

Abstract
Resistance to chemotherapy drugs remains a significant problem for the treatment of many types of cancer. Fascin-1 (FSCN-1) is an actin-bundling protein involved in the invasion and metastasis of a variety of tumors. However, its involvement in drug resistance in hepatocellular carcinoma (HCC) remains unclear. The present study aimed to investigate the function of FSCN-1 in HCC resistance to doxorubicin (DOX). FSCN-1 expression was increased in DOX-resistant HCC cell lines (SNU449 and SNU387) compared with DOX-sensitive cell lines (Huh7 and Hep3B). The resistance of HCC cells to DOX was decreased following FSCN-1 knockdown with small interfering RNA. FSCN-1 knockdown also significantly altered the expression of key markers of epithelial-mesenchymal transition (EMT). Notably, vimentin expression was reduced and epithelial-cadherin expression was increased. Furthermore, when EMT was suppressed through knockdown of Twist, an essential pathway of DOX-induced EMT, the viability of HCC cells following treatment with DOX was not affected by FSCN-1 expression. Furthermore, FSCN-1 knockdown eliminated hypoxia-induced doxorubicin resistance and EMT. The results of the present study indicated that FSCN-1 expression increased DOX resistance in HCC cells via the promotion of EMT, and this phenomenon was maintained in a hypoxic environment. FSCN-1 potentially represents a novel target to overcome resistance to DOX in HCC.
Keywords: hepatocellular carcinoma, fascin-1, epithelial-mesenchymal transition, doxorubicin
Introduction
Hepatocellular carcinoma (HCC) is a common malignancy with an increasing global incidence rate, and was reported to be the second leading cause of cancer-associated mortality worldwide in 2012 (1). Surgical intervention, including resection and liver transplantation, is the most effective strategy for improving 5-year survival rates of patients with HCC (2). Unfortunately, the proportion of patients with HCC who receive surgery for treatment remains at ~22% (3). This is primarily because numerous patients with HCC are not candidates for resection, due to insufficient liver function at the time of diagnosis. Infection with the hepatitis B virus (HBV) is not only responsible for almost half of all new HCC cases around the world each year (4), but also leads to serious liver cirrhosis in patients with underlying HCC. In patients with unresectable HCC, chemotherapy is the main strategy used to prevent tumor progression at present.
Doxorubicin (DOX) is a cytotoxic anthracycline antibiotic with susbtantial treatment potential in a broad variety of solid neoplasms (5). The major mechanism underlying the tumor-inhibiting activity of DOX is intercalation into primary double-stranded DNA, which blocks the interaction between DNA and RNA polymerases and results in impaired protein synthesis (6). In 1975, Olweny et al (7) first reported that 11 of 14 treated patients responded, with three patients showing complete tumor regression following two, three and five courses of DOX, respectively (7). Although DOX is considered a standard cytostatic agent for the treatment of HCC, response rates are typically low (8). Furthermore, DOX treatment may cause multidirectional cytotoxic effects, including cardiotoxicity, fever, urticarial and vomiting. Recently, several clinical studies have presented favorable safety profiles for transarterial chemoembolization (TACE) using DOX-eluting beads, compared with conventional TACE or oral chemotherapy. In 2002, Llovet et al (9) revealed that TACE combined with DOX improved the survival of certain patients with HCC, while decreasing the rate of cardiotoxic effects (9). TACE combined with DOX-eluting beads has become a common treatment method in patients with primary and metastatic liver cancer. The advantage of DOX-eluting beads is the limited toxicity to the normal liver parenchyma and other organs. Indeed, Forner et al (10) reported that the objective tumor response rate in patients with HCC following TACE with DOX-eluting beads was >80%. Furthermore, the 1, 3, 4, and 5-year survival rates were 89.7%, 67.8%, 50.8%, and 33.9% in patients with Barcelona clinic liver cancer (BCLC) stage A (11), respectively (and 88.2%, 64.4%, 47.3%, and 39.4% patients with BCLC stage B, respectively) following treatment with DOX-eluting beads (11).
However, DOX resistance still is a major challenge faced during the treatment of HCC. Therefore, the identification of novel molecular target is essential to improve the efficacy of Dox chemotherapy. Epithelial-mesenchymal transition (EMT) is known to be a primary mechanism underlying resistance to chemotherapy in HCC. During EMT, expression of the desmosome protein desmoplakin, the cell-adhesion molecule epithelial (E-)cadherin and the tight junction protein claudin-1 is inhibited in HCC cells, while neural-cadherin, several matrix metalloproteinases (MMPs), the intermediate filament protein vimentin and a number of transcription factors, including snail family transcription repressor 1, snail family transcription repressor 2, zinc finger E-box binding homeobox (ZEB)1, ZEB2 and Twist are overexpressed (12,13). Furthermore, EMT triggers the activation of the transforming growth factor (TGF)-β/SMAD, Wnt/β-catenin, mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK), phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) and Notch signaling pathways (14). These EMT-induced characteristics significantly increase the resistance of HCC cells to apoptosis and chemotherapy.
Fascin-1 (FSCN-1) is an actin bundling protein that serves key functions in cell-cell interactions, adhesion and motility via regulating the function of filopodial protrusions and microfilaments (15). Aberrant FSCN-1 expression has been observed in various types of cancer, and has been demonstrated to be associated with a more aggressive clinical course. Yoder et al (16) revealed that FSCN-1 is primarily overexpressed in estrogen receptor-negative breast cancer tissues, and that positive FSCN-1 expression is associated with decreased mean tumor-free survival and overall survival time (16). Likewise, in colorectal adenocarcinomas, strong and diffuse FSCN-1 expression is associated with disease progression and reduced survival (17). Furthermore, similar prognostic significance of FSCN-1 expression has been confirmed in renal cell carcinoma (18), gastric adenocarcinoma (19), laryngeal squamous cell carcinoma (20) and ovarian carcinoma (21). There is also evidence suggesting that FSCN-1 may be involved in regulating the EMT process. During vertebrate development, FSCN-1 is principally expressed throughout the embryonic and mesenchymal tissues, as well as in the developing nervous system (22). Suppression of microRNA (miR)-145 in breast cancer regulates cell migration by targeting FSCN-1 and inhibiting EMT (23). Furthermore, FSCN-1 expression was associated with suppressed E-cadherin expression and increased invasiveness, thus serving as a promoter of cancer aggressiveness in HCC (24). Therefore, investigation of FSCN-1 expression patterns and its association with EMT in HCC is necessary.
Furthermore, previous investigations have revealed that FSCN-1 promotes cancer metastasis and recurrence primarily through increasing cell motility (25). Furthermore, several studies have demonstrated that FSCN-1 knockdown suppresses cellular proliferation and cloning efficiency (17,26). FSCN-1 expression has also been implicated in the production of MMP9, which is induced by activation of the tumor necrosis factor-α signaling pathway in cholangiocarcinoma (27). However, up to now, no studies have investigated the associations between FSCN-1 and tumor drug resistance in HCC cells.
In the present study, our group investigated FSCN-1 expression patterns and the association between FSCN-1 and DOX resistance in HCC cell lines. HCC cells with increased expression of FSCN-1 were more resistant to DOX. Knockdown of FSCN-1 by small interfering (si)RNA reduced the resistance of HCC to DOX. In addition, FSCN-1 promoted EMT in HCC cells. FSCN-1 siRNA reversed hypoxia-induced EMT and DOX resistance, while suppressing EMT using Twist siRNA blocked the effect of FSCN-1 siRNA. In conclusion, the results of the present study demonstrated that FSCN-1 conferred doxorubicin resistance in HCC cells through the promotion of EMT.
Materials and methods
Cell lines and reagents
Five human HCC cell lines (SNU387, Huh7, Hep3B, and SNU449) were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Huh7 cells were grown in Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), Hep3B cells were cultured in minimum essential medium, and SNU387, SNU449 cells were incubated in RPMI-1640 complete medium (Gibco; Thermo Fisher Scientific, Inc.). All cell media were supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 100 U/ml streptomycin and penicillin. All cell lines were incubated at 37°C in 5% CO2. DOX and DAPI were purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The following antibodies were used: FSCN-1 (1:1,000, ab126772), while antibodies against β-actin (1:1,000, ab8227), Twist (1:1,000, ab50581), E-cadherin (1:1,000, ab40772), Vimentin (1:1,000, ab8978) and horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibodies (1:2,000, ab6721), all of which were purchased from Abcam (Cambridge, MA, USA), whereas HRP-conjugated goat anti-mouse secondary antibodies (1:2,000, 7076) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
siRNA transfection
Untreated SNU387, Huh7, Hep3B and SNU449 cells were plated at a density of 1×105/well in 2 ml of their corresponding media. Once the cells reached 20-30% confluence, 50 nM siRNA (targeting FSCN-1 or Twist) or a negative control siRNA (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) combined with 50 μl Lipofectamine 2000 transfection reagent (Invitrogen; Thermo Fisher Scientific, Inc.) were added into each well, following the manufacturer's protocol. The siRNA sequences were as follows: FSCN-1, sense 5′-GCU GCUACUUUGACAUCGATT-3′, antisense 5′-UCGAUGUC AAAGUAGCAGCTT-3′; Twist1, sense 5′-GGUACAUCGACUUCCUCUATT-3′, antisense 5′-UAGAGGAAGUCGAUGUACCTT-3′; negative control siRNA, sense 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense 5′-ACGUGACACGUUCGGAGAATT-3′. The transfection medium (Opti-MEM; Gibco; Thermo Fisher Scientific, Inc.) was replaced with complete medium 6 h following transfection, and the cells were incubated for 2 h. All treatments were initiated 24 h after transfection.
Cell viability assay
Following siRNA transfection, SNU387, Huh7, Hep3B and SNU449 cells were added to 96-well plates at a density of 3×103/well and incubated for 24 h. Following administration of DOX [half maximal inhibitory concentration (IC50) value] or the same amount of phosphate-buffered saline (PBS), cell viability was measured using Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer's protocol at an optical density of 450 nm using a microplate reader (Elx800; BioTek Instruments, Inc., Winooski, VT, USA) following incubation for 24, 48, and 72 h at 37°C. To induce hypoxia, the HCC cells were incubated in an atmosphere of 5% CO2, 1% O2 and 94% N2 at 37°C for 12 h.
Protein extraction and western blotting
The protein from HCC cells was harvested using cell lysis buffer (10X) (Cell Signaling Technology, Inc.) at 4°C following the manufacturer's protocol. Western blot analysis was conducted according to a standard protocol. Briefly, a BCA Protein assay kit (Pierce; Thermo Fisher Scientific, Inc.) was utilized to detect protein concentrations using a Bradford assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA). A total of 40 μg/10 μl protein was added per lane and separated by 10% SDS-PAGE at 80-120 V. Subsequently, the protein was transferred to 0.45 μm polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA) at 350 mA for 90 min. Following incubating with 5% non-fat milk prepared with Tris-buffered saline containing 0.05% Tween-20 (TBST) for 1 h at room temperature, the membrane was incubated with the primary antibodies overnight at 4°C. Following washing three times with TBST, the membrane was further incubated with the secondary antibodies for 1 h at room temperature. Finally, the protein concentrations on the membrane were determined using SuperSignal West Pico Chemiluminescent Substrate (Pierce; Thermo Fisher Scientific, Inc.).
EDU assay
A total of 1×105 cells were plated into 24-well plates and incubated for 24 h. Following treatment as aforementioned, EdU (50 μM; Invitrogen; Thermo Fisher Scientific, Inc.) was added into the cultures. Following 2-h incubation at 37°C, the cells were fixed with 4% methanol-free formaldehyde in PBS for 20 min at 4°C, followed by treatment with 0.1% Triton X-100 in PBS for 20 min.
Immunofluorescence
HCC cells were seeded into 48-well plates at a density of 5×103 cells/well. Cells were fixed with 4% formaldehyde for 15 min at 37°C, washed with PBS, blocked with 5% bovine serum albumin (Sangon Biotech Co., Ltd., Shanghai, China) for 30 min at room temperature, and incubated with anti-E-cadherin (1:200, 3195) or anti-human vimentin (1:200, 5741) primary antibodies (Cell Signaling Technology, Inc.) at 4°C overnight. The cells were then incubated with foat anti-rabbit immunoglobulin G H&L (Alexa Fluor® 488) antibodies (Abcam; 1:1000, ab150077) at 4°C for 2 h. Nuclear staining was performed with DAPI (Sigma-Aldrich; Merck KGaA) at room temperature for 2 min. Following washing with PBS three times, cells were observed using an inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Statistical analysis
Experimental data is presented as the mean with standard deviation (SD) or frequency. Two groups were compared using an unpaired Student's t-tests and multiple groups were performed using one-way analysis of variance followed by Tukey's post hoc test. All statistical analysis was performed with the SPSS software (version 19.0; IBM Corp., Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
FSCN-1 expression is positively associated with DOX resistance in HCC cell lines
The sensitivity of five HCC cell lines (SNU387, Huh7, Hep3B and SNU449) to DOX was estimated by the change in cell viability following drug administration for 48 h, using a CCK-8 assay. The IC50 of DOX from high to low (thus, from least sensitive to most sensitive) was SNU449, SNU387, Hep3B and Huh7 (Fig. 1A). Western blot analysis revealed that the basal level of FSCN-1 expression presented a similar trend to the IC50 of DOX (Fig. 1B).
Figure 1.
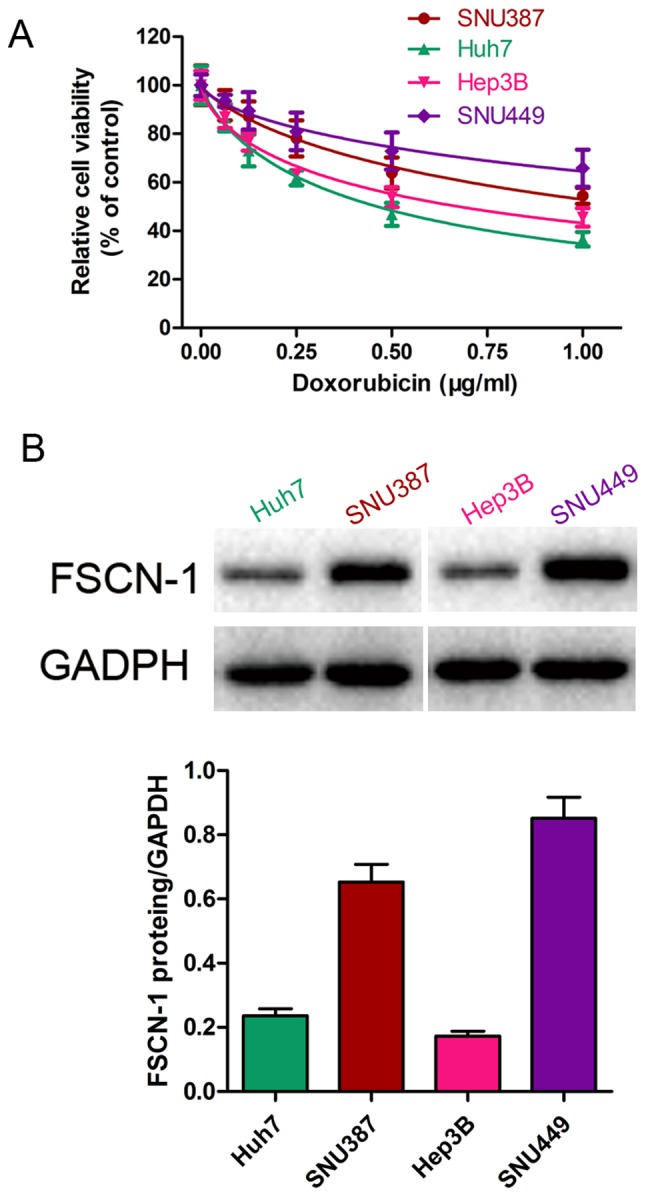
Expression of FSCN-1 is associated with DOX resistance in hepatocellular carcinoma cell lines. (A) Cell viability of SNU449, SNU387, Hep3B and Huh7 cells in the presence of different concentrations of DOX was assessed using Cell Counting Kit-8. (B) Basal level of FSCN-1 expression in SNU449, SNU387, Hep3B and Huh7 cells were assessed using western blotting. FSCN-1, Fascin-1; DOX, doxorubicin.
To further investigate the relationship between FSCN-1 expression and DOX resistance, the expression of FSCN-1 was knocked down with siRNA (Fig. 2A). The results of the cell viability (CCK-8) assays and EDU staining assays revealed that HCC cells with decreased FSCN-1 expression had lower cell viability (Fig. 2B–E) and inhibitory DNA copies compared with the control groups (negative siRNA; Fig. 2F–I). These results suggested that FSCN-1 expression is positively associated with DOX resistance in HCC cell lines.
Figure 2.

FSCN-1 expression is associated with DOX resistance in HCC cell lines. (A) Western blot analysis of FSCN-1 expression in HCC cell lines following siRNA transfection. Cell viability following FSCN-1 knockdown or transfection with negative siRNA in (B) Hep3B, (C) Huh7, (D) SNU387 and (E) SNU449 cells in the presence of different concentrations of DOX was assessed using Cell Counting Kit-8. EDU staining assays (DNA copy number) following DOX treatment and FSCN-1 knockdown in (F) Hep3B, (G) Huh7, (H) SNU387 and (I) SNU449 cells compared with the control (magnification, ×200). DOX concentrations (μg/ml) were as follows: Hep3B, 0.6751; Huh7, 0.4620; SNU387, 1.154; SNU449, 2.402. *P<0.05 and **P<0.01 vs. DOX. FSCN-1, Fascin-1; DOX, doxorubicin; HCC, hepatocellular carcinoma; siRNA, small interfering RNA.
FSCN-1 promotes EMT of HCC cells
Next, the expression of EMT markers in siRNA-FSCN-1 transfected HCC cell lines was detected. Following inhibition of FSCN-1 expression, vimentin expression was significantly suppressed while E-cadherin expression was increased (Fig. 3A). These results were confirmed by the immunofluorescence experiments (Fig. 3B). Notably, E-cadherin expression was higher in Huh7 and Hep3B cells, which were sensitive to DOX, compared with SNU387 and SNU449 cells with DOX resistance, while the opposite pattern was observed for vimentin expression (Fig. 3A). These data indicated that EMT is associated with DOX resistance in HCC cells.
Figure 3.

EMT detection with or without FSCN-1 knockdown. (A) Western blot showing the expression of EMT markers (vimentin and E-cadherin) in the hepatocellular carcinoma cell lines with or without FSCN-1 siRNA transfection. (B) Confocal immunofluorescence images revealing the expression of E-cadherin and vimentin (magnification, ×200). EMT, epithelial-mesenchymal transition; FSCN-1, Fascin-1; siRNA, small interfering RNA; E-cadherin, epithelial-cadherin.
FSCN-1 increases HCC resistance to DOX by promoting EMT
The change in expression of EMT markers was further investigated in HCC cells following DOX treatment. While vimentin expression was increased, E-cadherin expression was suppressed (Fig. 4A). Following FSCN-1 knockdown, DOX-induced EMT of HCC cells was inhibited (Fig. 4A). These results were further verified through immunofluorescence confocal experiments (Fig. 4B). Twist has been reported to play an vital role in adriamycin (DOX) induced EMT (28). HCC cells were transfected with Twist siRNA alone or combined with FSCN-1 siRNA, and then treated with DOX. There was no difference in cell viability between these two groups (Fig. 5C-F). FSCN-1 protein expression was detected by western blotting in order to verify knockdown efficiency (Fig. 5A and B). These data indicated that FSCN-1 enhances DOX resistance via promotion of the EMT process in HCC cell lines.
Figure 4.

Knockdown of FSCN-1-reverses doxorubicin-induced EMT in different HCC cell lines. (A) Western blot analysis of the expression of EMT markers (vimentin and E-cadherin) in the various HCC cell lines, with or without FSCN-1 knockdown, and in the presence or absence of DOX. (B) Confocal immunofluorescence images showing the expression of EMT markers (magnification, ×200). DOX concentrations (μg/ml) were as follows: Hep3B, 0.6751; Huh7, 0.4620; SNU387, 1.154; SNU449, 2.402. FSCN-1, Fascin-1; EMT, epithelial-mesenchymal transition; HCC, hepatocellular carcinoma; E-cadherin, epithelial-cadherin; DOX, doxorubicin; siRNA, small interfering RNA.
Figure 5.

FSCN-1 increases the resistance of HCC to DOX by promoting epithelial-mesenchymal transition. Western blot analysis shows the effective knockdown of (A) Twist and (B) FSCN-1 in HCC cells. Cell viability following DOX treatment was assessed in (C) Huh7, (D) Hep3B, (E) SNU387 and (F) SNU449 cells following Twist knockdown or Twist + FSCN-1 knockdown with Cell Counting Kit-8. FSCN-1, Fascin-1; HCC, hepatocellular carcinoma; DOX doxorubicin; siRNA, small interfering RNA.
Knockdown of FSCN-1 reverses EMT and resistance to DOX of HCC under hypoxic conditions
Hypoxia induces drug resistance and EMT in vitro and in vivo. Indeed, cells cultured under hypoxia were more resistant to DOX compared with normoxia (control group; Fig. 6A and B). Western blot analysis and immunofluorescence confocal assay results revealed that hypoxia decreased E-cadherin expression and upregulated vimentin expression, indicating that hypoxia induced the EMT process in HCC cells (Fig. 6C and D). However, HCC cells under hypoxia were more sensitive to DOX treatment following FSCN-1 knockdown by siRNA compared to the hypoxia group, similar to the control group (Fig. 6A and B). The expression of E-cadherin was increased and vimentin was downregulated, while the cells were transfected with FSCN-1 siRNA (Fig. 6C and D). These results indicated that FACN-1 reversed hypoxia-induced drug resistance and EMT.
Figure 6.

FSCN-1 knockdown reverses hypoxia-induced drug resistance and EMT. Cell viability was assessed following DOX treatment in (A) Huh7 and (B) Hep3B cells with or without FSCN-1 knockdown, in a hypoxic environment, using Cell Counting Kit-8. (C) Western blot analysis of vimentin and E-cadherin expression in HCC cells following hypoxia treatment. (D) Confocal immunofluorescence images showing the expression of EMT markers (magnification, ×200). FSCN-1, Fascin-1; EMT, epithelial-mesenchymal transition; DOX, doxorubicin; siRNA, small interfering RNA.
Discussion
As the prevalence of unresectable cases of HCC increases, so too does the demand for nonsurgical and minimally invasive alternatives to surgery to limit disease progression. In the present study, our group investigated whether the mechanism behind the resistance to the chemotherapy agent DOX was associated with FSCN-1 expression, a key protein involved in cell-cell interaction and motility. FSCN-1 expression was relatively increased in HCC cells with DOX resistance compared with DOX sensitive cells. FSCN-1 knockdown further suppressed the viability of HCC cells in the presence of DOX compared with control cells. Furthermore, investigation of the underlying mechanisms revealed that FSCN-1 promoted the EMT process and increased HCC cell resistance to DOX even under hypoxia. These results suggest that FSCN-1 overexpression is primarily responsible for DOX resistance in HCC.
Typically, FSCN-1 expression is low or absent in normal epithelia, but high in mesenchymal tissues. Previous studies have demonstrated that FSCN-1 is overexpressed in response to TGF-β1-mediated activation of the JNK and ERK signaling pathways (29). In addition, Zhao et al (23) demonstrated that inhibition of miR-145, a main regulatory miRNA for FSCN-1 expression, significantly increased EMT in breast cancer cells. These previous data suggested that upregulation of FSCN-1 expression increased HCC resistance to DOX, potentially through the induction of EMT. As expected, FSCN-1 suppression significantly suppressed vimentin expression and increased E-cadherin expression.
EMT in tumor cells allows them to gain metastatic features via the induction of cell-cell disconnection, cell depolarization and transitioning to an elongated, fibroblast-like morphology. However, certain controversies concerning the involvement of EMT in the promotion of tumor distant metastasis have occurred. In breast cancer and pancreatic ductal adenocarcinoma, EMT was revealed to be unessential for tumor distant dissemination or metastasis (30,31). Nonetheless, these two studies demonstrated that EMT is closely associated with chemotherapy resistance and immune escape. In the present study, our group observed that HCC cells that survived following treatment with DOX had increased expression of EMT markers. Meanwhile, Twist knockdown reduced the viability of HCC cells in the presence of DOX. These data confirmed that EMT was contributing to increased resistance of HCC cells to DOX.
Ischemia is a common phenomenon in HCC tissues during TACE, and is associated with metastatic capacity, resistance to chemotherapy and tumor progression, as well as poor patient prognosis (32). Sridharan et al (33) developed a Boolean network model using targeted drug intervention to mimic persistent hypoxia in a cell, and revealed that hypoxia regulated the p53/mouse double minute 2 homolog, PI3K/Akt/mechanistic target of rapamycin and the glycolysis/tricarboxylic acid cycle pathways to influence cell energy production, apoptosis and survival. Furthermore, the hypoxic microenvironment of tumors is known to enhance stemness features and EMT (34). Therefore, our group detected the effect of hypoxia on the viability of HCC cells following DOX administration. Hypoxia increased the survival of HCC cells following DOX treatment via the promotion of EMT. The effect of hypoxia on DOX resistance was inhibited in HCC cells following knockdown of FSCN-1. Tumor ischemia may also stimulate angiogenesis to support HCC development (35). Sorafenib, a tyrosine kinase inhibitor, is considered to be standard treatment for patients with advanced HCC. This drug not only directly suppresses HCC cell proliferation, but also significantly inhibits angiogenesis. In a large, randomized and double blind clinical trial, concurrent treatment of DOX-eluting beads and sorafenib presented a manageable safety and tolerability (36).
Finally, hypoxia-inducible factor (HIF)-1α has previously been demonstrated to promote autophagy (37). Upregulation of autophagy is a crucial mechanism of resistance to multiple antitumor drugs, including DOX (38). In addition, Zhao et al (39) revealed that the mechanisms underlying HIF-1α-mediated increased invasion and metastasis of pancreatic ductal adenocarcinoma was dependent on the upregulation of FSCN-1 expression. Therefore, the relationship between FSCN-1 and autophagy is worthy of further investigation.
In conclusion, the present study demonstrated that that FSCN-1 serves a critical role in doxorubicin resistance in HCC under normoxic and hypoxic conditions through the promotion of EMT. Therefore, FSCN-1 is potentially a novel target to overcome HCC resistance to DOX. Further studies, including animal experiments and clinical specimen analysis, should be performed in the future to verify the results of the present study.
Acknowledgments
Not applicable.
Footnotes
Funding
The present study was funded by the National Health and Family Planning Research Fund-Major Technology Plan of Medical and Health Science and Technology Project (grant no. WKJ-ZJ-1602), the Medical and Health Science and Technology Project of Zhejiang Province (grant no. 2016KYB025) and the Science and Technology Department Public Welfare Project of Zhejiang Province (grant no. GF18H160058).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
WY, WC and WW designed the study; YuaZ, JS, YL and CZ performed the experiments; YC, DH and YuhZ analyzed the data; WY wrote the manuscript. All authors have read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma: Recent trends in the United States. Gastroenterology. 2004;127(Suppl 1):S27–S34. doi: 10.1053/j.gastro.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 3.Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology. 2015;61:191–199. doi: 10.1002/hep.27388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 5.Piska K, Koczurkiewicz P, Bucki A, Wójcik-Pszczoła K, Kołaczkowski M, Pękala E. Metabolic carbonyl reduction of anthracyclines - role in cardiotoxicity and cancer resistance. Reducing enzymes as putative targets for novel cardioprotective and chemosensitizing agents. Invest New Drugs. 2017;35:375–385. doi: 10.1007/s10637-017-0443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 7.Olweny CL, Toya T, Katongole-Mbidde E, Mugerwa J, Kyalwazi SK, Cohen H. Treatment of hepatocellular carcinoma with adriamycin. Preliminary communication. Cancer. 1975;36:1250–1257. doi: 10.1002/1097-0142(197510)36:4<1250::AID-CNCR2820360410>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 8.Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012;56:686–695. doi: 10.1016/j.jhep.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 9.Llovet JM, Real MI, Montaña X, Planas R, Coll S, Aponte J, Ayuso C, Sala M, Muchart J, Solà R, et al. Barcelona Liver Cancer Group Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: A randomised controlled trial. Lancet. 2002;359:1734–1739. doi: 10.1016/S0140-6736(02)08649-X. [DOI] [PubMed] [Google Scholar]
- 10.Forner A, Ayuso C, Varela M, Rimola J, Hessheimer AJ, de Lope CR, Reig M, Bianchi L, Llovet JM, Bruix J. Evaluation of tumor response after locoregional therapies in hepatocellular carcinoma: Are response evaluation criteria in solid tumors reliable? Cancer. 2009;115:616–623. doi: 10.1002/cncr.24050. [DOI] [PubMed] [Google Scholar]
- 11.Burrel M, Reig M, Forner A, Barrufet M, de Lope CR, Tremosini S, Ayuso C, Llovet JM, Real MI, Bruix J. Survival of patients with hepatocellular carcinoma treated by transarterial chemoembolisation (TACE) using Drug Eluting Beads. Implications for clinical practice and trial design. J Hepatol. 2012;56:1330–1335. doi: 10.1016/j.jhep.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De, Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 14.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vignjevic D, Kojima S, Aratyn Y, Danciu O, Svitkina T, Borisy GG. Role of fascin in filopodial protrusion. J Cell Biol. 2006;174:863–875. doi: 10.1083/jcb.200603013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoder BJ, Tso E, Skacel M, Pettay J, Tarr S, Budd T, Tubbs RR, Adams JC, Hicks DG. The expression of fascin, an actin-bundling motility protein, correlates with hormone receptor-negative breast cancer and a more aggressive clinical course. Clin Cancer Res. 2005;11:186–192. [PubMed] [Google Scholar]
- 17.Hashimoto Y, Skacel M, Lavery IC, Mukherjee AL, Casey G, Adams JC. Prognostic significance of fascin expression in advanced colorectal cancer: An immunohistochemical study of colorectal adenomas and adenocarcinomas. BMC Cancer. 2006;6:241. doi: 10.1186/1471-2407-6-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin JS, Yu CP, Sun GH, Lin YF, Chiang H, Chao TK, Tsai WC, Sheu LF. Increasing expression of fascin in renal cell carcinoma associated with clinicopathological parameters of aggressiveness. Histol Histopathol. 2006;21:1287–1293. doi: 10.14670/HH-21.1287. [DOI] [PubMed] [Google Scholar]
- 19.Tsai WC, Jin JS, Chang WK, Chan DC, Yeh MK, Cherng SC, Lin LF, Sheu LF, Chao YC. Association of cortactin and fascin-1 expression in gastric adenocarcinoma: Correlation with clinicopathological parameters. J Histochem Cytochem. 2007;55:955–962. doi: 10.1369/jhc.7A7235.2007. [DOI] [PubMed] [Google Scholar]
- 20.Zou J, Yang H, Chen F, Zhao H, Lin P, Zhang J, Ye H, Wang L, Liu S. Prognostic significance of fascin-1 and E-cadherin expression in laryngeal squamous cell carcinoma. Eur J Cancer Prev. 2010;19:11–17. doi: 10.1097/CEJ.0b013e32832f9aa6. [DOI] [PubMed] [Google Scholar]
- 21.Lin CK, Su HY, Tsai WC, Sheu LF, Jin JS. Association of cortactin, fascin-1 and epidermal growth factor receptor (EGFR) expression in ovarian carcinomas: Correlation with clinicopathological parameters. Dis Markers. 2008;25:17–26. doi: 10.1155/2008/284382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Arcangelis A, Georges-Labouesse E, Adams JC. Expression of fascin-1, the gene encoding the actin-bundling protein fascin-1, during mouse embryogenesis. Gene Expr Patterns. 2004;4:637–643. doi: 10.1016/j.modgep.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 23.Zhao H, Kang X, Xia X, Wo L, Gu X, Hu Y, Xie X, Chang H, Lou L, Shen X. miR-145 suppresses breast cancer cell migration by targeting FSCN-1 and inhibiting epithelial-mesenchymal transition. Am J Transl Res. 2016;8:3106–3114. [PMC free article] [PubMed] [Google Scholar]
- 24.Hayashi Y, Osanai M, Lee GH. Fascin-1 expression correlates with repression of E-cadherin expression in hepatocellular carcinoma cells and augments their invasiveness in combination with matrix metalloproteinases. Cancer Sci. 2011;102:1228–1235. doi: 10.1111/j.1349-7006.2011.01910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim SJ, Choi IJ, Cheong TC, Lee SJ, Lotan R, Park SH, Chun KH. Galectin-3 increases gastric cancer cell motility by up-regulating fascin-1 expression. Gastroenterology. 2010;138:1035–1045. e1031–1032. doi: 10.1053/j.gastro.2009.09.061. [DOI] [PubMed] [Google Scholar]
- 26.Fu H, Wen JF, Hu ZL, Luo GQ, Ren HZ. Knockdown of fascin1 expression suppresses the proliferation and metastasis of gastric cancer cells. Pathology. 2009;41:655–660. doi: 10.3109/00313020903273100. [DOI] [PubMed] [Google Scholar]
- 27.Onodera M, Zen Y, Harada K, Sato Y, Ikeda H, Itatsu K, Sato H, Ohta T, Asaka M, Nakanuma Y. Fascin is involved in tumor necrosis factor-alpha-dependent production of MMP9 in cholangiocarcinoma. Lab Invest. 2009;89:1261–1274. doi: 10.1038/labinvest.2009.89. [DOI] [PubMed] [Google Scholar]
- 28.Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q, Tang F, Chen ZQ, Liu XP, Xu ZD. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res. 2009;15:2657–2665. doi: 10.1158/1078-0432.CCR-08-2372. [DOI] [PubMed] [Google Scholar]
- 29.Fu H, Hu Z, Wen J, Wang K, Liu Y. TGF-beta promotes invasion and metastasis of gastric cancer cells by increasing fascin1 expression via ERK and JNK signal pathways. Acta Biochim Biophys Sin (Shanghai) 2009;41:648–656. doi: 10.1093/abbs/gmp053. [DOI] [PubMed] [Google Scholar]
- 30.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sergio A, Cristofori C, Cardin R, Pivetta G, Ragazzi R, Baldan A, Girardi L, Cillo U, Burra P, Giacomin A, et al. Transcatheter arterial chemoembolization (TACE) in hepatocellular carcinoma (HCC): The role of angiogenesis and invasiveness. Am J Gastroenterol. 2008;103:914–921. doi: 10.1111/j.1572-0241.2007.01712.x. [DOI] [PubMed] [Google Scholar]
- 33.Sridharan S, Varghese R, Venkatraj V, Datta A. Hypoxia stress response pathways: Modeling and targeted therapy. IEEE J Biomed Health Inform. 2017;21:875–885. doi: 10.1109/JBHI.2016.2559460. [DOI] [PubMed] [Google Scholar]
- 34.Prasad P, Mittal SA, Chongtham J, Mohanty S, Srivastava T. Hypoxia-mediated epigenetic regulation of stemness in brain tumor cells. Stem Cells. 2017;35:1468–1478. doi: 10.1002/stem.2621. [DOI] [PubMed] [Google Scholar]
- 35.Myung SJ, Yoon JH. Hypoxia in hepatocellular carcinoma. Korean J Hepatol. 2007;13:9–19. In Korean. [PubMed] [Google Scholar]
- 36.Lencioni R, Llovet JM, Han G, Tak WY, Yang J, Guglielmi A, Paik SW, Reig M, Kim DY, Chau GY, et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: The SPACE trial. J Hepatol. 2016;64:1090–1098. doi: 10.1016/j.jhep.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 37.Wang P, Long M, Zhang S, Cheng Z, Zhao X, He F, Liu H, Ming L. Hypoxia inducible factor-1α regulates autophagy via the p27-E2F1 signaling pathway. Mol Med Rep. 2017;16:2107–2112. doi: 10.3892/mmr.2017.6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan Q, Joshua AM, Wang M, Bristow RG, Wouters BG, Allen CJ, Tannock IF. Up-regulation of autophagy is a mechanism of resistance to chemotherapy and can be inhibited by pantoprazole to increase drug sensitivity. Cancer Chemother Pharmacol. 2017;79:959–969. doi: 10.1007/s00280-017-3298-5. [DOI] [PubMed] [Google Scholar]
- 39.Zhao X, Gao S, Ren H, Sun W, Zhang H, Sun J, Yang S, Hao J. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014;74:2455–2464. doi: 10.1158/0008-5472.CAN-13-3009. [DOI] [PubMed] [Google Scholar]