

Abstract
Disordered coagulation contributes to death in sepsis and lacks effective treatments. Existing markers of disseminated intravascular coagulation (DIC) reflect its sequelae rather than its causes, delaying diagnosis and treatment. Here we show that disruption of the endothelial Tie2 axis is a sentinel event in septic DIC. Proteomics in septic DIC patients revealed a network involving inflammation and coagulation with the Tie2 antagonist, angiopoietin-2 (Angpt-2), occupying a central node. Angpt-2 was strongly associated with traditional DIC markers including platelet counts, yet more accurately predicted mortality in 2 large independent cohorts (combined N = 1,077). In endotoxemic mice, reduced Tie2 signaling preceded signs of overt DIC. During this early phase, intravital imaging of microvascular injury revealed excessive fibrin accumulation, a pattern remarkably mimicked by Tie2 deficiency even without inflammation. Conversely, Tie2 activation normalized prothrombotic responses by inhibiting endothelial tissue factor and phosphatidylserine exposure. Critically, Tie2 activation had no adverse effects on bleeding. These results mechanistically implicate Tie2 signaling as a central regulator of microvascular thrombus formation in septic DIC and indicate that circulating markers of the Tie2 axis could facilitate earlier diagnosis. Finally, interventions targeting Tie2 may normalize coagulation in inflammatory states while averting the bleeding risks of current DIC therapies.
Keywords: Hematology, Vascular Biology
Keywords: Coagulation, Platelets, endothelial cells
Introduction
Death from severe infection is a global health threat (1–3). Of all acute illnesses, the public health burden of sepsis may be the most pressing — patients spend longer in intensive care units (ICUs) and hospitals and more commonly suffer long-term health impairments compared with all other admission diagnoses (1). Severe sepsis is frequently complicated by a distinct and lethal syndrome in which coagulation occurs indiscriminately throughout the circulation. Exuberant formation of microthrombi consumes platelets and circulating coagulation factors from the blood. Platelet dysfunction arises and inhibitors of coagulation are depleted. These complex changes often render the patient susceptible to bleeding (4–6).
Termed disseminated intravascular coagulation (DIC), this complication of sepsis is extraordinarily difficult to treat for several reasons. First, the microthrombi of DIC critically impair organ perfusion, thereby accelerating the multiorgan dysfunction that precedes death from sepsis. Second, by the time septic DIC is recognized, individual patients may simultaneously exhibit manifestations of tissue ischemia from elevated microthrombi and spontaneous bleeding. Third, because the diagnosis of DIC is based on hematologic abnormalities that develop as a consequence rather than cause of excessive thrombus generation and breakdown — e.g., reduced platelets or elevated D-dimers (7) — clinical recognition of the pathophysiology underlying DIC may be delayed. Finally, the treatment of DIC is completely empirical. Platelets and coagulation factors are administered to treat excessive bleeding, and anticoagulants are given when thrombotic manifestations are dominant. As a result, treatment of DIC can itself inflict harm by triggering excess thrombus formation or bleeding. Improved understanding of the underlying mechanisms in septic DIC may therefore aid the development of new strategies to recognize and manage this complex syndrome.
Current models of DIC in sepsis have focused on interactions among platelets, leukocytes, and coagulation factors. Platelets can be activated in systemic inflammation either directly by leukocyte-derived cytokines and bacterial products (8, 9) or indirectly — e.g., tissue factor (TF) expression resulting in the generation of thrombin, which is a potent platelet agonist (4). Targeting upstream cytokines or bacterial products in sepsis has thus far been unsuccessful despite numerous attempts with potent biologics (refs. 10–12 and summarized in 13). Impairment of endogenous anticoagulation systems can lead to dysfunctional coagulation and promote microthrombi. Yet, pivotal late-stage clinical trials to restore physiological anticoagulants that become diminished in sepsis, such as antithrombin III, TF pathway inhibitor, and protein C, have not shown benefit, and instead may even increase adverse bleeding events (14–16).
In contrast, understanding of how the endothelium, which undergoes profound changes in response to systemic inflammation, contributes to septic DIC remains poorly developed. The endothelium provides a critical surface for activation of coagulation and recruitment of platelets (17), and homeostatic endothelial signaling pathways are markedly altered in human sepsis (18). Whether this disruption contributes directly to abnormal coagulation in sepsis is an open question. Similarly, it is unknown whether normalizing endothelial signaling would reverse the prothrombotic influence of systemic infection.
Described below, we undertook an unbiased proteomics approach in patients with septic DIC in order to identify causal factors. Whereas the clinical criteria for DIC relate solely to platelets and coagulation parameters, network analysis of the proteomics strongly implicated the endothelium, including alterations in the endothelial regulatory angiopoietin-1 (Angpt-1)/Tie2 signaling axis. Tie2 is a receptor that is highly enriched in the endothelium and actively signals vascular quiescence (19–22). Tie2 is stimulated by Angpt-1, a protein secreted by periendothelial cells and platelets. In the context of inflammation, a paralog of Angpt-1 called Angpt-2 competitively inhibits Tie2 (23–32). While several groups have shown that the Angpt/Tie2 pathway becomes dysregulated in human sepsis (reviewed in ref. 33), it is not known whether disrupted Angpt/Tie2 signaling in the endothelium exerts a direct mechanistic effect on the thrombotic complications. The present studies suggest that Tie2 signaling disruption is an early and pathogenic event in septic DIC, driving a fundamental phenotypic switch in the endothelium that promotes excessive fibrin deposition. The results identify a direct molecular mechanism by which the inflamed endothelium precipitates thrombus formation and propose new ways to inform the recognition and treatment of DIC in septic patients.
Results
Discovery proteomics in septic DIC implicate endothelium and Angpt-2.
To identify novel regulators of coagulation in sepsis, we performed unbiased proteomic analysis on plasma from matched patients in the ICU with sepsis (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI97488DS1) characterized as non-DIC or DIC. Protein-specific, slow-off-rate-modified DNA aptamers (SOMAmers) enabled simultaneous and sensitive detection of approximately 1,300 plasma proteins. Two hundred and two analytes were differentially expressed in septic patients with DIC versus septic non-DIC patients (P ≤ 0.05, Figure 1, A and B). These included both known markers of DIC — e.g., anticoagulant protein C, coagulation factor II (thrombin), coagulation factor XI — and potentially novel proteins. In order to elucidate candidate regulators among the top analytes, we conducted network and cluster analysis in STRING, a database of protein-protein interactions curated across major repositories of experimental results (34, 35). One hundred and seventy-three of the 202 hits in sepsis DIC (P value cutoff ≤ 0.05, Figure 1A) formed distinct interacting protein clusters that were highly enriched in pathways associated with coagulation, vascular function, complement, and endothelial inflammation (Figure 1C). Angpt-2 occupied a central node in this analysis linking vascular function, endothelial inflammation, and coagulation, and was itself highly increased in plasma of septic patients with DIC compared with those without (Figure 2A, 4.98-fold enhancement, P = 0.003). A fall in the ratio of Angpt-1/Angpt-2 is considered a peripheral indicator of reduced Tie2 signaling and is associated with adverse manifestations of diverse severe infections (36–38). We analyzed SOMAScan Angpt-1 levels (Figure 2B) and confirmed this ratio was significantly lower in patients with DIC compared with non-DIC sepsis (Figure 2C). Angpt-2, Angpt-1, and the Angpt-1/2 ratio detected by SOMAScan were confirmed by commercial ELISA (Figure 2D and Supplemental Figure 1, A and B) with a high degree of agreement between the 2 methods for Angpt-2 (r2 = 0.85, P < 0.0001; Figure 2E) and Angpt-1 (r2 = 0.63, P < 0.002, Supplemental Figure 1C). Plasma Angpt-2 discriminated between DIC and non-DIC septic patients within the selected cohort (area under the receiver operating characteristic [ROC] curve = 0.88, P = 0.007, Figure 2F). Using clinical laboratory tests, we found that Angpt-2 was inversely correlated with parameters that decline in DIC, including platelet count (r2 = 0.37, Figure 2G) and fibrinogen (r2 = 0.81, Figure 2H), the plasma glycoprotein cleaved by thrombin in the coagulation cascade to form fibrin in clots.
Figure 1. Discovery proteomics in septic DIC implicate endothelium and Angpt-2.

(A) Heatmap representation of SOMAScan plasma analytes in severe sepsis with DIC (n = 7) and sepsis (n = 7). Color scale indicates relative expression. (B) Volcano plot showing analytes that were increased or decreased in sepsis DIC compared with sepsis without DIC (non-DIC) with P values less than 0.05 indicated in red. (C) STRING analysis and visualization of high-confidence interaction clusters (k-means = 9 clusters indicated by node color) formed by plasma proteins and labeled on related functional categories. Solid line represents within-cluster, dashed gray line represents between-cluster interactions.
Figure 2. Angiopoietin levels in septic DIC.

Plasma SOMAScan levels of (A) Angpt-2, (B) Angpt-1, and (C) within-patient Angpt-1 to Angpt-2 ratio. **P < 0.005, Mann-Whitney U test. (D) Plasma Angpt-2 levels by ELISA and (E) scatter plot correlating ELISA versus SOMAScan (r2 = 0.84, P < 0.0001, n = 14). (F) Receiver operating characteristic (ROC) curve for plasma Angpt-2 to predict sepsis DIC from nonsevere sepsis within the selected cohort. Scatter plot and linear regression analysis between ELISA Angpt-2 and laboratory measurements of (G) platelet count (r2 = 0.37, P = 0.0208, n = 14) and (H) fibrinogen (r2 = 0.81, P = 0.0009, n = 9).
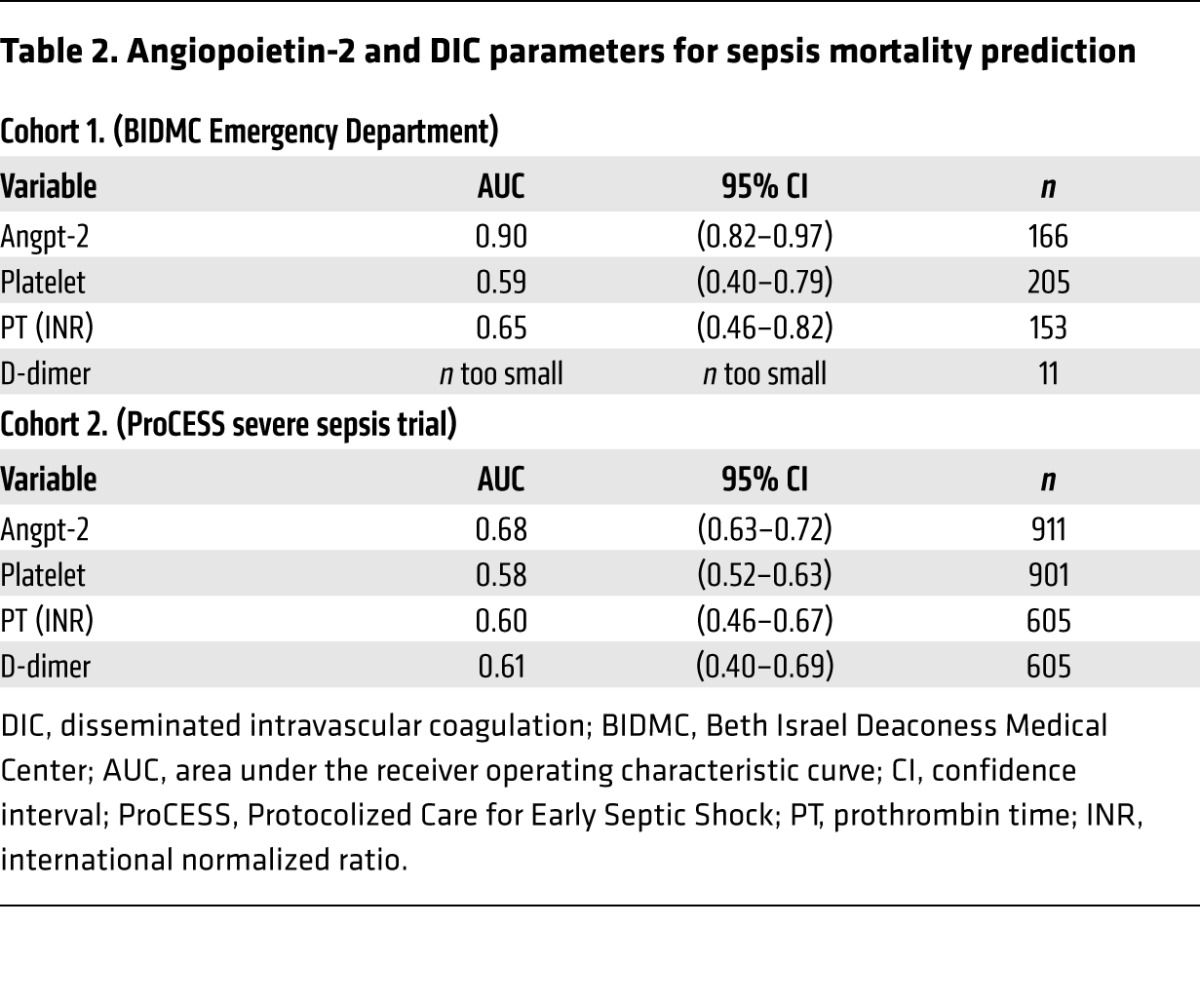
To validate the performance of Angpt-2 in differentiating patients with DIC risk factors in an independent cohort (n = 221; see ref. 27), a cutoff value (10 ng/ml) was derived from the ROC curve (Figure 2F) and Youden index analysis (0.6, sensitivity, 85.7; specificity, 71.43). Patients in the high Angpt-2 group (>10 ng/ml) had significantly prolonged clotting times (PTs) and lower platelet counts (Table 1), indicators of DIC. Although D-dimer tests were only ordered for 4% of patients in this cohort leading to a limited sample, D-dimer levels were significantly elevated in the high Angpt-2 group (>10 ng/ml). PT values were more likely to be prolonged by more than 3 seconds in the high versus the low Angpt-2 group (odds ratio 7.69; 95% CI 2.944 to 20.088; P < 0.0001). Similarly, low platelet counts (<100 × 109/l) were more common in the high Angpt-2 group (odds ratio 6.62; 95% CI 3.093 to 14.18; P < 0.0001). We further confirmed the relationship of high Angpt-2 with several traditional DIC indicators in a second, larger independent cohort with severe sepsis (n = 911, Table 1; see ref. 39). Finally, in both validation cohorts, Angpt-2 levels more accurately predicted mortality (area under the curve [AUC] = 0.90 and 0.68) than individual DIC markers (Table 2). Collectively, these results implicated the endothelium, via the Angpt/Tie2 pathway, as a candidate regulator of disordered coagulation in septic DIC leading to an increased risk of death.
Table 1. DIC parameters based on angiopoietin-2 levels.
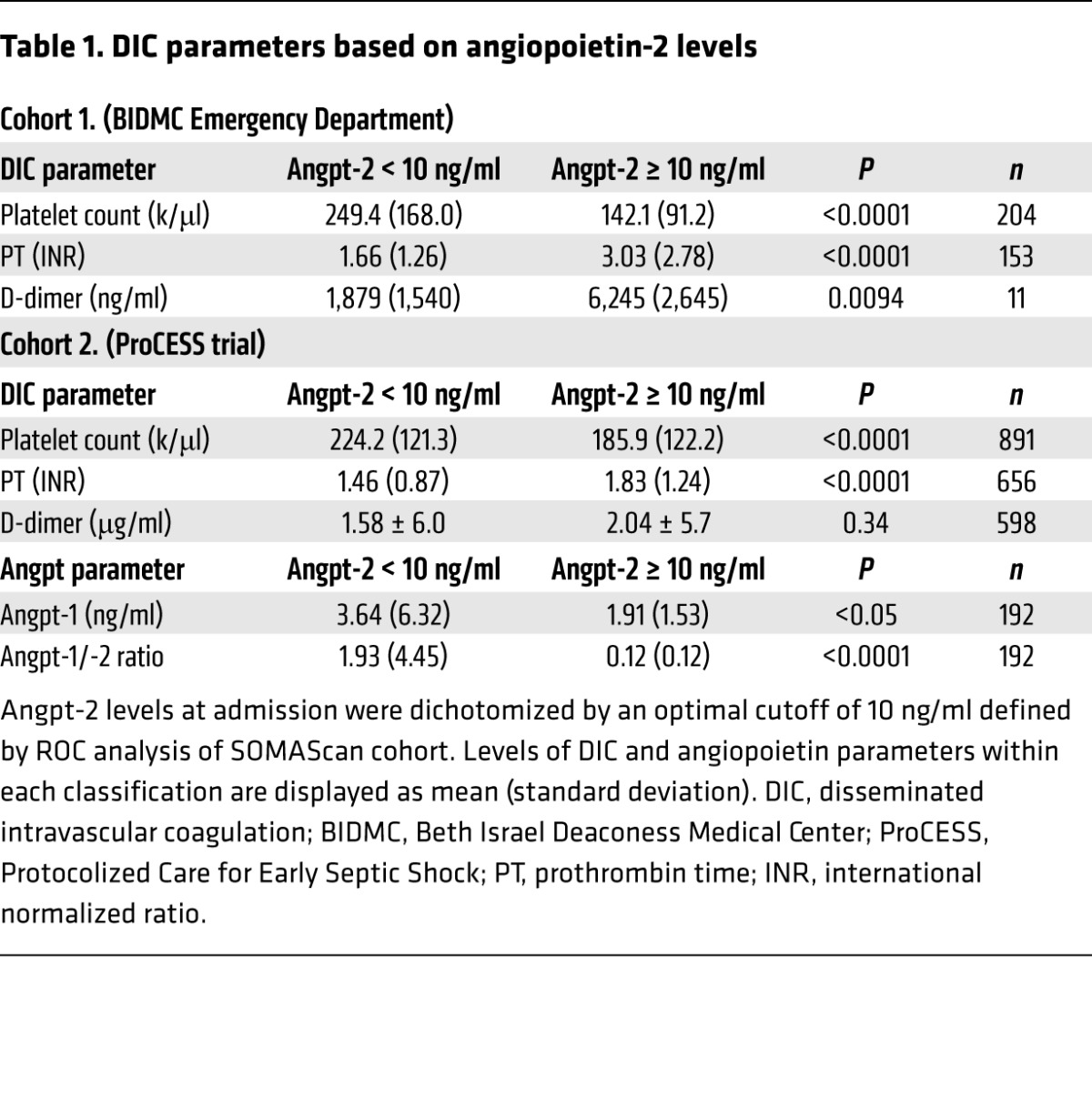
Table 2. Angiopoietin-2 and DIC parameters for sepsis mortality prediction.
Angpt/Tie2 axis and thrombotic response are disrupted prior to overt septic DIC.
We next tested the role of aberrant Angpt/Tie2 signaling in thrombus formation during sepsis. To do this, we utilized Gram-negative bacterial endotoxin lipopolysaccharide (LPS), an agent that induces features of DIC (40). We assessed consensus DIC hematological markers including platelet count, ex vivo clotting time, and fibrin-degradation products, i.e., D-dimer at baseline, 3, or 12 hours after LPS injection in order to determine the kinetics of disease progression. Whereas classical markers of DIC were all present only at the late 12-hour stage (Figure 3, A–D), the model yielded a progressive increase in sepsis severity (Figure 3E), suggesting the possibility of earlier events contributing to disease. Prothrombotic manifestations, evidenced by elevated thrombin-antithrombin (TAT) complex levels, a marker for intravascular thrombin generation, were present by the early 3-hour time point (Figure 3F). Levels of phosphorylated (active) Tie2 were significantly decreased in endotoxemic mice at the early 3-hour time point compared with saline controls (Figure 3, G and H). The temporal profile of the Angpt-1/Angpt-2 ratio in circulation (Supplemental Figure 2A) suggested that an early decline in Angpt-1 (Figure 3I) contributed to reduced Tie2 signaling, which in turn, derepressed Angpt-2 (Figure 3J), consistent with prior studies (19, 41–44). To assess whether the loss of vascular quiescence indicated by reduced Tie2 signaling was more broadly manifest, we analyzed a panel of endothelial activation markers in plasma collected at 3 hours after LPS; PAI-1, von Willebrand factor (VWF), and soluble forms of endothelial adhesion molecules (sE-selectin and sVCAM-1) were all elevated at the early 3-hour time point (Supplemental Figure 2, B–E).
Figure 3. Disruption of the Angpt/Tie2 axis and endothelial activation precede the onset of DIC in endotoxemic mice.

Markers of DIC, including (A) whole-blood platelet count, (B) plasma D-dimer levels, (C) prothrombin time (PT), and (D) activated partial thromboplastin time (aPTT) depicted as time to clot (seconds). (E) Disease severity signs (murine sepsis score) and (F) levels of circulating thrombin-antithrombin (TAT) complex were measured in C57BL/6J mice challenged with LPS (10 mg/kg) or saline at specific time points. (G) Immunoblots for p-Tie2 (p-Tie2Y992) and total Tie2 from lung tissue isolated at 3 hours after LPS challenge. (H) Densitometric analysis of p-Tie2 relative to total Tie2 from G (LPS = red, n = 7) normalized to saline control from 3 independent Western blots. (I and J) Plasma Angpt-1 and Angpt-2 kinetics following LPS determined by ELISA (n ≥ 5 per group). Data represent the mean ± SEM (n ≥ 5 per group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Mann-Whitney U test (2 groups) or 1-way ANOVA (3 groups).
In order to study sepsis-induced thrombus formation in the complex in vivo environment, we monitored real-time platelet accumulation and fibrin formation in microvessels following focal laser injury within 3 hours after LPS exposure. The early time window enabled us to focus on prothrombotic events that precede overt coagulopathy. Platelet deposition and fibrin accumulation were elevated in both arterioles (Figure 4A) and venules (Supplemental Figure 3) of endotoxemic mice compared with vehicle-treated animals. Median fluorescence intensities and AUCs of individual thrombi for platelet (Figure 4, B and C) and fibrin (Figure 4, D and E) signal were significantly increased in mice exposed to LPS. However, when removed from the context of the vessel wall interaction, platelets from endotoxemic mice showed no change in propensity to aggregate (Figure 4, F and G; mean aggregation ± SD: 97% ± 7.02%, n = 5 for saline vs. 99.8% ± 11.2%, n = 9 for LPS) at subthreshold and increasing concentrations of classical platelet agonists (Supplemental Figure 4). Furthermore, the exuberant thrombus formation during early endotoxemia was not attributable to changes in platelet activation, as assessed by platelet-factor 4 levels (Figure 4H). Together, these results implicate an early and coordinated response of the endothelium to systemic inflammation that promotes thrombus formation before the onset of overt DIC.
Figure 4. Endotoxemic mice develop increased thrombotic response before the onset of overt DIC.

(A) Representative binarized images of thrombus formation in response to laser injury in cremaster arterioles of mice exposed to saline or LPS (10 mg/kg) for 1–3 hours. Platelet accumulation (red, Dylight 649) and fibrin generation (green, Dylight 488) were visualized for 180 seconds following injury. Scale bar: 10 μm. Median integrated fluorescence intensity and area under the curve (AUC) were calculated for platelets (B and C) and fibrin (D and E). (C and E) Data are represented as the median AUC of individual thrombi (saline n = 36; LPS n = 34). *P < 0.05; ***P < 0.001, Mann-Whitney U test. PAR4-mediated platelet aggregation was measured in platelet-rich plasma obtained from C57BL/6J mice 3 hours after saline or LPS injection showing (F) quantification of aggregation and (G) a representative aggregation experiment of 3 independent experiments (saline = gray tracing; LPS = red tracing). (H) Platelet-factor 4 (PF4) levels were measured in plasma of C57BL/6J mice at 0 or 3 hours after LPS (10 mg/kg) (n = 9–11). Mann-Whitney U test.
Tie2 deficiency alone recapitulates sepsis-associated thrombotic response.
As dysregulation of the Angpt/Tie2 pathway was associated with a prothrombotic state, we next studied microvascular thrombus formation in Tie2+/– mice. Heterozygous mice are indistinguishable from wild-type (WT) littermates at baseline, but exhibit increased susceptibility to inflammation and infections (19, 37, 45). Unexpectedly, partial Tie2 deficiency alone was sufficient to enhance fibrin deposition independent of LPS (Figure 5, A–E). The enhanced fibrin deposition in Tie2+/– mice compared with Tie2+/+ mice was not attributable to differences in platelet accumulation at the injury site (Figure 5, B and C), circulating platelet count (mean [platelets/μl × 105] ± SD: 6.9 ± 0.7, n = 8 for Tie2+/+ vs. 6.7 ± 0.8, n = 7 for Tie2+/–), ex vivo coagulation parameters (mean PT [seconds] ± SD: 11.57 ± 0.94, n = 7 for Tie2+/+ vs. 11.14 ± 0.52, n = 15 for Tie2+/– and mean activated partial thromboplastin time [aPTT, in seconds] ± SD: 33.91 ± 5.34, n = 7 for Tie2+/+ vs. 31.77 ± 3.83, n = 13 for Tie2+/–) or baseline differences in circulating white cells (Supplemental Figure 5). However, when challenged with LPS, Tie2+/– mice exhibited an exacerbated response and progression to DIC-type complications, with increased PT and elevated thrombin generation, as indicated by increased TAT compared with Tie2+/+ controls (Supplemental Figure 6), supporting the suggestion that Tie2 is important for regulation of thrombus formation on the vessel wall and loss of Tie2 function may have detrimental effects on excessive thrombosis.
Figure 5. Tie2 deficiency alone recapitulates sepsis-associated thrombotic response.

(A–E) Platelet and fibrin accumulation was monitored in Tie2 heterozygous mice (Tie2+/–) and wild-type littermate controls (Tie2+/+) at the site of laser injury using anti-platelet Dylight 649–labeled and anti-fibrin Dylight 488–labeled antibodies. (A) Representative binarized images of a single thrombus (platelet = red; fibrin = green) at time of laser injury (0 seconds) and 30 to 120 seconds following laser injury. Median integrated fluorescence intensities and AUC were calculated for platelets (B and C) and fibrin (D and E). (C and E) Data are represented as the median for individual thrombi (Tie2+/–, n = 41; Tie2+/+, n = 40). (F–J) Platelet and fibrin generation was monitored in wild-type mice infused with eptifibatide (10 μg/g body weight) and exposed to saline or 10 mg/kg LPS. (F) Representative binarized images of thrombus formation, at time of laser injury (0 seconds) and up to 120 seconds following laser injury. Median integrated fluorescence intensities were calculated for platelets (G) and fibrin (I). (H and J) Data are represented as median individual thrombi (saline, n = 35; LPS n = 32). **P < 0.01, Mann-Whitney U test. Scale bars: 10 μm.
To evaluate the role for platelets within this context, we returned to the endotoxemia model in WT C57BL/6 mice in which we applied the platelet glycoprotein αIIbβ3 inhibitor eptifibatide to inhibit platelet aggregation at the injury site. Despite virtual absence of platelet accumulation (Figure 5, F–H), fibrin accumulation was increased in endotoxemic mice (Figure 5F, I and J), analogous to both the endotoxemia model without eptifibatide (Figure 4, A, D, and E) and the nonendotoxemic Tie2+/- mice (Figure 5, A, D, and E). These results suggest that the Tie2 signaling deficiency arising in sepsis is sufficient to provoke fibrin deposition on the vessel wall.
Tie2 activation suppresses prothrombotic actions of LPS on the endothelium.
To evaluate the role of the endothelium and Tie2 directly, we applied LPS and human plasma to primary human endothelial cells, then monitored the generation of factor Xa (FXa) and thrombin. The sequential action of these serine proteases converts fibrinogen to fibrin. LPS enhanced FXa and thrombin generation on the cultured endothelium; these effects were significantly attenuated by Angpt-1 application (Figure 6, A–D), with a more pronounced effect on thrombin generation even when Angpt-1 was applied to cells 2 hours after LPS exposure (Supplemental Figure 7).
Figure 6. Tie2 activation suppresses prothrombotic actions of LPS on the endothelium.

FXa (A and B) and thrombin (C and D) generation was determined on human umbilical vein endothelial cells (HUVECs) incubated with Angpt-1 (200 ng/ml 30 minutes prior to LPS), anti–tissue factor antibody (α-TF Ab, 10 μg/ml) or lactadherin (Lact., 100 nM) and exposed to LPS complex (LPS [100 ng/ml], sCD14 [100 ng/ml], and LBP [10 ng/ml]) or vehicle control for 3 hours. Representative experiments (mean ± SEM) are depicted as absorbance (405 nm) or arbitrary fluorescence units (RFU) as a function of time. (B and D) The rate of the reaction for FXa and thrombin generation converted to units/ml and normalized to controls. Immunoblot analysis (E) and quantification (F) of TF expression relative to GAPDH (n = 3) in lysates of LPS-stimulated HUVECs with and without Angpt-1. Flow cytometric analysis of (G) TF exposure, quantified from dot plots of FITC fluorescence intensity versus forward scatter and quantified as percentage of TF-positive cells relative to control and (H) phosphatidylserine (PtdSer) exposure, quantified as percentage of annexin V–FITC–positive and PI-negative cells (lower right quadrant of I). (I) Representative flow cytometric quadrant analysis of PtdSer exposure (annexin V–FITC) and cell death (PI) (n = 4–6 per group). *P < 0.05, **P < 0.01, ***P < 0.001 by 1-way ANOVA with Bonferroni’s posttest.
Since endothelial TF exposure was necessary for LPS-dependent FXa and thrombin generation (Figure 6, B and D), we next determined the effect of Angpt-1 on endothelial TF. Studies using a TF-specific antibody (Supplemental Figures 8, A and B) demonstrated that preapplication of Angpt-1 reduced both the level and surface exposure of TF induced by LPS (Figure 6, E–G), suggesting that constitutive Tie2 activation may lead to TF downregulation in quiescent endothelial cells. When we forced TF expression, Angpt-1’s ability to attenuate FXa generation was abrogated (Supplemental Figure 8, C and D), supporting a protective role for the Angpt-1/Tie2 pathway, in part, by regulating TF expression. Endothelial PI3K and Akt are activated by Tie2, and in nonendothelial cells, this signaling cascade suppresses TF (46, 47). Using Tie2 RNAi (Supplemental Figure 9, A and B) and a pharmacological inhibitor of PI3K to block Akt phosphorylation, we confirmed that Angpt-1 required intact Tie2 expression and downstream PI3K/Akt signaling in order to counteract LPS-induced FXa generation (Supplemental Figure 9, C–E).
To explain the effect of Angpt-1 on thrombin generation delivered after LPS exposure, we next found that LPS-treated HUVECs mimic the externalization of the anionic phospholipid phosphatidylserine (PtdSer) observed on activated platelets (48), with LPS-dependent FXa and thrombin generation reduced by lactadherin, a stereospecific blocker of PtdSer (Figure 6, B and D). LPS increased the population of FITC-labeled annexin V–positive HUVECs compared with controls, indicative of increased PtdSer exposure, and Angpt-1 effectively counteracted this effect (Figure 6, H and I). Cumulatively, these results demonstrate that Angpt-1 counteracts the LPS-dependent conversion of the endothelial surface to a procoagulant state.
Tie2 activation normalizes the septic thrombotic response to injury without increasing bleeding risk.
We next administered Angpt-1 via gene transfer in endotoxemic mice (Supplemental Figure 10). Angpt-1 normalized fibrin deposition at sites of vascular injury in LPS-exposed mice to levels observed in control-virus-treated mice (Figure 7, A, D, and E), with a trend towards lower platelet accumulation (Figure 7, A–C). This was not attributable to baseline differences in coagulation parameters or the potential to form thrombi in mice receiving adenoviral vector injections (Supplemental Figure 11). The ability of Angpt-1 delivery to normalize the thrombotic response was confirmed in an independent model of polymicrobial infection (cecal ligation and puncture, CLP; Supplemental Figure 12), complementing the findings from endotoxemia that normalization of Tie2 signaling may counteract septic DIC. Despite being stored and released by activated platelets (Supplemental Figure 13, A and B), increased systemic Angpt-1 levels did not affect platelet aggregation ex vivo in the absence of vessel wall interactions (Supplemental Figure 13, C and D). Importantly, Angpt-1 did not increase the risk of bleeding complications, a problem limiting the use of current clinical antithrombotic therapies for DIC. In fact, Angpt-1 treatment restored normal bleeding time and lowered the number of rebleeding events in endotoxemic mice (Figure 7, F and G) without a direct effect on total platelet count (Supplemental Figure 13E).
Figure 7. Tie2 activation normalizes the septic thrombotic response to injury without increasing bleeding risk.

Representative binarized images of thrombus formation (A) following laser injury in mice injected with control adenovirus (CtlAdv) (top) or an adenovirus expressing Angpt-1 (AdAngpt-1) after LPS injection (1–3 hours) (platelets = red; fibrin = green). Scale bar: 10 μm. Median integrated platelet (B) and fibrin (D) fluorescence intensities and AUC (C and E) were calculated for individual thrombi, CtlAdv (n = 2; 33 thrombi), CtlAdv plus LPS (n = 3; 37 thrombi), AdAngpt-1 (n = 3; 38 thrombi), and Angpt-1 plus LPS (n = 3; 37 thrombi). (F) Time to bleeding cessation (seconds) in CtlAdv and AdAngpt-1 mice 3 hours after LPS challenge (n = 18 or 19 mice). (G) The number of re-bleeding events during 10-minute observation window stratified as follows: 0, 1–3, or 4 or more events. Representative immunoblot (H) and quantification (I) of fibrin in liver isolates relative to pan cadherin. (J) Relative change in TAT complex levels at 3 hours after LPS challenge in CtlAdV- and AdAngpt-1–treated mice. Angpt-2 levels in (K) plasma and (L) lungs of CtlAdV or AdAngpt-1 mice after LPS (n = 5 per group). *P < 0.05, **P < 0.01 by 1-way ANOVA. (M) Representative immunoblots of p-Tie2 (p-Tie2Y992) and total Tie2 levels in lung isolates.
In agreement with our in vitro observations, delivery of Angpt-1 reduced spontaneous in situ fibrin accumulation triggered by LPS (Figure 7, H and I). Angpt-1 also reduced thrombin generation in response to LPS compared with generation observed in control-vector-treated endotoxemic mice, indicated by measuring the fold change in TAT complex levels (Figure 7J). Furthermore, Angpt-1 significantly reduced Angpt-2 expression (Figure 7, L and M), heightened levels of which were associated with DIC in our human sepsis cohorts (Figure 1 and Tables 1 and 2), via maintenance of Tie2 activation (Figure 7M). These results extend the cellular findings of Figure 6 to show that Angpt-1 normalizes thrombus formation in vivo during systemic inflammation without increasing bleeding risk.
Discussion
The present work demonstrates a pivotal and specific role for endothelial homeostasis signaled through Tie2 to combat disordered thrombus formation arising from systemic inflammation. Whereas DIC research has traditionally focused on platelets, coagulation factors, and leukocytes, a role for the endothelium, and more specifically the Angpt/Tie2 pathway, was suggested by unbiased discovery in human subjects that demonstrated a coordinated activation of this understudied single cell layer in septic DIC. These results were sufficiently robust to be reaffirmed in more than 1,000 additional patients from 2 independent cohorts, demonstrating that high Angpt-2, released by activated endothelial cells, is not only linked to markers of DIC, but also an accurate determinant of patient outcome. Studies in a mouse model of sepsis-induced DIC coupled with cellular experiments showed a mechanistic involvement of the endothelial Tie2 pathway in regulating thrombus formation.
These results support the concept that the endothelium is a pivotal contributor to inflammatory thrombus formation. Endothelial activation, including dysfunction of the Tie2 axis, temporally preceded the measurable involvement of platelets and coagulation factors. Moreover, this endothelial dysfunction could be targeted in a highly precise molecular fashion, using an adenoviral construct expressing the vascular protective protein Angpt-1 to maintain Tie2 signaling, both to normalize the initial fibrin deposition response of early sepsis, without adversely interfering with hemostasis, and to abrogate the elevated thrombin levels in DIC. Antecedent involvement of the endothelium and phenotypic reversal with an endothelium-targeted intervention therefore strongly implicate the endothelium in the pathogenesis of septic DIC.
An argument for the endothelium’s role in septic DIC is further strengthened by several additional lines of evidence. First, platelets are considered the primary surface for coagulation at sites of vascular injury. Nonetheless, our results indicate that the increased fibrin deposition at sites of injury associated with early sepsis occurs even when platelets are inhibited (Figure 5, F–J). Although this does not exclude a role for platelets in septic fibrin generation, the present results are consistent with previous in vivo studies showing that activated endothelial cells can support assembly of the prothrombinase complex required for thrombin and fibrin generation (17, 49). Second, while Tie2 signaling has long been known to decline in sepsis, here we found that genetically reduced Tie2 alone was sufficient to mimic the excessive fibrin deposition of sepsis (Figure 5, A–E). The small fraction of monocytes that express Tie2 are unlikely to account for this fibrin response (50); furthermore, Angpt-1 gene transfer did not significantly impact levels of red or white blood cells, including monocytes (Supplemental Figure 14). Third, the cellular studies directly identify endothelial inflammation as a sufficient provocation to catalyze fibrin formation. These experiments revealed that LPS upregulates parallel inducers of the coagulation cascade, TF and PtdSer, a finding that affirms and extends previous studies of the endothelium in vivo and in vitro (17, 49) and that Angpt-1 exerts actions to blunt this response. While our in vitro studies using HUVECs have limitations inherent to studies of coagulation on cultured cells, it is a well-established model (51–53). Finally, in endotoxemic mice, Angpt-1 gene transfer improved both the early prothrombotic response of systemic inflammation and the late fibrin accumulation, suggesting an action most consistent with a normalization effect. And unlike existing DIC therapies, this normalization was achieved without increasing bleeding events. Together, the results suggest that endothelial Tie2 disruption is necessary for endothelium-mediated thrombosis in early sepsis and that Tie2 activation is sufficient to ameliorate this process.
The striking normalization effect achieved by exogenous Angpt-1 on thrombus formation may unveil an adaptive endogenous process supported by platelets (54). Platelets may contain substantial amounts of Angpt-1 (55), so their aggregation at nascent thrombi may release Angpt-1 to restore endothelial homeostasis. In sepsis, platelet-derived Angpt-1 could also restore other aspects of endothelial quiescence such as barrier function and antiinflammation (56). The present results conversely suggest that the fall in circulating Angpt-1 during endotoxemia may contribute to DIC, raising questions about the major source(s) of Angpt-1 during health and its turnover in sepsis that will require further study.
The present results also advance an important contrast against vascular endothelial growth factor (VEGF). As major control pathways for the endothelium, both the Angpts and VEGF regulate angiogenesis, barrier function, and inflammation — Angpt-2 and VEGF both weaken the vascular barrier and promote inflammation, actions that Angpt-1 counteracts. Yet their deficiencies result in starkly divergent defects in clot formation. Our data show that Tie2 deficiency yielded fibrin-dominant lesions at sites of vascular injury, whereas VEGF deficiency is known to promote the platelet-rich thrombi found in thrombotic microangiopathy (TMA) (57–59). Since both injury-induced and spontaneous fibrin deposition during endotoxemia were attenuated by excess Angpt-1 (Figure 7, D and H) yet thrombocytopenia was not prevented by Angpt-1 (Supplemental Figure 13E), the results collectively suggest that the effects of sepsis on platelet abundance may not only be driven by consumption, but also additional mechanisms such as pulmonary sequestration and even blunted megakaryopoiesis (60–62). Regarding the status of the endothelium, just as certain forms of TMA are responsive to restored VEGF signaling (63), septic DIC may be similarly responsive to restored Tie2 signaling. With a ligand-independent activator of Tie2 and antibodies to inhibit Angpt-2 all in clinical development, there is significant translational potential for Tie2 stimulation (43, 64, 65). Therefore, the mechanisms underlying these phenotypic differences should be further investigated.
While the endothelium has long been considered a contributor to septic DIC, an easily measurable, mechanistic, and pharmacologically targetable response has been elusive. Therapeutic strategies such as Angpt-1 targeting the endothelium may reduce the risks of iatrogenic harm from the therapeutic overshoots associated with current empiric DIC treatments. Our results suggest that Tie2 stimulation after the onset of sepsis may still be beneficial against fibrin-rich clot formation (Supplemental Figure 7), in line with recent results demonstrating a survival benefit in septic mice even with delayed Tie2 stimulation (32). Application of Tie2 axis markers may aid the identification of individuals with pathomechanisms amenable to Tie2-stimulating therapies, as suggested by an earlier study of Angpt-2 in septic DIC (66) and augmented by the present investigation of more than 1,000 subjects. Considered together, these early human studies suggest the potential for diagnosing DIC risk by measuring its causal mechanisms rather than its consequences on platelet counts and other parameters. Beyond septic DIC, disordered coagulation and hemostasis occurs in many severe infections, ranging from Ebola virus disease to cerebral malaria. Clinical data and experimental models link disturbances in the Angpt/Tie2 pathway to several of these disease states (37, 38, 67–71), raising the possibility that defense of endothelial homeostasis can counteract prothrombotic mechanisms arising from diverse infections.
In summary, the present work demonstrates a pivotal and specific role for endothelial homeostasis signaled through Tie2 to combat disordered thrombus formation arising during systemic inflammation. These findings may open new avenues to study the role of the endothelium in diseases of abnormal coagulation, and also to develop new markers and therapies for individuals affected by septic DIC.
Methods
Human samples and patient selection for SOMAScan.
Samples for SOMAScan analysis were collected from the Molecular Epidemiology of ARDS (MEARDS) prospective cohort study (ClinicalTrials.gov Identifier: NCT00006496), which recruited patients at the ICUs of Massachusetts General Hospital (MGH) and Beth Israel Deaconess Medical Center (BIDMC) between 1998 and 2014. Study population and procedures were described previously (19). For the purpose of the current study, sepsis and sepsis DIC groups were defined based on the International Society of Thrombosis and Hemostasis (ISTH) DIC Scoring system (72), determined by laboratory tests for the following parameters: low platelet count, prolonged clotting time, and increased D-dimer and/or decreased fibrinogen.
Proteomic analysis using slow-off-rate-modified aptamer SOMAScan platform.
EDTA-plasma from 7 patients with sepsis and seven patients with sepsis DIC were analyzed using DNA-aptamer-based recognition on the SOMAScan platform (SomaLogic) at the BIDMC Genomics, Proteomics, Bioinformatics and Systems Biology Center. Samples were prepared and run using the SOMAScan Assay Kit for human plasma, 1.3k (catalog 900-00011), according to the standard protocols from SomaLogic, as described previously (73). Five pooled plasma controls and 1 no-protein buffer control were run in parallel with the samples. Median normalization and calibration of the data was performed according to the standard quality control protocols at SomaLogic. All samples passed the established quality control criteria. P values were determined by t test.
Network, clustering, and functional enrichment analysis.
Volcano plot was generated using binary fold-change and –log2 P value for plasma analytes measured by SomaScan. Proteins with significant differences in abundance between groups were selected based on a fold-change greater than 1.2 and P value of 0.05 or less and included in subsequent network analysis. The selected proteins were searched against the STRING database version 10.5 for protein-protein interactions and displayed as a functional network (35). Interactions were considered with a STRING confidence score of 0.7 or higher (high to highest confidence) culled from the “experimental” and “databases” categories. Nodes with no associations to other proteins in the network were removed. A k-means clustering algorithm was performed to identify densely connected regions (k-means = 9). Functional description of clusters was assigned based on a manual curated evaluation of enriched KEGG pathway, Gene Ontology (GO) terms, and PubMed literature search.
ELISA validation of SOMAScan.
EDTA-plasma concentrations of Angpt-2 and Angpt-1 were measured by ELISA (R&D Systems), according to the manufacturer’s instructions.
Clinical validation of SOMAScan.
A secondary analysis of patients who participated in 2 published studies was undertaken: (a) the Protocolized Care for Early Septic Shock (ProCESS) trial, a patient-level randomized, multicenter interventional trial of alternative resuscitation strategies in Emergency Department Sepsis (ClinicalTrials.gov Identifier: NCT00793442) (39); and (b) an IRB-approved prospective cohort study of 221 adult patients age 18 years and older presenting to the Emergency Department of BIDMC with suspected infection (27). Blood was collected upon enrollment, immediately centrifuged, and then stored at –80°C for subsequent analysis. PT, international normalized ratio (INR), aPTT, and platelet counts were measured using clinically available laboratory testing. D-dimers were measured using a latex agglutination assay (Diagnostica Stago). All assays were performed according to the manufacturer’s specifications. Ethics committee approval was received from participating sites, University of Pittsburgh and BIDMC Committee for Clinical Investigations.
Mice.
C57BL/6J mice (male, 8–12 weeks old) were purchased from The Jackson Laboratory. Heterozygous Tie2 mice (Tie2+/–) and littermate (Tie2+/+) controls were generated on a CD1 background as described previously (45). Mice were anesthetized through intraperitoneal (i.p.) injection of a ketamine (125 mg/kg) and xylazine (12.5 mg/kg) mixture in sterile saline. For laser-injury thrombosis experiments, pentobarbital (5 mg/kg) was administered through a jugular vein cannula to maintain anesthesia. For retro-orbital blood collection, mice were sedated using isoflurane.
Endotoxemia model.
Mice were administered LPS from E. coli serotype O111:B4 (Sigma-Aldrich; 10 mg/kg body weight) via i.p. injection. Disease severity was assessed using a 7-parameter scoring system for murine sepsis, as described previously (74).
Adenoviral gene transfer.
Mice were injected with an adenovirus expressing human Angpt-1 (AdAngpt-1; 2 × 1010 viral particles, Qbiogene Inc.) or GFP (Ad-CMV-GFP, control; 2 × 1010 viral particles, Qbiogene Inc. and Vector Biosystems) via intravenous (i.v.) tail injection in saline 72 hours prior to saline or LPS administration (75). Levels of Angpt-1 in plasma were confirmed by DuoSet ELISA against human Angpt-1 (R&D Systems). Mice that were assigned to receive AdAngpt-1 but did not have elevated Angpt-1 levels (above the control adenovirus group) were excluded from analysis.
Platelet aggregation.
Platelet-rich plasma (PRP) from citrated whole blood (3.2% sodium citrate) obtained by retro-orbital plexus puncture using glass capillaries was prepared by differentiated centrifugation with HEPES Tyrode buffer (20 mM HEPES, 134 mM NaCl, 0.34 mM NaH2PO4, 2.9 mM KCl, 12 mM NaHCO3, 1 mM MgCl2, 5 mM glucose; pH 7.3) as described previously (76). PRP was diluted to a final platelet count of 200,000 platelets per μl. Platelet aggregation was initiated using PAR4-agonist (AYPGKF; Sigma-Aldrich) or collagen (385, ChronoLog), at indicated concentrations, and measured using the ChronoLog 680 aggregometer. An agonist dose curve was run for each independent experiment to determine the lowest dose at which aggregation was obtained.
Endothelial and platelet activation markers.
Levels of soluble E-selectin, soluble VCAM, serpin E1/PAI-1, and platelet factor 4 (PF4) were measured in plasma using ELISA kits (Quantikine and DuoSet Immunoassays, R&D Systems), according to the manufacturer’s protocols. VWF levels were measured using an in-house-developed ELISA as described previously (77). Angpt-2 and TAT were measured using mouse ELISA kits, as per the manufacturer’s protocol (Abcam).
Coagulation assays.
PT (Neoplastin Cl Plus), aPTT, and D-dimer (Asserachrom D DI kit) were measured in plasma according to the manufacturer’s instructions (Diagnostica Stago Inc.). Hematological analysis of total blood count was performed using a Hemavet 850FS (Drew Scientific) for platelet, white blood cell (WBC), monocyte (MO), lymphocyte (LY) and red blood cell (RBC) counts, and hemoglobin (Hb) and hematocrit (HCT).
Laser-injury thrombosis model.
Thrombus formation in response to laser injury was measured as described previously (49, 78). Briefly, cremaster arterioles and venules were injured using a MicroPoint Laser system (Photonics Instruments). Platelet and fibrin accumulation was measured by infusion of Dylight 647–labeled anti-platelet antibodies (CD42b; 0.1 mg/g body weight; Emfret Analytics) and Dylight 488–labeled (Thermo Fisher Scientific) anti-fibrin antibodies (59D8; 0.5 mg/g body weight) through a jugular vein catheter. Data acquisition was done prior and subsequent to laser injury in 2 channels (488/520 nm and 640/670 nm). Images were captured for 180 seconds at 0.5 frames/second using a CCD camera (Hamamatsu). Data were analyzed using Slidebook 6.0 (Intelligent Imaging Innovations). Data from 30 to 40 thrombi were used to determine the median value of the integrated fluorescence intensity to account for the variability of thrombus formation at any given set of experimental conditions. AUC was calculated for individual thrombi to evaluate statistical significance. For experiments with eptifibatide, mice were injected i.v. with eptifibatide acetate salt (10 μg/g body weight; Bachem) dissolved in saline every 10–15 minutes.
Tail bleed assay.
Mice were anesthetized with a ketamine (125 mg/kg) and xylazine (12.5 mg/kg) mixture in sterile saline via i.p. injection prior to a surgical dissection of the tail (3 mm from tip). Amputated tails were immediately immersed in 50 ml buffered saline prewarmed to 37°C and the time to bleeding cessation was recorded within 10 minutes (including start and stop time). The number of times bleeding stopped and re-started within the 10-minute observation window were recorded and categorized into bins as follows: 0 events, 1–3 rebleed events, or 4 or more events.
Endothelial cell culture.
HUVEC (passage 1–5, pooled donors obtained from Lonza) monolayers cultured in EGM-2 media (Lonza) on gelatin- or collagen-coated culture vessels were treated with a cocktail of LPS (100 ng/ml), LPS-binding protein (LBP; R&D Systems; 10 ng/ml), and sCD14 (R&D Systems; 100 ng/ml) or vehicle (sCD14 and LBP) for 3 hours at 37°C. Pre- and postincubation with rAngpt-1 (R&D Systems; 200 ng/ml) or inhibitors was performed at indicated concentrations and time periods. A stable Ea.hy926 cell line expressing TF (Ea.hy629-TF) was made by transfecting Ea.hy926 cells with pLX304 vector expressing the F3 gene (NM_001993.4, clone HsCD00413770) using Lipofectamine 2000 (Life Technologies) and selecting positive colonies with blasticidin (6 μg/ml). A cell line with empty pLX304 vector was used as a control. Ea.hy926 cells were cultured in DMEM with 10% FBS and 5 μg/ml blasticidin.
siRNA transfection.
HUVECs grown to 60%–70% confluence were transfected with Silencer Select (Thermo Fisher Scientific) negative control siRNA or a validated TEK (Tie-2) siRNA (s13984) using HiPerfect transfection reagent (Qiagen). A Cells-to-Ct 1-Step Taqman kit (Ambion/Life Technologies) and Taqman gene expression assay probe (Hs00945155-m1 TEK) were used to confirm reduction of gene expression following 48 hours of transfection. Quantitative reverse transcription PCR (qRT-PCR) was performed according to the manufacturer’s instructions (Thermo Fisher Scientific) at the BIDMC Molecular Medicine core.
Thrombin and FXa generation assays.
Confluent HUVECs grown in 96-well plates were incubated with vehicle or LPS. For thrombin generation experiments, wells with confluent HUVECs (treated as above in “Endothelial cell culture”) were incubated with a mixture composed of 80 μl pooled human plasma (4 or 5 donors) to supply coagulation factors, the fibrin polymerization inhibitor H-Gly-Pro-Arg-Pro-OH (GPRP, 5 mM), 20 μl HEPES-buffered saline, pH 7.4 (HBS) and 1.5 to 5 mM CaCl2. Thrombin levels were measured using the SN-20 fluorogenic substrate (Haematologic Technologies). Fluorescence was measured every minute for 20 minutes using the Synergy 4 plate reader (BioTek). For FXa generation experiments, washed cells were incubated in HBS-BSA buffer supplemented with FX (100 nM; Haematologic Technologies), FVIIa (0.67 nM; Haematologic Technologies), calcium (5 mM), and the chromogenic substrate CS-11(22) (200 μM; BIOPHEN). Absorbance at 405 nm was measured every minute for 1.5 hours using the SpectraMax spectrophotometer. Thrombin and FXa levels were determined by converting reaction rate to units/ml. For experiments with anti-TF antibody and lactadherin, endothelial cells were incubated with anti-TF antibody (4509, Sekisui Diagnostics) or lactadherin (Haematologic Technologies) for 15 minutes prior to addition of plasma or buffer containing substrates.
Flow cytometry.
Confluent HUVECs (treated as above in “Endothelial cell culture”) were harvested using Accutase (Sigma-Aldrich) and pelleted at 300 g for 5 minutes. For analysis of TF exposure, cell pellets were resuspended in PBS containing 0.5% BSA and labeled with FITC-conjugated mouse anti–human TF or isotype control (100 μg/ml final concentration) for 30 minutes at 4°C. Washed samples were run in a Gallios flow cytometer (Beckman Coulter) and analyzed using Kaluza software. Dot plots of FITC fluorescence intensity versus forward scatter were derived to determine the staining profile of control and treated cells. The percentage of TF-positive cells was calculated relative to each study control set at 100%. Annexin V–FITC was used to detect Ca2+-dependent PtdSer exposure using the FITC Annexin V/Dead Cell Apoptosis kit (Invitrogen). HUVECs cultured and stimulated with LPS with and without Angpt-1 (described in “Endothelial cell culture”) and exposed to HEPES buffer containing 2.5 mM CaCl2 for 20 minutes were harvested using Accutase (Sigma-Aldrich) and washed in PBS. Cells (~1 × 106) were suspended in HEPES buffer (pH 7.4) containing 140 mM NaCl, 2.5 mM CaCl2, 5 μl annexin V–FITC, and 1 μg/ml propidium iodide (PI) and incubated in the dark for 15 minutes at room temperature. Events (10,000) were acquired in a BD FACSCanto and analyzed with BD FACSDiva Software. PtdSer-exposing cells were defined as the percentage of the parent population contained within quadrant Q4 (positive for FITC with low PI staining, a marker of cell death).
Preparation of protein lysates and Western blotting analysis.
HUVECs and tissue lysate (40 μg) protein suspensions extracted with RIPA buffer (Boston BioProducts) supplemented with protease and phosphatase inhibitor tablets (Roche Diagnostics), 1 mM EDTA, 1 mM Na3VO4, and 1 mM NaF were resolved by SDS-PAGE (4%–12% gradient gel, Invitrogen) under reducing conditions (NuPage SDS sample buffer containing β-mercaptoethanol). After transfer to a nitrocellulose membrane, targeted protein detection was accomplished using the following antibodies: polyclonal rabbit anti–phospho-Tie2/TEK (epitope to protein kinase domain, Tyr992, ABF131; Millipore) and monoclonal mouse anti–Tie2/TEK (clone Ab33; Millipore), Human/Mouse Phospho-Tie-2 (Tyr992, AF2720, R&D Systems), and goat anti–mouse/rat Tie2 (AF762, R&D Systems), phosphor-Akt (Ser473, 9271, Cell Signaling Technology), Akt (9272, Cell Signaling Technology), sheep anti-TF (Haematologic Technologies), an in-house anti-fibrin antibody made in a 59D8 hybridoma cell line, and anti-GAPDH (Cell Signaling Technology) or pan cadherin (AB6529, Abcam). Immunoblots were developed with SuperSignal West Pico/Femto Chemiluminescent Substrate (Thermo Fisher Scientific), and visualized with Syngene BioImage and GeneSnap image acquisition software. Protein phosphorylation levels were determined relative to the intensity of total protein level using antibodies from the same vendor.
Statistics.
Statistical calculations were performed with GraphPad Prism version 6.0. Sample size determination was based on the expected effect size and variability from previous observations of similar readouts in the investigators’ labs. Statistical significance for binary comparisons was assessed by Student’s t test, unless the data did not pass the Shapiro-Wilk test for normality in which case differences between groups were analyzed by Mann-Whitney U test. For comparison of more than 2 groups, 1-way ANOVAs were used, according to the experimental design, with Bonferroni’s multiple comparison test. P values of less than 0.05 were considered statistically significant and marked by asterisks. For the clinical cohort, SAS Version 9.3 was used to perform statistical analyses. The degree of diagnostic accuracy was determined by calculating the AUC with 95% confidence intervals. Unless otherwise indicated, data are represented as the mean ± SEM.
Study approvals.
Samples for SOMAScan analysis were collected from the Molecular Epidemiology of ARDS (MEARDS) prospective cohort study (ClinicalTrials.gov Identifier: NCT00006496), which recruited patients at the ICUs of MGH and BIDMC between 1998 and 2014. The study was reviewed and approved by IRBs of Harvard School of Public Health, MGH, and BIDMC. All participants or their surrogate care providers gave written informed consent. A secondary analysis of patients who participated in 2 published studies was undertaken: (a) the Protocolized Care for Early Septic Shock (ProCESS) trial, a patient-level randomized, multicenter interventional trial of alternative resuscitation strategies in Emergency Department Sepsis (ClinicalTrials.gov Identifier: NCT00793442) (39); and (b) an IRB-approved prospective cohort study of 221 adult patients age 18 years and older presenting to the Emergency Department of BIDMC with suspected infection (27). Ethics committee approval was received from participating sites: the University of Pittsburgh and the BIDMC Committee for Clinical Investigations. Mouse care and experimental procedures were performed in accordance with and under the approval of the BIDMC Institutional Animal Care and Use Committee.
Author contributions
SJH and KDC designed, conducted, and analyzed experiments. SAC, XC, SS, and SL conducted experiments. XG, JAK, TAL, NIS, and DCC designed, conducted, and analyzed human subjects studies. The manuscript was written by SJH, KDC, RF, and SMP with input from all authors.
Supplementary Material
Acknowledgments
We are grateful to S. Ananth Karumanchi for critical discussion, to Glenn Merrill-Skoloff for assistance with the microscopy, and to the ProCESS investigators for their contribution to subject enrollment and sample acquisition. This work was supported by the American Heart Association (grant 16POST31200017 to S.J. Higgins) and the NIH (T32HL007917 to K. De Ceunynck, R01HL091757 to J.A. Kellum and N.I. Shapiro, P50GM076659 to J.A. Kellum and N.I. Shapiro, R35HL135775 to R. Flaumenhaft, R01HL093234/R35HL139424 to S.M. Parikh, and R01HL125275 to R. Flaumenhaft and S.M. Parikh).
Version 1. 01/23/2018
In-Press Preview
Version 2. 03/05/2018
Electronic publication
Version 3. 03/19/2018
Scale bars clarified in Figures 5 and 7
Version 4. 04/02/2018
Print issue publication
Footnotes
Conflict of interest: SMP is listed as an inventor on a patent (WO2007033216A3) filed by Beth Israel Deaconess Medical Center. SJH is listed as an inventor on a patent (WO2015066426A3) pertaining to the use of angiopoietin-based interventions for treating complications of a disease or disorder associated with dysfunction of the Angpt/Tie2 pathway held by Regeneron Pharmaceuticals Inc./University Health Network.
Reference information: J Clin Invest. 2018;128(4):1471–1484.https://doi.org/10.1172/JCI97488.
Contributor Information
Sarah J. Higgins, Email: sjhiggin@bidmc.harvard.edu.
Karen De Ceunynck, Email: kdeceuny@bidmc.harvard.edu.
Xiuying Chen, Email: xchen6@bidmc.harvard.edu.
Xuesong Gu, Email: xgu@bidmc.harvard.edu.
Sharjeel A. Chaudhry, Email: sharjeel@gwmail.gwu.edu.
Sol Schulman, Email: sschulm1@bidmc.harvard.edu.
Towia A. Libermann, Email: tliberma@bidmc.harvard.edu.
Shulin Lu, Email: slu1@bidmc.harvard.edu.
Nathan I. Shapiro, Email: nshapiro@bidmc.harvard.edu.
David C. Christiani, Email: DCHRIS@hsph.harvard.edu.
Robert Flaumenhaft, Email: rflaumen@bidmc.harvard.edu.
Samir M. Parikh, Email: sparikh1@bidmc.harvard.edu.
References
- 1.Daniels R. What next for sepsis? Lancet Infect Dis. 2015;15(5):499–501. doi: 10.1016/S1473-3099(15)70147-7. [DOI] [PubMed] [Google Scholar]
- 2.Kissoon N, et al. World Federation of Pediatric Intensive Care and Critical Care Societies: Global Sepsis Initiative. Pediatr Crit Care Med. 2011;12(5):494–503. doi: 10.1097/PCC.0b013e318207096c. [DOI] [PubMed] [Google Scholar]
- 3.Seymour CW, Rea TD, Kahn JM, Walkey AJ, Yealy DM, Angus DC. Severe sepsis in pre-hospital emergency care: analysis of incidence, care, and outcome. Am J Respir Crit Care Med. 2012;186(12):1264–1271. doi: 10.1164/rccm.201204-0713OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers. 2016;2:16037. doi: 10.1038/nrdp.2016.37. [DOI] [PubMed] [Google Scholar]
- 5.Hawiger J, Veach RA, Zienkiewicz J. New paradigms in sepsis: from prevention to protection of failing microcirculation. J Thromb Haemost. 2015;13(10):1743–1756. doi: 10.1111/jth.13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esmon CT, Esmon NL. The link between vascular features and thrombosis. Annu Rev Physiol. 2011;73:503–514. doi: 10.1146/annurev-physiol-012110-142300. [DOI] [PubMed] [Google Scholar]
- 7.Wada H, et al. Guidance for diagnosis and treatment of DIC from harmonization of the recommendations from three guidelines. J Thromb Haemost. 2013;11(4):761–767. doi: 10.1111/jth.12155. [DOI] [PubMed] [Google Scholar]
- 8.Clark SR, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 9.Zimmerman GA, McIntyre TM, Prescott SM, Stafforini DM. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit Care Med. 2002;30(5 Suppl):S294–S301. doi: 10.1097/00003246-200205001-00020. [DOI] [PubMed] [Google Scholar]
- 10.Fisher CJ, et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med. 1996;334(26):1697–1702. doi: 10.1056/NEJM199606273342603. [DOI] [PubMed] [Google Scholar]
- 11.Abraham E, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb Sepsis Study Group. JAMA. 1995;273(12):934–941. [PubMed] [Google Scholar]
- 12.Opal SM, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309(11):1154–1162. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- 13.Russell JA. Management of sepsis. N Engl J Med. 2006;355(16):1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- 14.Warren BL, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286(15):1869–1878. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- 15.Abraham E, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290(2):238–247. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 16.Ranieri VM, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366(22):2055–2064. doi: 10.1056/NEJMoa1202290. [DOI] [PubMed] [Google Scholar]
- 17.Ivanciu L, Krishnaswamy S, Camire RM. New insights into the spatiotemporal localization of prothrombinase in vivo. Blood. 2014;124(11):1705–1714. doi: 10.1182/blood-2014-03-565010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shapiro NI, et al. The association of endothelial cell signaling, severity of illness, and organ dysfunction in sepsis. Crit Care. 2010;14(5):R182. doi: 10.1186/cc9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh CC, et al. Gene control of tyrosine kinase TIE2 and vascular manifestations of infections. Proc Natl Acad Sci USA. 2016;113(9):2472–2477. doi: 10.1073/pnas.1519467113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong AL, Haroon ZA, Werner S, Dewhirst MW, Greenberg CS, Peters KG. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res. 1997;81(4):567–574. doi: 10.1161/01.RES.81.4.567. [DOI] [PubMed] [Google Scholar]
- 21.Thurston G, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6(4):460–463. doi: 10.1038/74725. [DOI] [PubMed] [Google Scholar]
- 22.Thurston G, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286(5449):2511–2514. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 23.Benest AV, et al. Angiopoietin-2 is critical for cytokine-induced vascular leakage. PLoS One. 2013;8(8):e70459. doi: 10.1371/journal.pone.0070459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiedler U, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12(2):235–239. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- 25.Ziegler T, et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest. 2013;123:3436–3445. doi: 10.1172/JCI66549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clajus C, et al. Angiopoietin-2 is a potential mediator of endothelial barrier dysfunction following cardiopulmonary bypass. Cytokine. 2012;60(2):352–359. doi: 10.1016/j.cyto.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.David S, et al. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Crit Care Med. 2012;40(11):3034–3041. doi: 10.1097/CCM.0b013e31825fdc31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parikh SM, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. 2006;3(3):e46. doi: 10.1371/journal.pmed.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stiehl T, et al. Lung-targeted RNA interference against angiopoietin-2 ameliorates multiple organ dysfunction and death in sepsis. Crit Care Med. 2014;42(10):e654–e662. doi: 10.1097/CCM.0000000000000524. [DOI] [PubMed] [Google Scholar]
- 30.Yuan HT, Khankin EV, Karumanchi SA, Parikh SM. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol. 2009;29(8):2011–2022. doi: 10.1128/MCB.01472-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mueller SB, Kontos CD. Tie1: an orphan receptor provides context for angiopoietin-2/Tie2 signaling. J Clin Invest. 2016;126(9):3188–3191. doi: 10.1172/JCI89963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han S, et al. Amelioration of sepsis by TIE2 activation-induced vascular protection. Sci Transl Med. 2016;8(335):335ra55. doi: 10.1126/scitranslmed.aad9260. [DOI] [PubMed] [Google Scholar]
- 33.Parikh SM. Dysregulation of the angiopoietin-Tie-2 axis in sepsis and ARDS. Virulence. 2013;4(6):517–524. doi: 10.4161/viru.24906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li YR, et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat Med. 2015;21(9):1018–1027. doi: 10.1038/nm.3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szklarczyk D, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA, NHLBI ARDS Network Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med. 2012;40(6):1731–1737. doi: 10.1097/CCM.0b013e3182451c87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghosh CC, et al. Impaired function of the Tie-2 receptor contributes to vascular leakage and lethality in anthrax. Proc Natl Acad Sci U S A. 2012;109(25):10024–10029. doi: 10.1073/pnas.1120755109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeo TW, et al. Angiopoietin-2 is associated with decreased endothelial nitric oxide and poor clinical outcome in severe falciparum malaria. Proc Natl Acad Sci USA. 2008;105(44):17097–17102. doi: 10.1073/pnas.0805782105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Angus DC, Yealy DM, Kellum JA, ProCESS Investigators Protocol-based care for early septic shock. N Engl J Med. 2014;371(4):386. doi: 10.1056/NEJMc1406745. [DOI] [PubMed] [Google Scholar]
- 40.Berthelsen LO, Kristensen AT, Tranholm M. Animal models of DIC and their relevance to human DIC: a systematic review. Thromb Res. 2011;128(2):103–116. doi: 10.1016/j.thromres.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Daly C, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1) Genes Dev. 2004;18(9):1060–1071. doi: 10.1101/gad.1189704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh CC, et al. Drug repurposing screen identifies Foxo1-dependent angiopoietin-2 regulation in sepsis. Crit Care Med. 2015;43(7):e230–e240. doi: 10.1097/CCM.0000000000000993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim M, et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J Clin Invest. 2016;126(9):3511–3525. doi: 10.1172/JCI84871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korhonen EA, et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J Clin Invest. 2016;126(9):3495–3510. doi: 10.1172/JCI84923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kugathasan L, Ray JB, Deng Y, Rezaei E, Dumont DJ, Stewart DJ. The angiopietin-1-Tie2 pathway prevents rather than promotes pulmonary arterial hypertension in transgenic mice. J Exp Med. 2009;206(10):2221–2234. doi: 10.1084/jem.20090389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277(35):32124–32132. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 47.Schabbauer G, Tencati M, Pedersen B, Pawlinski R, Mackman N. PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24(10):1963–1969. doi: 10.1161/01.ATV.0000143096.15099.ce. [DOI] [PubMed] [Google Scholar]
- 48.Shi J, Pipe SW, Rasmussen JT, Heegaard CW, Gilbert GE. Lactadherin blocks thrombosis and hemostasis in vivo: correlation with platelet phosphatidylserine exposure. J Thromb Haemost. 2008;6(7):1167–1174. doi: 10.1111/j.1538-7836.2008.03010.x. [DOI] [PubMed] [Google Scholar]
- 49.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8(10):1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 50.De Palma M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8(3):211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 51.Naji A, et al. Induction of tissue factor expression on human umbilical vein endothelial cells by cell-specific HLA class I antibody: preliminary data. Transplant Proc. 2005;37(6):2892–2893. doi: 10.1016/j.transproceed.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Rao TP, Okamoto T, Akita N, Hayashi T, Kato-Yasuda N, Suzuki K. Amla (Emblica officinalis Gaertn.) extract inhibits lipopolysaccharide-induced procoagulant and pro-inflammatory factors in cultured vascular endothelial cells. Br J Nutr. 2013;110(12):2201–2206. doi: 10.1017/S0007114513001669. [DOI] [PubMed] [Google Scholar]
- 53.Coll E, Robles-Carrillo L, Reyes E, Francis JL, Amirkhosravi A. Assessment of protein C anticoagulant pathway by thrombin generation assay in the presence of endothelial cells. J Thromb Haemost. 2013;11(10):1916–1919. doi: 10.1111/jth.12353. [DOI] [PubMed] [Google Scholar]
- 54.Xiang B, et al. Platelets protect from septic shock by inhibiting macrophage-dependent inflammation via the cyclooxygenase 1 signalling pathway. Nat Commun. 2013;4:2657. doi: 10.1038/ncomms3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satchell SC, Harper SJ, Mathieson PW. Angiopoietin-1 is normally expressed by periendothelial cells. Thromb Haemost. 2001;86(6):1597–1598. [PubMed] [Google Scholar]
- 56.Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10(3):165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- 57.Eremina V, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111(5):707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maynard SE, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111(5):649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Venkatesha S, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12(6):642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 60.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood. 2005;106(7):2417–2423. doi: 10.1182/blood-2005-03-0916. [DOI] [PubMed] [Google Scholar]
- 61.Stohlawetz P, et al. Effects of endotoxemia on thrombopoiesis in men. Thromb Haemost. 1999;81(4):613–617. [PubMed] [Google Scholar]
- 62.Cremer M, Weimann A, Szekessy D, Hammer H, Bührer C, Dame C. Low immature platelet fraction suggests decreased megakaryopoiesis in neonates with sepsis or necrotizing enterocolitis. J Perinatol. 2013;33(8):622–626. doi: 10.1038/jp.2013.21. [DOI] [PubMed] [Google Scholar]
- 63.Eremina V, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358(11):1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shen J, et al. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J Clin Invest. 2014;124(10):4564–4576. doi: 10.1172/JCI74527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saharinen P, Eklund L, Alitalo K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov. 2017;16(9):635–661. doi: 10.1038/nrd.2016.278. [DOI] [PubMed] [Google Scholar]
- 66.Jesmin S, Wada T, Gando S, Sultana SS, Zaedi S. The dynamics of angiogenic factors and their soluble receptors in relation to organ dysfunction in disseminated intravascular coagulation associated with sepsis. Inflammation. 2013;36(1):186–196. doi: 10.1007/s10753-012-9534-6. [DOI] [PubMed] [Google Scholar]
- 67.Rasmussen AL, et al. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science. 2014;346(6212):987–991. doi: 10.1126/science.1259595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Higgins SJ, et al. Dysregulation of angiopoietin-1 plays a mechanistic role in the pathogenesis of cerebral malaria. Sci Transl Med. 2016;8(358):358ra128. doi: 10.1126/scitranslmed.aaf6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turner L, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 2013;498(7455):502–505. doi: 10.1038/nature12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hwang JY, Park JW, Hong SY, Park HS. Reduced expression of angiopoietin-1 in Hantaan virus-infected human umbilical vein endothelial cell increases their permeability. Acta Virol. 2009;53(1):7–13. doi: 10.4149/av_2009_01_07. [DOI] [PubMed] [Google Scholar]
- 71.Yacoub S, et al. Association of microvascular function and endothelial biomarkers with clinical outcome in dengue: an observational study. J Infect Dis. 2016;214(5):697–706. doi: 10.1093/infdis/jiw220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Toh CH, Hoots WK, SSC on Disseminated Intravascular Coagulation of the ISTH. The scoring system of the Scientific and Standardisation Committee on Disseminated Intravascular Coagulation of the International Society on Thrombosis and Haemostasis: a 5-year overview. J Thromb Haemost. 2007;5(3):604–606. doi: 10.1111/j.1538-7836.2007.02313.x. [DOI] [PubMed] [Google Scholar]
- 73.Gold L, Walker JJ, Wilcox SK, Williams S. Advances in human proteomics at high scale with the SOMAscan proteomics platform. N Biotechnol. 2012;29(5):543–549. doi: 10.1016/j.nbt.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 74.Shrum B, et al. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res Notes. 2014;7:233. doi: 10.1186/1756-0500-7-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghosh CC, et al. Impaired function of the Tie-2 receptor contributes to vascular leakage and lethality in anthrax. Proc Natl Acad Sci U S A. 2012;109(25):10024–10029. doi: 10.1073/pnas.1120755109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Graham GJ, Ren Q, Dilks JR, Blair P, Whiteheart SW, Flaumenhaft R. Endobrevin/VAMP-8-dependent dense granule release mediates thrombus formation in vivo. Blood. 2009;114(5):1083–1090. doi: 10.1182/blood-2009-03-210211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aisiku O, et al. Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood. 2015;125(12):1976–1985. doi: 10.1182/blood-2014-09-599910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jasuja R, et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest. 2012;122(6):2104–2113. doi: 10.1172/JCI61228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.