic target for pharmacoresistant epilepsies as it is already under investigation for cancers, Alzheimer disease and other diseases.47
mTOR, TGF-beta and metalloproteinases as a targets for treatment
(J. A. Gorter, D. Kaufer, M.O. Poulter)
Recent observations show that inhibition of the mTOR pathway affects SE-induced epileptogenesis in rat models of temporal lobe epilepsy.53; 54 Testing the hypothesis that the immunosuppressant rapamycin can also reduce brain inflammation after SE, Gorter reported that a 6 week treatment with rapamycin (6 mg/kg, i.p, 1st injection starting at 4 h post-SE induction) reduced seizure frequency during treatment and resulted in decreased neuronal death, mossy fiber sprouting and BBB leakage. Remarkably, hippocampal microglia and astroglia activation, which was used as an indicator of inflammation, was not altered compared to vehicle-injected rats, suggesting that the suppressive effect on seizures was not directly linked to reduction of inflammation.54 In order to get more insights into the time course of BBB damage and hippocampal inflammation, magnetic resonance imaging (MRI) was performed in rats using gadobutrol as a contrast agent, and, in a separate group of rats, RT-qPCR of hippocampal tissue was performed to measure markers of inflammation.54; 55 These experiments showed that both BBB leakage and inflammation were initially increased during rapamycin treatment in SE-rats compared to vehicle-treated rats, but recovered to a greater extent after the first week following SE in drug-than in vehicle-treated rats. The fact that inflammation markers were not significantly altered at 6 weeks after SE in rapamycin-treated rats as compared to vehicle animals, implies that the drug reduces inflammation over time (unpublished observations). Moreover, the data suggest that rapamycin can modulate BBB integrity depending on the physiological condition of BBB during epileptogenesis.
In order to find out whether rapamycin reduces seizure frequency via an anti-seizure effect or an anti-epileptogenic effect, additional experiments were performed.56 In the first experiment, rapamycin treatment was stopped 3 weeks post-SE and subsequent seizure development was monitored using hippocampal EEG recordings. It was found that the daily seizure reduction measured during treatment was lost after discontinuation of rapamycin. Nevertheless, the rate of seizure development with the “stop-treatment” protocol was slower than in rats that had received a vehicle treatment or historical post-SE groups, suggesting a seizure-modifying effect of this treatment. (54)
In the second experiment, rats that were treated with rapamycin during the chronic epilepsy phase, showed a reduction in the number of daily spontaneous seizures. Accordingly, inhibition of mTOR by rapamycin has been shown to suppress established chronic seizures in the pilocarpine rat model of epilepsy.57 Elimination of rapamycin from the bloodstream was extremely slow, with the drug being detectable still 1 month after the last injection in post-SE rats.56 Interestingly, in mice, elimination of rapamycin from the brain has been shown to be much slower than from blood/plasma.58
Overall, these data suggest that the effect of rapamycin on seizure development could be due at least in part to a symptomatic seizure suppressing effect. During rapamycin treatment in post-SE rats, the rat’s body weight was strongly reduced vs vehicle, even when rapamycin dose was reduced (3 mg/kg). Moreover, brain size was also reduced by rapamycin, as detected by MRI (6 mg/kg). Thus, alternative mTOR inhibitors with a safer profile should be considered for therapeutic drug development in epilepsy.
Gorter, Kaufer, Friedman and colleagues have contributed to establish that BBB dysfunction occurs in epilepsy as in multiple neurological disorders including stroke and trauma. Consequently, blood-borne molecules enter the brain and induce an injury response leading to pathological changes.59-61 In particular, Kaufer reported evidence in collaboration with Friedman that albumin, the most abundant protein in the blood, serves as a signaling molecule to trigger the inflammatory injury response.59-62 Upon entering the brain after vascular damage, albumin is sensed by astrocytes, apparently via binding to transforming growth factor beta receptors (TGFβR), inducing the inflammatory TGFβ signaling pathway. This sets in motion a cascade of events that represent the earliest stages of post-traumatic pathology. Astrocytes become reactive and proliferate, releasing TGFβ1 and additional pro-inflammatory cytokines that amplify the injury response throughout the brain. In turn, subsequent widespread inflammatory signaling modulates neural physiology and network connectivity, causing changes that enhance neural excitability and ultimately lead to epilepsy. It has been shown that albumin activates TGFβR in astrocytes, causing a strong pro-inflammatory response and variety of changes within the neurovascular unit, including: reactive gliosis, reduced K+ and glutamate buffering, increased excitatory synaptogenesis and aberrant neurogenesis. These changes recapitulate the major hallmarks of epileptogenesis that have been extensively documented across animal models of epilepsy and in tissue samples from patients, to provide a “missing link” for how injury triggers these pathological changes. Using bioinformatics transcriptome analysis following five diverse clinical scenarios where BBB is disrupted (stroke, trauma, chemically-induced BBB disruption, and infusion of serum albumin or TGFβ), a common transcriptional signature was discovered dependent on albumin-induced activation of TGFβ signaling. Extracellular matrix (ECM) related-genes emerged as a common transcriptional response, predicting consequent degradation of ECM.62 Indeed, exposure of brain environment to albumin led to persistent degradation of perineuronal nets (PNNs, a protective ECM structure that provide synaptic stability and restrict reorganization of inhibitory interneurons) via activation of TGFβ signaling.
Exposure of the brain environment to either albumin or TGFβ1 was sufficient to induce both epileptiform activity and delayed spontaneous seizures. Importantly, blocking TGF signaling prevented most albumin-induced hallmarks of epileptogenesis including TGF signaling, transcriptome-wide changes, synaptogenesis, neurogenesis, perineuronal net (PNN) degradation, hyperexcitability and seizures.59-61; 63; 64 Ongoing research evaluates the efficacy of multiple small molecules TGFβ signaling inhibitors in blocking or reversing the effects of BBB disruption on neuropathology, excitability, epilepsy development and cognitive functions.
The integrity and stability of PNNs in a cortical network is essential for proper network function.65 Loss of interneuron synaptic stability and precise organization can lead to disruptions in the excitation/inhibition balance, a characteristic of epilepsy.66; 67 Recent research by Poulter’s group supports the role of alterations to the GABAergic interneuron network in the piriform cortex after kindling-induced seizures.68 Immunohistochemistry was used to mark PNNs and interneuron nerve terminals in control and kindled tissue. PNNs were found to be significantly decreased around parvalbumin-positive interneurons after the induction of experimental epilepsy. Additionally, a layer-specific increase was detected in GABA release sites originating from calbindin, calretinin, and parvalbumin interneurons, implying that there is a re-wiring of the interneuronal network. This increase in release sites was matched by an increase in GABAergic postsynaptic densities. The observations suggest that the breakdown of the PNN could be due to the activity of matrix metalloproteinases (MMPs) and that the prevention of PNN breakdown may reduce the rewiring of interneuronal circuits and suppress seizures. To test this hypothesis, doxycycline (DOX) a broad spectrum MMP inhibitor, was used to stabilize PNNs in kindled rats. DOX was found to prevent PNN breakdown, re-organization of the inhibitory innervation, and seizure genesis. These observations indicate that PNN degradation may be necessary for the development of seizures by facilitating interneuron plasticity and increased GABAergic activity. Another hypothesis is that activation of MMP after kindling may also be responsible for the conversion of pro-BDNF to BDNF. Furthermore, BDNF has been shown to induce the upregulation of the voltage gated potassium channel Kv1.6. This channel has also been shown to be upregulated in a kindling model causing a profound change in the excitability of parvalbumin positive interneurons.69 In order to see if MMP inhibition altered expression of BDNF, immunohistochemistry was performed in brain slices from kindled rats treated with DOX. Interestingly, BDNF expression was significantly less in this group in comparison to saline-treated kindled rats. It has also been observed that BDNF treatment of cultured cortical neurons increased the expression of Kv1.6. In kindled tissue DOX also normalized Kv1.6 expression.
In sum, these data show that upregulation of MMP activity after kindling has wide ranging effects that alter innervation of interneurons as well as the excitability of a subpopulation neurons that are important regulators of network activity.
Chemokines
(P. Louboutin, M. Caleo, Y. Bozzi, E. Palma)
These inflammatory molecules play a pivotal role in leukocyte migration across the BBB during neuroinflammation and other neuropathological processes.70; 71 When interacting with endothelial surfaces, some chemokines initiate integrin clustering, arrest leukocytes at sites of injury and guide them from vascular lumen into the brain. Once within the brain, these cells, together with microglia and astrocytes, contribute to endothelial cell activation and further chemokine secretion. CCL2 (MCP-1), CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) induce chemotaxis of T lymphocytes and macrophages. In vitro CCL4 and CCL5 increase T cell adhesion to endothelial cells. CCL2 binding to CCR2 receptor stimulates efficient migration of monocytes/macrophages across brain vasculature, and CCR1 and CCR5 receptors facilitate CCL5-driven movement of blood mononuclear cells (PBMC) across brain endothelium. CCR5 is a member of the CC-chemokine receptor family that binds several chemokines, including CCL3, CCL4 and CCL5. It is reportedly increased in epilepsy, as are its ligands. CCR5 is also expressed by microglia.
Louboutin and colleagues71-73 investigated the role of CCR5 in a rat model of seizures provoked by intraperitoneal administration of kainic acid (KA). Four months before KA injection, adult rats were given femoral intramarrow inoculations of SV(RNAiR5-RevM10.AU1), which carries an interfering RNA (RNAi) against CCR5, plus a marker epitope (AU1), or its monofunctional RNAi-carrying homolog, SV(RNAiR5). This treatment lowered expression of CCR5 in circulating PBMC. Controls received unrelated SV(BUGT) vector. In control rats, seizures increased the expression of CCR5 ligands MIP-1α and RANTES in the microvasculature and induced BBB leakage as well as CCR-positive cells, inflammation, neuronal loss and gliosis in the hippocampi. Animals given either the bi- or the monofunctional vector were largely protected from KA-induced seizures, neuroinflammation and BBB damage, and exhibited a decreased production of MIP-1α and RANTES, as well as reduced neuron loss. Brain CCR5 mRNA was reduced in these rats. Rats receiving RNAiR5-bearing vectors showed far greater repair responses with increased neuronal proliferation. Thus, inhibition of CCR5 in circulating cells strongly protected rats from seizures, BBB leakage, CNS injury and inflammation, and facilitated neurogenic repair. These results suggest that inhibition of CCR5 in circulating PBMC can decrease their interaction with endothelial cells, thus reducing leukocyte migration across the BBB, and consequently neuroinflammation and deleterious related events.
The chemokine CCL2 is elevated in brain tissue from patients with pharmacoresistant epilepsy.74; 75 Caleo and colleagues74; 76 showed a crucial role for CCL2 and its receptor CCR2 in seizure control. Mice with spontaneous seizures and neuropathology resembling mesial temporal lobe epilepsy were systemically injected with lipopolysaccharide (LPS) to mimic a peripheral inflammatory challenge. LPS was found to increase seizure frequency and up-regulate the brain expression of many inflammatory proteins, including CCL2. To test the potential role of CCL2 in seizure exacerbation, either a CCL2 transcription inhibitor (bindarit) or a selective antagonist of the CCR2 receptor (RS102895) was administered systemically. Interference with CCL2 signaling potently suppressed LPS-induced seizure worsening.76 Intracerebral administration of anti-CCL2 antibodies also abrogated LPS-mediated seizure enhancement in chronically epileptic animals. These results reveal that CCL2 is a key mediator that link peripheral inflammation with neuronal hyperexcitability.
Palma reported that cytokines and chemokines can both influence GABAA receptor function, although they can exert opposite effects on neuronal synaptic transmission. For example the chemokine fractalkine (CX3CL1) is responsible for a positive modulation of GABAA receptor in human TLE brain because it slows the GABAA use-dependent desensitization (i.e. rundown).75 Differently, IL-1β was found to reduce GABAA-mediated current amplitude by activating IRAK1 and protein kinase C.77 In both cases, the effects on GABA currents were intrinsic characteristic of the epileptic brain tissue, since such effects were absent in non-epileptic controls. This observation is explained, at least in part, by an upregulation of the cytokine/chemokine receptors. Interestingly, while in the fractalkine case the receptor’s upregulation may represent an attempt to reduce changes induced by epileptic insults, the IL-1β pathway actively contributes to ictogenesis through the impairment of GABA-mediated neurotransmission.77; 78 Altogether, these data indicate that GABAergic system is significantly modulated by inflammatory mediators released in epileptic foci, thus opening a wide scenario of novel therapeutic opportunities for controlling neuronal network hyperexcitability in epileptic patients.
The role of peripheral immune cells
(S. Koh, R. Dingledine)
Koh’s laboratory has pioneered the characterization of inflammatory cell infiltrates in the surgically removed fresh brain samples from pediatric patients in search for novel therapeutic targets. They used an unbiased flow cytometric analysis of inflammatory leukocytes in resected brain tissues from pediatric patients with genetic (focal cortical dysplasia, FCD) or acquired (encephalomalacia) epilepsy. They detected functionally activated lymphocytes within the epileptogenic lesion of both patients and experimental animals. Brain infiltration of inflammatory myeloid cells and memory CD4+ and CD8+ T cells was observed (unpublished observations).77 This is in line with previous observations in both focal cortical dysplasia type IIb and cortical tubers in Tuberous Sclerosis Complex patients.78-81 In particular, T cells are concentrated in the epileptogenic lesion and their numbers positively correlate with disease severity, whereas numbers of regulatory T cells (Tregs) inversely correlate (unpublished observations). T cell-deficient mice systemically injected with KA showed reduction in SE severity while Treg depletion heightened seizure severity. These data support a role for peripherally-derived innate and adaptive immune responses in the pathogenesis of intractable pediatric epilepsy favoring the clinical development and testing of novel immunotherapeutic drugs.
Steroids that are known for their anti-inflammatory and immuno-suppressive properties have shown efficacy in many types of drug-resistant epilepsies. However, the severe side effects of steroids have prevented long-term or widespread use of these drugs. Koh and colleagues used a two-hit model of epileptogenesis to design novel therapies to treat epilepsy using immunomodulatory approaches independent of broadly-acting immunosuppressive agents. They hypothesize that dampening ongoing inflammation in the brain could effectively reduce unprovoked recurrent seizures in the absence of systemic immunosuppression. Previous work by Miller’s lab documented the success of using biodegradable nanoparticles formulated from the FDA-approved biopolymer poly(lactide-co-glycolide) (PLG) to treat a variety of inflammatory immune-mediated disease in animal models.82 Miller’s lab has also demonstrated that supplementation of autologous natural regulatory T cells (nTregs) significantly reduces disease severity in multiple animal models of multiple sclerosis 79; 80. In the two-hit model of early-life seizures, treatment with PLG nanoparticles induced leukocyte sequestration in the spleen, thereby reducing their brain infiltration, and prevented the priming effect of early-life seizures for heightened seizure susceptibility in adulthood. Similarly, infusion of nTregs after the first seizure blocked the priming effect of early-life seizures (unpublished observations). Inhibition of long-term effects of early seizures priming was attained also by directly blocking inflammatory microglia in the hippocampus with minocycline 81. The characterization of the adaptive immunity responses in epilepsy and the effects of restriction of brain infiltration by inflammatory leukocyte subsets on seizures represent major steps forward in our understanding of epilepsy pathophysiology.
Dingledine and colleagues examined the cellular components of innate immune inflammation in the early days following kainate- or pilocarpine-induced SE in adult mice by discriminating microglia versus brain-infiltrating monocytes.86 CCR2+ monocytes invade hippocampus between one and three days after SE. In contrast, at odds with early-life seizures only occasional CD3+ T-lymphocytes were encountered three days post-SE in adult animals. The chemokine CCL2, a ligand for CCR2, was expressed in perivascular macrophages and microglia one day after SE. Four days after SE the induction of the pro-inflammatory cytokine IL-1β was greater in Fluorescence activated cell sorting (FACS) -isolated microglia than in brain monocytes. However, in WT naive mice the mRNA levels of IL-1β were 125-fold higher in circulating blood monocytes than in resting microglia, and TNFα levels were 486-fold greater in blood monocytes. Ccr2 KO mice displayed greatly reduced monocyte recruitment into brain and reduced levels of the pro-inflammatory cytokine IL-1β in hippocampus after SE. This was explained by high level of the cytokine in circulating monocytes in wild-type mice which were prevented from entering the brain after SE in Ccr2 KO mice. These findings indicate that microglia and monocytes respond differently to SE. Whereas brain-invading monocytes show little induction of IL-1β and TNFα mRNA in response to SE, both IL-1β and CCL2 are significantly induced in activated microglia. However, these results also indicate that the level of IL-1β and TNFα in circulating monocytes is much higher than that of activated microglia. Therefore, preventing monocyte recruitment into the brain may reduce the extent of neuroinflammation. Mice with impaired monocyte recruitment showed accelerated weight regain, reduced BBB leakage, and attenuated neuronal damage following SE.82 These findings identify brain-infiltrating monocytes as a myeloid cell subclass that contributes substantially to the extent of neuroinflammation and morbidity after SE. Inhibiting brain invasion of CCR2+ monocytes could represent a viable method for alleviating the deleterious consequences of SE.
Conclusion and Future directions
Several inflammatory targets and pathways discussed above provide examples of new potential avenues for future therapeutic approaches in epilepsy. Importantly, these studies indicate that immunomodulatory treatments alone might not be sufficient to counteract epileptogenesis efficiently, rather that it is likely necessary to interfere with the complex pathophysiological underpinnings of epileptogenesis at multiple levels and in various cell types (e.g., with drug combinations or complementary treatment approaches). miRNAs as master regulators of immune responses could also represent interesting targets for therapy. miRNA formulation, delivery methods into the CNS for bypassing the BBB and specifically targeting the seizure focus remain major challenges in the clinical translation.
Despite the achievements and significant progress reported during the IIE2016 meeting, a major challenge is represented by the translation of these experimentally successful anti-ictogenic or disease modifying targets into clinically effective human therapies. This translation requires intense and collaborative efforts by both preclinical and clinical scientists as well as industry partners. Future success of drug development should be based on a more coordinated experimental and clinical trial designs guided by relevant biomarkers that may be used to monitor epileptogenesis.
Table 1.
Overview of targets and treatments validated in experimental models
| Target/pathway | Experimental models | Treatment/drugs | Effect | References |
|---|---|---|---|---|
| Cox-2 | Pilocarpine-induced SE Sprague-Dawley rats Electrically-induced SE Sprague-Dawley rats Electrically-induced SE Pilocarpine (+LPS)-induced SE Sprague-Dawley rats Wistar rats Absence seizures in WAG/Rij rats Pilocarpine-induced SE C57BL/6 mice |
COX-2 inhibitor celecoxib COX-2 inhibitor SC58236 COX-2 inhibitor parecoxib COX-2 inhibitor CAY 10404 COX-2 inhibitor etericoxib COX-2 neuronal conditional KO C57BL/6 mice |
Antiepileptogenic, disease-modifying No antiepileptogenic Disease-modifying No antiepileptogenic Disease-modifying |
9 10 11 12 13 8 |
| Cox 2 + IL-1β | Pilocarpine (+LPS)-induced SE Wistar rats |
COX-2 inhibitor CAY 10404+ recombinant Interleukin-1 antagonist | Disease-modifying | 12 |
| EP2/PGE2 receptor | Organophosphorus induced SE Sprague-Dawley rats Pilocarpine-induced SE C57BL/6 mice |
EP2 competitive antagonist TG6-10-1 EP2 competitive antagonist TG6-10-1 |
Disease-modifying Disease-modifying |
19; 20 18 |
| Monoacylglycerol lipase | Intraamygdala kainic acid induced SE C57BL/6 mice | CPD-4645 (MAGL inhibitor) | Disease-modifying | Terrone, Vezzani et al., unpublished observations |
| IL-1β | Bicuculline induced seizures C57BL/6 mice Pilocarpine-induced SE Sprague-Dawley rats Rapid kindling model Electrically-induced SE/pilocarpine-induced SE Wistar rats/Sprague-Dawley rats Genetic absence epilepsy (GAERS rats); kainic acid–induced seizures; electrically-induced SE/pilocarpine-induced SE; kindling model Sprague-Dawley rats |
Human recombinant IL-1 receptor antagonist (anakinra) VX-765 (caspase-1 inhibitor) |
Anti-convulsive Anti-convulsive Antiepileptogenic No antiepileptogenic or disease-modifying Anti-convulsive/Antiepileptogenic |
83 84 85 86 86-90 |
| TLR4 or HMGB1 | Kainic acid–induced seizures C57BL/6 mice |
HMGB1 (BoxA) and TLR4 (Lps-Rs) antagonists | Anti-convulsive | 91 |
| IL-1β/HMGB1 | Kainic acid–induced seizures C57BL/6 mice |
Anakinra (IL-1 receptor antagonist), BoxA (HMGB1 antagonist) | Disease-modifying | 25 |
| IL-1β/miR146 | Kainic acid–induced seizures Intraamygdala kainic acid induced SE C57BL/6 mice |
miR146a-mimic | Anti-convulsive and Disease-modifying | 25; 92; 93. |
| P2X7 receptor | intra-amygdala kainic acid-induced status epilepticus C57BL/6 mice |
P2X7 receptor antagonist JNJ-47965567 | Disease-modifying | 37 |
| Immunoproteasome | 4-aminopyridine-induced seizure-like events/Wistar rats | β5i immuno-subunit inhibitor | Anti-convulsive | 50 |
| mTOR | Kainic acid–induced SE Electrically-induced SE/Kainic acid–induced SE |
Rapamycin | Anti-convulsive/Antiepileptogenic Anti-convulsive and Disease-modifying |
94 54-56 |
| TGF β | rat model of vascular injury/albumin-induced seizures Wistar rats |
Losartan (TGF-β signaling blocker) | Anti-convulsive/Antiepileptogenic | 64 |
| Metalloproteinases | kindling-induced seizures Sprague-Dawley rats |
Doxycycline (broad spectrum MMP inhibitor) | Anti-ictogenic | 68 |
| Chemokines | Kainic acid–induced SE Sprague-Dawley rats Kainic acid–induced SE + LPS C57BL/6 mice |
RNAi against CCR5 CCR2 antagonist (RS102895); transcription inhibitor (bindarit) |
Disease-modifying Anti-convulsive |
71 76 |
| Peripheral immune cells | Acquired and genetic epilepsy models C57BL/6 mice |
PLG nanoparticles combined with autologous natural regulatory T cells Inhibiting brain invasion of CCR2+ monocytes |
Effects on epileptogenesis: to be determined Effects on epileptogenesis: to be determined |
Koh et al., unpublished observations 82 |
SE, status epilepticus. Celecoxib, Parecoxib, Etoricoxib: COX-2 inhibitors, nonsteroidal anti-inflammatory drug used for pain management; SC5823, CAY 10404: COX-2 inhibitors with anti-inflammatory properties. Human recombinant IL-1 receptor antagonist (anakinra): nonsteroidal anti-inflammatory drug used to treat autoinflammatory diseases. TG6-10-1: prostaglandin E2 receptor 2 competitive antagonist with anti-inflammatory properties. CPD-4645: monoacylglycerol lipase (MAGL) inhibitor with anti-inflammatory properties. VX-765: caspase-1 inhibitor (ICE/caspase-1 subfamily) with anti-inflammatory properties. BoxA, High-Mobility Group Box-1 (HMGB1) antagonist of Lps-Rs, toll-like receptor 4 (TLR4) antagonist with anti-inflammatory properties. miR146a-mimic: small, chemically modified double-stranded RNA that mimic endogenous miR146a with anti-inflammatory properties. JNJ-47965567: high affinity P2X7 antagonist with anti-inflammatory properties. β5i/LMP7 immuno-subunit inhibitor: immunomodulatory properties. Rapamycin: mammalian target of rapamycin (mTOR) inhibitor with immunosuppressant/immunomodulatoryproperties, originally applied as an immunosuppressant, blocking T-cell activation, and has been in use to prevent kidney graft rejection; mTOR inhibithors are used in the cancer therapy 95 and in the management of patient with Tuberous Sclerosis Complex (TSC;96). Losartan: selective, competitive angiotensin II receptor type 1 a commonly used anti-hypertensive, also shown to antagonize TGF-β signaling. Doxycycline: a broad-spectrum antibiotic of the tetracycline used to treat bacterial infections, which has been shown to have anti-inflammatory properties and to act as broad spectrum metalloproteinases (MMP) inhibitor. CCR5 (CC Chemokine Receptor). RS102895: a spiropiperidine compound that acts as a CCR2 (CC Chemokine Receptor) antagonist with anti-inflammatory properties. Bindarit: inhibitor of CCL2 synthesis with anti-inflammatory properties. Biodegradable nanoparticles formulated from the FDA-approved biopolymer poly(lactide-co-glycolide) (PLG) with anti-inflammatory properties.
Bullet points.
Inflammatory molecules have been identified in experimental models of epilepsy and surgically resected brain tissue from treatment-resistant epilepsy
Targeting these pathways results in anti-ictogenesis, anti-epileptogenesis and/or disease-modifying effects
Therapeutic approaches against inflammatory mechanisms could represent novel avenues for drug development in epilepsy
Acknowledgments
Additional contributors to the studies described in this manuscript are: Valentina Iori and Teresa Ravizza (IRCCS-Mario Negri Institute for Pharmacological Research, Milano, Italy); Jackelien van Scheppingen, Cato Drion and Erwin van Vliet (Amsterdam Medical Center, Amsterdam, The Netherlands); Tobias Engel (Royal College of Surgeons in Ireland, Dublin, Ireland).
The authors acknowledge their sources of support: Epitarget (FP7/2007-2013) under grant agreement n°602102 (AV, EA, JAG); EPISTOP (grant agreement no. 602391; EA); Epilepsiefonds “Power of the Small” and the Hersenstichting Nederland (EF-13-1; EF-14-08); Health Research Board Ireland (HRA-POR/2010/123, HRA-POR/2012/56) and Science Foundation Ireland (13/IA/1891) (to D.C.H.); AICE-FIRE and UCB Pharma Bruxelles (EP). NIH awards UO1 NS058158, R21 NS093364 and R01 NS097776 (RD).
Footnotes
Disclosure
The authors declare that they have no conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Aronica E, Crino PB. Inflammation in epilepsy: clinical observations. Epilepsia. 2011;52(Suppl 3):26–32. doi: 10.1111/j.1528-1167.2011.03033.x. [DOI] [PubMed] [Google Scholar]
- 2.Vezzani A, French J, Bartfai T, et al. The role of inflammation in epilepsy. Nature reviews Neurology. 2011;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aronica E, Ravizza T, Zurolo E, et al. Astrocyte immune responses in epilepsy. Glia. 2012;60:1258–1268. doi: 10.1002/glia.22312. [DOI] [PubMed] [Google Scholar]
- 4.Vezzani A, Friedman A, Dingledine RJ. The role of inflammation in epileptogenesis. Neuropharmacology. 2013;69:16–24. doi: 10.1016/j.neuropharm.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vezzani A, Aronica E, Mazarati A, et al. Epilepsy and brain inflammation. Exp Neurol. 2013;244:11–21. doi: 10.1016/j.expneurol.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 6.Vezzani A, Lang B, Aronica E. Immunity and Inflammation in Epilepsy. Cold Spring Harb Perspect Med. 2016;6:a022699. doi: 10.1101/cshperspect.a022699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rojas A, Jiang J, Ganesh T, et al. Cyclooxygenase-2 in epilepsy. Epilepsia. 2014;55:17–25. doi: 10.1111/epi.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serrano GE, Lelutiu N, Rojas A, et al. Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J Neurosci. 2011;31:14850–14860. doi: 10.1523/JNEUROSCI.3922-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung KH, Chu K, Lee ST, et al. Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neurobiol Dis. 2006;23:237–246. doi: 10.1016/j.nbd.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Holtman L, van Vliet EA, van Schaik R, et al. Effects of SC58236, a selective COX-2 inhibitor, on epileptogenesis and spontaneous seizures in a rat model for temporal lobe epilepsy. Epilepsy Res. 2009;84:56–66. doi: 10.1016/j.eplepsyres.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 11.Polascheck N, Bankstahl M, Loscher W. The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. Exp Neurol. 2010;224:219–233. doi: 10.1016/j.expneurol.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Kwon YS, Pineda E, Auvin S, et al. Neuroprotective and antiepileptogenic effects of combination of anti-inflammatory drugs in the immature brain. J Neuroinflammation. 2013;10:30. doi: 10.1186/1742-2094-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Citraro R, Leo A, Marra R, et al. Antiepileptogenic effects of the selective COX-2 inhibitor etoricoxib, on the development of spontaneous absence seizures in WAG/Rij rats. Brain Res Bull. 2015;113:1–7. doi: 10.1016/j.brainresbull.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 14.Bhala N, Emberson J, Merhi A, et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769–779. doi: 10.1016/S0140-6736(13)60900-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang X, Wu L, Wang Q, et al. Function of COX-2 and prostaglandins in neurological disease. J Mol Neurosci. 2007;33:94–99. doi: 10.1007/s12031-007-0058-8. [DOI] [PubMed] [Google Scholar]
- 16.Rojas A, Gueorguieva P, Lelutiu N, et al. The prostaglandin EP1 receptor potentiates kainate receptor activation via a protein kinase C pathway and exacerbates status epilepticus. Neurobiol Dis. 2014;70:74–89. doi: 10.1016/j.nbd.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganesh T, Jiang J, Dingledine R. Development of second generation EP2 antagonists with high selectivity. Eur J Med Chem. 2014;82:521–535. doi: 10.1016/j.ejmech.2014.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang JX, Quan Y, Ganesh T, et al. Inhibition of the prostaglandin receptor EP2 following status epilepticus reduces delayed mortality and brain inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:3591–3596. doi: 10.1073/pnas.1218498110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rojas A, Ganesh T, Lelutiu N, et al. Inhibition of the prostaglandin EP2 receptor is neuroprotective and accelerates functional recovery in a rat model of organophosphorus induced status epilepticus. Neuropharmacology. 2015;93:15–27. doi: 10.1016/j.neuropharm.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rojas A, Ganesh T, Manji Z, et al. Inhibition of the prostaglandin E2 receptor EP2 prevents status epilepticus-induced deficits in the novel object recognition task in rats. Neuropharmacology. 2016;110:419–430. doi: 10.1016/j.neuropharm.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu Y, Yang MS, Jiang J, et al. EP2 Receptor Signaling Regulates Microglia Death. Mol Pharmacol. 2015;88:161–170. doi: 10.1124/mol.115.098202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quan Y, Jiang J, Dingledine R. EP2 receptor signaling pathways regulate classical activation of microglia. J Biol Chem. 2013;288:9293–9302. doi: 10.1074/jbc.M113.455816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Ruden EL, Bogdanovic RM, Wotjak CT, et al. Inhibition of monoacylglycerol lipase mediates a cannabinoid 1-receptor dependent delay of kindling progression in mice. Neurobiology of Disease. 2015;77:238–245. doi: 10.1016/j.nbd.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Nomura DK, Morrison BE, Blankman JL, et al. Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iori V, Iyer AM, Ravizza T, et al. Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol Dis. 2016;99 doi: 10.1016/j.nbd.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 26.O’Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- 27.He X, Jing Z, Cheng G. MicroRNAs: new regulators of Toll-like receptor signalling pathways. Biomed Res Int. 2014;2014:945169. doi: 10.1155/2014/945169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorter JA, Iyer A, White I, et al. Hippocampal subregion-specific microRNA expression during epileptogenesis in experimental temporal lobe epilepsy. Neurobiol Dis. 2014;62:508–520. doi: 10.1016/j.nbd.2013.10.026. [DOI] [PubMed] [Google Scholar]
- 29.Iyer A, Zurolo E, Prabowo A, et al. MicroRNA-146a: a key regulator of astrocyte-mediated inflammatory response. PLoS One. 2012;7:e44789. doi: 10.1371/journal.pone.0044789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prabowo AS, van Scheppingen J, Iyer AM, et al. Differential expression and clinical significance of three inflammation-related microRNAs in gangliogliomas. J Neuroinflammation. 2015;12:97. doi: 10.1186/s12974-015-0315-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Scheppingen J, Iyer AM, Prabowo AS, et al. Expression of microRNAs miR21, miR146a, and miR155 in tuberous sclerosis complex cortical tubers and their regulation in human astrocytes and SEGA-derived cell cultures. Glia. 2016;64:1066–1082. doi: 10.1002/glia.22983. [DOI] [PubMed] [Google Scholar]
- 32.Volonte C, Apolloni S, Skaper SD, et al. P2X7 receptors: channels, pores and more. CNS Neurol Disord Drug Targets. 2012;11:705–721. doi: 10.2174/187152712803581137. [DOI] [PubMed] [Google Scholar]
- 33.Takenouchi T, Sugama S, Iwamaru Y, et al. Modulation of the ATP-lnduced release and processing of IL-1beta in microglial cells. Crit Rev Immunol. 2009;29:335–345. doi: 10.1615/critrevimmunol.v29.i4.40. [DOI] [PubMed] [Google Scholar]
- 34.Engel T, Jimenez-Pacheco A, Miras-Portugal MT, et al. P2X7 receptor in epilepsy; role in pathophysiology and potential targeting for seizure control. Int J Physiol Pathophysiol Pharmacol. 2012;4:174–187. [PMC free article] [PubMed] [Google Scholar]
- 35.Henshall DC, Diaz-Hernandez M, Miras-Portugal MT, et al. P2X receptors as targets for the treatment of status epilepticus. Front Cell Neurosci. 2013;7:237. doi: 10.3389/fncel.2013.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jimenez-Pacheco A, Mesuret G, Sanz-Rodriguez A, et al. Increased neocortical expression of the P2X7 receptor after status epilepticus and anticonvulsant effect of P2X7 receptor antagonist A-438079. Epilepsia. 2013;54:1551–1561. doi: 10.1111/epi.12257. [DOI] [PubMed] [Google Scholar]
- 37.Jimenez-Pacheco A, Diaz-Hernandez M, Arribas-Blazquez M, et al. Transient P2X7 Receptor Antagonism Produces Lasting Reductions in Spontaneous Seizures and Gliosis in Experimental Temporal Lobe Epilepsy. J Neurosci. 2016;36:5920–5932. doi: 10.1523/JNEUROSCI.4009-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei YJ, Guo W, Sun FJ, et al. Increased Expression and Cellular Localization of P2X7R in Cortical Lesions of Patients With Focal Cortical Dysplasia. J Neuropathol Exp Neurol. 2016;75:61–68. doi: 10.1093/jnen/nlv003. [DOI] [PubMed] [Google Scholar]
- 39.Groettrup M, Kirk CJ, Basler M. Proteasomes in immune cells: more than peptide producers? Nature Reviews Immunology. 2010;10:72–77. doi: 10.1038/nri2687. [DOI] [PubMed] [Google Scholar]
- 40.Liepe J, Holzhutter HG, Bellavista E, et al. Quantitative time-resolved analysis reveals intricate, differential regulation of standard- and immuno-proteasomes. Elife. 2015;4 doi: 10.7554/eLife.07545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mishto M, Liepe J, Textoris-Taube K, et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. European Journal of Immunology. 2014;44:3508–3521. doi: 10.1002/eji.201444902. [DOI] [PubMed] [Google Scholar]
- 42.Zanker D, Chen WS. Standard and immunoproteasomes show similar peptide degradation specificities. European Journal of Immunology. 2014;44:3500–3503. doi: 10.1002/eji.201445272. [DOI] [PubMed] [Google Scholar]
- 43.Groettrup M, Kirk CJ, Basler M. Proteasomes in immune cells: more than peptide producers? Nat Rev Immunol. 2010;10:73–78. doi: 10.1038/nri2687. [DOI] [PubMed] [Google Scholar]
- 44.Mishto M, Bellavista E, Santoro A, et al. Immunoproteasome and LMP2 polymorphism in aged and Alzheimer’s disease brains. Neurobiology of Aging. 2006;27:54–66. doi: 10.1016/j.neurobiolaging.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 45.Mishto M, Bellavista E, Ligorio C, et al. Immunoproteasome LMP2 60HH Variant Alters MBP Epitope Generation and Reduces the Risk to Develop Multiple Sclerosis in Italian Female Population. Plos One. 2010;5 doi: 10.1371/journal.pone.0009287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diaz-Hernandez M, Hernandez F, Valera AG, et al. Neuronal induction of the immunoproteasome in Huntington’s disease. Testing the ups hypothesis of neurodegeneration in vivo. Journal of Neurochemistry. 2007;102:100–100. [Google Scholar]
- 47.Fukasawa H, Kaneko M, Niwa H, et al. Circulating 20S Proteasome Is Independently Associated with Abdominal Muscle Mass in Hemodialysis Patients. Plos One. 2015;10 doi: 10.1371/journal.pone.0121352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mishto M, Ligorio C, Bellavista E, et al. Immunoproteasome expression is induced in mesial temporal lobe epilepsy. Biochem Biophys Res Commun. 2011;408:65–70. doi: 10.1016/j.bbrc.2011.03.117. [DOI] [PubMed] [Google Scholar]
- 49.van Scheppingen J, Broekaart DW, Scholl T, et al. Dysregulation of the (immuno)proteasome pathway in malformations of cortical development. J Neuroinflammation. 2016;13:202. doi: 10.1186/s12974-016-0662-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mishto M, Raza ML, de Biase D, et al. The immunoproteasome beta5i subunit is a key contributor to ictogenesis in a rat model of chronic epilepsy. Brain Behav Immun. 2015;49:188–196. doi: 10.1016/j.bbi.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 51.Basler M, Mundt S, Muchamuel T, et al. Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. Embo Molecular Medicine. 2014;6:226–238. doi: 10.1002/emmm.201303543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dianzani C, Bellavista E, Liepe J, et al. Extracellular proteasome-osteopontin circuit regulates cell migration with implications in multiple sclerosis. Sci Rep. doi: 10.1038/srep43718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galanopoulou AS, Gorter JA, Cepeda C. Finding a better drug for epilepsy: the mTOR pathway as an antiepileptogenic target. Epilepsia. 2012;53:1119–1130. doi: 10.1111/j.1528-1167.2012.03506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Vliet EA, Forte G, Holtman L, et al. Inhibition of mammalian target of rapamycin reduces epileptogenesis and blood-brain barrier leakage but not microglia activation. Epilepsia. 2012;53:1254–1263. doi: 10.1111/j.1528-1167.2012.03513.x. [DOI] [PubMed] [Google Scholar]
- 55.van Vliet EA, Otte WM, Wadman WJ, et al. Blood-brain barrier leakage after status epilepticus in rapamycin-treated rats I: Magnetic resonance imaging. Epilepsia. 2016;57:59–69. doi: 10.1111/epi.13246. [DOI] [PubMed] [Google Scholar]
- 56.Drion CM, Borm LE, Kooijman L, et al. Effects of rapamycin and curcumin treatment on the development of epilepsy after electrically induced status epilepticus in rats. Epilepsia. 2016;57:688–697. doi: 10.1111/epi.13345. [DOI] [PubMed] [Google Scholar]
- 57.Huang XX, Zhang HL, Yang J, et al. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiology of Disease. 2010;40:193–199. doi: 10.1016/j.nbd.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abs E, Goorden SM, Schreiber J, et al. TORC1-dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann Neurol. 2013;74:569–579. doi: 10.1002/ana.23943. [DOI] [PubMed] [Google Scholar]
- 59.Friedman A, Kaufer D, Heinemann U. Blood-brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res. 2009;85:142–149. doi: 10.1016/j.eplepsyres.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heinemann U, Kaufer D, Friedman A. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia. 2012;60:1251–1257. doi: 10.1002/glia.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim SY, Buckwalter M, Soreq H, et al. Blood-brain barrier dysfunction-induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia. 2012;53(Suppl 6):37–44. doi: 10.1111/j.1528-1167.2012.03701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cacheaux LP, Ivens S, David Y, et al. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci. 2009;29:8927–8935. doi: 10.1523/JNEUROSCI.0430-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim SY, Porter BE, Friedman A, et al. A potential role for glia-derived extracellular matrix remodeling in postinjury epilepsy. J Neurosci Res. 2016;94:794–803. doi: 10.1002/jnr.23758. [DOI] [PubMed] [Google Scholar]
- 64.Bar-Klein G, Cacheaux LP, Kamintsky L, et al. Losartan prevents acquired epilepsy via TGF-beta signaling suppression. Ann Neurol. 2014;75:864–875. doi: 10.1002/ana.24147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McRae PA, Porter BE. The perineuronal net component of the extracellular matrix in plasticity and epilepsy. Neurochem Int. 2012;61:963–972. doi: 10.1016/j.neuint.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dityatev A, Schachner M. The extracellular matrix and synapses. Cell Tissue Res. 2006;326:647–654. doi: 10.1007/s00441-006-0217-1. [DOI] [PubMed] [Google Scholar]
- 67.Thalhammer A, Cingolani LA. Cell adhesion and homeostatic synaptic plasticity. Neuropharmacology. 2014;78:23–30. doi: 10.1016/j.neuropharm.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 68.Pollock E, Everest M, Brown A, et al. Metalloproteinase inhibition prevents inhibitory synapse reorganization and seizure genesis. Neurobiol Dis. 2014;70:21–31. doi: 10.1016/j.nbd.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 69.Ashraf MN, Gavrilovici C, Shah SU, et al. A novel anticonvulsant modulates voltage-gated sodium channel inactivation and prevents kindling-induced seizures. J Neurochem. 2013;126:651–661. doi: 10.1111/jnc.12352. [DOI] [PubMed] [Google Scholar]
- 70.Fabene PF, Bramanti P, Constantin G. The emerging role for chemokines in epilepsy. J Neuroimmunol. 2010;224:22–27. doi: 10.1016/j.jneuroim.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 71.Louboutin JP, Strayer DS. Relationship between the chemokine receptor CCR5 and microglia in neurological disorders: consequences of targeting CCR5 on neuroinflammation, neuronal death and regeneration in a model of epilepsy. CNS Neurol Disord Drug Targets. 2013;12:815–829. doi: 10.2174/18715273113126660173. [DOI] [PubMed] [Google Scholar]
- 72.Louboutin JP, Chekmasova A, Marusich E, et al. Role of CCR5 and its ligands in the control of vascular inflammation and leukocyte recruitment required for acute excitotoxic seizure induction and neural damage. FASEB J. 2011;25:737–753. doi: 10.1096/fj.10-161851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marusich E, Louboutin JP, Chekmasova AA, et al. Lymphocyte adhesion to CCR5 ligands is reduced by anti-CCR5 gene delivery. J Neurol Sci. 2011;308:25–27. doi: 10.1016/j.jns.2011.06.039. [DOI] [PubMed] [Google Scholar]
- 74.Bozzi Y, Caleo M. Epilepsy, Seizures, and Inflammation: Role of the C-C Motif Ligand 2 Chemokine. DNA Cell Biol. 2016;35:257–260. doi: 10.1089/dna.2016.3345. [DOI] [PubMed] [Google Scholar]
- 75.de Vries EE, van den Munckhof B, Braun KP, et al. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci Biobehav Rev. 2016;63:177–190. doi: 10.1016/j.neubiorev.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 76.Cerri C, Genovesi S, Allegra M, et al. The Chemokine CCL2 Mediates the Seizure-enhancing Effects of Systemic Inflammation. J Neurosci. 2016;36:3777–3788. doi: 10.1523/JNEUROSCI.0451-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roseti C, van Vliet EA, Cifelli P, et al. GABAA currents are decreased by IL-1beta in epileptogenic tissue of patients with temporal lobe epilepsy: implications for ictogenesis. Neurobiol Dis. 2015;82:311–320. doi: 10.1016/j.nbd.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 78.Roseti C, Fucile S, Lauro C, et al. Fractalkine/CX3CL1 modulates GABAA currents in human temporal lobe epilepsy. Epilepsia. 2013;54:1834–1844. doi: 10.1111/epi.12354. [DOI] [PubMed] [Google Scholar]
- 79.Podojil JR, Liu LN, Marshall SA, et al. B7-H4Ig inhibits mouse and human T-cell function and treats EAE via IL-10/Treg-dependent mechanisms. J Autoimmun. 2013;44:71–81. doi: 10.1016/j.jaut.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang H, Podojil JR, Luo X, et al. Intrinsic and induced regulation of the age-associated onset of spontaneous experimental autoimmune encephalomyelitis. J Immunol. 2008;181:4638–4647. doi: 10.4049/jimmunol.181.7.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abraham J, Fox PD, Condello C, et al. Minocycline attenuates microglia activation and blocks the long-term epileptogenic effects of early-life seizures. Neurobiology of Disease. 2012;46:425–430. doi: 10.1016/j.nbd.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Varvel NH, Neher JJ, Bosch A, et al. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc Natl Acad Sci U S A. 2016;113:E5665–5674. doi: 10.1073/pnas.1604263113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vezzani A, Moneta D, Conti M, et al. Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc Natl Acad Sci U S A. 2000;97:11534–11539. doi: 10.1073/pnas.190206797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marchi N, Fan Q, Ghosh C, et al. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis. 2009;33:171–181. doi: 10.1016/j.nbd.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Auvin S, Shin D, Mazarati A, et al. Inflammation induced by LPS enhances epileptogenesis in immature rat and may be partially reversed by IL1RA. Epilepsia. 2010;51(Suppl 3):34–38. doi: 10.1111/j.1528-1167.2010.02606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Noe FM, Polascheck N, Frigerio F, et al. Pharmacological blockade of IL-1beta/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol Dis. 2013;59:183–193. doi: 10.1016/j.nbd.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 87.Akin D, Ravizza T, Maroso M, et al. IL-1beta is induced in reactive astrocytes in the somatosensory cortex of rats with genetic absence epilepsy at the onset of spike-and-wave discharges, and contributes to their occurrence. Neurobiol Dis. 2011;44:259–269. doi: 10.1016/j.nbd.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 88.Maroso M, Balosso S, Ravizza T, et al. Interleukin-1beta biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics. 2011;8:304–315. doi: 10.1007/s13311-011-0039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ravizza T, Lucas SM, Balosso S, et al. Inactivation of caspase-1 in rodent brain: a novel anticonvulsive strategy. Epilepsia. 2006;47:1160–1168. doi: 10.1111/j.1528-1167.2006.00590.x. [DOI] [PubMed] [Google Scholar]
- 90.Ravizza T, Noe F, Zardoni D, et al. Interleukin Converting Enzyme inhibition impairs kindling epileptogenesis in rats by blocking astrocytic IL-1beta production. Neurobiol Dis. 2008;31:327–333. doi: 10.1016/j.nbd.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 91.Maroso M, Balosso S, Ravizza T, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–419. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- 92.Iori V, Iyer A, Aronica E, et al. Book microRNA146a-mediated epigenetic control of brain inflammation: new therapeutic opportunities in epilepsy Montreal. Canada: 2013. microRNA146a-mediated epigenetic control of brain inflammation: new therapeutic opportunities in epilepsy. Editor (Ed)ˆ(Eds) [Google Scholar]
- 93.Ravizza T, Iori V, Iyer AM, et al. miR146a-Mediated Control Of Brain Inflammation: A New Therapeutic Opportunity In Epilepsy. Book miR146a-Mediated Control Of Brain Inflammation: A New Therapeutic Opportunity In Epilepsy. 2013 Editor (Ed)ˆ(Eds) [Google Scholar]
- 94.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. Journal of Neuroscience. 2009;29:6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xie J, Wang X, Proud CG. mTOR inhibitors in cancer therapy. F1000Res. 2016:5. doi: 10.12688/f1000research.9207.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sadowski K, Kotulska K, Schwartz RA, et al. Systemic effects of treatment with mTOR inhibitors in tuberous sclerosis complex: a comprehensive review. J Eur Acad Dermatol Venereol. 2016;30:586–594. doi: 10.1111/jdv.13356. [DOI] [PubMed] [Google Scholar]