

Abstract
Clinical molecular testing has been available for 22q11.2 deletion syndrome (22q11.2DS) for over two decades yet under-recognition and diagnostic delays are common. To characterize the “diagnostic odyssey” in 22q11.2DS we studied 202 well-characterized unrelated adults, none ascertained through an affected relative. We used a regression model to identify clinical and demographic factors associated with length of time to molecular diagnosis. Kaplan-Meier analysis compared time to diagnosis for the molecular testing era (since 1994) and earlier birth cohorts. The results showed that the median time to molecular diagnosis of the 22q11.2 deletion was 4.7 (range 0-20.7) years. Palatal and cardiac anomalies, but not developmental delay/intellectual disability, were associated with a shorter time to molecular diagnosis. Non-European ethnicity was associated with longer time to diagnosis. Inclusion of a cohort from another 22q11.2DS center increased power to observe a significantly earlier diagnosis for patients born in the molecular testing era. Nonetheless, only a minority were diagnosed in the first year of life. On average, patients were seen in seven (range 2-15) different clinical specialty areas prior to molecular diagnosis. The findings indicate that even for those born in the molecular testing era, individuals with 22q11.2DS and their families face a diagnostic odyssey that is often prolonged, particularly in the absence of typical physical congenital features or for those of non-European ancestry. The results support educational efforts to improve clinical recognition and testing, and ultimately newborn screening as a means of maximizing early detection that would provide the best opportunity to optimize outcomes.
Keywords: Developmental delay, feeding difficulties, recurrent infections, neurology, seizures, scoliosis, psychosis proneness, surgical complications, DiGeorge syndrome, velocardiofacial syndrome
INTRODUCTION
22q11.2 deletion syndrome (22q11.2DS) (MIM #192430/188400), previously known as DiGeorge or velocardiofacial syndrome, is the most common microdeletion syndrome, with an estimated prevalence of over 1 in 4000 live births [Goodship et al., 1998; McDonald-McGinn et al., 2015; Oskarsdottir et al., 2004]. Associated manifestations include classic developmental features such as birth defects and intellectual disability, and later onset features such as schizophrenia [Bassett et al., 2011; Fung et al., 2015; McDonald-McGinn et al., 2015]. Variable expression across the lifespan involves multiple organ systems, with substantial morbidity and mortality, beginning prenatally [Bales et al., 2010; Bassett et al., 2009; Bassett et al., 2005; Bassett et al., 2017; Bassett et al., 2011; Cheung et al., 2014; Costain et al., 2012; Fung et al., 2015; Gerdes et al., 2001; McDonald-McGinn et al., 2015]. Penetrance for one or more of the many associated features is high; typical 22q11.2 deletions are not found in large population control samples [Cooper et al., 2011].
Molecular testing has been available for over 20 years using fluorescence in situ hybridization (FISH) and a probe targeted at the typical deletion 22q11.2 locus [Driscoll et al., 1993]. More recently, genome-wide methods, e.g., microarray, have become the clinical standard, recommended for all individuals with developmental delay/intellectual disability, multiple congenital anomalies, and/or autism [Miller et al., 2010]. Despite widespread availability of molecular testing, multiple factors including under-recognition of 22q11.2DS could limit implementation of these best practices, contributing to diagnostic delays, or “odysseys”, for patients and their families [Bassett et al., 2011; Fung et al., 2015; McDonald-McGinn et al., 2015].
Information about the pathways to a diagnosis of 22q11.2DS may inform strategies to reduce time to molecular testing, and facilitate the identification of individuals who remain undiagnosed. In this study, we examined the “diagnostic odyssey” of a well-characterized cohort of 202 unrelated Canadian adults with 22q11.2DS who came to attention as the first in the family to receive a molecular diagnosis. We aimed to identify factors associated with time to molecular diagnosis. We hypothesized that, in addition to the presence of major congenital or developmental features, i.e., congenital heart disease (CHD), palatal anomalies and developmental delay/intellectual disability (DD/ID), availability of molecular testing at birth would be associated with a significantly shorter time to molecular diagnosis compared with individuals where the emergence of molecular testing capability became available during childhood or adulthood. We also evaluated the relationship between the number of specialities seen and the length of time to diagnosis. To enhance power for the main analysis, we subsequently obtained data from a comparably characterized cohort of unrelated individuals with 22q11.2DS from another major center.
METHODS
Subjects and clinical data
Canadian sample
The main sample comprised 202 adults with 22q11.2DS (n=98 male, 48.5%; median molecular diagnostic age 17.3 years, range 0.1-59.4 years) who met the following inclusion criteria with respect to molecular diagnosis: 1) typical 22q11.2 deletion detectable by a standard probe using FISH (n=191, 94.6%) or clinical genome-wide microarray (n=11, 5.4%) [Bassett et al., 2005; Bassett et al., 2008], and 2) diagnosis made prior to that of any other affected family member. Ethnicity was characterized as European (n=159, 78.7%) or non-European descent, the latter comprising Asian (n=14, 6.9%), African (n=7, 3.5%), or other (n=22, 10.9%) including mixed ethnicity. All patients were ascertained through a specialty clinic for adults with 22q11.2DS (The Dalglish Family 22q Clinic for Adults, or the Clinical Genetics Research Program, Toronto, Canada). Ascertainment of this sample followed active screening for 22q11.2DS at an adult congenital cardiac clinic (n=75) or referrals through genetics (n=68), psychiatric (n=40), or other (n=19) sources [Bassett et al., 2007; Bassett et al., 2005; Bassett et al., 2008; Cheung et al., 2014]. Molecular diagnosis originated in Ontario for most (n=180, 89.1%) subjects; the remainder were from other provinces. Informed consent was obtained in writing, and the study was approved by local research ethics boards.
To assess the impact of molecular testing availability for the 22q11.2 deletion on time to diagnosis, we divided the sample into three birth-cohort subgroups based on when molecular testing for the typical 22q11.2 deletion using FISH and a targeted probe became widely available [Driscoll et al., 1993], i.e., in 1994. These three birth cohorts comprised Group 1 (n=53, 26.2%) born before 1977 (i.e., testing became available in adulthood years), Group 2 (n=108, 53.5%) born 1977-1993 (i.e., testing became available in childhood years), and the index group, Group 3 (n=41, 20.3%) born 1994 to 1997 inclusive (i.e., born in the molecular testing era).
For this Canadian sample, comprehensive data on demographic and clinical features, and on the genetic diagnostic pathway, were recorded from birth to molecular diagnosis from available lifetime medical records collected by our program, and extensive clinical histories obtained from the patient and collateral sources (typically family members) at multiple time points [Bassett et al., 2007; Bassett et al., 2005; Bassett et al., 2008; Cheung et al., 2014]. We used previously described standard methods to classify congenital cardiac and palatal anomalies [Bassett et al., 2005; Billett et al., 2008], and to assess and classify ID, considered here as a proxy for clinically relevant DD/ID [Butcher et al., 2012; Chow et al., 2006; Van et al., 2015]. There were 82 subjects with CHD of moderate to severe complexity (e.g., tetralogy of Fallot) and 34 with mild complexity (e.g., ventricular or atrial septal defect) [Billett et al., 2008]. There were 87 subjects with velopharyngeal insufficiency (VPI), including 66 with VPI plus another palatal anomaly, 15 subjects with submucous cleft or short palate only, and none with overt cleft palate only. DD/ID was at the mild (n=94) or moderate/severe (n=20) level; the other subjects (n=88) were at the borderline to average intellectual level [Butcher et al., 2012; Chow et al., 2006; Van et al., 2015].
Philadelphia (US) sample
We obtained additional data from 112 comparably characterized individuals with 22q11.2DS followed at a major US center, the Children’s Hospital of Philadelphia. An additional 51 adults (n=19 male, 37.3%; median diagnostic age 2.9, range 0-17.6 years) brought the sample size of Group 3 to n=92. A further 61 subjects comprised a new pediatric comparison subgroup (Group 4: born in 1998 and thereafter).
For the US adult cohort (n=51), there were 18 subjects with CHD of moderate to severe complexity and 13 with mild complexity [Billett et al., 2008]; 33 subjects had VPI, including 9 with VPI plus another palatal anomaly, 7 had submucous cleft or short palate only and 1 overt cleft palate only; DD/ID in 21 subjects was at the mild (n=15) or moderate (n=6) level; the other subjects (n=9) were at the borderline to average intellectual level.
Statistical analyses
We constructed a multivariable linear regression model to examine potential contributors to time to molecular diagnosis of 22q11.2DS (i.e., “diagnostic delay”), defined as the number of years to molecular diagnosis from either 1994 (the year clinical FISH testing was widely available to the samples studied) or year of birth (given the importance of early diagnosis [Bales et al., 2010; Bassett et al., 2017; Cheung et al., 2014; Costain et al., 2012; McDonald-McGinn et al., 2015]), whichever came later. The variables selected, in addition to the birth-cohort subgroups, ethnicity and sex, were three classic developmental features of 22q11.2DS (cardiac anomaly, palatal anomaly, and DD/ID). We opted not to include dysmorphic features due to inconsistent documentation in medical records of these often subtle features in 22q11.2DS [Liu et al., 2014; Veerapandiyan et al., 2011].
Kaplan-Meier curves were constructed to assess time to diagnosis for the different birth-cohort subgroups. To enhance the power to determine whether being born in the molecular era reduced diagnostic delay in the regression and Kaplan-Meier analyses we used the additional adult and pediatric data from the US center (total North American sample n=314, n=253 for Groups 1-3). In a second analysis, a log rank test (uncorrected for other factors) was used to compare the Kaplan-Meier curves.
To better examine the clinical pathway to a molecular diagnosis of 22q11.2DS, we used a representative subset of the Canadian sample (total n=100; n=52 male; median diagnostic age 14.3 years, range 0.1-58.2 years) to examine detailed data available on the clinical services involved prior to molecular diagnosis. The three birth-cohort subgroups were matched on demographic and clinical data for the three classic developmental features used in the main regression analysis. Proportionate representation (Group 1, n=26, 26.0%; Group 2, n=53, 53.0%; Group 3, n=21, 21.0%) was comparable to that for the total Canadian sample (n=202). To examine the relationship of the number of different speciality areas to the time to molecular diagnosis, we comprehensively recorded additional 22q11.2DS-associated features [Bassett et al., 2009; Bassett et al., 2011; Fung et al., 2015; McDonald-McGinn et al., 2015] noted in medical records prior to molecular diagnosis, the number and type of clinical specialty areas (both medical and allied health) in which the patient was seen for these features, any other consults related to these specialty areas, and any other genetic syndromes that were documented as being suspected. Specialty areas recorded included pediatric and adult medical and surgical subspecialties, and the specialty area of the clinician who ordered the molecular diagnostic test. Family doctors and pediatricians were not included in the medical specialties count as it was assumed that all individuals would have been seen by these primary care clinicians in the Canadian medicare system. Also excluded were emergency room physicians and the areas of radiology and anaesthesia, although most patients would have had contact with these specialty areas. The regression analysis for diagnostic delay was repeated for this subset of 100 subjects, adding the number of clinical specialties seen prior to molecular diagnosis as a new factor.
All statistical analyses were two-tailed with statistical significance defined as P values < 0.05, performed with SAS 9.4 software (SAS Institute, Cary, NC).
RESULTS
The median time to molecular diagnosis for the 202 Canadian adult subjects with 22q11.2DS was 4.7 (range 0.0 to 20.7) years. Most adults (n=191; 94.6%) had one or more classic developmental feature of 22q11.2DS: cardiac or palatal anomalies or DD/ID. The median time to molecular diagnosis for the representative subset of 100 Canadian adult subjects was similar (4.5, range 0.0-19.5, years).
Demographic and selected clinical features associated with time to molecular diagnosis
Table 1 shows the selected clinical and demographic features of the sample and their relationship to the time to molecular diagnosis of 22q11.2DS. The regression model for time to molecular diagnosis for the Canadian sample was significant (adjusted R2=0.16, df=6, P<0.0001). Consistent with our hypothesis, palatal and cardiac anomalies were significantly associated with a shorter time to molecular diagnosis, however DD/ID was not a significant factor (Table 1). Also, non-European ethnicity was significantly associated with a longer time to molecular diagnosis (Table 1).
Table 1.
Demographic and selected clinical factors and their association with length of time to molecular diagnosis of 22q11.2 deletion syndrome (22q11.2DS)
| Canadian 22q11.2DS cohort (n=202) | Combined North American 22q11.2DS cohort (n=253) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Demographic Factors | n | % | Linear regression analysisa | n | % | Linear regression analysis | ||||||
| B | 95% CI | p | B | 95% CI | p | |||||||
| Male sex | 98 | 48.5 | 0.33 | −1.10 | 1.77 | 0.6487 | 117 | 46.3 | 0.49 | −0.74 | 1.72 | 0.4305 |
| European ethnicity | 159 | 78.7 | −2.11 | −3.96 | −0.27 | 0.0250 | 208 | 82.2 | −2.23 | −3.88 | −0.57 | <0.0085 |
| Born in the era of molecular testing | ||||||||||||
| Birth-cohort (group 3)b | 41 | 20.3 | −0.75 | −1.86 | 0.35 | 0.1776 | 92 | 36.4 | −1.04 | −1.89 | −0.19 | 0.0162 |
| Clinical features | ||||||||||||
| Palatal anomaly | 102 | 50.5 | −3.57 | −5.06 | −2.08 | <0.0001 | 143 | 56.5 | −3.13 | −4.44 | −1.83 | <0.0001 |
| Cardiac anomaly | 116 | 57.4 | −2.06 | −3.53 | −0.60 | 0.0060 | 145 | 57.3 | −2.63 | −3.87 | −1.40 | <0.0001 |
| Developmental delay /intellectual disability | 114 | 56.4 | −0.30 | −1.78 | 1.19 | 0.6943 | 135 | 53.4 | −0.14 | −1.39 | 1.11 | 0.8233 |
B, regression coefficient; CI, confidence interval; p value for regression coefficient post hoc t-tests; bold values indicate statistical significance (p < 0.05). The regression model for time to molecular diagnosis for the Canadian sample was significant (adjusted R2=0.16, df=6, P<0.0001). The regression model for time to molecular diagnosis for the combined North American sample was significant (adjusted R2= 0.20, df=6, p < 0.0001).
Multivariable linear regression model for length of time to molecular diagnosis of 22q11.2DS, defined as the number of years from the earliest opportunity for molecular diagnosis (i.e., number of years from 1994 or year of birth, whichever came later, to the year of molecular diagnosis.
Group 3 = index group born 1994 to 1997 inclusive (i.e., born in the molecular testing era), compared with Groups 1 (n=53, 26.2%, born before 1977, i.e., testing became available in adulthood years) and 2 (n=108, 53.5%, born 1977-1993, i.e., testing became available in childhood years).
The log rank test for the Kaplan-Meier curves (Figure 1) for the three Canadian birth-cohort subgroups (n=202) showed a non-significant trend towards a shorter time to diagnosis for the group born during the molecular testing era (Group 3) (p=0.082; Figure 1A) compared with the other two subgroups.
Figure 1. Length of time to molecular diagnosis of 22q11.2 deletion syndrome (22q11.2DS) for three sub-groups in (A) a Canadian and (B) in a combined North American cohort.
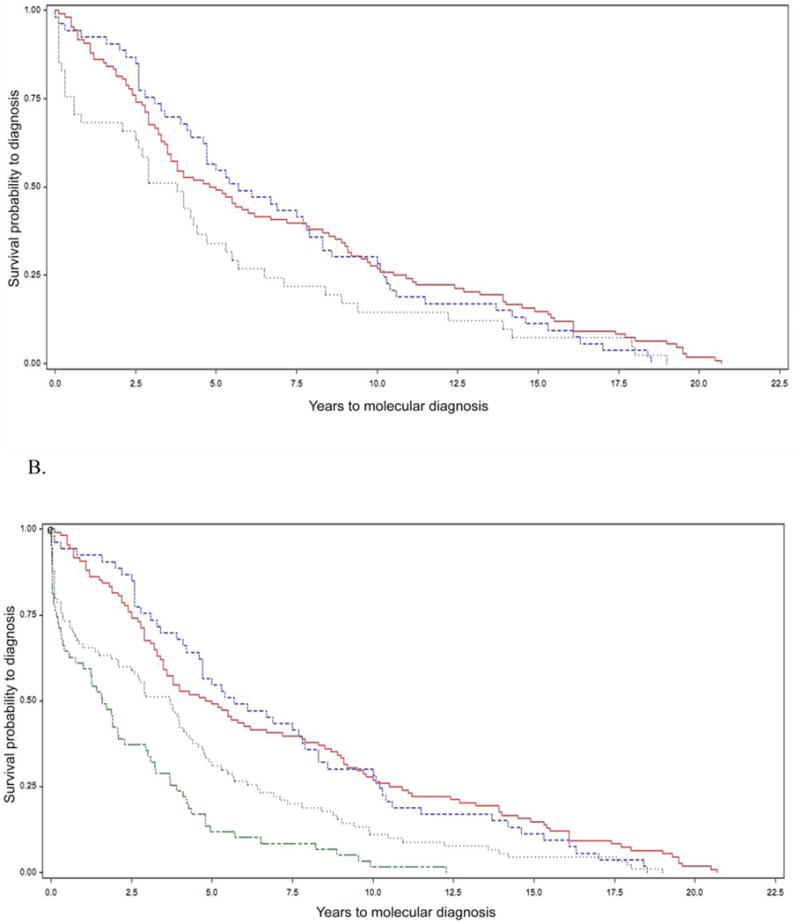
Kaplan-Meier curves showing time to molecular diagnosis of 22q11.2DS for individuals born in the era of molecular testing (group 3, grey dotted line), i.e., 1994 to 1997 inclusive, and for individuals who were children (group 2, red solid line) or were adults (group 1, blue dashed line) in the molecular era.
A. The median time to diagnosis was 3.8 years (range 0.1-19; 95% CI 2.1-4.7 years) for the 41 subjects born in the era of molecular testing (Group 3), the median diagnostic delay was 4.9 years (range 0.1-20.7 years; 95% CI 3.5 to 6.7 years) for those who were children (Group 2), and 5.7 years (range 0-18.5 years; 95% CI 4.2 to 7.9 years) for those who were adults (Group 1). The log rank test showed there was no significant difference between these curves (p=0.082). There were no censored values.
B. After adding 51 US subjects to Group 3, the median diagnostic delay was 3.3 years (range 0-19.0 years; 95% CI 2.1 to 4.2) for the total 92 subjects born in the era of molecular testing. Also shown are Group 4 (n=61 pediatric US subjects born in 1998 and thereafter, green double dash line), with a median diagnostic delay of 1.6 years (range 0-12.3 years; 95% CI 0. 6 to 2.3 years). There were two censored values each in group 3 and 4 where the ‘event’ of interest, a molecular diagnosis of 22q11.2DS, occurred at birth (i.e. diagnostic age 0). The log rank test revealed significant differences overall when comparing the survival curves of the four groups (p<0.0001). Subsequent comparisons of these four curves were made with Bonferroni’s correction for multiple comparisons. Significant differences were found between groups 1 and 3 (p=0.0043), 1 and 4 (p<0.0001), 2 and 3 (p=0.0011) and 2 and 4 (p<0.0001). There were no significant differences between groups 1 and 2 (p=0.99) and 3 and 4 (p=0.63).
For the combined North American sample (n=253), results using the same regression model were similar, but with the enhanced power afforded by the additional Group 3 subjects, a significantly shorter time to molecular diagnosis for this group became evident (p=0.016, Table 1). The log rank test for the Kaplan-Meier analysis comparing the survival curves of the three groups also became significant (p=0.0005), with significant between-group differences for groups 1 and 3 (p=0.0095) and groups 2 and 3 (p=0.0009). The overall Kaplan-Meier analysis was also significant (p<0.0001, Figure 1B) after the further addition of Group 4 (61 US pediatric subjects born 1998 and thereafter), with significant additional between-group differences for groups 1 and 4 (p<0.0001), and 2 and 4 (p<0.0001).
Notably, only a minority of patients at either center born in the molecular era were diagnosed in their first year of life (13/41 (31.7%) and 20/51 (39.2%) for the Canadian and US adult birth cohorts (Group 3), respectively; 26/61 (42.6%) for the US pediatric birth cohort (Group 4); Supplemental Figure 1). Even for those with CHD (with or without palatal anomalies or DD/ID), a substantial minority had diagnostic delays of over two years (10/22, 45.5%) and (23/68, 33.8%) in the Canadian and US molecular era (Group 3) cohorts, respectively). For those born in the molecular era with palatal anomalies and/or DD/ID, but without CHD, the majority had diagnostic delays of over two years, (13/14, 92.9% and 25/34, 73.5% in the Canadian and US Group cohorts, respectively).
Clinical pathway to molecular diagnosis
The detailed data available for the Canadian cohort subset (n=100) indicated that in addition to the classic developmental features, there were a total of 22 other clinical features of 22q11.2DS documented prior to molecular diagnosis (Table 2) at visits that involved a total of 26 specialty areas (Supplemental Table 1). The median number of specialty areas seen per subject prior to molecular diagnosis was 7 (range 2-15). The regression model remained significant with the additional variable of specialty areas seen (adjusted R2=0.22, df=7, p<0.0001). Results for the other variables were similar to those for the entire sample (Supplemental Table 2). Contrary to expectations, the number of specialty areas seen before diagnosis was significantly associated with a longer time to molecular diagnosis of 22q11.2DS (B=0.46, p=0.0088; Supplemental Table 2).
Table 2.
Lifetime clinical features (in addition to cardiac anomalies, palatal anomalies and developmental delay/intellectual disability) documented prior to molecular diagnosis in a subset of 100 individuals with 22q11.2DS studied in detail.a
| Lifetime clinical featuresb | n | % |
|---|---|---|
| Recurrent infectionsc | 66 | 66.0 |
| Cardiac anomalies | 64 | 64.0 |
| Developmental delay/intellectual disability | 56 | 56.0 |
| Infantile feeding difficultiesd | 48 | 48.0 |
| Palatal anomalies | 45 | 45.0 |
| Spinal deformitye | 32 | 32.0 |
| Seizuref | 30 | 30.0 |
| Hearing impairment | 25 | 25.0 |
| Psychotic disorderg | 24 | 24.0 |
| Hernia (any type) | 24 | 24.0 |
| Hypernasal speechh | 19 | 19.0 |
| Vision problemsi | 19 | 19.0 |
| Symptoms of depression and/or anxietyj | 19 | 19.0 |
| Hypocalcemia | 18 | 18.0 |
| Short stature | 14 | 14.0 |
| Asthma | 13 | 13.0 |
| Thrombocytopenia | 11 | 11.0 |
| Skin problemsk | 10 | 10.0 |
| Tonsillectomy and/or adenoidectomy | 9 | 9.0 |
| Movement abnormality/disorderl | 8 | 8.0 |
| Renal abnormalitiesm | 7 | 7.0 |
| Hypothyroidism | 5 | 5.0 |
| Hypoparathyroidismn | 4 | 4.0 |
| Imperforate anus | 2 | 2.0 |
The subset of 100 adults was selected from the Canadian cohort of 202 adults with 22q11.2DS studied. See manuscript text for details.
Percentages reported as documented by physicians prior to molecular testing and if two or more adults were noted to have the feature. Numbers may be influenced by ascertainment.
Otitis media, n=29; upper respiratory tract infection, n=25; bronchitis/pneumonia, n=22; sinusitis, n=11. Recurrent defined as two or more of any infection or documented in records as “recurrent” or “frequent” of any infection.
Included poor suck, nasal regurgitation, “sweaty/distressed” when feeding or if “feeding difficulties” were noted in records.
Included scoliosis, kyphosis, lordosis.
One or more seizures.
Schizophrenia (n = 23), psychotic mood disorder (n = 1).
Excluded subjects with palatal anomalies.
Included myopia, strabismus, keratoconus, glaucoma, amblyopia, astigmatism, and those with unspecified vision problems.
Symptoms of both depression and anxiety (n=6), anxiety only (n=6) or depression only (n=6) were noted by a professional, though a clinical diagnosis of these affective disorders was not made in all cases prior to molecular diagnosis of 22q11.2DS.
Included severe acne, psoriasis, seborrheic dermatitis.
Included gait/posture abnormalities, tremors, Parkinson’s disease.
Included solitary kidney, renal insufficiency/failure, kidney stones.
Included without hypocalcemia also noted prior to diagnosis of 22q11.2DS.
Molecular testing that led to a diagnosis of 22q11.2DS was ordered primarily by genetics specialists (n=62, 62%) followed by cardiologists (n=15, 15%), paediatricians/family doctors (n=12, 12%) and other specialists (Supplemental Table 1). For 26 of these 100 subjects, one or more of 9 other genetic syndromes was documented as being considered prior to the molecular diagnosis of 22q11.2DS: Fragile X (n=9), cri-du-chat (n=2), Down (n=2), Rubinstein-Taybi (n=2), Turner (n=2), and one each of Marfan, Noonan, Pierre-Robin, and Williams; five individuals were said to have an “indeterminate” genetic condition.
DISCUSSION
This study provides novel in-depth data on the pathways to molecular diagnosis for the current generation of individuals living with 22q11.2DS. Despite its prevalence and high penetrance for major associated features requiring medical attention [Bassett et al., 2011; Fung et al., 2015; McDonald-McGinn et al., 2015], 22q11.2DS has historically been a significant diagnostic challenge for clinicians [Fung et al., 2015; Veerapandiyan et al., 2011]. The results of the current study indicate that this remains the case, even in major 22q11.2DS centers of expertise and over 20 years after molecular testing for the typical associated 22q11.2 deletion became widely available. Together with previous studies [Bales et al., 2010; Cheung et al., 2014; McDonald-McGinn et al., 2015], the findings support the need for improved clinical recognition of and testing for the syndrome, and for the implementation of newborn screening for 22q11.2DS.
The results are consistent with previous findings of earlier diagnosis in the presence of physical congenital anomalies of the heart and/or palate that are considered typical of 22q11.2DS [Cancrini et al., 2014; Gerdes et al., 2001; Grassi et al., 2014; McDonald-McGinn et al., 2015]. However, our results also indicated that many individuals with classic features of 22q11.2DS still experienced notable delays in molecular diagnosis. Given the highly variable expression of 22q11.2DS and the results of this and other studies [Fung et al., 2015; Liu et al., 2014; Veerapandiyan et al., 2011], it would appear that over-reliance on the presence of “classic” features may be inadequate for optimizing diagnostic practice for 22q11.2DS. Our findings indicate this may be especially the case for those of non-European ancestry, consistent with previous studies of African-American and Chinese patient samples [Liu et al., 2014; Veerapandiyan et al., 2011]. The results for ethnicity also provide indirect support for evidence from other studies that relying on the presence of “characteristic” minor dysmorphic features and/or facial gestalt is insufficient for optimal decision-making about genetic testing or referral in this highly variable syndrome [Becker et al., 2004; Liu et al., 2014; McDonald-McGinn et al., 2015; Veerapandiyan et al., 2011].
Findings from this study show that, although there is evidence of some improvement over time, there continue to be notable delays even for those born in the era of molecular testing and followed at prominent medical centers. Only a minority received a diagnosis in the first few years of life. There was no significant difference observed in length of time to molecular diagnosis between adult and pediatric (i.e., younger) subjects born in the molecular era. Collectively, the results support a need for further advances in reducing diagnostic delays. Newborn screening has been proposed for 22q11.2DS [Bassett et al., 2011; Fung et al., 2015; McDonald-McGinn et al., 2015]. Although multiple criteria must be met prior to implementation, newborn screening would remove the identification bias related to classic features, including those obvious prenatally or at delivery, and ensure timely care and preventive measures for all individuals with 22q11.2DS and their families. The findings of the current study support the necessity of newborn screening to achieve such aims.
Contrary to our expectations, the presence of DD/ID in our sample was not associated with a shorter time to molecular diagnosis of 22q11.2DS. Under-recognition of 22q11.2DS as a common cause of all types of developmental delays [Bassett et al., 2011; McDonald-McGinn et al., 2015] may be a factor. Clinical microarrays are now the first line investigation recommended for those with DD/ID and other major developmental conditions [Miller et al., 2010], becoming broadly available in Ontario in 2009 (though available at the SickKids Hospital for many years before that, and requiring an extra form specifying that clinical criteria are met). One could imagine that genome-wide microarrays, not requiring an index of suspicion for a specific syndrome, should eventually change the landscape of molecular diagnosis, as implied by the results from the recent US pediatric cohort. However, the findings from the current study support those of other studies indicating that clinical uptake remains poor for individuals with DD/ID [Lowther et al., 2017; McKay et al., 2017; Mordaunt et al., 2014]. These DD/ID results thus provide further support for newborn screening.
To our knowledge, the results of this study provide the first evidence that having more clinical specialty areas involved in patient care could actually be associated with a longer time to molecular genetic diagnosis. Tautology (e.g., that the more challenging patients with delayed diagnosis see more specialties) appears unlikely to explain all cases, given the high numbers of specialty areas for most patients. It is possible that some specialists were uncertain as to how to refer for diagnostic testing. The finding that genetic specialists most commonly ordered diagnostic testing suggests that some of the diagnostic delay could be in the time to be referred to and be seen by a geneticist. Genetics referrals could also indicate unfamiliarity among other specialities with directly ordering genetic tests [Dominguez-Carral et al., 2017; Haga et al., 2013; Press et al., 2016; Salm et al., 2014], even though access to genetic testing for 22q11.2 deletions by licensed physicians in Ontario is universal. In many jurisdictions, insurance and other barriers, including additional forms and requirements, may limit genetic test capabilities and uptake. It is possible that some specialists had no suspicion of a genetic condition. Awareness of 22q11.2DS by pediatricians and teachers, including its associated physical, cognitive, and behavioral features, is reported to be lower than that for other genetic syndromes [Lee et al., 2005]. One could also speculate about other factors that could contribute to the finding. There are general issues with clinicians operating within “silos” of medical expertise [De Hert et al., 2011; Horvitz-Lennon et al., 2006], and the many terms (e.g. velocardiofacial and DiGeorge syndrome) for 22q11.2DS that may contribute to confusion regarding molecular diagnosis, best practice guidelines, and access to information and support services [Busse et al., 2011]. Misconceptions or incorrect assumptions are possible, e.g., that someone else is responsible for ordering molecular diagnostic testing, that a negative karyotype means no genetic etiology, or that the absence of one or more classic features (or the presence of non-classic features) rules out 22q11.2DS. Also, specialists may not have the means to call patients back when diagnostic practices change, e.g., if re-referral is required. There may also be reliance on special interest clinicians with experience in 22q11.2DS to take the lead (a Toronto example would include experienced speech language pathologists who for decades have initiated genetic referrals at the Hospital for Sick Children). Regardless of the underlying factors, the results support a recommendation of further education across all specialities, including pediatricians [McKay et al., 2017; Mordaunt et al., 2014], and family physicians and other primary care providers who could “put the puzzle pieces together” for undiagnosed patients.
Study advantages and limitations
We had access to a well-characterized cohort of adults with 22q11.2DS with long-term phenotypic data and were able to obtain comparable data from another major 22q11.2DS center. Such systematically collected data are rare. However, in addition to the sample size and related power issues, our findings are limited by patient ascertainment strategies, referral biases and unavoidable restrictions of available data from the retrospective design. Differing healthcare practices including referral and genetic testing availability in various jurisdictions across Canada and the US and changes over time could have affected the results. However the Canadian and US results were similar, and the fact that the majority of the subjects were from major 22q11.2DS centers would make it likely that the findings are conservative, close to best case with respect to availability and implementation of molecular testing. Generalizability to other jurisdictions with slower roll-out or barriers such as additional requirements before testing (e.g., medical genetics referral, specific features) therefore would likely tend to be in the same direction of the findings. This would also be expected to be the case where ascertainment bias was greater, e.g., even higher prevalence of major congenital features such as CHD than in the current study. Testing and subsequent diagnosis of 22q11.2DS remains heavily reliant on identification of classic features of the syndrome, likely influencing the findings. This would also pertain to dysmorphic features not included in the current study.
Using intellectual disability as a proxy for clinically relevant developmental delay has limitations given that an ID diagnosis is made after the newborn period. However, nearly all individuals with an ID also have documented developmental delays [Doherty et al., 2013] and only a small proportion of the cohort had ID/DD but no major physical congenital anomalies. Also, we found that a diagnosis of ID/DD for any subgroup (pediatric to adult), did not shorten time to diagnosis. Time of molecular diagnosis was defined as that recorded in the available medical records confirmed with lab results. The length of time to “disclose” the diagnosis of 22q11.2DS to patients and families may be longer than the time to molecular confirmation reported here, e.g., related to lack of follow-up by treating clinicians in some cases and missed appointments by these relatively complex patients. Strategies for disclosing and discussing a diagnosis of 22q11.2DS have been previously described [Bassett et al., 2011; Cancrini et al., 2014; Costain et al., 2012; Fung et al., 2015].
Conclusions
The variable phenotype of 22q11.2DS continues to present a significant diagnostic challenge to clinicians, more than 20 years after the introduction of molecular testing. Diagnostic delays can correspond to delayed access to appropriate anticipatory care, early interventions, and access to resources and services including genetic counselling and family planning [Cotter et al., 2016; Pfundt et al., 2016; Press et al., 2016]. Implementation of 22q11.2DS as the single diagnostic label, provision of timely confirmatory molecular testing, and increased use of clinical microarrays as a first-tier test for those with developmental delays/intellectual disabilities and/or congenital anomalies may improve confidence in ordering genetic testing and help to reduce diagnostic confusion while eliminating missed or prolonged diagnosis for patients with 22q11.2DS and their families [Sagoo et al., 2015; Trakadis and Shevell 2011]. Understanding and preventing delays in diagnosis may reduce the burden of disease and improve symptom management, patient long-term outcome, and lower overall health care costs. Prenatal detection is also likely to decrease morbidity and even neonatal mortality [Grati et al., 2015]. Despite availability of genetic testing, the diagnostic odyssey faced by patients with 22q11.2DS and their family members continues prolonged in many cases. Increased awareness and education, but especially the advent of newborn screening, could prevent these unnecessary diagnostic odysseys.
Supplementary Material
Acknowledgments
Supported by the Canadian Institutes of Health Research (ASB, MOP #97800, MOP#111238), Dalglish Chair (ASB) and Dalglish Fellow (EB) awards, Canada Research Chairs program (ASB), a Brain Canada Mental Health Training Award (NJB), and a Frederick Banting and Charles Best Canada Graduate Scholarship (NJB), a McLaughlin Centre Accelerator Grant, and National Institute of Mental Health grant U01 MH101723-01(3/5).
Footnotes
Conflict of interest: None declared (ASB, EB, LP, NJB, TH, AG, KAH, EWCC).
References
- Bales AM, Zaleski CA, McPherson EW. Patient and family experiences and opinions on adding 22q11 deletion syndrome to the newborn screen. Journal of Genetic Counseling. 2010;19:526–534. doi: 10.1007/s10897-010-9306-0. [DOI] [PubMed] [Google Scholar]
- Bassett AS, Caluseriu O, Weksberg R, Young DA, Chow EW. Catechol-O-methyl transferase and expression of schizophrenia in 73 adults with 22q11 deletion syndrome. Biological Psychiatry. 2007;61:1135–1140. doi: 10.1016/j.biopsych.2006.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, Husted J, Hodgkinson KA, Oechslin E, Harris L, Silversides C. Premature death in adults with 22q11.2 deletion syndrome. Journal of Medical Genetics. 2009;46:324–330. doi: 10.1136/jmg.2008.063800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA. Clinical features of 78 adults with 22q11 Deletion Syndrome. American Journal of Medical Genetics Part A. 2005;138:307–313. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Costain G, Marshall CR. Neuropsychiatric aspects of 22q11.2 deletion syndrome: considerations in the prenatal setting. Prenatal Diagnosis. 2017;37:61–69. doi: 10.1002/pd.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Marshall CR, Lionel AC, Chow EW, Scherer SW. Copy number variations and risk for schizophrenia in 22q11.2 deletion syndrome. Human Molecular Genetics. 2008;17:4045–4053. doi: 10.1093/hmg/ddn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J, International 22q11.2 Deletion Syndrome C Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediat. 2011;159:332–339. doi: 10.1016/j.jpeds.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker DB, Pilgram T, Marty-Grames L, Govier DP, Marsh JL, Kane AA. Accuracy in identification of patients with 22q11.2 deletion by likely care providers using facial photographs. Plast Reconstr Surg. 2004;114:1367–1372. doi: 10.1097/01.prs.0000138591.20999.f1. [DOI] [PubMed] [Google Scholar]
- Billett J, Cowie MR, Gatzoulis MA, Vonder Muhll IF, Majeed A. Comorbidity, healthcare utilisation and process of care measures in patients with congenital heart disease in the UK: cross-sectional, population-based study with case-control analysis. Heart. 2008;94:1194–1199. doi: 10.1136/hrt.2007.122671. [DOI] [PubMed] [Google Scholar]
- Busse T, Graham JM, Jr, Feldman G, Perin J, Catherwood A, Knowlton R, Rappaport EF, Emanuel B, Driscoll DA, Saitta SC. High-Resolution genomic arrays identify CNVs that phenocopy the chromosome 22q11.2 deletion syndrome. Human Mutation. 2011;32:91–97. doi: 10.1002/humu.21395. [DOI] [PubMed] [Google Scholar]
- Butcher N, Chow E, Costain G, Karas D, Ho A, Bassett A. Functional outcomes of adults with 22q11.2 deletion syndrome. Genetics in Medicine. 2012;14:836–843. doi: 10.1038/gim.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancrini C, Puliafito P, Digilio MC, Soresina A, Martino S, Rondelli R, Consolini R, Ruga EM, Cardinale F, Finocchi A, Romiti ML, Martire B, Bacchetta R, Albano V, Carotti A, Specchia F, Montin D, Cirillo E, Cocchi G, Trizzino A, Bossi G, Milanesi O, Azzari C, Corsello G, Pignata C, Aiuti A, Pietrogrande MC, Marino B, Ugazio AG, Plebani A, Rossi P, Italian Network for Primary Immunodeficiencies Clinical features and follow-up in patients with 22q11.2 deletion syndrome. J Pediatr. 2014;164:1475–1480. doi: 10.1016/j.jpeds.2014.01.056. [DOI] [PubMed] [Google Scholar]
- Cheung EN, George SR, Andrade DM, Chow EW, Silversides CK, Bassett AS. Neonatal hypocalcemia, neonatal seizures, and intellectual disability in 22q11.2 deletion syndrome. Genetics in Medicine. 2014;16:40–44. doi: 10.1038/gim.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow EW, Watson M, Young DA, Bassett AS. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophrenia Research. 2006;87:270–278. doi: 10.1016/j.schres.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, Abdel-Hamid H, Bader P, McCracken E, Niyazov D, Leppig K, Thiese H, Hummel M, Alexander N, Gorski J, Kussmann J, Shashi V, Johnson K, Rehder C, Ballif BC, Shaffer LG, Eichler EE. A copy number variation morbidity map of developmental delay. Nature Genetics. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costain G, Chow EW, Ray PN, Bassett AS. Caregiver and adult patient perspectives on the importance of a diagnosis of 22q11.2 deletion syndrome. Journal of Intellectual Disability Research. 2012;56:641–651. doi: 10.1111/j.1365-2788.2011.01510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter M, Archibald AD, McClaren BJ, Burgess T, Francis D, Hills L, Martyn M, Oertel R, Slater H, Cohen J, Metcalfe SA. Clinical audit of genetic testing and referral patterns for fragile X and associated conditions. American Journal of Medical Genetics Part A. 2016;170:1439–1449. doi: 10.1002/ajmg.a.37603. [DOI] [PubMed] [Google Scholar]
- De Hert M, Cohen D, Bobes J, Cetkovich-Bakmas M, Leucht S, Ndetei DM, Newcomer JW, Uwakwe R, Asai I, Moller HJ, Gautam S, Detraux J, Correll CU. Physical illness in patients with severe mental disorders. II. Barriers to care, monitoring and treatment guidelines, plus recommendations at the system and individual level. World Psychiatry. 2011;10:138–151. doi: 10.1002/j.2051-5545.2011.tb00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty E, O’Connor R, Zhang A, Lim C, Love JM, Ashton F, Claxton K, Gregersen N, George AM, Love DR. Developmental delay referrals and the roles of Fragile X testing and molecular karyotyping: a New Zealand perspective. Mol Med Rep. 2013;7:1710–1714. doi: 10.3892/mmr.2013.1386. [DOI] [PubMed] [Google Scholar]
- Dominguez-Carral J, Lopez-Pison J, Macaya A, Bueno Campana M, Garcia-Perez MA, Natera-de Benito D. Genetic testing among Spanish pediatric neurologists: Knowledge, attitudes and practices. European Journal of Medical Genetics. 2017;60:124–129. doi: 10.1016/j.ejmg.2016.11.007. [DOI] [PubMed] [Google Scholar]
- Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, Emanuel BS. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. Journal of Medical Genetics. 1993;30:813–817. doi: 10.1136/jmg.30.10.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung WL, Butcher NJ, Costain G, Andrade DM, Boot E, Chow EW, Chung B, Cytrynbaum C, Faghfoury H, Fishman L, Garcia-Minaur S, George S, Lang AE, Repetto G, Shugar A, Silversides C, Swillen A, van Amelsvoort T, McDonald-McGinn DM, Bassett AS. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genetics in Medicine. 2015;17:599–609. doi: 10.1038/gim.2014.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes M, Solot C, Wang PP, McDonald-McGinn DM, Zackai EH. Taking advantage of early diagnosis: preschool children with the 22q11.2 deletion. Genetics in Medicine. 2001;3:40–44. doi: 10.1097/00125817-200101000-00009. [DOI] [PubMed] [Google Scholar]
- Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child. 1998;79:348–351. doi: 10.1136/adc.79.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi MS, Jacob CM, Kulikowski LD, Pastorino AC, Dutra RL, Miura N, Jatene MB, Pegler SP, Kim CA, Carneiro-Sampaio M. Congenital Heart Disease as a Warning Sign for the Diagnosis of the 22q11.2 Deletion. Arq Bras Cardiol. 2014;103:382–390. doi: 10.5935/abc.20140145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grati F, Molina Gomes D, Ferreira J, Dupont C, Alesi V, Gouas L, Horelli-Kuitunen N, Choy KW, Garcia-Herrero S, de la Vega AG, Piotrowski K, Genesio R, Queipo G, Malvestiti B, Herve B, Benzacken B, Novelli A, Vago P, Piippo K, Leung TY, Maggi F, Quibel T, Tabet AC, Simoni G, Vialard F. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat Diagn. 2015;35:801–809. doi: 10.1002/pd.4613. [DOI] [PubMed] [Google Scholar]
- Haga SB, Burke W, Agans R. Primary-care physicians’ access to genetic specialists: an impediment to the routine use of genomic medicine? Genetics in Medicine. 2013;15:513–514. doi: 10.1038/gim.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz-Lennon M, Kilbourne AM, Pincus HA. From silos to bridges: meeting the general health care needs of adults with severe mental illnesses. Health Affairs. 2006;25:659–669. doi: 10.1377/hlthaff.25.3.659. [DOI] [PubMed] [Google Scholar]
- Lee TH, Blasey CM, Dyer-Friedman J, Glaser B, Reiss AL, Eliez S. From research to practice: teacher and pediatrician awareness of phenotypic traits in neurogenetic syndromes. American Journal of Mental Retardation. 2005;110:100–106. doi: 10.1352/0895-8017(2005)110<100:FRTPTA>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Liu AP, Chow PC, Lee PP, Mok GT, Tang WF, Lau ET, Lam ST, Chan KY, Kan AS, Chau AK, Cheung YF, Lau YL, Chung BH. Under-recognition of 22q11.2 deletion in adult Chinese patients with conotruncal anomalies: Implications in transitional care. Eur J Med Genet. 2014;57:306–311. doi: 10.1016/j.ejmg.2014.03.014. [DOI] [PubMed] [Google Scholar]
- Lowther C, Merico D, Costain G, Waserman J, Boyd K, Noor A, Speevak M, Stavropoulos D, Wei J, Lionel A, Marshall C, Scherer SW, Bassett A. Impact of IQ on the diagnostic yield of chromosomal microarray in a community sample of adults with schizophrenia. Genome Medicine Under review. 2017 doi: 10.1186/s13073-017-0488-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JAS, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS. 22q11.2 deletion syndrome. Nat Rev Dis Prim. 2015;19:15071. doi: 10.1038/nrdp.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay V, Efron D, Palmer EE, White SM, Pearson C, Danchin M. Current use of chromosomal microarray by Australian paediatricians and implications for the implementation of next generation sequencing. J Paediatr Child Health. 2017;53:650–656. doi: 10.1111/jpc.13523. [DOI] [PubMed] [Google Scholar]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American Journal of Human Genetics. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordaunt D, Gabbett M, Waugh M, O’Brien K, Heussler H. Uptake and Diagnostic Yield of Chromosomal Microarray in an Australian Child Development Clinic. Children (Basel) 2014;1:21–30. doi: 10.3390/children1010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsdottir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Archives of Disease in Childhood. 2004;89:148–151. doi: 10.1136/adc.2003.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfundt R, Del Rosario M, Vissers LE, Kwint MP, Janssen IM, de Leeuw N, Yntema HG, Nelen MR, Lugtenberg D, Kamsteeg EJ, Wieskamp N, Stegmann AP, Stevens SJ, Rodenburg RJ, Simons A, Mensenkamp AR, Rinne T, Gilissen C, Scheffer H, Veltman JA, Hehir-Kwa JY. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genetics in Medicine. 2016 doi: 10.1038/gim.2016.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press KR, Wieczorek L, Hoover-Fong J, Bodurtha J, Taylor L. Overview: referrals for genetic evaluation from child psychiatrists. Child Adolesc Psychiatry Ment Health. 2016;10:7. doi: 10.1186/s13034-016-0095-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagoo GS, Mohammed S, Barton G, Norbury G, Ahn JW, Ogilvie CM, Kroese M. Cost Effectiveness of Using Array-CGH for Diagnosing Learning Disability. Appl Health Econ Health Policy. 2015;13:421–432. doi: 10.1007/s40258-015-0172-7. [DOI] [PubMed] [Google Scholar]
- Salm M, Abbate K, Appelbaum P, Ottman R, Chung W, Marder K, Leu CS, Alcalay R, Goldman J, Curtis AM, Leech C, Taber KJ, Klitzman R. Use of genetic tests among neurologists and psychiatrists: knowledge, attitudes, behaviors, and needs for training. J Genet Couns. 2014;23:156–163. doi: 10.1007/s10897-013-9624-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakadis Y, Shevell M. Microarray as a first genetic test in global developmental delay: a cost-effectiveness analysis. Dev Med Child Neurol. 2011;53:994–999. doi: 10.1111/j.1469-8749.2011.04080.x. [DOI] [PubMed] [Google Scholar]
- Van L, Butcher NJ, Costain G, Ogura L, Chow EWC, Bassett AS. Fetal growth and gestational factors as predictors of schizophrenia in 22q11.2 deletion syndrome. Genetics in Medicine. 2015 doi: 10.1038/gim.2015.84. [DOI] [PubMed] [Google Scholar]
- Veerapandiyan A, Abdul-Rahman OA, Adam MP, Lyons MJ, Manning M, Coleman K, Kobrynski L, Taneja D, Schoch K, Zimmerman HH, Shashi V. Chromosome 22q11.2 deletion syndrome in African-American patients: a diagnostic challenge. Am J Med Genet A Part A. 2011;155A:2186–2195. doi: 10.1002/ajmg.a.34226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.