

Abstract
Breast cancer metastasis remains a clinical challenge, even within a single patient across multiple sites of the disease. Genome-wide comparisons of both the DNA and gene expression of primary tumors and metastases in multiple patients could help elucidate the underlying mechanisms that cause breast cancer metastasis. To address this issue, we performed DNA exome and RNA sequencing of matched primary tumors and multiple metastases from 16 patients, totaling 83 distinct specimens. We identified tumor-specific drivers by integrating known protein-protein network information with RNA expression and somatic DNA alterations and found that genetic drivers were predominantly established in the primary tumor and maintained through metastatic spreading. In addition, our analyses revealed that most genetic drivers were DNA copy number changes, the TP53 mutation was a recurrent founding mutation regardless of subtype, and that multiclonal seeding of metastases was frequent and occurred in multiple subtypes. Genetic drivers unique to metastasis were identified as somatic mutations in the estrogen and androgen receptor genes. These results highlight the complexity of metastatic spreading, be it monoclonal or multiclonal, and suggest that most metastatic drivers are established in the primary tumor, despite the substantial heterogeneity seen in the metastases.
Keywords: Oncology
Keywords: Breast cancer
Introduction
Breast cancer remains the second leading cause of cancer-related death in women in the United States and is typically caused by metastasis. Significant genetic heterogeneity exists both within a single primary breast cancer (1–3) and across patients (4–7). Despite this intratumor heterogeneity, the intrinsic gene expression features of the primary tumor as measured by RNA can predict future sites of recurrence (8, 9), response to therapy (10), and overall survival (8, 11). Few studies have compared both the RNA and DNA sequencing of multiple distant metastases within a patient, or across multiple subtypes of breast cancer from larger cohorts of patients.
Previous studies of breast cancer metastatic evolution heavily emphasized mutational genetic drivers, as defined by previous large-scale sequencing projects (6, 7, 12, 13). Because copy number variations (CNVs) cause many genes to be altered at once, potential genetic drivers from CNVs are often difficult to identify. Computational approaches integrating known protein-protein interaction networks, with gene expression from RNA-sequencing (RNA-seq) data and DNA-based somatic alterations have demonstrated the power of this approach for identifying unique driver sets beyond somatic mutations alone (14–16). This integrative approach could be used to identify shared drivers of breast cancer metastasis across patients and subtypes.
Multiple efforts to understand the genetic evolution of metastasis by sequencing matched primary tumors and metastases via a single matched pair, or single-cell sequencing, revealed both linear expansion of a single clone from the primary tumor to a metastasis (17–21), branched evolution of metastasis (22, 23), and cross-seeding of metastases (24). Few of these studies, however, span multiple subtypes of breast cancer. Thus, it remains unknown how dynamic these methods of metastatic seeding are, both within a patient or across multiple subtypes of breast cancer.
Here, we analyzed the metastatic evolutionary process and computationally predicted drivers of breast cancer metastasis in a panel of matched primary tumors and multiple metastases. Using the Rapid Autopsy Program established at the UNC at Chapel Hill, we collected matched primary and metastatic breast cancers from 16 individuals and performed RNA-seq and DNA whole-exome sequencing on the primary tumor, 67 matched metastases (2–7 per patient), and a matched normal tissue comparator for each patient. We examined computationally predicted metastatic drivers by integrating known protein-protein networks with gene expression and DNA-seq data and the clonal evolution of metastasis within each patient.
Results
Patient and sequencing characteristics
To explore metastatic evolution and identify drivers of breast cancer metastasis, we performed DNA whole-exome sequencing and RNA-seq on 16 primary invasive breast cancers and 67 matched metastases (Figure 1, Supplemental Figures 1 and 2, and Supplemental Tables 1 and 2; supplemental material available online with this article; https://doi.org/10.1172/JCI96153DS1). This cohort of metastatic patients had a median age of 45.5 years at the diagnosis of breast cancer, a median time to relapse of 14.5 months, and an overall survival of 36.5 months (Table 1). These patients all received at least 1 chemotherapeutic agent prior to death, and all but 1 patient received radiation, predominantly to the breast and/or brain (Supplemental Table 1).
Figure 1. Overview of the study methods.

Primary tumors and 68 metastases from 16 patients who died of metastatic breast cancer were sequenced with both DNA whole-exome sequencing and RNA sequencing. DNA somatic mutations, somatic copy number alterations, and RNA gene expression were called. Biologic subtype was determined with the PAM50 predictor. Clonality was determined from the DNA mutations. Genetic drivers were predicted using the DawnRank driver analysis tool, integrating RNA expression, DNA mutations, and copy number.
Table 1. Clinical characteristics of the study population (n = 16).
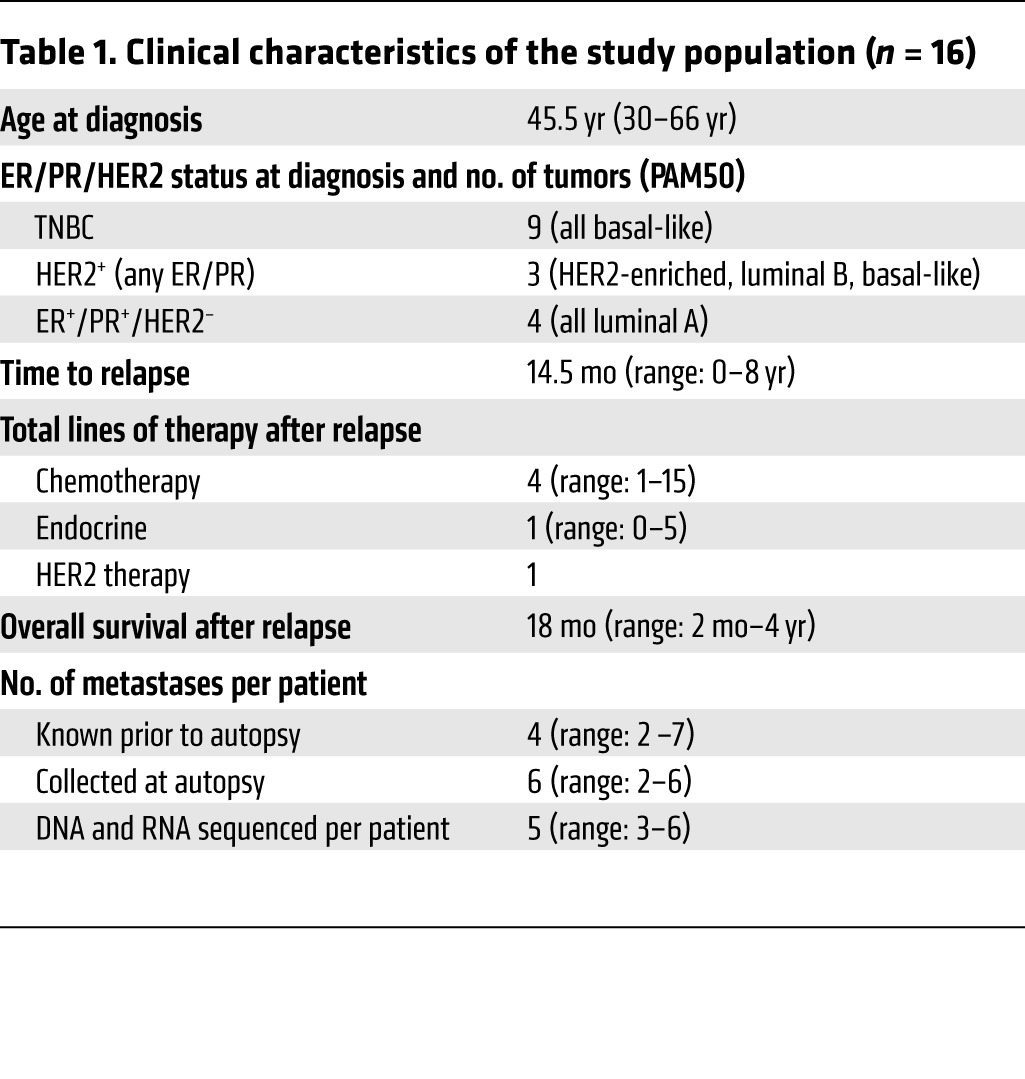
Of the primary tumors sequenced, 6 of 16 were therapy naive, 5 of 16 received neoadjuvant chemotherapy, and 5 of 16 received both neoadjuvant chemotherapy and radiation therapy (Supplemental Table 1). The median overall coverage of DNA exome sequencing was 108× (75×–250×; Supplemental Table 2). An established DNA variant pipeline (25), an RNA-seq variant (26), and an RNA-seq gene expression pipeline (27) were used to call variants in the DNA and the RNA, as well as to determine gene expression levels in all 83 samples (see Methods and Supplemental Figure 2). Of the DNA single base mutations with at least 5 reads of coverage at that position in the RNA, 83% were also identified in the RNA-seq data. Additionally, droplet PCR confirmation of all estrogen receptor 1 (ESR1) mutations demonstrated high accuracy and sensitivity of our variant calling pipeline (Supplemental Figure 3).
We next examined the clinical features and molecular subtypes of each of the primary tumors and their matched metastases. We applied the PAM50 subtype predictor (28) to determine the intrinsic molecular subtype (Supplemental Table 2). Breast tumors from 4 patients were positive for estrogen receptor (ER) expression, but negative for human epidermal growth factor receptor 2 (HER2) overexpression (ER+/HER2–) according to standard clinical assays on the primary tumor at diagnosis. According to subtyping based on the primary tumor, 1 of these patients had luminal A subtype, 1 had luminal B subtype, and 2 had “normal-like” tumors; these 2 primary tumors were both formalin-fixed, paraffin-embedded (FFPE) and of low tumor cellularity and were thus excluded from gene expression analyses but included for DNA-based analyses. Of the primary breast tumors from 4 patients who were clinically HER2+, 1 was of the HER2-enriched subtype, 2 were of the luminal A subtype, and 1 was of the basal-like subtype. Breast tumors from 9 patients were triple-negative (negative on clinical assays for the ER, the progesterone receptor [PgR], and HER2), with 6 patients’ tumors classified as the basal-like subtype and 3 as normal-like, but next-closest to the basal-like centroid, and all metastases from these 9 patients were classified as basal-like (Supplemental Table 2); note that none of these normal-like FFPE samples were included in subsequent gene expression analyses.
As reported previously (8, 22), the intrinsic gene expression profiles in tumors from an individual patient are typically highly correlated with one another (Supplemental Figure 4). This result was recapitulated in our sample set when we excluded the 5 “normal-like” specimens. We performed hierarchical clustering analysis using the intrinsic gene list of Parker et al. (28) and noted that all tumors from 10 of the 16 patients were clustered together, with 5 of 16 patients’ samples categorized into 2 subgroups, although all tumors were in the same overall subtype cluster, and 1 of the 16 patients’ samples was clustered into 2 different subtype dendrogram locations. By PAM50 centroid–based subtyping, 12 of 16 tumors had the same subtype calls, including all basal-like tumors, 2 of 16 had mixed luminal A/B subtypes, and 2 of 16 had multiple samples with different subtype calls (luminal A, luminal B, and HER2-enriched).
Computationally predicted drivers of breast cancer metastasis.
Many mutations and copy number alterations (CNAs) are probably passenger alterations without functional biologic consequences. We therefore used a computational tool called DawnRank (14) to identify genetic drivers. DawnRank integrates DNA alterations, protein-protein interaction networks, and the expression of these networks via RNA gene expression data for each individual tumor. By evaluating the perturbation of the network through RNA gene expression data for each tumor, DNA alterations can be ranked in terms of the RNA networks’ expression in that tumor, and thus those DNA alterations with the greatest effects in terms of RNA network expression can be identified as genetic drivers in an individual tumor specimen (14).
DawnRank network analysis was applied to each tumor using upper-quantile–normalized RNA counts from RSEM software (29) that were normalized to the mRNA-seq platform (Supplemental Table 4). Varying the “driver” cutoff top ranks selected from 90% to 99.5% showed consistent numerical predominance of DNA copy number drivers as compared with somatic mutations (Supplemental Figure 5). Because DawnRank scores follow a normal distribution, for each individual tumor, genes with DawnRank network scores in the top 5% of more than 8,000 genes in predetermined networks (akin to P = 0.05) were overlapped with the somatic CNA and/or mutation profile from that tumor and were thus considered to be genetic drivers.
Timing of genetic alterations and drivers.
To understand when during the metastatic process potential “driver” somatic mutations (Supplemental Table 3) and CNAs (Supplemental Table 4) occurred across the cohort, we classified both somatic alterations and DawnRank drivers within each patient into 4 categories: founder mutations (present in all samples from a patient), subclonal primary-metastasis mutations (in the primary tumor and a metastasis but not in every tumor), metastasis-shared (not in the primary tumor), and metastasis-private (in only 1 metastasis) (Figure 2A).
Figure 2. Timing of somatic alterations and driver acquisition in metastases.

Somatic DNA alterations within a single patient classified into 4 categories on the basis of a hypothesized timing with which they were acquired during the development of metastasis. (A) Founder alterations established in the primary tumor and observed in all metastases (gray), shared in the primary tumor and metastases but not in all tumors (purple), shared in 2 metastases but not the primary tumor (pink), and private to 1 metastasis (blue). The distributions of all (B) somatic mutations, (C) somatic CNAs, (D) DawnRank predicted mutations, and (E) DawnRank CNAs within each patient. shared primary/mets, shared in the primary tumor and metastases but not in all tumors; shared met-met, shared in 2 metastases but not the primary tumor.
Of these categories, the majority of mutations and CNAs in the metastases were shared with at least 1 other tumor and were not private in a single tumor (mutations: 27% founder, 7.7% primary-metastasis shared, 26% metastasis-shared, 35% private; CNAs: 8.7% founder, 16.7% primary-metastasis shared, 34% metastases-shared, 24% metastasis-private) (Figure 2, B and C). Interestingly, when narrowing to only predicted DawnRank drivers, an enrichment for earlier events was noted, especially in the mutations (mutations: 43% founder, 2.5% primary-metastasis shared, 15% metastasis-shared, 22% private; CNAs: 18% founder, 18% primary-metastasis shared, 40% metastases-shared, 19% metastasis-private) (Figure 2, D and E).
In the 16 patients, 110 mutations were identified as drivers: 39 founder mutations, 11 subclonal primary-metastasis shared, 29 metastasis-shared, and 31 private (Supplemental Figure 6A). Of the 50 driver mutations observed in the primary tumors, 25 (50%) were at a variant allele frequency (VAF) of less than 10%; thus, filtering at a greater than 10% VAF would overestimate the number of metastasis-specific events.
Genetic drivers were more likely to be founders as compared with private mutations (Figure 2D, 1-sided t test P = 0.03, t estimate = 0.22). The only recurrent genetic driver mutation in more than half of the patients was TP53 (13 of 16 patients; Figure 3A). Beyond TP53, genetic drivers (ESR1 and PIK3CA) caused by a mutation were detected in only 3 of 16 patients (Figure 3A). All other mutation drivers were identified in only 1 or 2 patients in the data set, and many were uniquely observed in patients with basal-like tumors (Figure 3A, genes shown in red font).
Figure 3. Timing and frequency of predicted drivers in primary and metastatic breast cancers.

(A) DawnRank drivers from somatic mutations. (B) DawnRank copy number amplifications in at least 12 of 16 patients, and (C) deletions in at least 10 of 16 patients. Each alteration is characterized per patient as a founder alteration (gray), an alteration shared in the primary tumor and metastases (primary/mets) (purple), an alteration shared in 2 metastases but not in the primary tumor (pink), or an alteration private to 1 metastasis (blue). Copy number–altered drivers are annotated with the chromosomal cytoband location.
Many more genes were identified as drivers from CNAs rather than from mutations (Supplemental Figure 6B). In contrast to the low frequency of common mutational drivers in our data set, many copy number amplifications and deletions were consistently identified as drivers across most patients (Figure 3, B and C). Previously identified common regions of amplification in breast cancer (8q, 5p, and 1q) included the DawnRank hits ANGPT1, LYN, SDC2, SHC1, GDNF, and TERT, which were identified as drivers in 15 of 16 patients, with 6 of 10 of these events showing amplification in the primary tumor that was maintained in the metastases in those patients (Figure 3B, gray). Common copy number losses occurred in FAS, a critical member of the apoptosis cascade, in PIK3R1, the regulatory subunit of PIK3CA, and in AURKB, a central inhibitor of the cell-cycle pathway (Figure 3C).
In an analysis restricted to the subset of patients with basal-like tumors (n = 10), we collectively identified common copy number amplifications of genes involved in the cell cycle, specifically those involved in the G1/S transition including CCNE1, CUL1, and CDK5, as well as the chromatin-associated proteins RBBP4 and HDAC1 (Supplemental Figure 7). A gain in BCAN expression, specifically in the patients with basal-like tumors, has not been previously described in breast cancer, but this gene has been shown to be highly overexpressed in aggressive gliomas via STAT3 signaling (30). Basal-like copy number loss of the DNA damage cascade regulator RAD51 was also called as a common basal-specific driver.
To further test the robustness of DawnRank, we compared DawnRank results using the ratio of each metastasis to its matched primary tumor as the input, as opposed to the comparison of each tumor with the median of all tumors in The Cancer Genome Atlas (TCGA), as was done above (see Methods). This resulted in an overall similar number and identification of genetic mutational drivers per tumor (Supplemental Figure 8A) when compared with the median normalization method; in addition, the timing of when drivers were established did not change, regardless of the method of normalization (Supplemental Figure 8B).
Luminal-specific resistance to aromatase inhibitor therapy via ESR1 mutations
DawnRank analysis identified ESR1 mutations as genetic drivers specifically in the metastatic samples in 3 ER+ patients (Figure 3A). ESR1 mutations in the binding pocket of the ER have been previously described as effectors of resistance mechanisms to estrogen suppression by aromatase inhibitors (AIs) (31, 32). In this cohort, 5 patients had ER+ breast cancer and had received both a nonsteroidal AI (letrozole) and a steroidal AI (exemestane). Three of the 5 ER+ patients had ESR1 mutations in the metastases, but not in the primary tumor; all were called as drivers by DawnRank. Interestingly, the 3 ER+ patients who developed ESR1 mutations had primary tumors of the luminal subtype, while the 2 patients who did not develop ESR1 mutations did not have tumors of the luminal molecular subtype (patient A8 = HER2-enriched; patient A2 = mixed luminal/HER2-enriched). The ESR1 mutation was the only metastasis-specific mutation identified in more than 2 patients.
TP53 as a founder and subtype-agnostic driver event in metastatic breast cancer
We investigated whether there were founder mutations or pathways common either within or across subtypes of breast cancer. All 16 patients in our cohort harbored TP53 alterations identified by DawnRank as drivers: 13 of 16 patients’ primary tumors had a TP53 mutation that was in the primary tumor and every metastasis from that patient (Figure 3A), while tumors from the 3 remaining patients had copy number loss for TP53, also identified by DawnRank as a driver (Supplemental Table 5). TP53 mutations were observed in both basal-like and luminal subtype tumors: Patient A12’s luminal tumors harbored a 45-bp deletion between exons 4 and 5 incorporating the splice site; patient A8’s HER2-enriched tumors had a premature stop codon introduced at Arg306*, and 9 of 10 of the tumors from the patients with basal-like primary cancers had either nonsense or deleterious missense mutations (33). Our data suggest that disruption of TP53 is an early and typically founding event critical to the ability of a breast cancer to metastasize, regardless of subtype.
Shared transcriptional program in metastases
To identify genes differentially expressed in metastases versus primary cancers, a linear regression model was built comparing matched primary tumors to metastases. Briefly, for each gene, the RNA values for the primary tumor were compared with its matched metastases (Supplemental Figure 9, A and C). The t statistic defines how consistently altered the RNA gene expression values for each patient’s metastases are compared with the primary tumor. If one was to center the primary tumor at zero within each patient and adjust the gene expression values accordingly for the metastases, consistent alterations could be observed across our data set, despite the location of the metastases (Supplemental Figure 9, B and D). The labels defining primary tumors and metastases were randomized 100 times to calculate the FDR.
At an FDR of 0, we found that 333 genes from RNA-seq were differentially expressed (Supplemental Table 6). Hierarchical clustering of these significantly expressed genes across the data set clustered the primary tumors in 1 clade on the left and the metastases on the right (Figure 4A and Supplemental Table 7). Notably, within the metastases, the clades were defined by patient, despite a diverse range of metastatic sites represented by multiple liver, lung, and brain metastases (Figure 4A). These genes were consistently over- and underexpressed in the metastases as compared with expression levels in the primary tumors; additionally, the gene expression program was more similar within a patient than across sites.
Figure 4. Differential gene expression in metastases.

(A) Hierarchical clustering of median-centered RNA gene expression defined as significantly differentially expressed genes in metastases (Mets) as compared with matched primary tumors (Primaries) with the lmer function in R. Each color in the dendrogram identifies a different patient. Box plots of the mean signature score of (B) upregulated genes and (C) downregulated genes, comparing the following categories: TCGA normal breast tissue, TCGA luminal A/B primary tumors, UNC RAP luminal primary tumors, UNC RAP luminal metastases, TCGA HER2-enriched primary tumors, UNC RAP HER2-enriched primary tumors, UNC RAP HER2-enriched metastases, TCGA basal-like primary tumors, UNC RAP basal-like primary tumors, and UNC RAP basal-like metastases. ADR, adrenal gland; Ax-LN, axillary lymph node; Basal, basal-like; HER2E, HER2-enriched; Lum, luminal; LLL, left lower lobe; LUL, left upper lobe; MED, mediastinum MET, metastases; OVA, ovary; PRIM, primary tumor; RLL, right lower lobe; SKIN-L, left skin; SKIN-R, right skin ; SPIN, spinal.
Pathway analysis (34) revealed that the upregulated genes in the metastases as compared with those in the primary tumor were associated with the Gene Ontology (GO) terms hypoxia, cellular metabolism, and fatty acid β oxidation, consistent with previous research demonstrating a switch in metabolism in the metastatic setting (35). Additionally, genes associated with cell migration/motility and IP3 signaling were noted by the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis as also being more highly expressed in metastases. Significantly downregulated genes in the metastases relative to expression in the primary tumors were mostly nucleic acid–processing genes including both RNA- and DNA-processing genes.
A signature score, calculated as the mean gene expression value for the metastasis upregulated genes in both TCGA and this data set, was plotted by subtype. Interestingly, the basal-like TCGA primary tumors had increased expression compared with levels in the other tumor subtypes, with all primary tumors from this study having higher expression levels than did those in TCGA primary tumors (TCGA: ANOVA P = 0.0113, F = 3.714) (Figure 4B). A similar signature score for the downregulated genes was calculated in TCGA and this data set as the mean of the genes within the gene list defined by the linear model. Interestingly, we observed a slight upregulation of gene expression in our primary tumors as compared with expression in TCGA primary tumors, but a significant downregulation of gene expression in the metastases. Downregulated genes had significantly lower means in the basal-like tumors than in the other tumor subtypes (TCGA: ANOVA P = 7.7 × 10–16, F = 25.39) (Figure 4C). Last, neither of these signatures (the upregulated or downregulated signatures in metastases) was prognostic of survival when tested on multiple external data sets including that of the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) (5).
Genetic heterogeneity of the primary breast cancer is maintained in the metastases
Previous work by our group demonstrated that low-frequency subclones present at 1% to 5% in the primary tumor can be enriched to greater than 40% in the related metastases (22). In contrast, other groups have used VAF cutoffs such as 5% and 10% to exclude variants (17, 21). In order to minimize false-positives and still maintain high sensitivity, we used our clinically validated variant pipeline (25) to call high-quality somatic mutations, followed by computational reinterrogation. Computational reinterrogation was performed such that, for a given patient, high-quality somatic variants were initially called by comparing each tumor (be it the primary tumor or a metastasis) with its matched normal tissue, all variants were then combined into a single file, and the resulting somatic mutations were specifically requeried in all tumors from that particular patient; this greatly improved the detection of low-frequency variants in some specimens, typically the primary tumor.
To evaluate the clonal evolution of a metastasis, we calculated cancer cell fractions and subclonal structure with PyClone (36) and reconstructed evolutionary processes with Dollo parsimony, an R wrapper for PHYLIP (37). We classified patients into 3 categories on the basis of the evolutionary relationship: those with monoclonal primary tumors that led to a linear evolution of metastases, those with monoclonal primary tumors with multiclonal metastases, and those with multiclonal primary tumors with multiclonal metastases.
Of the 16 patients examined, 4 had monoclonal primary tumors and metastases (Figure 5, A and B, and Supplemental Figure 10, A–C), 3 patients had monoclonal primary tumors with multiclonal metastases (Supplemental Figure 10, D–E), and 7 patients had multiple clones in the primary tumor and seeding of each distant metastasis by at least 2 clones (Figure 5, C and D, and Supplemental Figure 11). As previously reported (22) and as again seen here, triple-negative breast cancer (TNBC) patients with a basal-like tumor subtype often had multiple clones in the primary tumor and metastases (Figure 5D and Supplemental Figure 11). Interestingly, multiclonal seeding of metastases from a multiclonal primary tumor was also observed in patient A12, who had an ER+, luminal A subtype cancer (Figure 5C).
Figure 5. Multiclonal and monoclonal metastatic seeding patterns in breast cancer patients.

(A–C) Dendograms depicting the overall relationship of the tumors in (A) patient A2, (B) patient A20, (C) patient A12, and (D) patient A15. Each subclone detected in a patient is represented as a separate color along the x axis for each primary tumor and metastasis on the y axis. The radius of each circle is proportionate to the mean cellular prevalence of that clone in each tumor. The total number of mutations per clone is indicated on the right, and the percentages of mutations detected in that clone in each tumor are plotted as a bar graph below each dendogram. Across subtypes, monoclonal seeding patterns in (A) patient A2 (ER+, luminal) and (B) patient A20 (ER–/PR–/HER2–, basal-like) and multiclonal patterns in both (C) luminal (patient A12) and (D) basal-like (patient A15) tumors are shown. LungL, left lung; LungR, right lung.
Last, a recent report on colon cancer metastases examined the relationship between primary tumors, lymph node metastases, and distant metastases and determine that lymph node metastases were not an obligate step in the spreading to distant organs (38). We analyzed axillary lymph node metastases in 2 patients (patients A1 and A23), and these were relatively distant from their primary tumors. In patient A1, the primary tumor shared 2 clones with the distant metastases, and these clones were not detected in the A1 axillary lymph node (Supplemental Figure 11B). In patient A23, the predominant clone in the brain metastases was observed in both the primary tumor and the A23 axillary lymph node; however, the close clustering of the primary tumor with the 3 brain metastases on 1 main branch in the dendrogram suggests a direct hematogenous spreading of the primary cancer to the brain rather than through the axillary lymph nodes (Supplemental Figure 1A). These findings suggest that seeding of the distant metastases in these 2 patients did not occur via an obligate seeding from the lymph node metastasis.
Discussion
The molecular mechanisms driving the metastatic process are critical to understand in order to better prevent and treat existing metastases. Using the UNC’s Rapid Autopsy Program and next-generation sequencing of multiple tumors from 16 breast cancer patients with aggressive disease courses, we showed that the majority of genetic drivers were established in the primary breast cancer and maintained throughout the metastatic process;we observed this in both luminal and basal-like breast cancers. We also showed that TP53 was the only mutation driver that was common across all subtypes of breast cancer metastases and that the majority of drivers were predominantly altered by virtue of somatic CNAs. Moreover, our data illustrate that metastasis is often a result of multiclonal seeding of breast cancer metastases, regardless of subtype.
Historically, point mutations or small intragenic insertions/deletions (in/del) have been regarded as the driving force behind oncogenesis. One of the unique aspects of this work was our use of DawnRank, which allows a more functional-based assessment of genetic drivers and integrates prior knowledge of protein interaction networks with mutations, CNAs, and RNA expression data, to refine our ability to identify drivers for each individual patient. By contrast, DNA-only–based methods can only identify drivers on the basis of correlations to previous data sets from which population-based enrichments of specific genes were determined. Our functional-based genetic driver approach demonstrated that the majority of drivers were, in fact, the result of CNAs. A challenge with interpreting and acting upon large-scale amplification/deletions in the genome is that often tens to hundreds of genes are altered; a strength of DawnRank is that it can further prioritize these many CNA candidate genes on the basis of functional data from individual patients, thus providing a means of finding the driver(s) present in these regions of large-scale changes.
TP53 mutations were seen repeatedly in both basal-like and luminal breast cancers, with the mutation always established in the primary tumor and maintained in every metastasis from that patient. This suggests that TP53 is one of the founding mechanisms of aggressive, lethal breast cancer. Beyond TP53, no other driver mutations were present in more than 3 of the 16 patients in this data set.
DawnRank analysis identified mutations in the binding pocket of ESR1 in 3 of 16 patients with luminal subtype breast cancers who received AIs, consistent with previous reports demonstrating this mechanism of resistance to AIs (31, 32). Interestingly, in 2 patients with clinically determined ER+ tumors who received AI therapy and did not have ESR1 mutations, those tumors were of the HER2-enriched subtype. Previous reports have described HER2 amplification as a known mechanism of resistance to endocrine therapy (39). Furthermore, preliminary evidence has shown that ER+ tumors that metastasize often have features of HER2 enrichment in them (40). This molecular diversity of ER+ tumors (16, 41) may explain the differential response of many patients’ metastases to AI therapy.
In studying the dynamic gene expression in the primary tumors compared with expression in the metastases, we were able to generate a unique signature showing consistent gene expression differences in the metastases, regardless of the site of metastasis. Previous research comparing single matched pairs of primary and metastatic tissues showed that hypoxia, proliferation, and dedifferentiation signatures were upregulated in the metastases (8). Other studies have evaluated groups of primary tumors and metastases from specific sites such as bone (42) and brain (43), but not multiple matched sites from the same patient. Here, we identify a unique set of genes that were upregulated and downregulated in metastases commonly across the cohort; however, we note that these signatures were not prognostic. This is consistent with many previous genomic predictors of future metastasis development (including OncotypeDX [ref. 44], Mammaprint [ref. 45], and Prosigna [ref. 46]), in which a large component of these signatures is proliferation. An important feature of our metastasis-associated signature was hypoxia and possibly an altered cellular metabolism, which likely reflects a type of Warburg effect seen when comparing metastases versus primary tumors; these features could potentially provide new therapeutic vulnerabilities, and strategies targeting metabolism in breast cancer, especially TNBC, is an area of extensive current research (47–49).
Previous whole-genome sequencing of tumors from 2 patients (A1 and A7) identified multiclonal seeding as a mechanism in breast cancer metastasis, with the majority of functional mutations established in the primary tumor and maintained throughout metastasis (22). Here, we build upon that small study, reanalyze these same 2 patients using DNA exomes and RNA-seq, include another 14 new patients, and demonstrate that, at least for basal-like tumors, multiclonal seeding is a common mechanism of metastasis. The short duration of progression-free survival seen in many of our patients has interesting implications for observation of the polyclonal seeding of metastases; importantly, whether this varies with the time to progression will need to be examined in larger data sets with more variability in the time to progression.
In patients in whom multiclonal seeding occurred, the metastasis formation may have occurred via a clump of cells containing more than 1 clone that left the primary tumor, which then traveled to the distant site and seeded this site. This has important clinical implications: if the metastasis “seed” is a collection of cells with distinct subclonal populations, then successful therapy to prevent metastasis focused on inhibiting individual cell migration/motility may have a reduced effect on tumors that use multiclonal metastatic seeding. This heterogeneous metastasis has been demonstrated in both HER2+ breast cancer metastasis (50) as well as in animal model studies showing growth factor crosstalk between 2 clones (51). Strategies to more comprehensively identify and inhibit the multiple subclones, which may have growth factor crosstalk (51), may be necessary in these tumors to effectively inhibit the process of metastases and/or primary tumor growth.
Last, though only 2 of 16 patients had axillary lymph nodes sequenced, our data suggest that the distant metastases were not necessarily seeded from the axillary lymph node metastasis. Patient A1 had 2 clones that were shared between the primary tumor and the distant metastases but were not found in the axillary lymph node metastasis, supporting the notion of seeding specifically via the vasculature rather than the lymphatic system. This could be a false-negative result as a result of this clone being missed in the sequencing process; however, these 2 triple-negative, basal-like breast cancers from our data set support the hypothesis that lymph node metastases may be markers rather than required seeding routes for distant metastases.
Our study had a number of limitations, most notably a small sample size of 16 patients, which inhibited our ability to identify recurrent somatic mutations common to the metastatic setting, although our sample size was large enough to identify the importance of TP53 and ESR1. A larger sample size will also be needed to identify site-specific (i.e., lung or brain) differences and adaptations. In addition, given that our analysis involved only 2 patients with HER2-enriched cancers, data on additional patients with this tumor subtype will be needed to confirm the clonality of the metastases and to understand the resistance mechanisms that develop specifically in HER2+ breast cancer. Technically speaking, the conclusions of clonality were based on assumptions of sampling. As previous researchers have revealed substantial heterogeneity of the primary breast cancer, it is difficult to know whether a subclone is definitively not present in a tumor without sequencing the entire tumor. Furthermore, 5 of 16 of the primary breast cancers in this data set were treated with neoadjuvant (preoperative) therapy prior to mastectomy. Thus, the conclusions drawn are a combination of both the natural history of the breast cancer and the resistance to therapy. Future studies comparing matched, therapy-naive, post-neoadjuvant therapy, axillary lymph nodes, liquid biopsies, and distant metastases will be needed to understand the multiple, complex steps of clonal evolution, particularly given the prevalence of multiclonal seeding. Finally, this study is overrepresented in terms of aggressive, late-stage breast cancers, making it more difficult to draw conclusions regarding more indolent metastases.
In summary, this study validates and further expands upon the compelling evidence of multiclonal seeding across multiple subtypes of breast cancer, especially for TNBC/basal-like tumors. Additionally, we show that most genetic drivers arise from CNAs. The mechanisms underlying the generation of genetic diversity are becoming known, including the consistency we observed across our patient cohort and previous literature suggesting that TP53 dysfunction is an early and critical event in the development of aggressive breast cancers. Despite the high degree of heterogeneity in primary breast cancers and metastases, our results also show that the majority of genetic drivers are established in the primary breast cancer and maintained throughout metastasis. This study provides hope that the therapeutic targeting of founding events driving the primary and metastatic tumor phenotype might both prevent and inhibit the progression of metastasis.
Methods
Patient consent and tissue processing
Tumor tissue was obtained from patients with metastatic breast cancer who consented to participation in the Rapid Autopsy Tumor Donation program (RAP) at the UNC. Primary tumor, metastatic tumor, and normal tissues were taken within 6 hours of death for all metastatic sites, both known and found, at the time of autopsy. Tissues were frozen in a –80°C freezer, and RNA and DNA were isolated from each tissue using QIAGEN RNAeasy and DNAeasy kits, respectively, according to the manufacturer’s protocols. Primary breast cancer tissues taken at diagnosis were also acquired when possible. Patients were selected if they had a primary tumor and at least 2 metastases available for analysis.
Archived tissues in FFPE tissues had total RNA isolated with a Roche High Pure RNA Paraffin Kit (catalog 03270289001) and DNA isolated with a Maxwell 16 FFPE Tissue LEV DNA Purification Kit. RNA quality was verified with an Agilent BioAnalyzer RNA 6000 Nano Kit. Sequencing data were deposited in the NCBI’s genotypes and phenotypes database (dbGaP) (accession number phs000676).
DNA whole-exome sequencing
DNA was prepared for sequencing using the Agilent Technologies SureSelect XT library protocol. Fresh-frozen tumors were processed according to the manufacturer’s protocol for 3 μg input, while FFPE tumors were processed with the low-volume input according to the manufacturer’s protocol for 200 ng input. DNA libraries were captured and amplified with Agilent Technologies SureSelect Human All Exon, version 5 or 6, according to the manufacturer’s protocol (Supplemental Table 2). The quality of both the DNA libraries and DNA exome capture and concentration were quantified with Agilent TapeStation DNA 1000 and High Sensitivity D1000, respectively.
Paired-end sequence data (2 × 100 bp) were generated using the Illumina HiSeq 2500 for each tumor or normal sample, with 3 samples per lane. Illumina reads were mapped to the NCBI Build 36 reference sequence with BWA 0.7.9a (52), realigned with ABRA, version 0.96 (53), and processed by biobambam2 (54). Viral alignments were counted with Samtools (55) and BEDTools-Version-2.15.0. Picard 1.92 (56) was used to calculate sequencing metrics. ISAAC (57) and Freebayes were used to call germline mutations with quality scores above 30. SnpSIFT, version 1.3.4, band SnpEFF (58) was used to annotate alterations with population-level frequencies. CADABRA SomaticLocusCaller was used for further filtration. Somatic variants were called with STRELKA (59) using strelka_config_bwa_default.ini. STRELKA filter -lane ‘if(/^#/) {next;} if(\$F[7]!~/QSS_NT=(\d+);?/) {next;} if(\$1>=10) {print;}’. UNCeqR v0.1.14 (26), Cadabra version 0.9 (53), and ENSEMBL (60) with Python 2.7.10.
We used minor allele frequencies of highly variable SNPs called by Freebayes in the general population for sample identity. All samples had an expected 87%–100% identity with both the tumor and matched normal tissue from the same patient.
Copy numbers were called with SynthEx (61) using 50,000-bp–sized bins and K nearest neighbors = 3 from the pool of 16 normal tissues available in dbGaP (accession number phs000676). Briefly, the ratio of on-target and off-target exome reads of the tumor were compared with a normal tissue selected from the data set by the highest degree of similarity by K-nearest neighbor (KNN) based on library size and fold enrichment. Segment-level ratios were calculated and log2 transformed (Supplemental Table 4). For DawnRank analyses, copy number levels greater than 0.25 were considered gains, and levels below –0.32 were considered losses (61).
RNA-seq
Fresh-frozen RNA was prepared for sequencing following the Illumina TruSeq polyA Select protocol. If libraries failed the protocol, they were then prepared with the Illumina TruSeq RiboZero Gold protocol according to the manufacturer’s instructions. FFPE RNA was prepared with the Illumina TruSeq FFPE RiboZero Gold protocol according to the manufacturer’s instructions. RNA libraries were sequenced as 2 × 50 bp paired-end reads with 2 samples per lane on Illumina HiSeq 2500 sequencers. Reads were aligned with MapSplice, version 0.7.4 or version 0.7.6 if 0.7.4 failed (62), and gene values were quantitated with RSEM (29) and counts upper-quartile normalized and log2 transformed for analysis.
Because of bias in the FFPE and total RNA-seq data as compared with the mRNA-seq data, a normalization vector was calculated: previously published matched samples of fresh and FFPE RNA (63) sequenced at the UNC using mRNA-seq and total RNA-seq, respectively, were used to find the mean difference for each gene across each platform. For example, the mean difference of fresh-frozen, ribo-zero total RNA-seq was calculated on a per-gene basis according to the same sample’s fresh-frozen mRNA-seq. Likewise, the mean difference of FFPE ribo-zero total RNA-seq was calculated on a per-gene basis according to the same sample’s fresh-frozen mRNA-seq. Each of these vectors was applied to samples sequenced with total RNA-seq and FFPE, respectively, giving a gene-by-gene adjustment to the mRNA-seq platform. Using these mRNA-normalized counts, the PAM50 score was calculated exactly as previously described (7). Briefly, a vector calculated from the 2015 TCGA Lobular Breast Cancer study (7) for adjustment of the UNC sequencers using fresh-frozen mRNA-seq, mapped with MapSplice (62) and quantified with RSEM (29), was used for the calibration parameter in Bioclassifier R (28).
Droplet PCR for ESR1 mutations
Digital droplet PCR for WT and 4 hotspot ESR1 alleles (D538G, Y537C, Y537S, and Y537N) was performed using Raindrop Source and Sense instruments (Raindance Technologies). Primers for a 75-bp amplicon that includes these hotspot mutations were used in conjunction with locked nucleic acid TaqMan probes for WT (conjugated to TET) or mutant (conjugated to FAM) ESR1 alleles, purchased from Integrated DNA Technologies (IDT). The multiplexed genotyping reaction was validated using synthesized 125-bp DNA fragments (gBlocks; IDT). The primer and probe sequences are listed in Supplemental Table 8. TaqMan Genotyping Master Mix (Applied Biosystems) was used for 10 to 100 ng Covaris-sheared genomic DNA in a 50-μl reaction volume. After PCR amplification in a thermocycler (C1000 Touch Thermal Cycler; Bio-Rad), the emulsion was analyzed with the Raindrop Sense instrument (RainDance Technologies) to measure the endpoint fluorescence signal from each droplet using the manufacturer’s standard protocols. The fluorescence intensity and duration for each droplet in the FAM and TET channels were analyzed using RainDrop Analyst Software II (RainDance Technologies). Two-dimensional (FAM and TET intensity) plots were made for each sample, and gates were used to define graphical areas with specific fluorescence properties. The number of droplet events specific for WT or mutant ESR1 alleles was used to calculate the mutation frequency.
Computational analyses
Linear model of gene expression.
Patients with fresh-frozen, polyA select primary tumors and metastases were compared using the linear mixed-effects model lme4 package in R (64). Using the log2-transformed, RSEM-normalized gene counts, each gene was tested for significantly differential expression in the primary tumors versus matched metastases, with the patient taken into account as a confounding variable: lmer(gene[i] ~ prim/met + (1|patient))
The labels of primary or metastasis were permutated 100 times to calculate an FDR for each gene (Supplemental Table 6 and see Supplemental Data File 2 for the code).
Gene signature score.
Genes identified in the linear model at an FDR of 0 were used to supervise the original data set. The fresh-frozen RAP tumors were median centered for each gene, clustered in Cluster (65), and viewed with Java TreeView (66). Genes were clustered into 2 distinct clusters: 1 upregulated cluster and 1 downregulated cluster. For each sample, the gene signature was calculated as the average log2-transformed, RSEM-normalized RNA-seq value for the genes in each cluster. The signature score was calculated from TCGA RNA-seq data from the 2015 Lobular Breast Cancer data set (7) using 1,098 primary breast cancers. Using the RNA-seq, RSEM-normalized, log2-transformed counts, the RNA-seq data were added to our mRNA-seq–normalized data from the primary tumors and metastases reported here. The entire data set was median centered, and the genes identified as significant at an FDR of 0 from the above linear model were averaged to calculate the gene signature. This gene signature was then box plotted according to previously reported PAM50 subtypes (7).
Hierarchical clustering of gene expression.
RNA-seq data from the 1,098 primary breast cancers from the 2015 TCGA Lobular Breast Cancer data set using RSEM-normalized RNA counts (7) were log2 transformed, merged with tissues from this study normalized to the mRNA-seq platform as described above, and median centered. Correlation-centered hierarchical clustering of the median-centered data set with the PAM50 genes (28) was performed with Cluster (65) and visualized with Java TreeView (66).
Computational reinterrogation of somatic mutations in related tumors.
Low read coverage or low tumor cell purity can cause the rigorous somatic mutation caller to miss mutations (26, 53). Thus, all of the high-confidence somatic mutations from every tumor taken from 1 patient were reinterrogated within the same tumors from that patient. First, all of the somatic mutations from the tumors within 1 patient were collapsed into 1 file, excluding any guanine-to-adenine or cytosine-to-thymine mutations from FFPE samples. For each mutation from a single patient, we then counted the mutant and reference alleles at that position from the original BAM file of each tumor from that patient. VAFs (alternate counts/total read counts × 100) were recalculated from the new calls. All mutations from the data set were interrogated in the normal sequence for all tumors in this data set to account for false-positives. Mutations with VAFs of greater than 20% in at least 2 normal tissues from unrelated patients were excluded from future analyses.
DawnRank.
We generated a binary matrix of 0, indicating no alteration, and 1, indicating any alteration (mutation or copy number) for genes published in DawnRank (14). We combined log2-transformed, normalized RNA-seq data from the 2015 TCGA Lobular Breast Cancer data set (7) with the mRNA-seq RAP-normalized data set, median centered the data for each gene, and further transformed scores to the absolute value. Thus, each gene’s expression was normalized to a representative cohort of breast cancers. DawnRank was then run for each individual tumor using the parameter mu = 3 (14). DawnRank scores were saved (Supplemental Table 5), and the top 5% of scores within each tumor were considered candidate drivers. These candidate drivers were then filtered by nonsilent mutations and CNAs, such that if an alteration was present within the top 5% of ranked genes (of a total of 8,000 in the networks), it was then further considered a driver.
An alternate analysis of DawnRank was performed by comparing the ratio of RNA-seq of each matched metastasis to its primary tumor tissue. Thus, for each metastasis, the RNA-seq value was the normalized RNA-seq value of the metastasis over the matched primary tumor. These values were then used to re-run DawnRank with mu = 3 and the same network as above. Again, the top 5% ranked genes were considered candidates and filtered on the basis of mutation and CNAs for each tumor. All drivers across the metastases in a given patient were then categorized as follows: “truncal,” meaning in the primary tumor and every metastasis from that patient; “shared primary-met,” meaning in the primary tumor and a metastasis but not in every tumor; “shared metastasis,” meaning shared in at least 2 metastases but not identified in the primary tumor; and “private,” meaning identified in only 1 tumor in the patient. Box plots were generated with R.
RNA interrogation of DNA mutations.
Using the “union” list of mutations for each patient, UNCeqR (26) was used to count the number of mutated reads from the RNA BAMs at each position within a patient. For example, for patient A1, all of the mutations from the tumors within that patient were measured in the binary alignment map generated from MapSplice from the RNA-seq data. Mutations within each tumor were only considered if at least 5 reads of that gene were detected within the RNA-seq. Genes for which the RNA gene expression of the gene was less than 5 were removed from the total number of DNA mutations in that tumor. UNCeqR was additionally run on the de novo mutation identification with default parameters.
Subclonal analysis.
PyClone (36) was applied to all related tumors for each patient using the mutation calls following computational reinterrogation, as described above, and copy numbers from SynthEx (61). The mean cellular prevalence (CP) of the mutations comprising each clone was then calculated per tumor. Circles were drawn, with the radius of the circle proportionate to the mean CP. Clustering was performed for each patient with Dollo parsimony, an R wrapper for PHYLIP (37), with the R code, where i is the index for each patient (see Supplemental Data File 2 for the code).
Statistics
For DawnRank analyses, a normal distribution was confirmed, and a cutoff of the top 5% ranked genes, corresponding to a P value of 0.05, was used to determine statistical significance. A linear model was described as above, with a permutation of ×100 based on randomization of the primary/metastasis label. An FDR was then calculated, and an FDR of 0 was used for RNA signature development. All plotting was performed using R, version 3.3.0, in RStudio (67).
Study approval
Patients provided consent prior to death for a rapid autopsy, in accordance with protocols of the UNC at Chapel Hill Office for Human Research Ethics and the US Department of Health and Human Services. Primary breast cancers in the form of fresh-frozen tissues were collected prior to autopsy under study ID number LCCC 9819 (ClinicalTrials.gov Identifier: NCT01000883). This study was approved by the IRB of the University of North Carolina.
Accession numbers and data sharing
Sample information for RNA-seq and DNA-seq fastQ runs, including the clinical information, were uploaded to the NCBI’s dbGAP (phs000676.v1.p1). RNA-seq RSEM upper-quantile–normalized counts were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE110590).
Author contributions
LAC, CMP, and MBS conceptualized the study. MBS, XH, AH, AEDVS, JSP, MC, SK, GPG, and KAH designed the study methodology. MBS and XH conducted experiments. MBS, KAH, AEDVS, ERM, CMB, LAC, and CKA wrote the manuscript. JBP, ALG, CKA, LAC, LBT, NK, and CL were responsible for project administration. VJM, CMB, ALG, JBP, XH, SK, GPG, LBT, NK, MBS, DM, and KAH were responsible for data curation. CMP, LAC, and CKA provided resources. ERM, JSP, CKA, LAC, and CMP supervised the study.
Supplementary Material
Acknowledgments
We dedicate this article to the memory of Minhthu Nguyen, and we also thank the patients in this study and their families, without whom this study would not have been possible. The authors also thank Mei Huang of the UNC’s Tissue Procurement Facility. This study was supported by funds from the following sources: Susan G. Komen (to LAC and CMP); National Cancer Institute (NCI), NIH, Breast Specialized Programs of Research Excellence (SPOREs) (P50-CA58223, to LAC and CMP; RO1-CA195754-01 and RO1-CA148761, to CMP; and K23-157728, to CKA); and the Breast Cancer Research Foundation (to 16-023, to LAC, and 17-124, to CMP). MBS has been supported by NCI grant T32 2-T32-CA071341-16 (UNC Cancer Cell Biology Training Program), NCI grant T32 GM008719 (UNC Medical Scientist Training Program), and NCI grant F30-CA200345.
Version 1. 02/26/2018
Electronic publication
Version 2. 04/02/2018
Print issue publication
Footnotes
Conflict of interest: CMP is an equity stock holder of BioClassifier LLC and University Genomics, and ERM, JSP, and CMP have filed a US patent on the PAM50 subtyping assay (US 12995459).
Reference information: J Clin Invest. 2018;128(4):1371–1383.https://doi.org/10.1172/JCI96153.
Contributor Information
Xiaping He, Email: xiaping_he@med.unc.edu.
Katherine A. Hoadley, Email: hoadley@med.unc.edu.
Alan Hoyle, Email: alanh@email.unc.edu.
Sunil Kumar, Email: sunil_kumar@med.unc.edu.
David Marron, Email: dmarron@cs.unc.edu.
Gaorav P. Gupta, Email: gaorav_gupta@med.unc.edu.
Niamh Kieran, Email: nkieran@novellaclinical.com.
Chad Livasy, Email: chad_livasy@med.unc.edu.
Mengjie Chen, Email: mengjiechen@uchicago.edu.
References
- 1.Yates LR, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21(7):751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah SP, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nik-Zainal S, et al. The life history of 21 breast cancers. Cell. 2012;149(5):994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 5.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciriello G, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163(2):506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrell JC, et al. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat. 2012;132(2):523–535. doi: 10.1007/s10549-011-1619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113(10):2638–2645. doi: 10.1002/cncr.23930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carey LA, et al. Molecular heterogeneity and response to neoadjuvant human epidermal growth factor receptor 2 targeting in CALGB 40601, a randomized phase III trial of paclitaxel plus trastuzumab with or without lapatinib. J Clin Oncol. 2016;34(6):542–549. doi: 10.1200/JCO.2015.62.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smid M, et al. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008;68(9):3108–3114. doi: 10.1158/0008-5472.CAN-07-5644. [DOI] [PubMed] [Google Scholar]
- 12.Dees EC, et al. Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res. 2012;18(17):4775–4784. doi: 10.1158/1078-0432.CCR-12-0589. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou JP, Ma J. DawnRank: discovering personalized driver genes in cancer. Genome Med. 2014;6(7):56. doi: 10.1186/s13073-014-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silva GO, et al. Cross-species DNA copy number analyses identifies multiple 1q21-q23 subtype-specific driver genes for breast cancer. Breast Cancer Res Treat. 2015;152(2):347–356. doi: 10.1007/s10549-015-3476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gatza ML, Silva GO, Parker JS, Fan C, Perou CM. An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat Genet. 2014;46(10):1051–1059. doi: 10.1038/ng.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brastianos PK, et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015;5(11):1164–1177. doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krøigård AB, et al. Clonal expansion and linear genome evolution through breast cancer progression from pre-invasive stages to asynchronous metastasis. Oncotarget. 2015;6(8):5634–5649. doi: 10.18632/oncotarget.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding L, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464(7291):999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savas P, et al. The subclonal architecture of metastatic breast cancer: results from a prospective community-based rapid autopsy program “CASCADE”. PLoS Med. 2016;13(12):e1002204. doi: 10.1371/journal.pmed.1002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoadley KA, et al. Tumor evolution in two patients with basal-like breast cancer: a retrospective genomics study of multiple metastases. PLoS Med. 2016;13(12):e1002174. doi: 10.1371/journal.pmed.1002174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murtaza M, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015;6:8760. doi: 10.1038/ncomms9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown D, et al. Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat Commun. 2017;8:14944. doi: 10.1038/ncomms14944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao X, et al. Combined Targeted DNA sequencing in non-small cell lung cancer (NSCLC) using UNCseq and NGScopy, and RNA sequencing using UNCqeR for the detection of genetic aberrations in NSCLC. PLoS One. 2015;10(6):e0129280. doi: 10.1371/journal.pone.0129280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilkerson MD, et al. Integrated RNA and DNA sequencing improves mutation detection in low purity tumors. Nucleic Acids Res. 2014;42(13):e107. doi: 10.1093/nar/gku489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parker JS, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Natesh K, et al. Oncostatin-M differentially regulates mesenchymal and proneural signature genes in gliomas via STAT3 signaling. Neoplasia. 2015;17(2):225–237. doi: 10.1016/j.neo.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4(6):1116–1130. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller CA, et al. Aromatase inhibition remodels the clonal architecture of estrogen-receptor-positive breast cancers. Nat Commun. 2016;7:12498. doi: 10.1038/ncomms12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouaoun L, et al. TP53 Variations in human cancers: New lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37(9):865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- 34.Chang JT, Nevins JR. GATHER: a systems approach to interpreting genomic signatures. Bioinformatics. 2006;22(23):2926–2933. doi: 10.1093/bioinformatics/btl483. [DOI] [PubMed] [Google Scholar]
- 35.Wright HJ, et al. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc Natl Acad Sci U S A. 2017;114(32):E6556–E6565. doi: 10.1073/pnas.1703791114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roth A, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11(4):396–398. doi: 10.1038/nmeth.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Felsenstein J. PHYLIP – Phylogeny inference package (ver 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 38.Naxerova K, et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science. 2017;357(6346):55–60. doi: 10.1126/science.aai8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9(9):631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 40.Cejalvo JM, et al. Intrinsic subtypes and gene expression profiles in primary and metastatic breast cancer. Cancer Res. 2017;77(9):2213–2221. doi: 10.1158/0008-5472.CAN-16-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ciriello G, et al. The molecular diversity of luminal A breast tumors. Breast Cancer Res Treat. 2013;141(3):409–420. doi: 10.1007/s10549-013-2699-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang XH, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16(1):67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bos PD, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paik S, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 45.Cardoso F, et al. 70-Gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med. 2016;375(8):717–729. doi: 10.1056/NEJMoa1602253. [DOI] [PubMed] [Google Scholar]
- 46.Wallden B, et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med Genomics. 2015;8:54. doi: 10.1186/s12920-015-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Briggs KJ, et al. Paracrine induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell. 2016;166(1):126–139. doi: 10.1016/j.cell.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shu S, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529(7586):413–417. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang X, et al. Cystine addiction of triple-negative breast cancer associated with EMT augmented death signaling. Oncogene. 2017;36(30):4379. doi: 10.1038/onc.2017.192. [DOI] [PubMed] [Google Scholar]
- 50.Priedigkeit N, et al. Intrinsic subtype wwitching and acquired ERBB2/HER2 amplifications and mutations in breast cancer brain metastases. JAMA Oncol. 2017;3(5):666–671. doi: 10.1001/jamaoncol.2016.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang M, et al. Intratumoral heterogeneity in a Trp53-null mouse model of human breast cancer. Cancer Discov. 2015;5(5):520–533. doi: 10.1158/2159-8290.CD-14-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mose LE, Wilkerson MD, Hayes DN, Perou CM, Parker JS. ABRA: improved coding indel detection via assembly-based realignment. Bioinformatics. 2014;30(19):2813–2815. doi: 10.1093/bioinformatics/btu376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tischler G, Leonard S. biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol Med. 2014;9:13. doi: 10.1186/1751-0473-9-13. [DOI] [Google Scholar]
- 55.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Picard. Broad Institute. http://broadinstitute.github.io/picard Accessed January 10, 2018.
- 57.Baier H, Schultz J. ISAAC - InterSpecies Analysing Application using Containers. BMC Bioinformatics. 2014;15:18. doi: 10.1186/1471-2105-15-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 60.Aken BL, et al. The Ensembl gene annotation system. Database (Oxford) 2016;2016:baw093. doi: 10.1093/database/baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Silva GO, et al. SynthEx: a synthetic-normal-based DNA sequencing tool for copy number alteration detection and tumor heterogeneity profiling. Genome Biol. 2017;18(1):66. doi: 10.1186/s13059-017-1193-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang K, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38(18):e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao W, He X, Hoadley KA, Parker JS, Hayes DN, Perou CM. Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genomics. 2014;15:419. doi: 10.1186/1471-2164-15-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67(1):48 [Google Scholar]
- 65.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20(9):1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 66.Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics. 2004;20(17):3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 67. RStudio. RStudio: integrated development environment for R. https://www.rstudio.com/ Accessed January 10, 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.