

Abstract
Coupling is the process that links bone resorption to bone formation in a temporally and spatially coordinated manner within the remodeling cycle. Several lines of evidence point to the critical roles of osteoclast-derived coupling factors in the regulation of osteoblast performance. Here, we used a fractionated secretomic approach and identified the axon-guidance molecule SLIT3 as a clastokine that stimulated osteoblast migration and proliferation by activating β-catenin. SLIT3 also inhibited bone resorption by suppressing osteoclast differentiation in an autocrine manner. Mice deficient in Slit3 or its receptor, Robo1, exhibited osteopenic phenotypes due to a decrease in bone formation and increase in bone resorption. Mice lacking Slit3 specifically in osteoclasts had low bone mass, whereas mice with either neuron-specific Slit3 deletion or osteoblast-specific Slit3 deletion had normal bone mass, thereby indicating the importance of SLIT3 as a local determinant of bone metabolism. In postmenopausal women, higher circulating SLIT3 levels were associated with increased bone mass. Notably, injection of a truncated recombinant SLIT3 markedly rescued bone loss after an ovariectomy. Thus, these results indicate that SLIT3 plays an osteoprotective role by synchronously stimulating bone formation and inhibiting bone resorption, making it a potential therapeutic target for metabolic bone diseases.
Keywords: Bone Biology
Keywords: Coupling factors
Introduction
Bone is a highly dynamic tissue that constantly undergoes changes and regeneration throughout adult life through a process called remodeling. This consists of 3 consecutive phases: the initiation of bone resorption by osteoclasts; the transition from catabolism to anabolism; and then bone formation by osteoblasts, followed by termination (1). Each of these phases is finely controlled by humoral factors or molecules mediating the communication among bone cells to maintain skeletal integrity (2). As an imbalance of bone remodeling leads to metabolic bone disorders such as osteoporosis (3), a better understanding of the molecular mechanisms regulating its various phases is crucial to the development of better approaches to the prevention and treatment of metabolic bone diseases.
Coupling is the process that links bone resorption to formation in a temporally and spatially coordinated manner within the remodeling cycle (4), and it has become clear that there are important coupling factors involved in preserving bone balance. For example, TGF-β and IGF-1, which are matrix-derived signals released during bone resorption, have been reported to contribute to coupling (5, 6). Interestingly, several lines of evidence, especially from human and mouse genetic studies, point to the critical roles of osteoclast-derived factors in the regulation of osteoblast performance (7). Patients with osteoclast-rich osteopetrosis caused by mutations in osteoclast a3-V-ATPase or chloride channel ClC-7 have unaltered or even increased bone formation despite reduced bone resorption (8), supporting the importance of osteoclasts themselves in the coupling process. In fact, several such “osteoclast-derived coupling factors” have been identified (9), and some were validated by studies using genetically altered mice, including cardiotrophin-1 (CT-1) (10), sphingosine-1-phosphoate (S1P) (11), WNT10B, BMP6 (12), collagen triple helix repeat containing 1 (CTHRC1) (13), and complement factor 3a (C3a) (14). However, since coupling is not as simple as can be explained by one key signal, there is still much to be learned regarding the crosstalk between bone cells.
As one of the efforts to further understand how osteoclasts talk to osteoblasts, we previously performed an in vitro study to investigate the coupling phenomenon, finding that the conditioned media (CM) of mature osteoclasts generated from both primary mouse bone marrow macrophages (BMMs) and RAW264.7 cells stimulated the migration of osteoblast lineages (15). By using fractionated secretomics to purify specific factors, we identified one of these clastokines as the axon-guidance molecule SLIT3. In the present study, we provide experimental evidence, including results from tissue-specific genetic deletion analyses and truncated recombinant protein therapy, that SLIT3 plays an osteoprotective role by synchronously inhibiting bone resorption and stimulating bone formation and is thus a therapeutic target for metabolic bone diseases.
Results
Increased SLIT3 production during osteoclast differentiation.
To identify factors playing a role in the bone remodeling process, we used a fractionated secretomic strategy (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI91086DS1) (15). Briefly, the CM from differentiated osteoclast-like cells and their undifferentiated cells (using RAW264.7 cells) were separated into 96 paired fractions according to molecular weight, and each fraction was tested for its ability to stimulate osteoblast migration. The paired fraction with the greatest difference in ability to stimulate osteoblast migration was selected, and the proteins in that fraction were identified through liquid chromatography–tandem mass spectrometry (LC-MS/MS) and use of the Mascot database (http://www.matrixscience.com/). We thereby identified 9 proteins with secretory features, i.e., afamin, ADAMTS-like protein 4, atrial natriuretic peptide, angiotensin-converting enzyme, SLIT3, laminin β2, Fras-1, collagen α-1 chain, and usherin (15). Among these factors, we focused specifically on SLIT3 because it is known to increase migration in various cell types (16, 17) and thus could potentially contribute to preosteoblast recruitment. Indeed, SLIT3 showed the most convincing results in our preliminary analyses. As shown in Supplemental Table 1, all 3 sequences identified by LC-MS/MS were unique to the SLIT3 protein and were not present in other SLIT family members, such as SLIT1 and SLIT2. The sequence similarity of mouse SLIT3 to SLIT1 and SLIT2 was 60% and 67%, respectively. An ELISA confirmed that the SLIT3 levels were significantly higher in CM obtained from mature mouse osteoclasts than in CM from preosteoclasts (Figure 1A). SLIT3 concentrations in mouse bone marrow fluid (76.77 ± 5.03 ng/ml) were markedly higher than in peripheral blood (2.95 ± 1.84 ng/ml) (Supplemental Figure 1B). SLIT3 was also found to be expressed in bone cells in vivo (Supplemental Figure 1, C and D), suggesting that this protein may play a critical role in the bone microenvironment.
Figure 1. Increased SLIT3 production during osteoclast differentiation.

(A) SLIT3 concentrations measured by ELISA in the CM from lineages of osteoclasts (OCs) and osteoblasts (OBs). The levels were normalized by the protein amount of each cell lysate. (B) Quantitative RT-PCR to measure Slit3 levels in RAW264.7 cells. Cells were treated with RANKL (15 ng/ml) for 4 days to induce osteoclast-like cells. Quantitative gene expression analysis was performed by RT-PCR using the LightCycler 480 system. 18S rRNA was used as an internal control. Ratios of Slit3 and 18S rRNA expression levels were calculated using the 2–ΔΔCT method. (C and D) Quantitative RT-PCR and luciferase assays of Slit3 expression and Slit3 promoter activity, respectively, after transfection with 50 ng cDNAs expressing Creb or NF-κB subunits p50 or p65 for 48 hours, in RAW264.7 cells. The values were normalized to the 18S rRNA level and β-galactosidase activity, respectively. (E and F) Quantitative RT-PCR and luciferase assays before and after pretreatment with inhibitors of NF-κB p50 and CREB (PDTC and KG501, respectively) in BMMs with M-CSF. (G and H) ChIP assay after IP with antibodies against NF-κB p50 and phosphorylated CREB in RAW264.7 cells to assess the activation of these factors at Slit3 promoter regions. RANKL (100 ng/ml) treatment duration was 1 hour. Data are presented as mean ± SEM of 3–4 independent experiments. *P < 0.05 vs. untreated or empty vector–transfected control using the Mann-Whitney U test or Kruskal-Wallis test followed by Bonferroni’s correction.
Western blot analysis revealed that SLIT3 expression was higher in the CM and cell lysates of osteoclasts differentiated by RANKL from primary mouse BMMs (Supplemental Figure 1E) and RAW264.7 cells (Supplemental Figure 1F) compared with CM and cell lysates from undifferentiated cells. In contrast, there was no significant difference found in the SLIT3 levels between the CM obtained from differentiated osteoblasts and that obtained from preosteoblasts (Figure 1A). Quantitative reverse-transcriptase PCR (RT-PCR) further revealed that Slit3 expression was significantly higher in mature osteoclasts than in preosteoclasts, whereas Slit2 expression was comparable (Supplemental Figure 1G). Slit1 was minimally expressed in both cell types.
The binding of RANKL to its receptor, RANK, is crucial for osteoclast differentiation. RANKL-RANK binding subsequently activates several signaling pathways, including those mediated by NF-κB, MAPK, and cAMP response element–binding protein (CREB). The promoter region of Slit3 contains multiple putative binding sites for NF-κB and CREB (Supplemental Figure 1H). Quantitative RT-PCR data indicated that treatment with RANKL increased Slit3 mRNA expression in RAW264.7 cells (Figure 1B). In addition, transfection of the NF-κB subunit p50 and Creb in RAW264.7 cells also increased Slit3 mRNA levels (Figure 1C) and Slit3 promoter activity (Figure 1D). Pretreatment with inhibitors of NF-κB p50 and CREB blocked the RANKL-induced increase in Slit3 mRNA expression and Slit3 promoter activity in RAW264.7 cells (Supplemental Figure 2, A and B, respectively) and primary mouse cells (Figure 1, E and F, respectively). ChIP analysis confirmed that the recruitment of NF-κB p50 and phosphorylated CREB to Slit3 promoter regions was induced by RANKL (Figure 1, G and H, respectively). These results suggest that the activation of NF-κB p50 and CREB mediates the increase in Slit3 expression during osteoclast differentiation.
SLIT3 stimulates the migration and proliferation of osteoblast lineages.
We assessed the effect of Slit3 silencing in mature osteoclasts upon osteoblast migration. When compared with CM from undifferentiated BMMs, CM from osteoclasts differentiated with RANKL increased the directional migration of murine preosteoblast MC3T3-E1 cells (Figure 2A). The silencing of Slit3 in differentiated osteoclasts abrogated the osteoclast-induced chemotaxis of preosteoblasts. When osteoblast lineages, such as calvaria osteoblasts, MC3T3-E1 cells, and human bone marrow stromal cells, were exposed to recombinant SLIT3 protein, the directional migration of these cells increased in a concentration-dependent manner (Figure 2B and Supplemental Figure 3, A and B, respectively). SLIT3 also stimulated the proliferation of mouse calvaria osteoblasts and human bone marrow stromal cells (Figure 2C and Supplemental Figure 3C, respectively).
Figure 2. SLIT3 stimulates osteoblast migration and proliferation.

(A) Quantitative RT-PCR and Western blot of SLIT3 after transfection with Slit3 siRNA for 24 hours in mouse mature osteoclasts. Mouse BMMs were differentiated into mature osteoclasts with 15 ng/ml RANKL and 15 ng/ml M-CSF for 3 days, and CM were collected during the subsequent 24 hours with or without Slit3 siRNA. Directional migration of MC3T3-E1 cells was assessed after treatment with the collected CM for 24 hours. (B and C) Directional migration (B) and proliferation (C) of mouse calvaria osteoblasts with SLIT3 for 24 hours and 48 hours, respectively. (D) Intrabone marrow mobilization of GFP-labeled MC3T3-E1 cells (n = 5 per group). (E) Western blot of β-catenin after IP with N-cadherin in mouse calvaria osteoblasts with 1.0 μg/ml SLIT3 for 60 minutes. The experiment was performed without WNTs. (F) TCF/LEF reporter assay with 1.0 μg/ml SLIT3 for 48 hours in MC3T3-E1 cells. (G) Quantitative RT-PCR and Western blot of β-catenin after transfection with β-catenin siRNA (Ctnnb1) for 24 hours in mouse calvaria osteoblasts. Directional migration and proliferation were assessed after treatment with 1.0 μg/ml SLIT3 for 24 hours and 48 hours, respectively. (H) Von Kossa staining of femur (upper) and lumbar spine (lower) of 7-week-old male Slit3–/– mice and WT littermates (n = 4–5 per group). Trabecular bone parameters in the femur were assessed by histomorphometric analyses. BV/TV, bone volume/tissue volume; Tb.Th, trabecular thickness; Tb.N, trabecular number; Tb.Sp, trabecular separation. Scale bars: 500 μm. Detailed information appears in the Supplemental Methods. Data are presented as mean ± SEM. In vitro experiments were performed 3–5 times independently. *P < 0.05 vs. untreated control or WT mice using the Mann-Whitney U test or Kruskal-Wallis test followed by Bonferroni’s correction.
To investigate whether SLIT3 stimulated preosteoblast migration in vivo, GFP-labeled MC3T3-E1 preosteoblasts with SLIT3 and PBS were injected into the right and left tibia, respectively. GFP-positive cells in the tibia injected with SLIT3 were found through the bone marrow cavity as the injected SLIT3 spread out. However, the cells injected with PBS were agglomerated and mainly found at the injected site, and thus they were scarce in the bone marrow cavity (Figure 2D and Supplemental Figure 3D). Collectively, these in vitro and in vivo results suggest that SLIT3 derived from mature osteoclasts stimulates the recruitment and proliferation of osteoblast lineages.
The binding of SLIT ligand to its receptor dissociates a multimolecular complex containing ROBO and N-cadherin and results in the release of cadherin-associated β-catenin (18). Activation of β-catenin (19, 20) and N-cadherin (21, 22) also plays an important role in osteoblast migration and proliferation. In our current experiments, SLIT3 treatment decreased the level of N-cadherin–associated β-catenin in mouse calvaria osteoblasts (Figure 2E), resulting in increased β-catenin activity (Figure 2F). The knockdown of β-catenin completely blocked SLIT3-stimulated osteoblast migration and proliferation (Figure 2G). These results suggest that the activation of β-catenin plays a crucial role in SLIT3-stimulated osteoblast migration and proliferation.
SLIT3 is essential for the regulation of bone remodeling.
Next, we assessed the effect of Slit3 deletion in vivo. Newborn Slit3–/– mice showed smaller skeletal sizes than their WT littermates (Supplemental Figure 4A). The histomorphometry of the femur revealed that Slit3–/– mice were severely osteopenic and had a significantly lower trabecular bone mass, trabecular thickness, and trabecular number and a higher trabecular separation than their WT littermates (Figure 2H). Histomorphometric analyses of the vertebrae produced similar results (data not shown). Micro-CT analyses of the femur also indicated that Slit3–/– mice had marked osteopenic phenotypes (Supplemental Figure 4B), which were consistent with the histomorphometric findings of the trabecular bone parameters (Supplemental Figure 4C). In addition, the thicknesses and diameters of cortical bones were lower in Slit3–/– mice than in WT (Supplemental Figure 4D).
In addition to its direct action on bone cells, it has been reported that SLIT3 functions as an angiogenic factor (23) and that type H endothelial cells, which are strongly positive for CD31 and endomucin, provide niche signals for perivascular osteoprogenitors in the bone microenvironment (24). The H type endothelium was found in our current analysis to be significantly reduced in Slit3–/– mice (Supplemental Figure 4E), suggesting that SLIT3 has direct effects not only on bone cells, but also on angiogenesis, which provides an osteoblast niche.
SLIT3 decreases bone resorption.
Dynamic histomorphometric analyses of the femur indicated that Slit3–/– mice exhibited markedly reduced bone formation rates, including osteoblast numbers, compared with WT mice (Figure 3A). Surprisingly, bone resorption parameters, such as the numbers of eroded surfaces and multinucleated osteoclasts, were significantly higher in Slit3–/– mice (Figure 3A). Compared with levels in WT, the serum level of bone-specific ALP, a bone formation marker, was lower in Slit3–/– mice, whereas the serum level of the C-terminal telopeptide of type I collagen (C-telopeptide), a bone resorption marker, was higher (Figure 3B). These data indicate that SLIT3 not only stimulates bone formation, but also suppresses bone resorption.
Figure 3. Regulation of bone resorption by SLIT3.

(A) Histomorphometric analyses including calcein double labeling of the femur of 7-week-old male Slit3–/– mice and WT littermates (n = 4–5 per group). BFR/BS, bone formation rate per bone surface; MAR, mineral apposition rate; N.Ob/BS, osteoblast number/bone surface; ES/BS, eroded surface/bone surface; OC/BS, multinucleated osteoclast number/bone surface. Scale bars: 10 μm. (B) Serum bone turnover markers in 7-week-old male Slit3–/– mice and WT littermates (n = 9–10 per group). (C) TRAP staining of mouse BMMs with 15 ng/ml M-CSF and 15 ng/ml RANKL for 4 days. (D) The same methods were performed in BMMs obtained from 6-week-old male or female Slit3–/– mice and WT littermates (n = 3 per group). (E) Semiquantitative RT-PCR of Trap, Ctr, and Dc-stamp in mouse BMMs with M-CSF and RANKL. (F) TRAP staining of mouse BMMs with M-CSF and RANKL for 2–3 days. The nuclei number per TRAP-positive cell was counted. (G) Intrabone marrow mobilization of red fluorescent protein–labeled (RFP-labeled) BMMs (n = 5 per group). (H) Western blot of RhoA, Rac, and Cdc42 following 1.0 μg/ml SLIT3 treatment for 15 minutes in mouse BMMs with M-CSF and RANKL. (I) Western blot of Rac GTPase after transfection with empty vector (pCMV5) or mutationally activated Rac1 (Rac1-V12) in mouse BMMs. TRAP staining was also performed at 4 days after transfection. (J) Directional migration of mouse BMMs with 1.0 μg/ml SLIT3 for 24 hours after transfection. Detailed information is supplied in the Supplemental Methods. Data are presented as mean ± SEM. In vitro experiments were performed 3–4 times independently. *P < 0.05 vs. untreated or empty vector–transfected control or between indicated groups using the Mann-Whitney U test or Kruskal-Wallis test followed by Bonferroni’s correction.
These unexpected findings led us to investigate the effect of exogenous SLIT3 protein on osteoclast differentiation. We found that, in the presence of RANKL, SLIT3 suppressed osteoclast formation in various types of osteoclast lineages, including primary mouse BMMs (Figure 3C), RAW264.7 cells (Supplemental Figure 5A), and human peripheral mononuclear cells (Supplemental Figure 5B). Consistently, the expression of osteoclast differentiation markers was significantly decreased by SLIT3 protein (Supplemental Figure 5C). To further confirm the autocrine role of SLIT3 on osteoclastogenesis, BMMs obtained from Slit3–/– mice and WT littermates were exposed to RANKL and macrophage CSF (M-CSF). Slit3–/– BMMs had a greater osteoclastogenic response than WT BMMs, and SLIT3 was found to suppress osteoclastogenesis even in Slit3–/– cells (Figure 3D). A coculture of osteoclasts and osteoblasts indicated that osteoclastogenesis was enhanced when Slit3 was deficient in either of these cell types (Supplemental Figure 5D). However, a Slit3 deficiency in both cell populations did not produce further enhancement of osteoclastogenesis. These results suggest that SLIT3 derived from any cells may be important in osteoclastogenesis, at least in an in vitro system.
Osteoclastogenesis proceeds through the proliferation of preosteoclasts, their fusion, and subsequent osteoclast differentiation. In our present analysis, RANKL exposure increased the expression of osteoclast differentiation markers, such as tartrate-resistant acid phosphatase (Trap) and calcitonin receptor (Ctr), in a time-dependent manner (Figure 3E). SLIT3 decreased the expression of these markers from the early stage of osteoclast differentiation (Figure 3E), and signaling pathways such as NF-κB, MAPK, and CREB were unaffected by SLIT3 (data not shown). Thus, we next investigated the effect of SLIT3 protein on the proliferation and fusion of preosteoclasts, finding that preosteoclast fusion decreased in the presence of SLIT3 (Figure 3F and Supplemental Figure 5E), but that preosteoclast proliferation was unaffected (Supplemental Figure 5F). Confirming this finding, SLIT3 exposure decreased the expression of dendritic cell–specific transmembrane protein (Dc-stamp), which plays an essential role in preosteoclast fusion (25) (Figure 3E). The suppressed fusion was more prominent at the lower than at the higher cell density (Supplemental Figure 5G), suggesting that the SLIT3-affected fusion may be mediated by impaired migration, at least in part. We further found in this regard that BMMs spread out less upon SLIT3 treatment in the bone marrow cavity (Figure 3G and Supplemental Figure 5H).
Small GTPases mediate osteoclastogenesis, including osteoclast fusion (26, 27), and the SLIT/ROBO system is one of their critical regulators (28). We thus assessed the effect of SLIT3 on the activity of small GTPases. SLIT3 suppressed Rac GTPase expression, but not that of RhoA or Cdc42 (Figure 3H). Since Rac1 has a more profound effect on osteoclastogenesis than Rac2 (27), we overexpressed Rac1 in mouse BMMs (Figure 3I). The overexpression of Rac1 almost completely blocked the SLIT3-mediated suppression of osteoclast differentiation (Figure 3I). Similarly, SLIT3 significantly decreased both preosteoclast migration on plastic discs (Figure 3J) and osteoclast migration on dentin discs (Supplemental Figure 5I), which was reversed by pretreatment with Rac1 overexpression. Although we found in our analysis that SLIT3 activated β-catenin in BMMs (Supplemental Figure 5J), the inhibition of β-catenin did not reverse SLIT3-suppressed osteoclastogenesis (Supplemental Figure 5, K and L), indicating that SLIT3-stimulated β-catenin is not a critical mediator of SLIT3-suppressed osteoclastogenesis. We further observed that SLIT3 did not affect actin ring formation (Supplemental Figure 5M). Collectively, these results suggest that osteoclast-derived SLIT3 inhibits bone resorption by decreasing osteoclast differentiation in an autocrine manner.
Robo1 deletion produces osteopenic phenotypes.
The SLIT receptor, ROBO, has 4 members: ROBO1-4 (29). ROBO1 and ROBO2 were found in our present experiments to be predominantly expressed in mouse calvaria osteoblasts (Figure 4A and Supplemental Figure 6A). A binding ELISA assay indicated that the amount of SLIT3-associated ROBO1 and ROBO2 increased with an increase in the amount of osteoblast lysate (Supplemental Figure 6B), indicating that SLIT3 had directly associated with ROBO1 and ROBO2 receptors in osteoblasts. In support of these findings, a knockdown of Robo1 and Robo2 completely reversed the osteoblast migration stimulated by SLIT3 (Figure 4B).
Figure 4. Osteopenic phenotypes in Robo1–/– mice.

(A) Expression of Robo mRNA in mouse calvaria osteoblasts using semiquantitative RT-PCR. Mouse brain and vascular tissues were used as positive controls (+) for Robo1–3 and Robo4, respectively. (B) Semiquantitative RT-PCR and Western blotting analysis of ROBO1 and ROBO2 after transfection with siRNA for 24 hours in mouse calvaria osteoblasts. The SLIT3-stimulated (1.0 μg/ml) directional migration of mouse calvaria osteoblasts with or without Robo1 siRNA or Robo2 siRNA was assessed using a Boyden chamber system. SLIT3 treatment was for 24 hours, after which the invaded cell numbers were counted. (C) Analysis of Robo mRNA levels in mouse BMMs using semiquantitative RT-PCR. (D) Semiquantitative RT-PCR and Western blotting of ROBO1 and ROBO3 after transfection with siRNA for 24 hours in mouse BMMs. The SLIT3-mediated (1.0 μg/ml) suppression of osteoclastogenesis with or without Robo1 siRNA or Robo3 siRNA was assessed. SLIT3 treatment was for 4 days with 15 ng/ml M-CSF and 15 ng/ml RANKL, and TRAP-positive cells with more than 3 nuclei were counted. (E) Histomorphometric analyses of the femurs of 25-week-old male Robo1–/– mice and WT littermates (n = 4–5 per group). Data are presented as mean ± SEM. In vitro experiments were performed 3–4 times independently. *P < 0.05 vs. untreated control or WT mice using the Mann-Whitney U test or Kruskal-Wallis test followed by Bonferroni’s correction.
In contrast, ROBO1 and ROBO3 were found to be predominantly expressed in primary mouse BMMs (Figure 4C and Supplemental Figure 6A). A binding ELISA experiment showed that ROBO1 and ROBO3 directly associated with SLIT3 (Supplemental Figure 6C), and a knockdown of these 2 molecules by siRNA blocked the SLIT3-mediated suppression of preosteoclast migration and osteoclastogenesis (Supplemental Figure 6D and Figure 4D, respectively). Taken together, these results suggest that the major receptors of SLIT3 are ROBO1 and ROBO2 in osteoblasts and ROBO1 and ROBO3 in osteoclasts.
We were unable to directly assess the effects of a Robo2 or Robo3 deletion in mice due to embryonic lethality (30, 31), and we therefore assessed the bone phenotypes of Robo1–/– mice (Supplemental Figure 6E). Robo1–/– mice showed a significantly reduced trabecular bone mass, trabecular thickness, and trabecular number and a higher trabecular separation than their WT littermates (Figure 4E). The bone formation rate was lower and bone resorption parameters were higher in Robo1–/– mice than in WT. The reduced bone mass in Robo1–/– mice was not as severe as that in Slit3–/– mice, suggesting that the other isotypes of ROBO may have functional redundancy in the SLIT3 pathway in bone cells. Furthermore, we cannot exclude the possibility that the bone phenotypes in Robo1–/– mice may be caused by the lack of a response to other SLIT members, such as SLIT2, which can bind to ROBO1 (32).
The importance of SLIT3 as a local determinant in bone metabolism.
Slit3 was found to be expressed in various organs, including bone and brain (Supplemental Figure 7A), and many lines of evidence have indicated that bone remodeling could be regulated by the nervous system (33, 34). Thus, we investigated whether neuron-derived SLIT3 may affect bone metabolism by deleting Slit3 in neurons. For these experiments, floxed Slit3 mice were generated (Supplemental Figure 7B) and then mated with transgenic mice expressing the nestin promoter (nestin-cre) (35), which can also target mesenchymal stem cells (MSCs) (36) to obtain conditional knockout mice (Slit3nestin–/–). Slit3nestin–/– mice (Supplemental Figure 7C) developed normally without any growth retardation or perinatal lethality. Micro-CT analyses of the femur indicated that Slit3nestin–/– mice had bone mass and trabecular bone parameters similar to those of their Slit3fl/fl littermates (Figure 5A). To dissect the relative role of SLIT3 derived from osteoblasts, we generated osteoblast-specific Slit3-deficient mice using a 2.3 kb promoter of 1(I) collagen-cre mice (37) (Supplemental Figure 7D). However, the bone phenotypes of the resulting Slit3col2.3–/– mice were not significantly different from those of their Slit3fl/fl littermates (Figure 5B). Next, to demonstrate the critical role of osteoclast-derived SLIT3 on bone mass in vivo, we generated osteoclast-specific Slit3-deficient mice using cathepsin K–cre mice (38). The Slit3 expression levels were specifically reduced in bones and osteoclasts of Slit3ctsk–/– mice (Supplemental Figure 8, A and B), and the cathepsin K–cre mice showed no changes in bone mass (Supplemental Figure 8C). Slit3ctsk–/– mice also developed normally without any gross abnormalities. Micro-CT (Figure 5C) and histological analyses (Figure 5D) of the femurs of Slit3ctsk–/– mice revealed markedly osteopenic phenotypes due to lower bone formation and higher bone resorption than the controls. Taken together, these results indicate that the lack of SLIT3 in osteoclasts, rather than in neurons, MSCs, and osteoblasts, could be the main cause of the bone abnormalities in Slit3–/– mice.
Figure 5. Osteoclast-derived SLIT3 is indispensable for normal bone mass in vivo.

(A) Micro-CT analyses of the femurs of 16-week-old male Slit3nestin–/– mice and their Slit3fl/fl littermates (n = 4–5 per group). (B) Micro-CT analyses of the femurs of 16-week-old male Slit3col2.3–/– mice and their Slit3fl/fl littermates (n = 4–6 per group). (C) Micro-CT analyses of the femurs of 16-week-old male Slit3ctsk–/– mice and their Slit3fl/fl littermates (n = 4–5 per group). (D) Von Kossa staining and histomorphometric analyses including calcein double-labeling of the femurs of 16-week-old male Slit3ctsk–/– mice and their Slit3fl/fl littermates (n = 4–5 per group). Scale bars: 500 μm (left panels); 10 μm (right panels). Data are presented as mean ± SEM. *P < 0.05 vs. littermate control using the Mann-Whitney U test.
SLIT3 is a therapeutic target for osteoporosis in humans.
To investigate the possible role of the SLIT/ROBO system in humans, we performed targeted deep sequencing of 7 genes, SLIT1–3 and ROBO1–4, in a cohort of postmenopausal women. The genetic study was performed in 2 stages. In the discovery stage, the 7 genes were sequenced in 501 control and 481 osteoporotic women (Supplemental Table 2). Two putative functional variants of SLIT2 and SLIT3 were found to be associated with the prevalence of osteoporosis (Supplemental Table 3) and were selected for further analysis in 3,895 additional participants. Only one genetic variant of SLIT3, but not of SLIT2, was found to be significantly associated with lumbar spine bone mineral density (BMD) (Table 1).
Table 1. Common functioning variants associated with BMD in postmenopausal women.

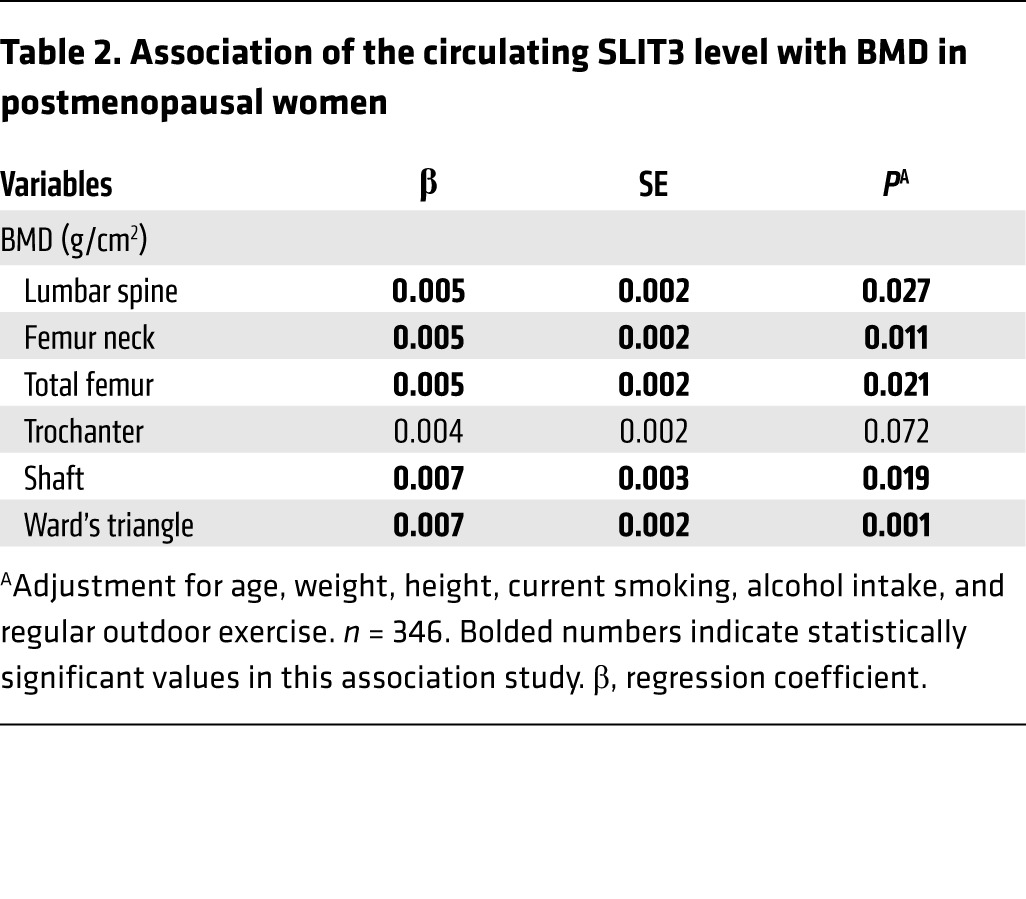
The plasma SLIT3 concentration was measured in a further 346 postmenopausal women, and the range was found to be 1.11 to 14.13 ng/ml (Supplemental Table 4). After adjustment for potential confounders, higher circulating SLIT3 levels were associated with higher BMD values at the lumbar spine and all measured proximal femur sites, except the trochanter, showing marginal significance (Table 2).
Table 2. Association of the circulating SLIT3 level with BMD in postmenopausal women.
SLIT proteins possess 4 leucine-rich repeat domains (LRRDs), and the second LRRD (LRRD2) of SLIT3 binds to its ROBO receptor (39–41). The sequence similarity of mouse and human SLIT3 LRRD2 is 98%. Human recombinant SLIT3 LRRD2, composed of 130 amino acids (Supplemental Table 5), stimulated the migration and proliferation of osteoblasts (Figure 6, A and B, respectively) to the same extent as full-length SLIT3 used at the same molar concentration. Similarly to its full length counterpart, LRRD2 also reduced N-cadherin–associated β-catenin (Supplemental Figure 9A). We found that LRRD2 increased N-cadherin–associated cables and β-catenin phosphorylated on Y489 in a sequential manner. The increase of β-catenin phosphorylated on Y489 was followed proportionally by the decrease in the association of total β-catenin and N-cadherin (Supplemental Figure 9A). This suggests that, when LRRD2 binds with ROBO, cables may be recruited and may phosphorylate β-catenin at Y489. The phosphorylated β-catenin showed a decreased affinity for N-cadherin, resulting in the release of β-catenin from N-cadherin and increased β-catenin activity (Supplemental Figure 9B).
Figure 6. Effect of LRRD2 of human SLIT3 on bone mass in OVX mice.

(A) Directional migration of mouse calvaria osteoblasts upon treatment with the same molar concentration of SLIT3 (1.0 μg/ml = 10 nM) and LRRD2 for 24 hours using a Boyden chamber system. The invaded cell numbers were counted. (B) Proliferation of mouse calvaria osteoblasts in the presence of SLIT3 or LRRD2 for 48 hours assessed using a BrdU incorporation assay. (C) TRAP staining of mouse BMMs exposed to 15 ng/ml M-CSF and 15 ng/ml RANKL in the presence of SLIT3 or LRRD2 for 4 days. TRAP-positive cells with more than 3 nuclei were counted. (D) Von Kossa staining and histomorphometric analyses including calcein double-labeling of the femur of sham-operated, OVX, and LRRD2-treated OVX mice (n = 7 per group). The female C57BL/6J mice were OVX at 8 weeks of age, and 2 μg LRRD2 was injected via the tail vein twice a day (mean 0.192 mg/kg/day) from 12 weeks of age for 4 weeks. The same volume of saline was injected in the other groups. Mice were then sacrificed for analyses at 16 weeks of age. Scale bars: 500 μm (left panels); 10 μm (right panels). Data are presented as mean ± SEM. In vitro experiments were performed 3 times independently. *P < 0.05 vs. untreated control or between the indicated groups using the Mann-Whitney U test or Kruskal-Wallis test followed by Bonferroni’s correction. †P < 0.05 vs. 5 nM-treated group using Mann-Whitney U test.
LRRD2 was further found to directly associate with ROBO1 and ROBO2 receptors in osteoblasts (Supplemental Figure 9C). The knockdown of Robo1 and Robo2 reversed the osteoblast migration stimulated by LRRD2 (Supplemental Figure 9D), and LRRD2-stimulated migration and proliferation were not noted in osteoblasts of Robo1–/– mice (Supplemental Figure 9E). The activation of β-catenin is known to stimulate osteoblastic differentiation, and we observed that LRRD2 increased the Alp and osteocalcin mRNA levels in osteoblasts of Robo1+/+ mice, but not in those of Robo1–/– mice (Supplemental Figure 9E). In addition, LRRD2 suppressed osteoclastogenesis to the same extent as full-length SLIT3 (Figure 6C), decreased Rac GTPase expression (Supplemental Figure 9F), and directly associated with ROBO1 and ROBO3 receptors in BMMs (Supplemental Figure 9G). Gene silencing with Robo1 or Robo3 siRNAs blocked the LRRD2-mediated suppression of osteoclastogenesis (Supplemental Figure 9H). Taken together, these results indicate that LRRD2 can exert its actions through ROBO1 and ROBO2 in osteoblasts and through ROBO1 and ROBO3 in osteoclasts and thus elicit the same downstream signaling events as full-length SLIT3.
To examine the in vivo effects of human SLIT3 LRRD2, we first compared its efficacy depending on the treatment frequency. Specifically, although the total intravenously injected doses were the same during the entire experiment period, mice treated with 2 μg LRRD2 twice a day showed a higher bone volume than those treated with 4 μg LRRD2 once a day or 28 μg LRRD2 once a week (Supplemental Table 6). We thus investigated the effect of SLIT3 LRRD2 on existing bone loss in an ovariectomized (OVX) mouse model of postmenopausal osteoporosis using twice-daily 2 μg LRRD2 treatment. Injection of SLIT3 LRRD2 for 4 weeks significantly rescued bone loss after ovariectomy both by promoting osteoblastic bone formation and by inhibiting osteoclastic bone resorption (Figure 6D).
Discussion
As bone remodeling occurs in different parts of the skeleton asynchronously and at different times, locally generated and regulated activities compromise important control mechanisms. In the present study, we determined that osteoclast-derived SLIT3 stimulates the migration and proliferation of osteoblast lineages via the activation of β-catenin. In particular, the robust secretion of SLIT3 from mature osteoclasts, followed by a positive effect on osteoblasts, suggests that this protein could be a non–bone-derived coupling factor that mainly plays a role in the early stage of reversal phase during bone remodeling as a way to prepare for bone formation. Furthermore, SLIT3 suppresses osteoclastogenesis in an autocrine manner.
SLIT proteins were originally discovered as chemorepellents that controlled axon crossing in the midline of the brain (42), and there is evidence that several axon-guidance molecules could be involved in the intercellular crosstalk among bone cells. Specifically, the bidirectional interaction between ephrin-B2 on osteoclasts and its receptor, Eph-B4, on osteoblasts links bone resorption and formation through direct cell-cell contact (43). However, it seems unlikely that this is a major contributor to the coupling process because of the lack of any bone abnormality in mice lacking ephrin-B2 in myeloid cells (43) and the rarity of cell-cell contact between mature osteoclasts and osteoblasts. Semaphorin 3A (SEMA3A), another axon-guidance molecule, is abundantly expressed by osteoblast lineage cells, and a global knockout of the Sema3A gene results in a low bone mass due to decreased bone formation (44). However, an osteoblast-specific deletion had no such effect, implying that SEMA3A indirectly regulates bone remodeling, possibly by modulating sensory nerve development (34). In this context, to obtain definitive evidence that osteoclast-derived SLIT3 acts as a local determinant that regulates coupling in vivo, we analyzed osteoclast- and neuron-specific conditional Slit3-knockout mice. Consequently, mice lacking Slit3 in osteoclasts (Slit3ctsk–/–) had a markedly low bone mass, whereas neuron-specific Slit3-deficient (Slit3nestin–/–) mice had a bone mass similar to that of their controls, indicating that SLIT3, as a clastokine, directly acts on bone metabolism. In addition, Slit3col2.3–/– mice do not develop an abnormal bone phenotype, suggesting that the function of SLIT3 derived from MSCs and osteoblasts is likely to be dispensable because of relatively low expressions of SLIT3 in these cells.
Because of the importance of osteoclast lineage itself in bone remodeling, much research has been devoted to understanding how osteoclasts talk to osteoblasts. Through transcription profiling of osteoclasts and macrophages, 3 candidates with anabolic potential, BMP6, WNT10B, and S1P, were found to be preferentially expressed in osteoclasts (11, 12, 45). CT-1 has been also proposed as a clastokine that promotes osteoblast differentiation (10). Recently, CTHRC1 (13) and C3a (14) have been suggested as potential osteoclast-derived coupling factors that stimulate osteoblast differentiation and/or migration. All of these investigations have greatly contributed to elucidating the local factors involved in coupling signals between osteoclasts and osteoblasts (7). However, the question of whether these factors can really play an important role in the human system still remains. A strength of our current study in this regard is that we included clinical data related to SLIT3 in addition to in vitro and animal studies. When we tested 7 SLIT/ROBO-related genes in 4,877 postmenopausal women, only a genetic variant of SLIT3 was found to be associated with bone parameters. In addition, subjects with higher plasma SLIT3 levels had a higher bone mass at all measured skeletal sites, even after adjustment for confounders. Although the exact source of circulating SLIT3 is still unknown, its expression pattern in each organ (Supplemental Figure 7A) suggests that bone may be one of its major sources. These findings are consistent with the results from experimental studies showing the beneficial effects of SLIT3, suggesting that the circulating SLIT3 level could be a potential biomarker for predicting bone health in humans. Further longitudinal studies are necessary to confirm this possibility.
The distinguishing feature of our current analysis, as compared with other clastokine studies, is that we have demonstrated that SLIT3 can synchronously regulate both resorption and formation. Antiresorptive drugs such as bisphosphonates have been the primary therapy for osteoporosis for many decades. However, they concomitantly suppress bone formation due to coupling, leading to limited efficacy and long-term adverse events (46, 47). Hence, the development of therapeutic agents that can dissociate resorption from formation would be desirable. In our present analysis, SLIT3 exerted an osteoprotective effect by both inhibiting bone resorption and promoting bone formation and could thus be one of the potential dual-action candidates for the treatment of metabolic bone diseases.
Human SLIT3 has a mass of approximately 170 kDa and consists of 1,523 amino acids. This large size could be disadvantageous for drug development; shorter recombinant fragments might be more easily administered and their production might be more cost effective. To allay this concern, we made a truncated small recombinant fragment of human SLIT3 of 130 amino acids, which is an LRRD2. Consequently, it produced activity similar to that of the whole SLIT3 protein in bone cells and efficiently stimulated bone formation and reduced bone resorption, resulting in the rescue of bone loss in OVX mice. However, LRRD2 treatment of OVX mice did not result in a remarkable increase in bone mass despite its synchronous effect of stimulating bone formation and suppressing bone resorption, as shown in Figure 6D. In addition, the more frequent injection of LRRD2, despite the same total dosage during the entire experiment period, showed better treatment efficacy in mice, indicating that the half-life of LRRD2 in vivo is likely to be short. This is a critical barrier in terms of possible clinical applications, as frequent injections are not desirable when treating patients. Hence, the development of long-acting LRRD2 using drug modification technologies, such as albumin binding, could maximize the therapeutic effects of an LRRD2-based agent and make the use of SLIT3 as an auspicious therapeutic target for osteoporosis more likely.
In summary, the results of our current in vitro and animal experiments have demonstrated that the SLIT3 secreted by differentiated osteoclasts functions as a potent local determinant of bone mass that regulates both osteoclasts and osteoblasts. Furthermore, our additional clinical data suggest the possible important role of SLIT3 in human bone heath. The results of our current study may thus provide a molecular basis for the development of a therapeutic agent with combined antiresorptive and bone-forming activities.
Methods
Animal care.
Slit3–/– mice (030759-Mu) and WT littermates were purchased from the Mutant Mouse Regional Resource Center (MMRRC) at the University of Missouri (Columbia, Missouri, USA). The generation of Slit3–/– mice has been described previously (48). As 58% of Slit3–/– mice die by 150 days, mainly as a result of lung compression and intestinal obstruction (48), studies using Slit3–/– mice and WT littermates were performed in 7-week-old animals. Cryopreserved Robo1–/– spermatozoa (0304032-Mu) were purchased from MMRRC and mated to C57BL/6N females (Orient) to generate Robo1+/– mice. The resultant heterozygotes were crossed to generate homozygous Robo1–/– mice. The Robo1–/– animals were viable and fertile and appeared grossly normal. Robo1–/– mice have also been described previously (49) and have been genotyped by PCR. Cathepsin K–cre (MGI:3764465), ColaI 2.3-cre (MGI: 3835846), and nestin-cre (MGI: 2176173) mice were purchased from the Jackson Laboratory and were crossed with floxed Slit3 mice to generate conditional knockout mice. All animals were sacrificed by cardiac puncture under anesthesia with an intraperitoneal injection of 40 mg/kg Zoletil 50 (Virbac) and 5.6 mg/kg Rompun (Bayer Korea). No specific inclusion or exclusion criteria were used in our animal studies.
Generation of mice carrying floxed Slit3 allele.
The Slit3 targeting vector was prepared by recombineering as previously described (50). Briefly, 12 kb of genomic DNA spanning Slit3 introns 6 to 8 was retrieved from RP24-287J5 obtained from the BACPAC Resources Center at the Children’s Hospital Oakland Research Institute (Oakland, California, USA). The first loxP site was inserted into intron 7 approximately 500 bp 5′ of exon 8, and the second loxP site together with the Frt-PGKneo-Frt cassette was inserted in intron 8 approximately 600 bp 3′ of exon 8. The final targeting vector contained 6 kb and 4.2 kb of 5′ and 3′ arms, respectively. The vector was linearized by NotI digestion and then electroporated into ES cells derived from F1 (129Sv/C57BL6J) blastocysts. Drug-resistant clones were screened by nested long-range PCR using primer pairs outside the arms and specific to the loxP site. Targeted ES cell clones were used to generate chimeric animals by aggregating ES cells with morula-stage embryos, as shown in Supplemental Figure 7B. Chimeric males were bred with ROSA26-FLPe mice (stock no. 009086; Jackson Laboratory) to remove the PGKneo cassette to generate the final floxed F1 pups. These mice were then backcrossed to C57BL/6J mice (Jackson Laboratory) at least 6 times and bred to generate homozygous flox (Slit3fl/fl) mice.
Skeletal preparations and cartilage and bone staining in newborn mice.
Cartilage and bone in whole newborn mice were visualized after staining with Alcian blue and Alizarin red S (Sigma-Aldrich). Newborn mice were fixed in 95% ethanol for 1 week, and 0.01% Alcian blue solution (4 mg Alcian blue, dissolved in 1.6 ml H2O, 32 ml absolute ethanol, and 8 ml glacial acetic acid) was then added for 3 days. Embryos were washed in 95% ethanol for 1 day and then stained in 0.0005% Alizarin red S solution (0.5 mg Alizarin red S in 100 ml 1% KOH) for 3 days. Specimens were then washed in 1% KOH, rinsed twice in glycerol, and stored in 100% glycerol.
Histological and histomorphometric analysis.
Undecalcified femurs and vertebrae were embedded in destabilized methyl methacrylate and sectioned at 6 μm. Sections were stained using Villanueva and Von Kossa procedures (51). For the assessment of dynamic histomorphometric indices, mice were injected with calcein at a dose of 30 mg/kg of body weight at 6 days and at 2 days before sacrifice. Histomorphometric analysis was conducted using the semiautomatic image analyzing system (Histometry RT Digitizer; System Supply Ltd.) and the BIOQUANT program. Standard bone histomorphometrical nomenclatures, symbols, and units were used as described by the American Society for Bone and Mineral Research Histomorphometry Nomenclature Committee (52).
Micro-CT analysis of femur.
For 3D morphological and histomorphometric analysis, femurs were scanned using the Skyscan 1172 system at 50 kV/200 μA with 6.48 μm pixel size and 0.5 Al filters. Reconstructions were performed with NRecon (Skyscan). For analysis of trabecular bone, regions of interest (ROIs) of cancellous bone were created within the endosteal envelope on the 2D slices. The ROIs extended 3 mm from the growth plate of each femur to the proximal metaphysis, and 3D algorithms were used to determine the relevant parameters. For analysis of the cortical bone, operator-drawn ROIs were created within the diaphyseal segment, extending 0.5 mm in length from the proximal end. All morphometric parameters were determined using CTan (Skyscan). The coefficient of variation (CV) of bone volume/tissue volume was 4.9%.
Tail-vein injection of LRRD2 in OVX mice.
Female C57BL/6J (Orient) mice were bilaterally OVX at 8 weeks of age, and 2 μg LRRD2 or saline was injected via the tail vein 2 times per day from 12 weeks of age for 4 weeks (7 mice per group). Mice were sacrificed at 16 weeks of age by cardiac puncture, and the ovariectomy success was confirmed by the absence of ovaries and atrophy of uteri. All treatment groups were weight matched and randomized to treatment at the initiation of an experiment. The researcher conducting the treatment was not blinded to the experimental groups, but the researcher assessing bone parameters was blinded to the analyses.
Cloning and expression of the second LRRD of human SLIT3.
The human SLIT3 LRRD2 domain was amplified from a cDNA library of fibroblast-like synoviocytes of rheumatoid arthritis patients using the following primers: 5′-GGGCTCGAGGCCTGCCCCACCAAGTG-3′ (sense) and 5′-GGGCTCGAGGGCATCGTCGAAATACGC-3′ (antisense). The product was cloned into the pET22b(+) expression vector carrying a His-tag with NotI and NdeI. Expression of LRRD2 was induced by 0.5 mM IPTG in an E. coli recombinant protein expression system. E. coli extracts were obtained by centrifugation after sonication. For the purification of recombinant LRRD2 with the His-tag, metal affinity chromatography was used (QIAGEN) according to the manufacturer’s instructions. The purified protein was assessed with Coomassie Brilliant Blue staining after SDS-PAGE and quantified by Bradford assay.
Sequence analysis.
Sequence alignments were generated using mouse SLIT1, -2, and -3 protein sequences (AAD44758, AAD44759, and AAD44760, respectively) and human SLIT1, -2, and -3 protein sequences (NP_003052, AAD25539, and AAQ89243, respectively) deposited in GenBank.
Statistics.
All in vitro and animal data were expressed as the mean ± SEM of at least 3 independent experiments conducted with triplicate measurements, unless otherwise specified. The significance of differences between 2 groups was assessed using the Mann-Whitney U test, whereas differences among 3 or more groups were tested using the Kruskal-Wallis test followed by Bonferroni’s correction. For animal studies, no statistical method was used to predetermine sample size.
For the genetic study in postmenopausal women, genetic variants were tested for their association with osteoporosis-related phenotypes using multiple regression analysis of SNPs with BMD at the lumbar spine and femur neck. Age, weight, and height were used as covariates. Participants with and without osteoporosis were analyzed in terms of genotype using logistic regression with adjustments for confounding variables.
For the clinical study assessing plasma SLIT3 concentrations in postmenopausal women, continuous and categorical variables are reported as mean ± SD and percentages, respectively. Associations of plasma SLIT3 levels with BMD values were assessed by multiple linear regression analysis after adjustment for confounders such as age, weight, height, current smoking, alcohol use, and regular outdoor exercise.
All statistical analyses were performed using GraphPad PRISM and SPSS statistical software, and P values of less than 0.05 were considered statistically significant.
Study approval.
All in vivo experiments and protocols were approved by the Institutional Animal Care and Use Committee of the Asan Institute for Life Sciences. Human studies were approved by the local Institutional Review Board of Asan Medical Center. All subjects enrolled in this study provided written informed consent.
Author contributions
BJK, YSL, and JMK designed the study, analyzed the data, and wrote the paper. SYL, SWK, KUL, and GSK performed the in vitro experiments. WYB, YJC, SAM, JEK, and JMK performed the animal experiments. SHL, SHA, and HK performed the human studies. EJC and EYK purified recombinant LRRD2 of human SLIT3. JY and SWK investigated the expression mechanisms of SLIT3. SHR performed the LC-MS/MS analyses. SKL and JAL designed the study and generated the knockout mice. All of the authors discussed the results and approved the manuscript.
Supplementary Material
Acknowledgments
We express our sincere gratitude to Siu-Pok Yee (Gene Targeting Transgenic Facility in the University of Connecticut Health Center) for his valuable assistance with generating the floxed Slit3 mice. This study was supported by grants from the Korean Health Technology R&D Project, Ministry of Health & Welfare, South Korea (project nos. HI14C2258 and HI13C1634) and by a grant from the Asan Institute for Life Sciences (project no. 2015-347).
Version 1. 03/05/2018
Electronic publication
Version 2. 04/02/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(4):1429–1441.https://doi.org/10.1172/JCI91086.
See the related Commentary at Opening windows for bone remodeling through a SLIT.
Contributor Information
Beom-Jun Kim, Email: umkbj@naver.com.
Young-Sun Lee, Email: carrotlet77@hanmail.net.
Sun-Young Lee, Email: quore07@hanmail.net.
Wook-Young Baek, Email: arikato83@naver.com.
Young Jin Choi, Email: jin2627@gmail.com.
Sung Ah Moon, Email: moon9792@naver.com.
Seung Hun Lee, Email: hun0108@amc.seoul.kr.
Jung-Eun Kim, Email: kjeun@knu.ac.kr.
Eun-Young Kim, Email: ssainokim76@gmail.com.
Jin Yoon, Email: yoonjin123@nate.com.
Seung-Whan Kim, Email: swkim7@amc.seoul.kr.
Sung Ho Ryu, Email: sungho@postech.ac.kr.
Sun-Kyeong Lee, Email: klee@uchc.edu.
Joseph A. Lorenzo, Email: jlorenzo@nso2.uchc.edu.
Seong Hee Ahn, Email: shahn1017@gmail.com.
Hyeonmok Kim, Email: ragnastone@naver.com.
Ki-Up Lee, Email: kulee@amc.seoul.kr.
Ghi Su Kim, Email: gskim3@amc.seoul.kr.
Jung-Min Koh, Email: jmkoh99@daum.net.
References
- 1.Matsuo K, Irie N. Osteoclast-osteoblast communication. Arch Biochem Biophys. 2008;473(2):201–209. doi: 10.1016/j.abb.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 2.Negishi-Koga T, Takayanagi H. Bone cell communication factors and Semaphorins. Bonekey Rep. 2012;1:183. doi: 10.1038/bonekey.2012.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng X, McDonald JM. Disorders of bone remodeling. Annu Rev Pathol. 2011;6:121–145. doi: 10.1146/annurev-pathol-011110-130203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hattner R, Epker BN, Frost HM. Suggested sequential mode of control of changes in cell behaviour in adult bone remodelling. Nature. 1965;206(983):489–490. doi: 10.1038/206489a0. [DOI] [PubMed] [Google Scholar]
- 5.Tang Y, et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009;15(7):757–765. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xian L, et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med. 2012;18(7):1095–1101. doi: 10.1038/nm.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henriksen K, Karsdal MA, Martin TJ. Osteoclast-derived coupling factors in bone remodeling. Calcif Tissue Int. 2014;94(1):88–97. doi: 10.1007/s00223-013-9741-7. [DOI] [PubMed] [Google Scholar]
- 8.Segovia-Silvestre T, et al. Advances in osteoclast biology resulting from the study of osteopetrotic mutations. Hum Genet. 2009;124(6):561–577. doi: 10.1007/s00439-008-0583-8. [DOI] [PubMed] [Google Scholar]
- 9.Sims NA, Martin TJ. Coupling signals between the osteoclast and osteoblast: how are messages transmitted between these temporary visitors to the bone surface? Front Endocrinol (Lausanne) 2015;6:41. doi: 10.3389/fendo.2015.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker EC, et al. Cardiotrophin-1 is an osteoclast-derived stimulus of bone formation required for normal bone remodeling. J Bone Miner Res. 2008;23(12):2025–2032. doi: 10.1359/jbmr.080706. [DOI] [PubMed] [Google Scholar]
- 11.Ryu J, Kim HJ, Chang EJ, Huang H, Banno Y, Kim HH. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. 2006;25(24):5840–5851. doi: 10.1038/sj.emboj.7601430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci U S A. 2008;105(52):20764–20769. doi: 10.1073/pnas.0805133106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeshita S, et al. Osteoclast-secreted CTHRC1 in the coupling of bone resorption to formation. J Clin Invest. 2013;123(9):3914–3924. doi: 10.1172/JCI69493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuoka K, Park KA, Ito M, Ikeda K, Takeshita S. Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J Bone Miner Res. 2014;29(7):1522–1530. doi: 10.1002/jbmr.2187. [DOI] [PubMed] [Google Scholar]
- 15.Kim BJ, et al. Afamin secreted from nonresorbing osteoclasts acts as a chemokine for preosteoblasts via the Akt-signaling pathway. Bone. 2012;51(3):431–440. doi: 10.1016/j.bone.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Geutskens SB, Hordijk PL, van Hennik PB. The chemorepellent Slit3 promotes monocyte migration. J Immunol. 2010;185(12):7691–7698. doi: 10.4049/jimmunol.0903898. [DOI] [PubMed] [Google Scholar]
- 17.Kramer SG, Kidd T, Simpson JH, Goodman CS. Switching repulsion to attraction: changing responses to slit during transition in mesoderm migration. Science. 2001;292(5517):737–740. doi: 10.1126/science.1058766. [DOI] [PubMed] [Google Scholar]
- 18.Rhee J, Buchan T, Zukerberg L, Lilien J, Balsamo J. Cables links Robo-bound Abl kinase to N-cadherin-bound beta-catenin to mediate Slit-induced modulation of adhesion and transcription. Nat Cell Biol. 2007;9(8):883–892. doi: 10.1038/ncb1614. [DOI] [PubMed] [Google Scholar]
- 19.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116(5):1202–1209. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonnet N, Conway SJ, Ferrari SL. Regulation of beta catenin signaling and parathyroid hormone anabolic effects in bone by the matricellular protein periostin. Proc Natl Acad Sci U S A. 2012;109(37):15048–15053. doi: 10.1073/pnas.1203085109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kashima T, et al. Overexpression of cadherins suppresses pulmonary metastasis of osteosarcoma in vivo. Int J Cancer. 2003;104(2):147–154. doi: 10.1002/ijc.10931. [DOI] [PubMed] [Google Scholar]
- 22.Haÿ E, Buczkowski T, Marty C, Da Nascimento S, Sonnet P, Marie PJ. Peptide-based mediated disruption of N-cadherin-LRP5/6 interaction promotes Wnt signaling and bone formation. J Bone Miner Res. 2012;27(9):1852–1863. doi: 10.1002/jbmr.1656. [DOI] [PubMed] [Google Scholar]
- 23.Zhang B, Dietrich UM, Geng JG, Bicknell R, Esko JD, Wang L. Repulsive axon guidance molecule Slit3 is a novel angiogenic factor. Blood. 2009;114(19):4300–4309. doi: 10.1182/blood-2008-12-193326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507(7492):323–328. doi: 10.1038/nature13145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yagi M, et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202(3):345–351. doi: 10.1084/jem.20050645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito Y, et al. Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. J Clin Invest. 2010;120(6):1981–1993. doi: 10.1172/JCI39650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Lebowitz D, Sun C, Thang H, Grynpas MD, Glogauer M. Identifying the relative contributions of Rac1 and Rac2 to osteoclastogenesis. J Bone Miner Res. 2008;23(2):260–270. doi: 10.1359/jbmr.071013. [DOI] [PubMed] [Google Scholar]
- 28.Hu H, et al. Cross GTPase-activating protein (CrossGAP)/Vilse links the Roundabout receptor to Rac to regulate midline repulsion. Proc Natl Acad Sci U S A. 2005;102(12):4613–4618. doi: 10.1073/pnas.0409325102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stein E, Tessier-Lavigne M. Hierarchical organization of guidance receptors: silencing of netrin attraction by slit through a Robo/DCC receptor complex. Science. 2001;291(5510):1928–1938. doi: 10.1126/science.1058445. [DOI] [PubMed] [Google Scholar]
- 30.Grieshammer U, Le M, Plump AS, Wang F, Tessier-Lavigne M, Martin GR. SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev Cell. 2004;6(5):709–717. doi: 10.1016/S1534-5807(04)00108-X. [DOI] [PubMed] [Google Scholar]
- 31.Sabatier C, et al. The divergent Robo family protein rig-1/Robo3 is a negative regulator of slit responsiveness required for midline crossing by commissural axons. Cell. 2004;117(2):157–169. doi: 10.1016/S0092-8674(04)00303-4. [DOI] [PubMed] [Google Scholar]
- 32.Brose K, et al. Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell. 1999;96(6):795–806. doi: 10.1016/S0092-8674(00)80590-5. [DOI] [PubMed] [Google Scholar]
- 33.Takeda S. Central control of bone remodelling. J Neuroendocrinol. 2008;20(6):802–807. doi: 10.1111/j.1365-2826.2008.01732.x. [DOI] [PubMed] [Google Scholar]
- 34.Fukuda T, et al. Sema3A regulates bone-mass accrual through sensory innervations. Nature. 2013;497(7450):490–493. doi: 10.1038/nature12115. [DOI] [PubMed] [Google Scholar]
- 35.Okada S, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12(7):829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- 36.Méndez-Ferrer S, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dacquin R, Starbuck M, Schinke T, Karsenty G. Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 2002;224(2):245–251. doi: 10.1002/dvdy.10100. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura T, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130(5):811–823. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 39.Morlot C, Hemrika W, Romijn RA, Gros P, Cusack S, McCarthy AA. Production of Slit2 LRR domains in mammalian cells for structural studies and the structure of human Slit2 domain 3. Acta Crystallogr D Biol Crystallogr. 2007;63(Pt 9):961–968. doi: 10.1107/S0907444907035470. [DOI] [PubMed] [Google Scholar]
- 40.Chen JH, Wen L, Dupuis S, Wu JY, Rao Y. The N-terminal leucine-rich regions in Slit are sufficient to repel olfactory bulb axons and subventricular zone neurons. J Neurosci. 2001;21(5):1548–1556. doi: 10.1523/JNEUROSCI.21-05-01548.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howitt JA, Clout NJ, Hohenester E. Binding site for Robo receptors revealed by dissection of the leucine-rich repeat region of Slit. EMBO J. 2004;23(22):4406–4412. doi: 10.1038/sj.emboj.7600446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kidd T, Bland KS, Goodman CS. Slit is the midline repellent for the robo receptor in Drosophila. Cell. 1999;96(6):785–794. doi: 10.1016/S0092-8674(00)80589-9. [DOI] [PubMed] [Google Scholar]
- 43.Zhao C, et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006;4(2):111–121. doi: 10.1016/j.cmet.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 44.Hayashi M, Nakashima T, Taniguchi M, Kodama T, Kumanogoh A, Takayanagi H. Osteoprotection by semaphorin 3A. Nature. 2012;485(7396):69–74. doi: 10.1038/nature11000. [DOI] [PubMed] [Google Scholar]
- 45.Lotinun S, et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest. 2013;123(2):666–681. doi: 10.1172/JCI64840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reid IR. Osteonecrosis of the jaw: who gets it, and why? Bone. 2009;44(1):4–10. doi: 10.1016/j.bone.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 47.Chavassieux PM, Arlot ME, Reda C, Wei L, Yates AJ, Meunier PJ. Histomorphometric assessment of the long-term effects of alendronate on bone quality and remodeling in patients with osteoporosis. J Clin Invest. 1997;100(6):1475–1480. doi: 10.1172/JCI119668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan W, Rao Y, Babiuk RP, Greer JJ, Wu JY, Ornitz DM. A genetic model for a central (septum transversum) congenital diaphragmatic hernia in mice lacking Slit3. Proc Natl Acad Sci U S A. 2003;100(9):5217–5222. doi: 10.1073/pnas.0730709100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Long H, et al. Conserved roles for Slit and Robo proteins in midline commissural axon guidance. Neuron. 2004;42(2):213–223. doi: 10.1016/S0896-6273(04)00179-5. [DOI] [PubMed] [Google Scholar]
- 50.Lee EC, et al. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73(1):56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- 51.Sugimoto M, Futaki N, Harada M, Kaku S. Effects of combined treatment with eldecalcitol and alendronate on bone mass, mechanical properties, and bone histomorphometry in ovariectomized rats: a comparison with alfacalcidol and alendronate. Bone. 2013;52(1):181–188. doi: 10.1016/j.bone.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 52.Dempster DW, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28(1):2–17. doi: 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.