

Abstract
As oncogenes drive carcinogenesis and promote cancer cell survival, they are highly attractive therapeutic targets, and oncogene-targeting small molecules have achieved some clinical success. While many oncogenes are presently considered to be “druggable,” tumors often acquire treatment resistance, and patients are rarely cured in response to oncogene-specific treatment. In this issue of the JCI, Veatch and colleagues describe a patient with metastatic acral melanoma who experienced a complete tumor response following infusion of tumor-infiltrating T cells that targeted multiple tumor antigens, including a BRAFV600E driver mutation. T cells genetically engineered to express an anti-BRAFV600E T cell receptor (TCR) from the patient demonstrated recognition of an epitope that spanned the BRAFV600E mutation. These findings suggest that BRAFV600E might be targeted therapeutically with adoptive transfer of anti-BRAFV600E T cells. This research supports the emerging therapeutic paradigm of targeting oncogenic drivers with T cell immunotherapy.
The intersection of molecular therapy and immune therapy
Molecularly targeted therapy aims to inhibit or kill cancer cells by interfering with specific molecular pathways that promote tumor growth and survival. In some cases, this can be achieved by a small molecule drug. The most successful example of a therapeutic small molecule is imatinib, which targets the BCR-ABL fusion protein, the product of the Philadelphia translocation of chronic myelogenous leukemia (CML). In CML patients, response rates to imatinib are high, and prolonged suppression of disease is possible. There have been other major advances for targeted therapies, but the success achieved in CML has not been replicated at the same level in other types of cancer. The lack of efficacy appears to be related to the ability of tumors to overcome molecular pathway blockade by a panoply of mechanisms, including upregulation of other pathways and acquisition of resistance mutations in the target protein–encoding gene (1, 2). The small molecule approach has been further constrained by a dearth of drugs that safely target some of the most promising oncogenes, such as mutant KRAS.
An alternative strategy to targeting driver oncogenes may be to use T cell receptor (TCR) gene–engineered T cells (TCR-T cells) (3) (Figure 1). For this approach, T cells are genetically engineered ex vivo to express a TCR that recognizes the complex of peptide (in this case one unique to a driver oncoprotein) bound to a patient HLA molecule. These engineered T cells are then administered systemically to the patient. The intent is to harness the physiological function of T cells in the elimination of cells that harbor intracellular pathogens and direct it to the destruction of tumor cells that harbor intracellular driver oncoproteins. This strategy may overcome some of the inherent limitations of small molecule–based treatments. For example, direct killing of tumors by T cells may provide less opportunity for upregulation of compensatory pathways or acquisition of secondary resistance mutations than indirect killing by pathway inhibition. In addition, TCR-T cell therapy can be specifically directed against a target that is only present in tumor cells (i.e., a protein encoded by a mutant proto-oncogene, an oncogenic fusion protein breakpoint, or a viral oncoprotein). Because of this specificity, TCR-T cell therapy may result in less toxicity to healthy tissues than treatment with molecularly targeted small molecules. Finally, TCR-T cell therapy has the potential to target so-called undruggable oncogenes, as TCRs can, in theory, be generated against any protein.
Figure 1. TCRs that recognize tumor antigen epitopes that encompass oncoprotein mutations have potential for potent immunotherapy.
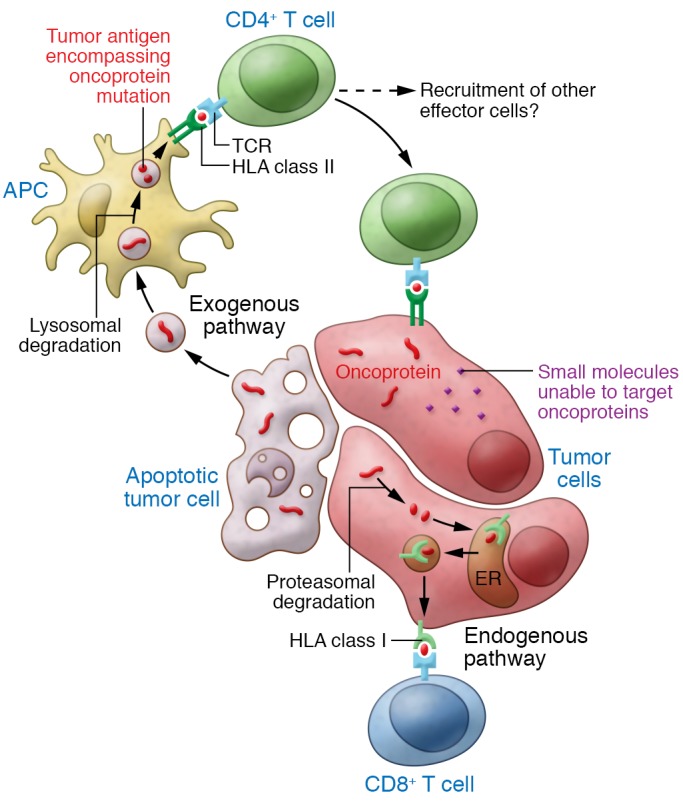
For many types of cancer, small molecule inhibitors to specific oncoproteins have not been identified or have limited efficacy due to off-target effects and acquisition of treatment resistance. In this issue, Veatch and colleagues characterized the T cell response in a patient who had a complete tumor response following infusion of tumor-infiltrating T cells that targeted multiple tumor antigens. TCRs from this patient recognized an epitope that encompassed the driver mutation within an oncoprotein. Targeting of oncoproteins with T cells, which kill tumor cells, rather than small molecules, which inhibit tumor cells, may permit the development of more effective treatments. Additionally, the highly specific oncoprotein targeting by TCRs may reduce the off-target toxicity of treatment.
Emerging evidence
Several lines of evidence suggest that oncogenic drivers, including some oncoproteins regarded as undruggable, might indeed be effectively targeted with TCR-T cell therapy. In one study, patients with metastatic cervical cancer received tumor-infiltrating lymphocytes (TILs) that possessed reactivity against HPV E6 and/or E7 oncoproteins. Two of 9 patients treated with HPV-targeting TCR-T cells experienced complete tumor responses (4); however, in this study, non-oncoprotein tumor antigens were also targeted, precluding definitive conclusions about which antigens were responsible for tumor regression (5). A recent clinical trial of TCR-T cells that target HPV-16 E6 has provided more direct evidence to suggest that an HPV oncoprotein can be targeted with TCR-T cells (6, 7). In this report, 2 of 12 patients with metastatic HPV+ cancers experienced tumor responses (2 of 9 patients treated at the highest dose of TCR-T cells). Further insight into the targeting of HPV oncoproteins with TCR-T cell therapy is expected from an ongoing clinical trial of T cells directed against HPV-16 E7 (https://clinicaltrials.gov/; NCT02858310) (8).
The potential of therapeutic T cells to target oncogenic drivers is also supported by a case report of a patient with metastatic colorectal cancer who experienced a partial response after treatment with TILs that predominately targeted mutant KRASG12D (9). The number of patients who underwent surgery for the generation of TIL cultures to achieve this single result was not reported, and the general applicability of the approach is uncertain. Nonetheless, this case supports the principle that adoptively transferred T cells can target oncoproteins that are not druggable with small molecules. A strategy for more consistent targeting of KRAS mutations, through TCR gene–engineered T cells directed against HLA-A*11:01-restricted epitopes of KRASG12V and KRASG12D, has also been described and may be more broadly applicable to other cancers (10).
In this issue, Veatch and colleagues have provided additional insight into the targeting of oncoproteins by therapeutic T cells (11). The authors describe a patient with metastatic acral melanoma who experienced a complete response to TIL therapy that targeted BRAFV600E and other tumor antigens. Interestingly, the patient had previously been treated with a checkpoint inhibitor that failed to induce complete tumor regression. The patient’s tumor harbored 29 nonsynonymous missense mutations, including BRAFV600E. Twenty mutations were selected (based on variant allele frequency and/or evidence of gene expression) and tested for recognition by TILs. Only the BRAFV600E mutation was recognized, and this recognition of was mediated by CD4+ T cells and restricted by the HLA-DQA1*03/DQB1*03 class II molecule, which is common in many populations (12). The sequences of the α- and β-chains of a BRAFV600E-targeted TCR from this patient were identified, and T cells that were genetically engineered to express this TCR targeted BRAFV600E in in vitro assays. These findings support BRAFV600E as a potential therapeutic target of T cells and demonstrate a possible first step in a strategy to target BRAFV600E with T cell therapy.
Challenges ahead
The concept of bringing T cell therapy to bear on the targeting of oncogenic pathways is appealing but has practical limitations. For example, each TCR-T cell treatment is restricted to a specific HLA molecule that presents the targeted oncoprotein epitope to the therapeutic T cells, thereby decreasing the number of patients who can be treated with any given TCR-T cell therapy. Additionally, application of this approach may be constrained to certain antigens, because some target epitopes may not bind well to HLA molecules with high allelic frequency. Another consequence of HLA restriction is that tumor loss of HLA expression might permit escape. HLA loss is a well-described phenomenon, but its frequency and impact on immunotherapy is increasingly appreciated. A recent study found HLA loss of heterozygosity (LOH) in 40% of non–small cell lung cancers and revealed genomic evidence that HLA LOH was linked to immune-driven selection pressures (13). HLA loss was also reported as the cause of tumor resistance in a patient with a partial response to a TIL infusion that targeted mutated KRAS (9). In addition to HLA loss, other mechanisms of tumor resistance to oncoprotein-targeted TCR-T cell therapy may be important in limiting efficacy. Intratumoral genomic heterogeneity may result in clones that do not express the targeted oncogene (14, 15). Mutations, deletions, and/or reduced expression of molecules involved in antigen processing and presentation by tumor cells may also contribute to resistance (16). Finally, some tumor cells may harbor mutations that blunt their sensitively to T cell effector molecules, such as IFN-γ (16–18).
Bringing the promise to patients
Personalized cancer therapy based on targeting the molecular drivers of an individual patient’s cancer are yielding new and effective treatments. TCR-T cell therapy adds further personalization to molecular-based strategies by defining a cancer by the oncogenic-driver and by the HLA type of the patient and by treating the cancer with a therapy derived from the patient’s own T cells. For this approach to be broadly applicable, a library of therapeutics that can be matched to a tumor antigen and to a patient HLA molecule may be needed. Discovery and development of treatment libraries may require new clinical trial and regulatory process paradigms. While much remains to be done, the work by Veatch and colleagues helps reveal a vision in which the strength of T cell therapy is combined with the precision targeting of oncogenic drivers for the benefit of patients with cancer.
Acknowledgments
The author thanks Stan Lipokitz for his thoughtful comments in manuscript preparation.
Version 1. 03/12/2018
Electronic publication
Version 2. 04/02/2018
Print issue publication
Footnotes
Conflict of interest: CSH is an inventor on NIH patents for cellular therapy, and his laboratory receives research funding through an NIH Cooperative Research and Development Agreement with Kite Pharma.
Reference information: J Clin Invest. 2018;128(4):1261–1263. https://doi.org/10.1172/JCI120386.
See the related article at Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma.
References
- 1.Ramos P, Bentires-Alj M. Mechanism-based cancer therapy: resistance to therapy, therapy for resistance. Oncogene. 2015;34(28):3617–3626. doi: 10.1038/onc.2014.314. [DOI] [PubMed] [Google Scholar]
- 2.De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12(6):594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 3.Hinrichs CS. Molecular pathways: breaking the epithelial cancer barrier for chimeric antigen receptor and T-cell receptor gene therapy. Clin Cancer Res. 2016;22(7):1559–1564. doi: 10.1158/1078-0432.CCR-15-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevanović S, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. 2015;33(14):1543–1550. doi: 10.1200/JCO.2014.58.9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevanović S, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. 2017;356(6334):200–205. doi: 10.1126/science.aak9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinrichs CS, et al. A phase I/II clinical trial of E6 T-cell receptor gene therapy for human papillomavirus (HPV)-associated epithelial cancers. J Clin Oncol. 2017;35(15 suppl):3009–3009. [Google Scholar]
- 7.Draper LM, et al. Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered T cells directed against E6. Clin Cancer Res. 2015;21(19):4431–4439. doi: 10.1158/1078-0432.CCR-14-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. National Cancer Institute. E7 TCR T cells with or without PD-1 blockade for human papillomavirus-associated cancers. NIH website. https://clinicaltrials.gov/ct2/show/NCT02858310 Accessed February 16, 2018.
- 9.Tran E, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375(23):2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, Yang JC. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol Res. 2016;4(3):204–214. doi: 10.1158/2326-6066.CIR-15-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veatch JR, et al. Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J Clin Invest. 2018;128(4):1563–1568. doi: 10.1172/JCI98689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez-Galarza FF, Christmas S, Middleton D, Jones AR. Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res. 2011;39(Database issue):D913–D919. doi: 10.1093/nar/gkq1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGranahan N, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171(6):1259–1271.e11. doi: 10.1016/j.cell.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res. 2010;16(3):790–799. doi: 10.1158/1078-0432.CCR-09-2446. [DOI] [PubMed] [Google Scholar]
- 16.Patel SJ, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548(7669):537–542. doi: 10.1038/nature23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaretsky JM, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]