

Abstract
Cellular senescence is a physiological phenomenon that has both beneficial and detrimental consequences. Senescence limits tumorigenesis and tissue damage throughout the lifetime. However, at the late stages of life, senescent cells increasingly accumulate in tissues and might also contribute to the development of various age-related pathologies. Recent studies have revealed the molecular pathways that preserve the viability of senescent cells and the ones regulating their immune surveillance. These studies provide essential initial insights for the development of novel therapeutic strategies for targeting senescent cells. At the same time they stress the need to understand the limitations of the existing strategies, their efficacy and safety, and the possible deleterious consequences of senescent cell elimination. Here we discuss the existing strategies for targeting senescent cells and upcoming challenges in translating these strategies into safe and efficient therapies. Successful translation of these strategies could have implications for treating a variety of diseases at old age and could potentially reshape our view of health management during aging.
Introduction
The world’s population is rapidly aging (1, 2). Living to a late age provides many opportunities but also presents a huge challenge, as it increases vulnerability to the development of chronic pathological conditions. In fact, aging is the leading risk factor for the world’s most prevalent pathologies, including cardiovascular diseases, cancer, and neurodegenerative diseases (3). Aging is heterogeneous, and some people function better than others at the same chronological age, exhibiting a longer period of good general health. Thus, a better understanding of common cellular and molecular pathways that drive the development of age-related multimorbidities is necessary. Treatment of age-related diseases based on such pathways could provide better therapies than treatment of each age-related disease individually. Recent discoveries have provided insights into the cellular and molecular events that play a role in biological aging (3, 4). One emerging factor is the accumulation of senescent cells in tissues.
Cellular senescence is an essentially irreversible cell cycle arrest that occurs in normal proliferating cells in response to various forms of cellular stress. Replicative exhaustion, oncogene activation, direct DNA damage, cell-cell fusion, and other forms of stress that elicit activation of the DNA damage response pathway can lead to senescence (5–8). Cellular senescence is a vital physiological response aimed at preventing propagation of damaged cells in the organism (9–11). It acts as a bona fide tumor suppression mechanism, limits tissue damage, and aids wound healing (12–16). Despite the protective role of cellular senescence as a cellular response to stress, studies in mouse models have shown that the long-term presence of senescent cells that form as a result of this response may be detrimental to the organism (17, 18). These cells secrete a plethora of proinflammatory factors that assist in their removal by the immune system (19, 20). Studies on diverse animal models indicate that multiple components of the immune system, including NK cells, T cells, and macrophages, are involved in controlling the presence of senescent cells in tissues (13, 21–25). The efficacy of this removal is variable among tissues and pathological conditions, and the mechanisms and rules regulating the homeostasis of senescent cells are yet to be fully understood.
At the late stages of life, senescent cells increasingly accumulate in tissues and contribute to the establishment of a chronic “sterile” inflammation that arises due to continuous secretion of proinflammatory cytokines (11, 26, 27). This condition, also known as “inflammaging,” is a pervasive feature of the majority of age-related diseases (28). Indeed, senescent cells are especially abundant at sites of age-related pathologies, and a growing body of evidence from mouse models demonstrates a causal role for senescent cells in the pathogenesis of age-related diseases including atherosclerosis, idiopathic lung fibrosis, osteoarthritis, bone loss, and hepatic steatosis (29–34). Furthermore, genetic approaches to promoting clearance of p16-expressing senescent cells in mice delay the onset of age-related deterioration of several organs and increase median survival of the mice (35, 36). Hence, elimination of senescent cells might be a promising approach for treatment and prevention of many age-related diseases, hopefully leading to healthy longevity (37–39).
Therapeutic strategies for targeting of senescent cells
There is growing interest in the possibility of targeting senescent cells therapeutically. Several promising approaches that focus on either clearance of senescent cells or prevention of their proinflammatory impact are in development (Figure 1). Current efforts are largely invested in the discovery of pharmacological agents that can induce cell death in senescent cells. These compounds are often termed “senolytic drugs” or “senolytics.” Research in this direction is mainly based on the biological pathways underlying senescent cell accumulation with age, and the therapies aim to utilize some of the unique molecular features that senescent cells display over other cells in the organism. One of the most prominent features of these cells is their relative resistance to apoptosis. Unlike normal cells, senescent cells are protected from both intrinsic and extrinsic proapoptotic signals, a property that allows them to persist and promote diverse biological processes under stress conditions (40–42). Targeting these apoptotic pathways preferentially in senescent cells, although different from a genetic approach in multiple respects (43), leads to selective death of these cells and prevents them from exerting their detrimental effects.
Figure 1. Strategies targeting cellular senescence.

Three principal approaches could be implemented to block deleterious effects of senescent cells. (A) The leading options are strategies that induce apoptosis selectively in senescent cells with the use of senolytic drugs, which block prosurvival pathways. (B) The second type of strategy is to potentiate an immune response against senescent cells in a way that would lead to their clearance from tissues. (C) A third strategy proposes specific blockade of SASP components by targeting of their upstream regulators and effectors. ADCC, antibody-dependent cell-mediated cytotoxicity; DDR, DNA damage response; GOS, galacto-oligosaccharides; ITAM, immunoreceptor tyrosine-based activation motif.
Blocking prosurvival pathways.
Several of the prosurvival pathways that have been identified can be used for directed elimination of senescent cells (44–49). These include the BCL-2 protein family, the p53/p21 axis, PI3K/AKT, receptor tyrosine kinases, and the HIF-1α and HSP90 proteins. Currently, most identified senolytics are directed against members of the BCL-2 protein family. Antiapoptotic proteins of the BCL-2 protein family are well studied on the molecular level and are potentially attractive drug targets (50). These proteins play multiple roles in cell death regulation through their effects on apoptosis and autophagy (40, 51). Studies across different cell types have demonstrated an upregulation of the BCL-2 family members BCL-2, BCL-W, and BCL-XL during senescence (46, 48). Silencing those proteins in senescent cells leads to the activation of programmed cell death.
The BCL-2 protein family is an extensively investigated target for pharmacological intervention in cancer, offering several well-characterized candidates as senolytics (50). ABT-737, for example, is a BH3 domain mimetic that blocks the interaction of antiapoptotic family members BCL-2, BCL-W, and BCL-XL with BH3 domain–containing proapoptotic proteins, allowing senescent cells to undergo apoptosis (52, 53). ABT-737 efficiently eliminates senescent cells that were induced by DNA damage in lungs of irradiated mice, as well as senescent cells formed by p14ARF induction in skin epidermis of transgenic mice (48). This study also suggested that elimination of senescent cells from the epidermis might positively affect hair growth, as it induced proliferation of hair follicle stem cells. ABT-263, also known as navitoclax, is a next-generation, orally available analog of ABT-737 (54). ABT-263 has demonstrated the capacity to eliminate senescent cells from sublethally irradiated mice and naturally aged mice, including senescent muscle stem cells and senescent hematopoietic stem cells (46). This action results in rejuvenation of those stem cell populations. ABT-263 also showed the capacity to eliminate senescent foam cell macrophages from atherosclerotic lesions, thereby halting progression of the disease (29).
Notably, treatment with these general inhibitors of BCL-2, BCL-W, and BCL-XL causes several mechanism-based hematological toxicities, such as neutropenia and thrombocytopenia (55). These safety issues will likely impede the development of BCL-2 family inhibitors into common clinical interventions, and emphasize the need for more specific inhibitors that might serve as superior candidates. These might include specific BCL-XL inhibitors, such as A1331852 and A1155463, which are expected to cause less toxicity to nonsenescent cells (56). A1331852 can induce apoptosis in senescent cholangiocytes and senescent fibroblasts in a genetic mouse model of biliary liver fibrosis, thereby reducing liver injury and fibrosis (57). Nonetheless, platelet survival is dependent on BCL-XL, so these compounds would likely still cause transient thrombocytopenia. They might also be less potent senolytics for senescent populations that express higher levels of BCL-2 or BCL-W (46, 48). Local administration of BCL-2 family inhibitors to the insulated sites of interest could limit those side effects. For instance, intra-articular injection of the BCL-2–targeting UBX0101 compound efficiently eliminated senescent cells in articular cartilage and synovium and reduced signs of osteoarthritis in aged mice (33). Overall, the available evidence suggests that although targeting the BCL-2 family of proteins is a molecularly validated mechanism-based approach for targeting senescent cells, its side effects will likely limit therapeutic use.
Senolytic combinations.
An alternative route to overcoming the cytotoxic side effects of BCL-2 protein family inhibitors might be to synergistically combine low doses with compounds that target other senescence-associated prosurvival pathways. Indeed, the first published senolytic was not a single agent, but a combination of the pan–tyrosine kinase inhibitor dasatinib and a naturally occurring flavonoid, quercetin (47). When given together, these two compounds were shown to selectively kill senescent primary preadipocyte cells and senescent human umbilical vein endothelial cells (HUVECs) in tissue culture, as well as reduce senescent cell signature in a set of pathological conditions (30, 31, 47, 58). Administration of the dasatinib/quercetin combination reduced aortic calcification and osteogenic signaling in aged and hypercholesterolemic mice (34). It also improved lung function during bleomycin-induced lung fibrosis in mice (30). In both systems, however, the combination failed to alleviate the fibrotic component of these conditions, suggesting that it may be more effective as a preventative intervention during early stages of diseases. Consistent with this suggestion, dasatinib/quercetin can prevent age-related bone loss and reduce overall hepatic steatosis (a prerequisite condition of nonalcoholic fatty liver diseases) (31, 32).
Dasatinib and quercetin were previously tested in clinical settings, making this combination a promising senolytic candidate. Nevertheless, some issues might impede its future use. First, both compounds target a long list of biological pathways, and the combination suffers from lack of understanding of the mechanisms underlying its senolytic actions. In addition, the effects observed following the combination treatment might be mediated through alterations in multiple target pathways in multiple tissues and organs, which could result in adverse effects after long exposures. Still, short-term administration of the combination is sufficient to eliminate senescent cells and provide benefit as observed in the above studies. Since the combination might not need to be present continuously in order to be effective, off-target complications could be avoided. In addition to quercetin, other natural compounds, including fisetin and piperlongumine, have been suggested to have senolytic effects (56, 59). These molecules, too, are expected to kill senescent cells by targeting multiple molecular pathways. Owing to a lack of well-defined mechanism of action and questionable pharmacokinetics, it is unlikely that they will find their way into the clinic.
Targeting senescence-specific pathways.
Recent studies highlight the p53/p21 axis as a promising target for development of novel senolytics (49, 60). p53, the most frequently mutated tumor suppressor gene in human cancer, is a core component of the senescence program (61). p53 is stabilized and accumulates in response to DNA damage (7, 62, 63). Its transcriptional activity controls a myriad of biological processes, positioning it on a decisional fork between transient cell cycle arrest, senescence, and apoptosis (61). Interfering in p53’s direct interaction with the transcription factor FOXO4 leads to the release of p53 from the nucleus and induction of cell-intrinsic apoptosis. Administration of a modified FOXO4/p53-interfering peptide was able to neutralize murine liver chemotoxicity in the setting of doxorubicin treatment and restore fitness, hair density, and renal function in progeroid and naturally aged mice (60). The senolytic potential of this pathway is not limited to the p53-FOXO4 interaction (64). p21 (also known as CDKN1A), a primary transcriptional target of p53, is necessary for survival and retention of senescent cells in tissues (48). Following DNA damage, it prevents NF-κB–mediated activation of JNK, caspase-3 cleavage, and subsequent cell death. In mice, p21 knockout leads to a reduction of senescent cells in fibrotic livers and alleviates liver fibrosis (49). Despite the fact that the p53/p21 axis can be indirectly modulated, for example by controlling ROS levels, there are no known drugs that can induce apoptosis in senescent cells by inhibition of either p53 or p21. Therefore, further research and development of new drug candidates is needed to fulfill the promise of the p53/p21 prosurvival pathway in targeting senescent cells. Currently, the senolytic effect of the FOXO4 peptide is a promising start, and the peptide or the peptide-mimetic molecules could be a basis for senolytic drug development.
The identification of molecular pathways that preserve the viability of senescent cells provided the initial insights for the development of senolytics. In addition to pathway-based senolytic development, high-throughput approaches can also be used to identify new targets. Lately, the development of such an approach identified HSP90 proteins as a novel class of targets (44). HSP90, a family of ubiquitously expressed molecular chaperones, can promote cell survival via stabilization of AKT or ERK (65, 66), members of signaling pathways that are upregulated during senescence (67, 68). Disruption of the HSP90-AKT interaction inhibited the PI3K/AKT pathway, resulting in a selective killing of senescent cells of different origins (44). In vivo, administration of the HSP90 inhibitor 17-DMAG to progeroid mice reduced the senescence signature and extended health span. These findings mark HSP90 inhibitors as potential constituents for future combinatorial senolytics.
Directed elimination of senescent cells.
Targeting of senescence-specific pathways is promising, but it is not the only way to develop senolytics. It is possible to eliminate senescent cells by exploiting other senescence-specific features. One well-described feature of senescent cells is senescence-associated β-galactosidase (SA β-gal) activity (26). A targeted delivery system using mesoporous silica nanoparticles coated with galacto-oligosaccharides was developed based on this feature (69). While the coated particles cannot be activated in nonsenescent cells, the coating is digested in senescent cells and the nanoparticle content can be released. It is conceivable that coated nanoparticles containing a cytotoxic drug could release it to the cytoplasm of senescent cells to induce apoptosis. Increased β-galactosidase activity is not a complete surrogate feature of senescent cells, as it is known that other cells in the body, e.g., macrophages, exhibit high β-galactosidase activity in some circumstances (70). In this case, it may be beneficial to increase selectivity by tethering the particles to antibodies against known surface markers for senescent cells (71). Senolytic approaches based on SA β-gal activity are yet to be tested in vivo, and more studies are needed to examine the outcomes of such treatments.
Immune-mediated interventions.
Apart from directed elimination of senescent cells based on their intrinsic properties, therapeutic interventions could be also mediated by the immune system. Senescent cells are immunogenic in nature and are subject to immune surveillance mechanisms (13, 20, 21, 23, 72, 73). In a model of hepatocellular carcinoma, induction of senescence by p53 restoration triggers an innate immune response consisting of infiltrating leukocytes, neutrophils, macrophages, and NK cells (21). In a model of liver fibrosis, senescent hepatic stellate cells promote their own NK cell–mediated elimination (13). Senescent cells upregulate the immune recognition molecule NKG2D, which is usually not present on the surface of normal cells (72). Thus, NKG2D expression is a potential asset in immunotherapy approaches. The mechanisms of senescence surveillance are context-dependent. Induction of senescence by expressing of NRASG12V in hepatocytes results in a mixed phenotype of both innate and adoptive immune response, as CD4+ T cells cooperate with monocytes/macrophages to execute the clearance of senescent hepatocytes (23). Accumulation of senescent cells in aged tissues could be attributed, at least in part, to an age-related decline in immune function. Therefore, repotentiating the immune system may result in successful clearance of senescent cells from aged tissues. In support of this notion, administration of the immune stimulator polyI:C facilitates NK cell–mediated clearance of senescent cells in fibrotic livers (13). However, in vivo treatment with such a strong immune stimulator would probably overwhelm the aging immune system, which is often chronically activated. Therefore, more precise immune modulators should be considered.
Adapting common immunotherapy practices from the field of cancer might contribute to this effort. For example, blockade of the immune checkpoint programmed death 1 (PD-1) is a powerful treatment for cancer that can also improve pathological symptoms in a murine model of Alzheimer’s disease (74). In addition to immune-boosting strategies, a growing arsenal of tools such as chimeric antigen receptor (CAR) T cells provide the option to redirect immune responses against senescent cells (75). The bottleneck in this and other immune-targeting approaches is a shortage of highly selective senescence markers. Very recently, however, novel senescence cell-surface antigens have been suggested. In one study, senescent fibroblasts were shown to express an oxidized form of membrane-bound vimentin on their cell surface (76). In another study, mass spectrometry analysis identified dipeptidyl peptidase 4 (DPP4) as a cell surface marker for senescent fibroblasts (77). The selective expression of DPP4 on the surface of senescent cells enables their preferential killing by NK cells in an antibody-dependent cell-mediated cytotoxicity (ADCC) assay (77). Senescent cells are partly protected from NK cell–mediated cytotoxicity because of their upregulation of decoy receptor 2 (DcR2) (41). DcR2 inhibits the activation of death receptors 4 and 5 (DR4/5) by TNF-related apoptosis–inducing ligand (TRAIL), confining the actual killing to perforin- and granzyme-mediated pathways. Finding ways to boost this process, e.g., by blocking DcR2, could render senescent cells more vulnerable to natural immune surveillance and immunotherapy-based targeting. Overall, while the findings regarding the immune targeting of senescent cells are quite preliminary and require further investigation, they highlight the potential in harnessing the immune system to eliminate senescent cells in the context of age-related pathologies. Identification of novel and specific cell surface markers of senescent cells will further facilitate the development of immune system–based senotherapies.
Strategies aimed to limit detrimental roles of senescent cells.
While elimination of senescent cells appears to be a favorable strategy, it might be unsuitable for some conditions in which senescent cells occur. In those cases, specific targeting of detrimental components of senescence could potentially serve as an alternative strategy. Senescent cells exhibit a profound proinflammatory secretory profile that is largely conserved between different senescent states and cell origins (19, 78–80). The senescence-associated secretory phenotype (SASP) is mainly composed of inflammatory chemokines and cytokines, growth factors, and matrix-remodeling proteases. The SASP promotes non–cell-autonomous tumor suppression by enforcing cell cycle arrest in damaged and cancer cells, and by recruiting immune cells to eliminate them. Nonetheless, it has also been strongly implicated in disruption of tissue homeostasis and development of age-related pathologies (81). Several signaling pathways that converge into activation of the NF-κB pathway and the C/EBPβ pathway orchestrate the complex secretome (78, 80, 82–84). These signaling pathways are well studied and can be modulated by a broad spectrum of drugs, some of which are approved by the FDA. For instance, IL-1α/IL-1 receptor signal transduction is upstream of NF-κB, and the use of neutralizing antibodies against either IL-1α or its receptor is sufficient to reduce NF-κB transcriptional activity (85). mTOR inhibitors, such as rapamycin and it analogs, can abolish SASP by reducing the expression of membrane-bound IL-1α (86). This action has been shown to ameliorate the ability of senescent fibroblasts to stimulate prostate tumor growth in mice. Metformin, a commercially approved drug for type 2 diabetes, blocks NF-κB translocation to the nucleus, thus restricting its transcriptional activity (87). Treating mice with metformin limits inflammatory processes, alleviates different age-related pathologies, and extends lifespan (88). Reports of similar effects in human diabetic patients have initiated investigations into metformin’s effects on aging in the general population (89, 90). C/EBPβ transcriptional activity can be induced by JAK/STAT signaling (91). JAK1/2 inhibitors, such as the FDA-approved drug ruxolitinib, can reduce systemic inflammation and improve fitness in aged mice (92). Therefore, there is evidence that existing inhibitors can dampen the effects of SASP in vivo and potentially reduce the deleterious effects of senescent cells.
While restricting inflammation is a potentially promising way to limit the non–cell-autonomous effects of senescent cells, treatment with strong antiinflammatory drugs can potentially cause a wide spectrum of side effects (93). These circumstances might not permit a prolonged therapeutic regimen, which is prerequisite to preserving their beneficial effects. The solution to this problem might come from new studies on epigenetic regulators, including mixed-lineage leukemia protein 1 (MLL1), bromodomain-containing protein 4 (BRD4), and high-mobility group box 2 (HMGB2), which are responsible for reshaping the epigenetic landscape of senescent cells in a manner that would dictate expression of SASP components (24, 94, 95). Experimental knockdown of these chromatin modifiers results in sustainable antiinflammatory effects without compromising cell cycle arrest. Relevant small-molecule modulators, such as the bromodomain and extra-terminal motif (BET) inhibitors, are available and should be further tested for this exciting possibility (96). Targeting specific components of SASP could also provide a safer way to mitigate the deleterious effects of SASP. Cytokines, well-defined components of SASP, such as IL-6, IL-8, and matrix-remodeling proteases such as ADAM17, could serve as possible targets. An attractive method to block these molecules is the application of neutralizing antibodies, which can be developed based on the plethora of commercially available monoclonal antibodies. For example, targeting of IL-6 or its receptor could be achieved by the approved drugs siltuximab or tocilizumab, blocking the cytokine or the receptor, respectively (97–99). However, these agents were not developed for use in the context of cellular senescence, and their effect on SASP and aging must be studied in detail before their effect as senescence modifiers can be exploited therapeutically.
Moving toward clinical trials
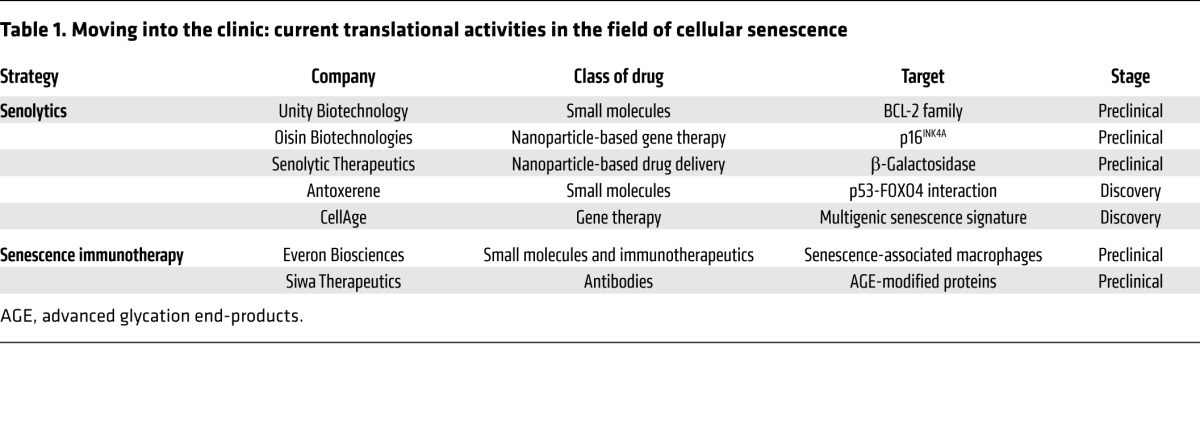
Successful translation of strategies aimed at targeting senescent cells into the clinic could have a vast impact on the treatment of a variety of diseases at old age and potentially revolutionize our view of aging. Several technologies aiming at senescent cells are now taking their first steps in the challenging, time-consuming, and expensive journey of drug development (Table 1). In order to maximize their chances of becoming common treatments, critical issues of efficacy, safety, and tolerability should be considered during the early stages of development.
Table 1. Moving into the clinic: current translational activities in the field of cellular senescence.
Upcoming challenges of strategies to target cellular senescence.
The main question that still remains in the field is the efficiency of different senotherapies in different tissues of human patients. Many of the present data on these treatments were obtained from experiments in cell culture, which might only partially represent the situation in vivo and are far from the context of a disease. Efficacy of compounds in vivo was often tested in mouse models, some of which are only partly relevant to aging and age-related pathologies. Therefore, the relevance to specific human conditions and pathologies must be properly investigated before senotherapies can be enrolled for clinical trials. Moreover, current strategies target a general population of senescent cells, whereas senescent cells are a highly heterogeneous population. Diversity in cell of origin, trigger of senescence induction, or pathological context can create disparate vulnerability of cell subpopulations to a certain treatment. For this reason, it would be rational to expect that some treatments would be more suitable than others for a certain tissue, organ, or age-related disease. Therefore, a current concept that considers senotherapies as pan-antisenescence treatments is somewhat concerning and needs to be carefully reevaluated. It will be necessary to investigate the influence of every treatment on diverse cell populations while using the most relevant model for each condition to which the treatment may be applied.
In using senescence-targeted treatments, safety issues might become a major challenge, owing to the physiological role of senescent cells and the side effects of the existing senolytics. Some senolytics, especially among BCL-2 family inhibitors, pose a cytotoxic threat to nonsenescent cells when administered systemically (55). In the case of immunotherapy, certain senescence epitopes might be also expressed to some extent in nonsenescent cells, such as stressed cells or secretory cells. There are several possible ways to overcome this problem. For instance, drugs could be administered directly to the area of interest. More importantly, combinatorial approaches based on senescent cell–specific delivery of senolytics could have a synergistic impact, thus allowing the use of lower, less toxic doses with higher efficacy and better safety. Another possibility to increase targeting specificity is to modify the agent to be active only after processing by an enzyme that is preferentially expressed by senescent cells. In addition to the side effects of the targeting moiety itself, compromised safety might also stem from possible interference with the beneficial roles of senescence. The senescence program contributes to embryogenesis, tissue repair, regeneration, and tumor suppression, but the effect of targeting senescence on these processes is still unexplored in the context of senolytic treatments (8, 100–105). Fortunately, existing senolytics induce apoptosis in senescent cells after short exposures, and their administration can be limited to short periods of time. Future treatments will have to take this notion into account and will have to be conducted in a spatiotemporally controlled manner.
Careful design of clinical studies would have a strong impact on their fate. Indications for first-wave senotherapies should be carefully chosen and could involve an uncommon age-related pathology with only a symptomatic rather than therapeutic treatment as standard of care. A causal role of cellular senescence in the pathogenesis of each disease or condition must be well ensured, and the mechanism of action should be clear. Availability of adequate preclinical models will help to evaluate the effect of each treatment’s risks and benefits. For successful clinical studies there is also a cardinal need for senescence-specific and disease-oriented biomarkers for prognostic and predictive use. Basic research is still constrained by the lack of satisfactory senescent markers, and quantitative methods are limited to ex vivo analysis of cells (106, 107). There is an urgent need to develop novel senescence biomarkers in order to reliably assess senescence burden in tissues in a noninvasive manner. Candidate patients must be considered according to their senescence burden. Careful consideration of the scientific evidence, experimental settings, and patient management will increase the chances of successful trial completion.
Overall, cellular senescence is a comprehensive phenomenon with both beneficial and detrimental consequences. Senotherapies hold promise to block detrimental effects and treat a large set of age-related pathologies, but they will have to be implemented in an accurate manner in order to assure both efficient and safe use. Understanding the mechanisms of possible side effects while designing the dosage and the regimen of senolytic treatments will help facilitate their way to the clinic and provide new hope for improvement of human health span and longevity.
Acknowledgments
The Krizhanovsky laboratory was supported by grants from the European Research Council under the European Union’s FP7 and H2020, the Israel Science Foundation, the Rising Tide Translational Research Fund, and the Bruce Kanter Fund. VK is the incumbent of the Carl and Frances Korn Career Development Chair in Life Sciences.
Version 1. 04/02/2018
Print issue publication
Footnotes
Conflict of interest: YO and VK are inventors on pending patents in the field (PCT/IL2014/050358, PCT/IL2016/050220, and PCT/IL2016/050702 for VK; and PCT/IL2016/050535 for VK and YO).
Reference information: J Clin Invest. 2018;128(4):1247–1254.https://doi.org/10.1172/JCI95149.
Contributor Information
Yossi Ovadya, Email: yossi.ovadya@weizmann.ac.il.
Valery Krizhanovsky, Email: valery.krizhanovsky@weizmann.ac.il.
References
- 1. World Health Organization. World Report on Ageing And Health. Geneva, Switzerland: World Health Organization; 2015. http://www.who.int/ageing/events/world-report-2015-launch/en/ Accessed February 15, 2018.
- 2. Bloom DE, Boersch-Supan A, McGee P, Seike A. Population aging: facts, challenges, and responses. Program on the Global Demography of Aging. No. 71. Harvard University Website. https://cdn1.sph.harvard.edu/wp-content/uploads/sites/1288/2013/10/PGDA_WP_71.pdf Accessed February 15, 2018.
- 3.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newgard CB, Sharpless NE. Coming of age: molecular drivers of aging and therapeutic opportunities. J Clin Invest. 2013;123(3):946–950. doi: 10.1172/JCI68833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8(21):2540–2551. doi: 10.1101/gad.8.21.2540. [DOI] [PubMed] [Google Scholar]
- 6.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14(4):501–513. doi: 10.1016/S1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 7.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 8.Chuprin A, et al. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013;27(21):2356–2366. doi: 10.1101/gad.227512.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burton DG, Krizhanovsky V. Physiological and pathological consequences of cellular senescence. Cell Mol Life Sci. 2014;71(22):4373–4386. doi: 10.1007/s00018-014-1691-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 11.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demaria M, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31(6):722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krizhanovsky V, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braig M, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436(7051):660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michaloglou C, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436(7051):720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 17.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 18.van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509(7501):439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coppé JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sagiv A, Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. 2013;14(6):617–628. doi: 10.1007/s10522-013-9473-0. [DOI] [PubMed] [Google Scholar]
- 21.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soriani A, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113(15):3503–3511. doi: 10.1182/blood-2008-08-173914. [DOI] [PubMed] [Google Scholar]
- 23.Kang TW, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 24.Tasdemir N, et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 2016;6(6):612–629. doi: 10.1158/2159-8290.CD-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eggert T, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30(4):533–547. doi: 10.1016/j.ccell.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimri GP, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ressler S, et al. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5(5):379–389. doi: 10.1111/j.1474-9726.2006.00231.x. [DOI] [PubMed] [Google Scholar]
- 28.Franceschi C, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 29.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354(6311):472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schafer MJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farr JN, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. 2017;23(9):1072–1079. doi: 10.1038/nm.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogrodnik M, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017;8:15691. doi: 10.1038/ncomms15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeon OH, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23(6):775–781. doi: 10.1038/nm.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roos CM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973–977. doi: 10.1111/acel.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baker DJ, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ovadya Y, Krizhanovsky V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology. 2014;15(6):627–642. doi: 10.1007/s10522-014-9529-9. [DOI] [PubMed] [Google Scholar]
- 39.Vécsei L. [Nature Reviews Drug Discovery: editorial article of neuroscientists from Szeged about kynurenine (IF: 33.078)] (article in Hungarian) Ideggyogy Sz. 2014;67(1–2):70. [PubMed] [Google Scholar]
- 40.Wang E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995;55(11):2284–2292. [PubMed] [Google Scholar]
- 41.Sagiv A, Biran A, Yon M, Simon J, Lowe SW, Krizhanovsky V. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene. 2013;32(15):1971–1977. doi: 10.1038/onc.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sasaki M, Kumazaki T, Takano H, Nishiyama M, Mitsui Y. Senescent cells are resistant to death despite low Bcl-2 level. Mech Ageing Dev. 2001;122(15):1695–1706. doi: 10.1016/S0047-6374(01)00281-0. [DOI] [PubMed] [Google Scholar]
- 43.Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. 2017;21:21–28. doi: 10.1016/j.ebiom.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuhrmann-Stroissnigg H, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017;8(1):422. doi: 10.1038/s41467-017-00314-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Y, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15(3):428–435. doi: 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22(1):78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Y, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644–658. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yosef R, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016;7:11190. doi: 10.1038/ncomms11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yosef R, et al. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017;36(15):2280–2295. doi: 10.15252/embj.201695553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 51.Levine B, Sinha SC, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4(5):600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 53.Mérino D, et al. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood. 2012;119(24):5807–5816. doi: 10.1182/blood-2011-12-400929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tse C, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 55.Cang S, Iragavarapu C, Savooji J, Song Y, Liu D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J Hematol Oncol. 2015;8:129. doi: 10.1186/s13045-015-0224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu Y, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XLinhibitors, A1331852 and A1155463. Aging (Albany NY) 2017;9(3):955–963. doi: 10.18632/aging.101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moncsek A, et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2–/–) mice. Hepatology. 2017;67(1):247–259. doi: 10.1002/hep.29464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lehmann M, et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J. 2017;50(2):1602367. doi: 10.1183/13993003.02367-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y, et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY) 2016;8(11):2915–2926. doi: 10.18632/aging.101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baar MP, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017;169(1):132–147.e16. doi: 10.1016/j.cell.2017.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170(6):1062–1078. doi: 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196(1):33–39. doi: 10.1016/0014-4827(91)90453-2. [DOI] [PubMed] [Google Scholar]
- 63.Arce VM, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444(7119):638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 64.Papismadov N, Gal H, Krizhanovsky V. The anti-aging promise of p21. Cell Cycle. 2017;16(21):1997–1998. doi: 10.1080/15384101.2017.1377500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karkoulis PK, Stravopodis DJ, Konstantakou EG, Voutsinas GE. Targeted inhibition of heat shock protein 90 disrupts multiple oncogenic signaling pathways, thus inducing cell cycle arrest and programmed cell death in human urinary bladder cancer cell lines. Cancer Cell Int. 2013;13(1):11. doi: 10.1186/1475-2867-13-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97(20):10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Astle MV, et al. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene. 2012;31(15):1949–1962. doi: 10.1038/onc.2011.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deschênes-Simard X, et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013;27(8):900–915. doi: 10.1101/gad.203984.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Agostini A, et al. Targeted cargo delivery in senescent cells using capped mesoporous silica nanoparticles. Angew Chem Int Ed Engl. 2012;51(42):10556–10560. doi: 10.1002/anie.201204663. [DOI] [PubMed] [Google Scholar]
- 70.Hall BM, et al. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 2016;8(7):1294–1315. doi: 10.18632/aging.100991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thapa RK, et al. Progressive slowdown/prevention of cellular senescence by CD9-targeted delivery of rapamycin using lactose-wrapped calcium carbonate nanoparticles. Sci Rep. 2017;7:43299. doi: 10.1038/srep43299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sagiv A, et al. NKG2D ligands mediate immunosurveillance of senescent cells. Aging (Albany NY) 2016;8(2):328–344. doi: 10.18632/aging.100897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Biran A, et al. Senescent cells communicate via intercellular protein transfer. Genes Dev. 2015;29(8):791–802. doi: 10.1101/gad.259341.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Baruch K, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat Med. 2016;22(2):135–137. doi: 10.1038/nm.4022. [DOI] [PubMed] [Google Scholar]
- 75.Grupp SA, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frescas D, et al. Senescent cells expose and secrete an oxidized form of membrane-bound vimentin as revealed by a natural polyreactive antibody. Proc Natl Acad Sci U S A. 2017;114(9):E1668–E1677. doi: 10.1073/pnas.1614661114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim KM, et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 2017;31(15):1529–1534. doi: 10.1101/gad.302570.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Acosta JC, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 79.Coppé JP, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One. 2010;5(2):e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kuilman T, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 81.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP) Cell Signal. 2012;24(4):835–845. doi: 10.1016/j.cellsig.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 83.Chien Y, et al. Control of the senescence-associated secretory phenotype by NF-ĸB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25(20):2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16(5):238–246. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106(40):17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Laberge RM, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049–1061. doi: 10.1038/ncb3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moiseeva O, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell. 2013;12(3):489–498. doi: 10.1111/acel.12075. [DOI] [PubMed] [Google Scholar]
- 88.Martin-Montalvo A, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Campbell JM, Bellman SM, Stephenson MD, Lisy K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: a systematic review and meta-analysis. Ageing Res Rev. 2017;40:31–44. doi: 10.1016/j.arr.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 90.Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a tool to target aging. Cell Metab. 2016;23(6):1060–1065. doi: 10.1016/j.cmet.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Niehof M, et al. Interleukin-6-induced tethering of STAT3 to the LAP/C/EBPβ promoter suggests a new mechanism of transcriptional regulation by STAT3. J Biol Chem. 2001;276(12):9016–9027. doi: 10.1074/jbc.M009284200. [DOI] [PubMed] [Google Scholar]
- 92.Xu M, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A. 2015;112(46):E6301–E6310. doi: 10.1073/pnas.1515386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017;168(1–2):37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Capell BC, et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 2016;30(3):321–336. doi: 10.1101/gad.271882.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aird KM, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol. 2016;215(3):325–334. doi: 10.1083/jcb.201608026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Karkera J, et al. The anti-interleukin-6 antibody siltuximab down-regulates genes implicated in tumorigenesis in prostate cancer patients from a phase I study. Prostate. 2011;71(13):1455–1465. doi: 10.1002/pros.21362. [DOI] [PubMed] [Google Scholar]
- 98.van Rhee F, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966–974. doi: 10.1016/S1470-2045(14)70319-5. [DOI] [PubMed] [Google Scholar]
- 99.Emery P, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67(11):1516–1523. doi: 10.1136/ard.2008.092932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Storer M, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155(5):1119–1130. doi: 10.1016/j.cell.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 101.Muñoz-Espín D, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155(5):1104–1118. doi: 10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 102.Lujambio A, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153(2):449–460. doi: 10.1016/j.cell.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mosteiro L, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016;354(6315):aaf4445. doi: 10.1126/science.aaf4445. [DOI] [PubMed] [Google Scholar]
- 104.Ritschka B, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31(2):172–183. doi: 10.1101/gad.290635.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Helman A, et al. p16Ink4a-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med. 2016;22(4):412. doi: 10.1038/nm.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Biran A, et al. Quantitative identification of senescent cells in aging and disease. Aging Cell. 2017;16(4):661–671. doi: 10.1111/acel.12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4(12):1798–1806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]