

Summary
The eukaryotic helicase is an 11-subunit machine containing an Mcm2-7 motor ring that encircles DNA, Cdc45 and the GINS tetramer, referred to as CMG (Cdc45, Mcm2-7, GINS). CMG is “built” on DNA at origins in two steps. First, two Mcm2-7 rings are assembled around duplex DNA at origins in G1 phase, forming the Mcm2-7 “double hexamer”. In a second step, in S phase Cdc45 and GINS are assembled onto each Mcm2-7 ring for production of two CMGs that ultimately form two replication forks that travel in opposite directions. Here we review recent findings about CMG structure and function. The CMG unwinds the parental duplex and is also the organizing center of the replisome; it binds DNA polymerases and other factors. EM studies reveal a 20-subunit core replisome with the leading Pol ε and lagging Pol α-primase on opposite faces of CMG, forming a fundamentally asymmetric replisome architecture. Structural studies of CMG at a replication fork reveal unexpected details about how CMG engages a DNA fork. The structures of CMG and the Mcm2-7 double hexamer on DNA suggest a completely unanticipated process about the production of bidirectional replication forks at origins.
Keywords: CMG helicase, replisome, origin initiation, DNA polymerase, DNA replication
1. Introduction
Cells have several DNA and RNA helicases that are involved in a variety of processes such as genome replication, transcription, translation, and repair. [1–4] In all cell types, the replicative helicase is a hexameric ring that encircles one strand of DNA and motors along it, acting as a wedge to separate the duplex (reviewed in [5, 6]). The circlular shape may have evolved to increase the grip on DNA for unwinding long chromosomes. This review focuses on the eukaryotic CMG helicase, the 11-subunit enzyme that unwinds genomic DNA for replication in eukaryotic cells.
The replicative helicases of bacteria and archaea are composed of six identical subunits (i.e. homohexamers), while the eukaryotic replicative helicase has six different, but homologous subunits referred to as Mcm2-7 [1, 2]. Mcm2-7 does not become an active helicase until it associates with Cdc45 and the GINS heterotetramer (Psf1,2,3 and Sld5), and the term “CMG” was coined by the Botchan lab to refer to the 11-subunit helicase (Cdc45, Mcm2-7, GINS) [7, 8]. CMG was first identified in the Drosophila melanogaster (D.m.) system [7, 8], and human and Saccharomyces cerevisiae (S.c.) CMG have also been confirmed to have helicase activity [9, 10]. CMG can be isolated intact from budding yeast with numerous other proteins that bind to it, referred to as the RPC (Replisome Progression Complex). The RPC includes CMG along with Ctf4, Pol α-primase, Mrc1, Tof1, Csm3, Mcm10, FACT, and TopI [11, 12].
The Mcm2-7 subunits of CMG and the archaeal MCM homohexamer track on ssDNA 3’-5’ and the ATP site is based on the AAA+ fold (ATPases Associated with various cellular Activities); in contrast bacterial helicases are homohexamers with ATP sites based on the RecA fold and track the opposite direction, 5’-3’ along DNA (reviewed in [5, 6]). In an interesting mystery of nature, sequence homology reveals that most of the replication apparatus of bacterial cells evolved independently from the apparatus of eukaryotic/archaeal cells, consistent with the opposite tracking directions of their replicative helicases [13, 14]. In contrast, the core machinery of transcription and translation are homologous in all domains of life.
The Mcm2-7 ring has the order of Mcm3, 5, 2, 6, 4, 7 [15]. The Cdc45 and GINS subunits are not catalytic and are proposed to bring the Mcm2-7 motors into proper configuration for activity [1, 8, 16]. The Mcm2-7 ring, like the homohexamer helicases, has an appearance of two stacked rings because each subunit is dumbbell shaped, composed of an N-terminal domain (NTD) and a C-terminal domain (CTD) (Figure 1a). We refer here to the ring formed by the 6 NTD domains as the N-tier, and the ring formed by the 6 CTDs as the C-tier. The AAA+ and RecA based ATPase motors are found in the C-tier of both bacterial and archaeal/eukaryotic replicative helicases.
Figure 1. Eukaryotic CMG helicase and orientation to other proteins in a replisome.
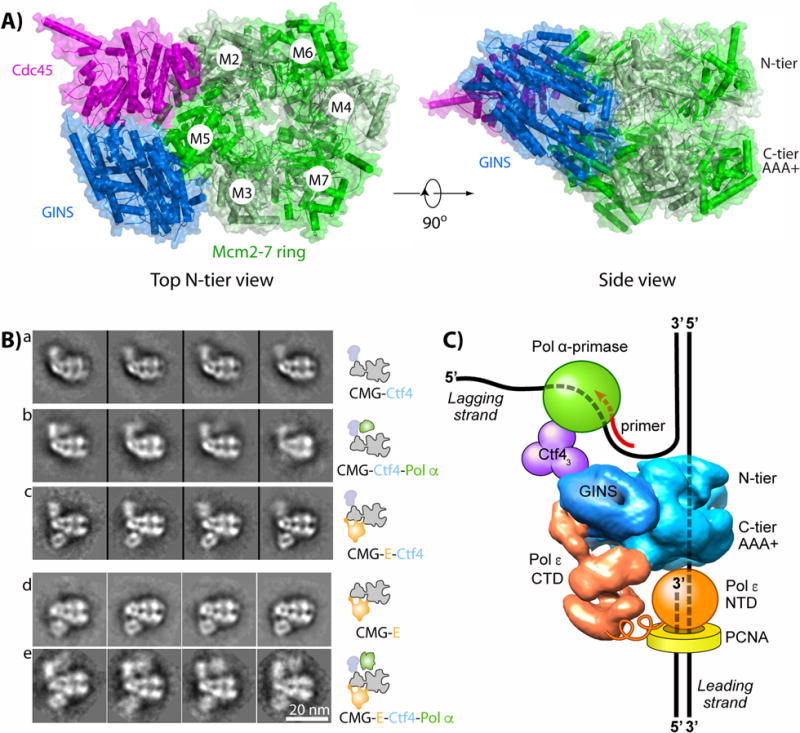
a) Top view (left) and side view (right) of CMG, indicating the Mcm2-7 motor ring and the Cdc45 and GINS accessory factors (PDB ID 3JC5). b) Selected EM 2D averages of CMG in side view, with additional proteins as indicated (adapted with permission from Fig. 5 in [39]). c) Illustration of the core eukaryotic replisome, see text for details.
The ATP sites of hexameric helicases are located at subunit interfaces and require residues from both subunits for catalysis [3, 4]. Interfacial ATP sites is a common feature of many ATP-driven ring-shaped oligomers as exemplified by clamp loaders that open and close polymerase processivity clamps onto DNA [17, 18], protein unfoldases like ClpX that feed protein chains into proteolytic chambers [19, 20], the Rho transcription terminator RNA translocase [21], and DNA packaging motors of phage [22].
CMG is the final product of a two-step process that first loads two Mcm2-7 rings onto an origin in G1 phase, forming a head-to-head inactive Mcm2-7 double hexamer (Mcm2-7 DH) [2, 23–27]. Then, in S phase, Cdc45 and GINS are assembled onto each Mcm2-7 to form two active CMG helicases for bidirectional replication forks [1, 2, 25, 28, 29]. The entire process requires many proteins and kinases that do not travel with replisomes [2, 23–27]. New Mcm2-7 rings can not be loaded in S phase, and therefore the separation of CMG assembly into two distinct cell cycle stages limits origin firing to only once per cell cycle. Cell biology studies and in vitro single-molecule techniques demonstrate that the two CMGs come apart to form two replication forks; each CMG travels 3’-5’ while encircling ssDNA [30–32]. The detailed mechanism of these origin initiation steps are still relatively unknown, but successful reconstitution of replication from an origin using recombinant proteins promises detailed mechanistic understanding in the near future [27, 33, 34]. This review focuses on advances in the last 3-4 years on the structure and function of CMG helicase.
2. CMG helicase is a central scaffold for replisome organization
Replicative helicases act in the context of a large machinery referred to as the “replisome”. Replisomes contain, at a minimum, a helicase, primase, DNA polymerases, sliding clamps, and a clamp loader in all cell types (reviewed in [1, 2, 14, 29, 35, 36]). While the E. coli replisome contains a helicase and identical DNA polymerases (Pol) for the two daughter strands [37–39], eukaryotes contain three replicative DNA Pols, Pol ε and Pol δ, for bulk chromosome replication, and Pol α-primase that functions to prime DNA synthesis by Pols ε and δ [35, 40]. Numerous biochemical and cell biology studies in the Kunkel and Burgers laboratories and their collaborators, have elegantly demonstrated that Pol δ functions mainly on the lagging strand and Pol ε functions mainly on the leading strand [40–47]. Biochemical assays using recombinant CMG have identified the mechanisms by which the three DNA Pols are targeted to their respective strands [9, 37, 48, 49]. In addition, recent studies have indicated that Pol δ sometimes extend the first leading strand primer at an origin before being taken over by Pol ε [50–52]. In vitro assays have identified the minimal protein requirements for both leading and lagging strand synthesis, and for replication fork rates on chromatized DNA consistent with in vivo observations [9, 34, 48, 52–54].
While some replisome protein contacts are transient, CMG forms sufficiently strong contacts to other proteins to reconstitute and isolate larger complexes in the absence of DNA [37–39]. Structural studies demonstrate that CMG forms a direct complex with Pol ε, referred to as CMGE [37, 39]. CMG also binds a homotrimeric Ctf4 (AND-1 in human) that cross-links CMGE to Pol α-primase [38, 39]. The protein organization of this core replisome, consisting of 20 different subunits, has been determined by EM as illustrated in Figure 1b,c [39]. Due to domain mobility, neither the full Pol ε nor Pol α-primase were visualized, but sufficiently large amounts of these Pols were observed to determine the overall organization of the core replisome. Cross-linking mass spectrometry and EM studies confirmed that Pol ε binds the C-tier of CMG while Pol α-primase-Ctf4 bind to the N-tier of CMG [39]. The DNA threading shown in Fig. 1 is demonstrated for S.c. CMG [55] and this model enables Pol ε-PCNA to extend the leading strand DNA as the unwound leading template strand emanates from the central channel of CMG. This arrangement is consistent with earlier footprinting results in the Xenopus system which predicted that Pol ε is right beneath CMG [30]. The DNA threading through CMG in Fig. 1 also provides Pol α-primase ready access to the unwound lagging strand at the “top” of CMG for repeated re-initiation of the numerous short (i.e. 100-200 bp) lagging strand fragments. A flexible joint in Pol ε appears to tether the NTD active domain to the CTD domain that binds CMG, possibly enabling the catalytic NTD of Pol ε to dynamically come on and off the 3’ terminus, thereby providing access of translesion polymerases or other enzymes to the 3’ terminus as needed [55–57].
3. Mcm2-7 is assembled onto dsDNA to form a Mcm2-7 double hexamer
EM studies of D.m. Mcm2-7, S.c. Mcm2-7-Cdt1 and Mcm2-7 of a parasitic eukaryote (Encephalitozoon cuniculi) reveal two major states of the Mcm2-7 ring (Figure 2a) [58–60]. In one state, the ring is essentially planar and the Mcm2/5 subunits connect to form a closed ring. The other state is a left-handed spiral and there is a gap between the Mcm2/5 subunits. These EM studies are supported by earlier biochemical studies in S. cerevisiae that predicted a gap between the Mcm2/5 subunits [61]. The presence of ATPγS favors the closed state, presumably due to ATP binding at the Mcm2/5 ATP site bringing the subunits together for catalysis. Opening at the Mcm2/5 interface is required for loading two Mcm2-7 hexamers onto dsDNA, a process that also involves 1-2 ORC (Origin Recognition Complex), Cdc6 and Ctd1 [23, 26, 31, 62–65].
Figure 2. The Mcm2-7 motor and Mcm2-7 double hexamer.
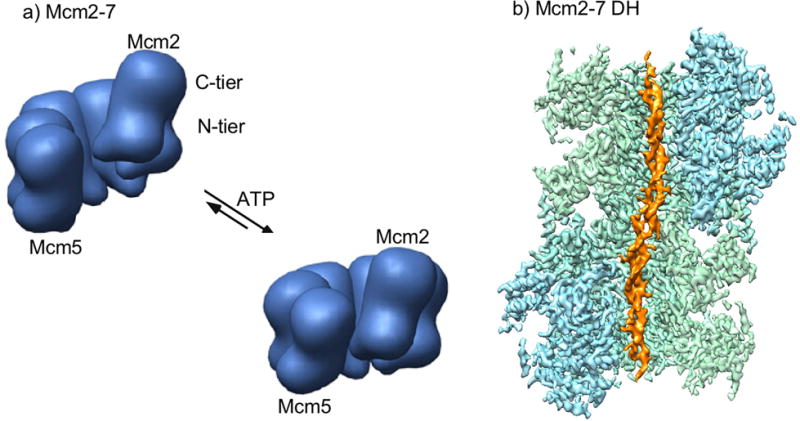
a) Illustration of the density shell from negagtive stain EM of Mcm2-7 from Encephalitozoon cuniculi (adapted with permission from Fig. 5 in [59]) b) Cut-open view of the 3D density map of Mcm2-7 double hexamer from S. cerevisiae loaded on 60 base pairs of double stranded DNA (adapted with permission from [67]).
EM studies of the Mcm2-7 DH in the absence of DNA reveal two closed Mcm2-7 rings that are locked together via the N-tiers, and the hexamers are slightly askew from one another which suggested that the dsDNA would be distorted [66]. Subsequent cryoEM studies of the Mcm2-7 DH bound to dsDNA demonstrate that the DNA zig zags through the Mcm2-7 DH, but the DNA is not unwound (Figure 2b) [67]. There are many interesting details about how the Mcm2-7 DH engages dsDNA, some of which will be described later in this review.
4. The mechanism of CMG translocation along DNA
Negative stain EM of D.m. CMG revealed the organization of the CMG subunits [58]. The Cdc45/GINS accessory proteins bind one side of Mcm2-7, bridging the Mcm2/5 and Mcm3/Mcm5 interfaces, and appear to form a small secondary channel (see Figure 1a). Cross-linking studies of CMG on DNA demonstrate that Cdc45 guards the Mcm2/5 gate and helps keep DNA inside Mcm2-7 during translocation [68]. The high resolution structure of S.c. CMG shows that the secondary channel, suggested to possibly bind one strand [39, 58], is filled-in by amino acid side chains instead [69].
Interestingly, both the S.c. and D.m cryoEM CMG structures reveal a rigid N-tier ring, apparently stabilized by Cdc45 and GINS, and a highly flexible C-tier motor ring with two major conformers [69, 70]. Deletion of the N-tier of the archaeal MCM homohexamer retains minimal helicase activity, indicating that the C-tier contains the intrinsic translocation process and that the N-tier may facilitate processivity [71, 72].
The two S.c. CMG helicase conformer structures in the resolution range of 3.7 Å to 4.8 Å show that the C-tier of one CMG conformer is planar and stacks parallel to the N-tier ring, referred to as the “compact” conformer of CMG (Fig. 3a, left) [69]. In the other CMG conformer, the CTD domains of all 6 subunits undergo rotatational and translational movements, with the CTDs of Mcm7,4,6,2 showing the largest movements and forming a C-tier in the shape of a right-handed partial spiral. This shape change leads to an expanded distance between the N- and C-tiers, and therefore this conformer of CMG is referred to as the “extended” conformer (Fig. 3a, right) [69]. It is important to note that in both conformers, the C-tier ring is formally closed because even in the extended conformer with the partial spiral C-tier, a domain-swapped alpha helix of Mcm5 clearly binds to the CTD of Mcm2. In the D.m. CMG helicase in the resolution range of 7.4 Å to 10.2 Å, the C-tier motor ring always stacks nearly parallel to the N-tier ring, with the CTD of Mcm2 packing against the CTD of Mcm5 either extensively (compact conformation) or with less surface area (relaxed conformation) [70]. Due to the lower resolution it is not clear whether the Mcm2/5 gate in the C-tier motor ring of the relaxed conformation is open or formally closed (e.g. like the domain swap in S.c. CMG). These different conformers of the C-tier motor ring of the CMG helicase suggest possible mechanisms for translocation along DNA, as illustrated in Figure 3b,c and discussed below.
Figure 3. Possible translocation processes.
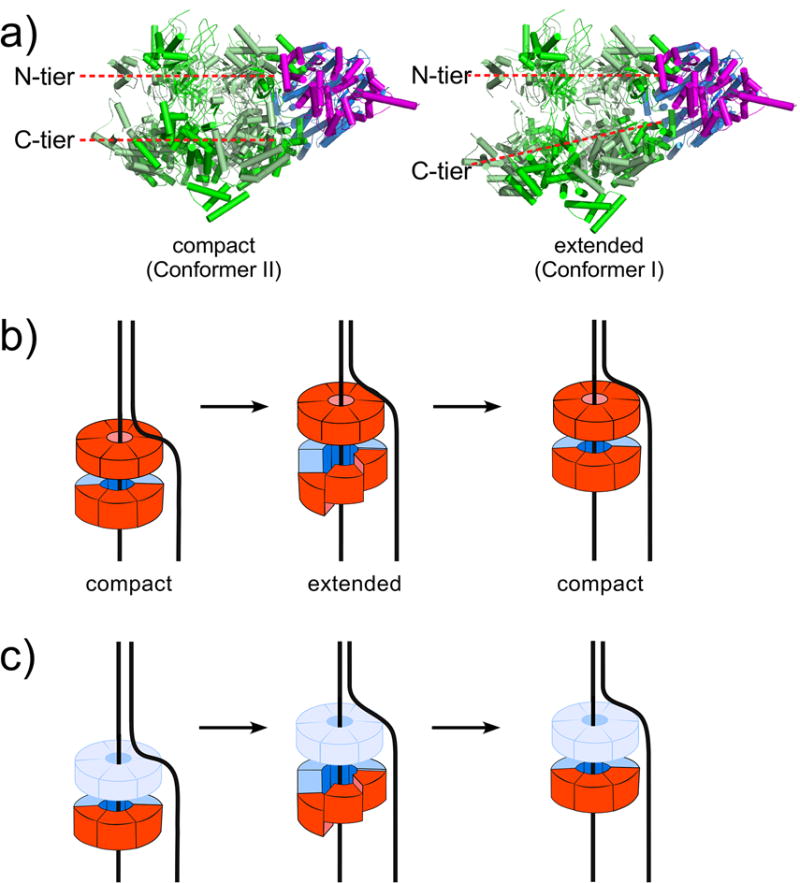
a) CMG alternates between the compact and extended conformers (PDB 3JC5, and 3JC7). b) Inchworm process proposed for movements between the N- tier and C-tier (N-tier in solid red, because it directly contributes to translocation by binding DNA). c) Inchworm process proposed for planar to spiral movments solely in the C-tier (N-tier shown in light blue, because it is a processivitiy ring and does not participate in inchworm translocation).
The conformational changes between the compact and extended conformers of S.c. CMG are proposed to be driven by ATP binding and hydrolysis and suggest either of two types of inchworm driven motions along DNA (Figures 3b and 3c) [69]. One inchworming hypothesis is driven by the distance changes between the N- and C-tiers (Figure 3b). This model requires cooperativity between N- and C-tiers, both of which bind DNA. DNA binding to both the N-tier and C-tier of S.c. CMG and an archaeal MCM has been demonstrated [55, 73, 74]. Interestingly, DNA is bound to the C-tier of D.m. CMG in the ATPγS state, but the DNA is shifted into the N-tier in the absence of ATPγS, implying a potential N- and C-tier cooperation during translocation and DNA twisting for origin initiation [70]. The other inchworming hypothesis is driven by the flat to spiral ring conformation changes in the C-tier (Figure 3c) [69]. Movements in the C-tier ring are also supported by cryoEM analysis of the S.c. Mcm2-7-Cdt1 complex, which shows both planar and spiral C-tier conformations in Mcm2-7 and has led to an inchworm proposal similar to that of Figure 3c [60]. In the case of Mcm2-7-Cdt1 there is a topological discontinuity in the C-tier ring (e.g. an opening between Mcm2 and Mcm5). However, we note that the open spiral conformation observed in Mcm2-7-Cdt1 occurs before Mcm2-7 loading onto DNA. As discussed earlier, the Mcm2/5 gate in CMG is either closed by a domain swap (S.c. CMG) or the gate status is unclear (D.m. CMG).
Replicative helicases of bacteria and of Bovine Papillopa Virus (BPV) E1 are homohexamer rings, and a very different translocation process is proposed [5, 6]. The crystal structure of BPV E1-ssDNA-ADP suggests a rotary ATP hydrolysis staircasing translocation process [5, 6, 75]. Co-crystal structures of E. coli Rho-ssRNA, a hexameric RNA translocase, and bacterial replicative helicase DnaB–ssDNA also suggest a rotary sequential ATP hydrolysis staircasing process [21, 76]. In the rotary sequential ATPase staircase mechanism, nucleic acid binding elements lining the inner channel of the hexamer ring move in response to ATP/ADP, and as ATP is hydrolysed sequentially around the ring, DNA is moved through the central pore escorted one subunit at a time with each ATP hydrolysis event [5, 75]. Neither the helicase nor the nucleic acid actually rotate. When one subunit hydroylzes ATP, its DNA binding elements come off DNA and travel a linear path over DNA to leapfrog the other five DNA binding subunits and then rebinds DNA, thereby going from one end of the staircase to the other. Although these leapfrog movements require further experimental support, the staircase hypothesis is compelling because simple permutation of each subunit conformer around the ring invokes the sequential staircasing process [5, 6, 21, 75].
Whether CMG translocates by inchworming or by staircasing will require further study. But the current information on CMG is hard to reconcile with a staircasing model. For example, not all the ATP sites of CMG are needed for helicase activity [7], while each site is needed for the T7 gp4 replicative helicase [62], as predicted for a strict sequential hydrolysis process. Stochastic models of ATP hydrolysis for CMG would eliminate this concern. However, mutational studies of individual ATP sites within CMG give highly asymmetric results. Only two individual ATP site mutants, Mcm 2/5 and Mcm5/3, eliminate helicase activity [7]. Individual mutations in the other ATP sites give only 20-50% reduction of helicase activity [7]. Interestingly, the two essential sites for helicase activity are nearest to the hinge motion between the compact and extended structures of CMG [69]. Asymmetry in ATP site action is accentuated by a recent CMG-forked DNA-ATP structure that shows only four subunits of the C-tier in CMG interact with the leading strand ssDNA (Mcm 3,5,2 and 6), and only three ATP sites are filled (Mcm6/2, Mcm2/5 and Mcm5/3) [55]. One may speculate that some of the ATP sites and DNA binding elements of CMG are utilized for another function besides helicase action (e.g. initiation at an origin).
5. CMG binds both dsDNA and ssDNA at a forked junction
Thus far, the structure of a helicase at a DNA forked junction has not been determined. However, biochemical studies of CMG, in both the S.c. and Xenopus systems, show that CMG functions by steric exclusion like other ring shaped replicative helicases [30, 77]. In the steric exclusion model the helicase ring encircles only one strand of DNA at a forked junction, excluding the other strand and acting as a moving wedge to melt the duplex [30, 78–83]. It is widely believed that during the steric exclusion process, the helicase ring has no specific or direct contact with the unwinding point. This stands in contrast with some earlier models that suggested that duplex DNA may enter half way through the helicase and then split, with one strand extruded out a side channel between the N- and C-tiers while the other strand continues through the central channel. But the biochemical studies cited above have largely discarded the “side channel extrusion” model.
To visualize a helicase at a forked structure, cryoEM studies of D.m. CMG and S.c. CMG have been performed on a forked DNA using either ATPγS or AMPPNP, respectively, but did not visualize the forked junction [55, 70]. It is possible that ATP hydrolysis is needed for CMG to translocate from the 3’ end of the leading strand to the forked junction. To test this possibility a forked junction with two biotin- streptavidin (SA) blocks on the duplex was constructed, enabling use of ATP while at the same time blocking helicase mediated unwinding as illustrated in Figure 4a. [55] Indeed, this strategy worked and yielded the first glimpse of how a helicase engages a replication fork (Figure 4a-d) [55]. CryoEM 2D averages of CMG-forked DNA side views showed that the N-tier abuts the SA blocks at the forked junction (Figure 4b). Thus CMG translocates on DNA N-tier first, pushed by the C-tier motors from behind. This N-tier first orientation is not unique, as it has been noted for the eukaryotic BPV E1 helicase [5, 75, 84]. Low resolution EM and FRET studies of D.m. CMG and archaeal MCM arrived at different conclusions and thus the orientation of CMG on DNA requires further study [16, 81]. Because S.c. CMG-forked DNA is the only helicase thus far to be visualized on a forked junction, this review will utilize the N-tier first orientation of CMG on DNA.
Figure 4. Structure of CMG on forked DNA.
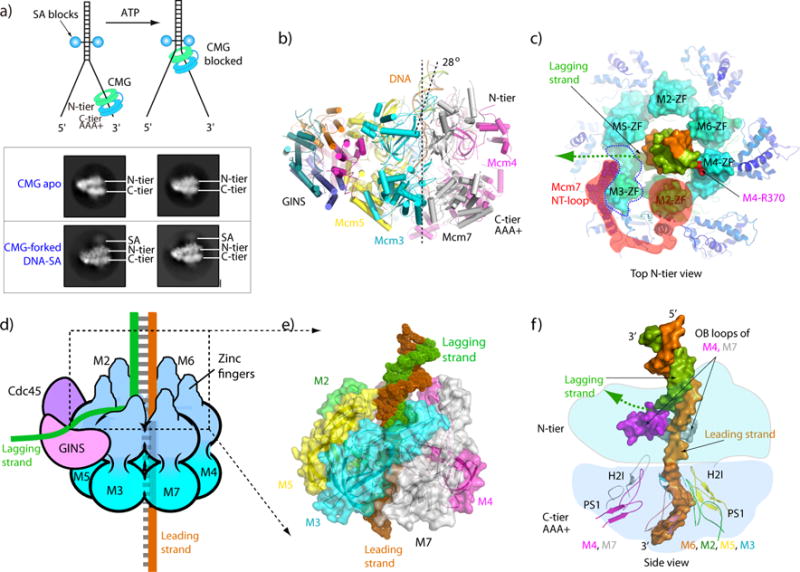
a) Top: Blocked fork strategy to determine the CMG-forked DNA structure with ATP. Bottom: 2D averages of CMG side views at the streptavidin blocks of the fork. b) Atomic model of CMG in cartoon view, showing dsDNA entering the N-tier at a 28° angle, and the ds/ssDNA junction going through the CMG structure. c) Top view of the zinc finger domains (ZF) of CMG encircling the dsDNA. Both Watson and Crick strands of the duplex are contacted by the ZF’s which enter the CMG ZF protrusions. A possible lagging strand exit path at the floor between the protruding ZFs of Mcm3 and Mcm5 is marked by a dashed green arrow. The N-terminal loop of Mcm7 is highlighted in red shadow, which extends to and wraps around Mcm3 ZF to pull it away from Mcm5 FZ and towards Mcm7 ZF. d) Cartoon illustration of CMG illustrating the ZF protrusions from the N-surface and that the DNA unwinding point resides inside CMG at the floor of the ZF protrusions. The position of the lagging strand is suggested by the gap between ZF3/5. e) Close up space filling view of the ZF and OB regions of Mcm2-7 within the CMG-forked DNA structure, showing how they encircle the dsDNA and the gap between ZF3/5. f) Cut out showing the OB loops of Mcm4,7 that approach the forked nexus within the N-tier, and the PS1 loops of Mcm 6,2,5,3 that bind the ssDNA in the C-tier. Panels a and b are adapted with permission from Fig. 6a,b and Fig. 7a of [55].
The S.c. CMG-forked DNA structure supports the steric exclusion model in overview, but shows that CMG binds the dsDNA at a forked junction, unexpected from traditional steric exclusion models [55]. Five of the six Mcms have a zinc finger (ZF) that have a bound zinc atom and these ZFs form protrusions at the top surface of the N-tier; the exception is Mcm3 which has a conserved sequence of the ZF region but does not have the cysteine residues to coordinate zinc. The dsDNA stem is contacted directly by the ZF domains of Mcm 7, 4, 6 (and maybe 2) which importantly bind both Watson and Crick strands (Figure 4c), placing the unwinding point just under these protrusions and holding the duplex at a 28° angle (Figure 4b,c). The duplex separation point is located on the floor of the ZF protrusions (Figure 4d), and while the lagging strand is not visualized in the structure, the lagging strand nucleotide at the base of the unwinding point is positioned to exit at a gap between the ZF domains of Mcm3/5 (Figure 4d,e). Because GINS bridges Mcm3/5 at the N-tier, this possible exit point would place the lagging ssDNA directly over the GINS, the position to which Pol α-primase locates and thus making strategic biological sense. The unwinding point is also in close proximity to two loops of the Mcm4 and Mcm7 OB domains, suggesting side chains are actively involved in unwinding the ss/ds junction (Figure 4f). It is interesting to note that the ZF region of Mcm3 (lacking zinc) is wrapped by the N-terminus of Mcm7, and there is an increased distance between the ZF protrusion of Mcm3 and Mcm5, relative to the ZF protrusions of the other Mcms. This arrangement creates a “shelf” at the bottom of the protruding ZFs between Mcm3 and Mcm5, and this might form the floor of an exit path for the lagging strand (Figure 4c).
Notably, ZF4 binds the future lagging strand in dsDNA, and therefore both strands of the duplex are engaged by CMG, and the unwinding point is at the top of the N-tier, but just beneath the protruding ZFs, which likely explains inhibition of S.c. CMG by a lagging strand dual SA block [77]. Interestingly, S. c. Mcm10 binds to CMG and enables S.c. CMG to rapidly traverse the dual SA lagging strand block by steric exclusion [54]. The exact mechanism by which Mcm10 promotes this bypass is not yet understood. In overview, the unwinding point is just beneath the surface envelope of CMG, at the floor of the ZF protrusions at the top surface of CMG (Figure 4d), and the unwound lagging strand might “glance off the top” of CMG through a cleft, just after the ZF’s of Mcm7,4,6 bind the duplex, supporting a steric exclusion mechanism as originally proposed for CMG by work in Xenopus extracts [30].
Observation of extensive interactions between the helicase and the ss/dsDNA junction were unanticipated by classic steric exclusion models, and brings up interesting questions regarding the mechaniochemical forces used to unwind DNA. Hexameric helicases are proposed to unwind DNA by either: 1) “passive unwinding”, in which the helicase simply translocates on ssDNA and takes advantage of spontaneous thermal DNA fraying, and 2) “active unwinding”, in which the energy of ATP hydrolysis is coupled to strand unwinding [85, 86]. Given that CMG has extensive interactions with the ss/DNA junction, certain residues might actively participate in destabilizing the duplex, but this remains untested.
Hexameric helicases have DNA binding loops within the C-tier motor that project into the central channel. In eukaryotic CMG and the related archaeal MCM, these loops are referred to as PSI and H2I. Mutational studies in archaeal MCM show that the H2I loops can be deleted without inhibiting helicase activity [71]. Furthermore, the Bovine Papilloma Virus E1 helicase contains the PS1 loops, but lacks H2I loops. Therefore we focus below on the PS1 loops of CMG, although the reader should keep in mind that little is known about the H2I loops in CMG, and their function may come to light in future studies.
After DNA unwinding at the top of the N-tier, the PS1 loops in the C-tier reach up to the nexus of the N- and C-tier to grasp the leading ssDNA where it binds in a right hand spiral to the PS1 loops of Mcm 2,3,5,6 (Figure 4d) [55]. It is noteworthy that each PS1 loop binds two phoshodiester bonds, and that only 4 of the 6 PS1 loops bind the ssDNA (PS1 loops of Mcms 2,4,5,6; and not Mcm 3 or 7). S.c. CMG has a wide central channel and can slide over dsDNA [77]. Given the size of the channel and interaction with only 4 PS1 loops, the DNA hugs one side of the central channel.
6. The C-tier motor ring engages the lagging strand DNA in the Mcm2-7 double hexamer but with the leading strand in CMG
A cryoEM structure of the S.c. Mcm2-7 DH loaded onto dsDNA has been determined to 3.9 Å [67] and shows unanticipated differences compared to the CMG-forked DNA structure. A 60-bp duplex is resolved in the central channel of the Mcm2-7 DH. The two hexamers are laterally offset by about 20 Å and are tilted, producing two 15° bends in the DNA in each of the two N-tiers (Figure 2b). These bends cause the dsDNA to zig zag through the Mcm2-7 DH, but the dsDNA does not unwind. Like CMG, the ATP sites of Mcm6/2, Mcm2/5 and Mcm5/3 are filled, but the Mcm2-7 DH has a fourth occupied site, at the Mcm3/7 interface.
An important distinction between the DH and CMG structures is that the PS1 loops bind the future lagging DNA strand in the Mcm2-7 DH, while PS1 loops of CMG only bind the leading strand (Figure 5). As with CMG, only 4 PS1 loops are involved in DNA binding in the Mcm2-7 DH, but surprisingly they are not the same set of PS1 loops as those that bind DNA in the CMG-forked DNA structure. In the Mcm2-7 DH, the PS1 loops of Mcm 3,7,4,6 bind the lagging strand and the PS1 loops of Mcm 2,5 do not engage DNA. This feature may have evolved in the Mcm2-7 DH to accommodate the need for the Mcm2/5 gate to open up for extrusion of the lagging strand DNA. In CMG the PS1 loops of 6,2,5,3 bind the leading strand and the PS1 loops of Mcm 4,7 do not engage DNA (the loops of the Mcm 2-7 DH and CMG are compared in Figure 5a-d). Hence, there appears to be a division of labor in that Mcm2, 3, 5, and 6 function to translocate on DNA (via their respective PS1 loops), and Mcm4 and 7 function to separate the duplex (via their respective OB loops). In another striking distinction with CMG, the Mcm2-7 DH PS1 loops each bind only one phosphodiester bond, compared to the CMG PS1 loops that each bind two phosphodiester bonds.
Figure 5. Comparison of DNA binding elements in the motors of the Mcm2-7 DH and CMG.
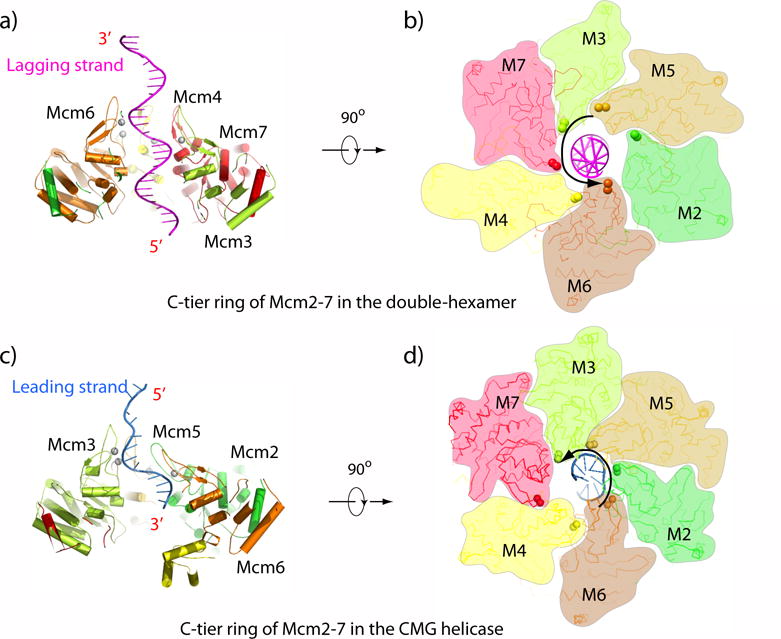
Panels a,b) Mcm2-7 DH PS1 loops of Mcm3,7,4,6 bind the DNA, which is the future lagging strand. Panels c,d) The CMG Mcm6,2,5,3 PS1 loops bind the leading strand. Adapted with permission from Figures 4g and 6d of [67].
7. The mplications of structural studies to origin function
The lagging strand is next to the Mcm2/5 gate in the Mcm2-7 DH, and assuming that the PS1 loops track 3’-5’ like in the active helicase, each Mcm2-7 hexamer would pull on the lagging strand to produce a ssDNA loop at the tight interface between them. This arrangement suggests a process for extruding the lagging strand to the outside of the Mcm2-7 ring, a preresquisite to form a helicase that functions by steric exclusion and encircles only the leading strand. We note that unwinding activity of the Mcm2-7 DH has not been examined or detected yet, and understanding the initial unwinding step remains an important goal. The location of the lagging strand near the Mcm2/5 gate in each of the Mcm2-7 hexamers of the Mcm2-7 DH aligns the lagging strand with the 2/5 gate, suggesting it may be expelled from the interior of Mcm2-7 through the Mcm2/5 gate, leaving the leading ssDNA inside. The transition of Mcm2-7 from encircling dsDNA to encircling ssDNA remains an unidentified and important step of replication initiation.
The Mcm2-7 DH structure indicates that the lagging strand is the tracking strand in the Mcm2-7 DH state. If pushing of the lagging strand creates ssDNA loops between the hexamers, as hypothesized in Figure 6a,b this may be the process by which the lagging strand is extruded out of the Mcm2/5 gate. When Cdc45 and GINS bind Mcm2-7 and block the 2/5 gate, the PS1 loops of Mcm2-7 may organize on the leading strand trapped inside, and 3’-5’ tracking will produce bidirectional forks (illustrated in Figure 6c-d). Mcm10 is reported to be involved in this process, although the exact details of Mcm10 action are not understood. One may presume that the transition from the Mcm 2-7 DH to two CMGs may require several intermediate conformational steps, a subject of intense future investigations [67].
Figure 6. Proposed model for origin initiation.
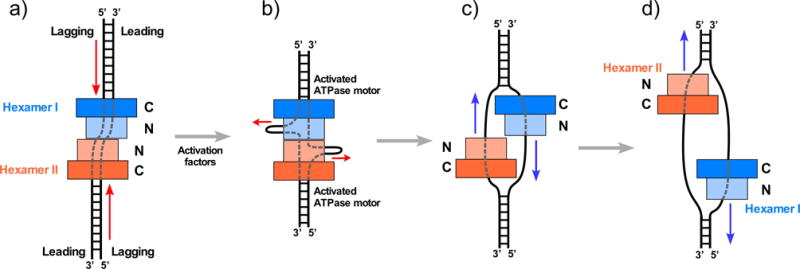
a) The Mcm2-7 hexamers are oriented head-to-head (N-to-N) at the origin. b) The PS1 loops of the Mcm2-7 DH track on the lagging strand and thus might pull it toward the central connection of the tight Mcm2-7 DH, producing ssDNA loops composed of the future lagging strand. c) The lagging ssDNA has been extruded outside the Mcm2-7 ring, and two CMGs have formed in S phase. d) the CMGs pass one another to leave the origin and produce bidirectional replication forks. Red arrows indicate the direction of DNA movement, and blue arrows indicate the direction of CMG movement. Adapted with permission from Figure 8 of [45] and Figure 6 of [67].
8. A quality control mechanism for bidirectional replication from the origin
Several in vivo and in vitro studies have demonstrated that two CMGs are formed before sufficient DNA unwinding occurs for RPA to bind the ssDNA [24, 87–89]. Given the CMG orientation in which the N-tier leads the C-tier, the two head-to-head CMGs at an origin are directed inward, toward one another. Therefore each CMG must pass the other CMG to leave the origin as illustrated in (Figure 6 c,d). Interestingly, the E. coli DnaB helicases have also been demonstrated to pass one another at the origin [90]. This architecture carries important implications for quality control at an origin. Specifically, if either CMG were to continue to encircle dsDNA, it would block movement of the other CMG that transitioned from dsDNA to encircling ssDNA. Thus, the head-to-head orientation ensures that both CMGs must transition to encircle opposite ssDNA strands before they can pass one another and leave the origin. This feature results in a previously unknown quality control checkpoint that ensures that each origin produces two bidirectional forks, or none at all.
9. Conclusions and prospects
The 11- subunit CMG is the most complex of the cellular replicative helicases and many recent advances have been made. Low resolution EM determined the overall organization of subunits within CMG, and cryoEM has determined atomic models of CMG from both Drosophila and yeast [69, 70, 91]. Furthermore, the structure of active CMG at a replication fork held many unexpected surprises revealing an intimate connection of the steric exclusion helicase with the ds and ss portions of the fork in the N-tier ring of the helicase [55]. The organization of the leading Pol ε and lagging Pol α-primase attached to CMG have also been determined, giving a more detailed view of how the helicase organizes a replisome than currently understood for any system [39]. Through its connections to other proteins CMG helps dictate polymerase asymmetry at the fork [9, 37, 39, 48], stimulates CMG unwinding to fork rates observed in vivo [49, 52, 92], and enables forks to displace nucleosomes [53].
Despite these advances, there are numerous questions that remain about CMG. Many of these questions are now within reach. The mechanism by which CMG translocates along and unwinds DNA needs to be studied by new techniques rather than only structural approaches, and the same could be said for homohexameric helicases. The structure of CMG on forked DNA shows an internal unwinding point with residues close to the surface, and whether CMG actively facilitates DNA unwinding using amino acid side chains should soon be forthcoming. Although CMG assembly at origins has been reconstituted in vitro [27, 33, 34], only the first two steps of this multi-step process have been elucidated [65, 67, 93]. Several key steps during helicase activation are largely unknown. But these mechanistic processes are now within grasp.
The replication process is highly interconnected with many processes of genomic integrity, including transcription, telomere replication, DNA damage checkpoints, interfacing with recombination intermediates, tracking through chromatized DNA while maintaining epigenetic information – all interweave with the replisome machinery. Considering that CMG leads the way at replication forks, and interacts with many proteins, the prospects are extremely bright for exciting new discoveries about the workings of this central and complex machine.
Acknowledgments
We are grateful to Lin Bai (Van Andel Research Institute), Nina Yao (Rockefeller University) and Lance Langston (Rockefeller University) for making some of the illustrations in this review. This work was supported by grants from the NIH (GM111472 and GM124170 (to H.L.) and GM115809 (to M.O.D.)), the Van Andel Research Institute (H.L.); and the Howard Hughes Medical Institute (M.O.D.).
Abbreviations
- CMG
complex of Ctd45, Mcm2-7, GINS
- CTD
C-terminal domain
- NTD
N-terminal domain
- Mcm2-7 DH
double hexamer of Mcm2-7
- RPC
replisome progression complex
- ORC
origin recognition complex
- Pol
DNA polymerase
- PCNA
proliferating nuclear antigen
- MCM
minichromosome maintence
- AAA+
ATPases Associated with various cellular Activities
- BPV
bovine papilloma virus
- SA
streptavidin
Footnotes
The authors have declared no conflict of interest.
References
- 1.Bell SD, Botchan MR. Cold Spring Harb Perspect Biol. 2013;5:a012807. doi: 10.1101/cshperspect.a012807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell SP, Labib K. Genetics. 2016;203:1027. doi: 10.1534/genetics.115.186452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eoff RL, Raney KD. Biochem Soc Trans. 2005;33:1474. doi: 10.1042/BST0331474. [DOI] [PubMed] [Google Scholar]
- 4.Singleton MR, Dillingham MS, Wigley DB. Annu Rev Biochem. 2007;76:23. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 5.Enemark EJ, Joshua-Tor L. Curr Opin Struct Biol. 2008;18:243. doi: 10.1016/j.sbi.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyubimov AY, Strycharska M, Berger JM. Curr Opin Struct Biol. 2011;21:240. doi: 10.1016/j.sbi.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ilves I, Petojevic T, Pesavento JJ, Botchan MR. Mol Cell. 2010;37:247. doi: 10.1016/j.molcel.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moyer SE, Lewis PW, Botchan MR. Proc Natl Acad Sci U S A. 2006;103:10236. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Georgescu RE, Langston L, Yao NY, Yurieva O, Zhang D, Finkelstein J, Agarwal T, O’Donnell ME. Nat Struct Mol Biol. 2014;21:664. doi: 10.1038/nsmb.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang YH, Galal WC, Farina A, Tappin I, Hurwitz J. Proc Natl Acad Sci U S A. 2012;109:6042. doi: 10.1073/pnas.1203734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, Labib K. Nat Cell Biol. 2006;8:358. doi: 10.1038/ncb1382. [DOI] [PubMed] [Google Scholar]
- 12.Gambus A, van Deursen F, Polychronopoulos D, Foltman M, Jones RC, Edmondson RD, Calzada A. K Labib, EMBO J. 2009;28:2992. doi: 10.1038/emboj.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leipe DD, Koonin EV, Aravind L. J Mol Biol. 2003;333:781. doi: 10.1016/j.jmb.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 14.Yao NY, O’Donnell ME. Crit Rev Biochem Mol Biol. 2016;51:135. doi: 10.3109/10409238.2015.1125845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davey MJ, Indiani C, O’Donnell M. J Biol Chem. 2003;278:4491. doi: 10.1074/jbc.M210511200. [DOI] [PubMed] [Google Scholar]
- 16.Costa A, Renault L, Swuec P, Petojevic T, Pesavento J, Ilves I, MacLellan-Gibson K, Fleck RA, Botchan MR, Berger JM. Elife. 2014:e03273. doi: 10.7554/eLife.03273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeruzalmi D, O’Donnell M, Kuriyan J. Cell. 2001;106:429. doi: 10.1016/s0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- 18.Kelch BA, Makino DL, O’Donnell M, Kuriyan J. BMC Biol. 2012;10:34. doi: 10.1186/1741-7007-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker TA, Sauer RT. Biochim Biophys Acta. 2012;1823:15. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madsen L, Seeger M, Semple CA, Hartmann-Petersen R. Int J Biochem Cell Biol. 2009;41:2380. doi: 10.1016/j.biocel.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 21.Thomsen ND, Berger JM. Cell. 2009;139:523. doi: 10.1016/j.cell.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hilbert BJ, Hayes JA, Stone NP, Duffy CM, Sankaran B, Kelch BA. Proc Natl Acad Sci U S A. 2015;112:E3792. doi: 10.1073/pnas.1506951112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. Proc Natl Acad Sci U S A. 2009;106:20240. doi: 10.1073/pnas.0911500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heller RC, Kang S, Lam WM, Chen S, Chan CS, Bell SP. Cell. 2011;146:80. doi: 10.1016/j.cell.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker MW, Botchan MR, Berger JM. Crit Rev Biochem Mol Biol. 2017;52:107. doi: 10.1080/10409238.2016.1274717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. Cell. 2009;139:719. doi: 10.1016/j.cell.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeeles JT, Deegan TD, Janska A, Early A, Diffley JF. Nature. 2015;519:431. doi: 10.1038/nature14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duderstadt KE, Berger JM. Curr Opin Struct Biol. 2013;23:144. doi: 10.1016/j.sbi.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Donnell M, Langston L, Stillman B. Cold Spring Harb Perspect Biol. 2013;5:a010108. doi: 10.1101/cshperspect.a010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Scharer OD, Walter JC. Cell. 2011;146:931. doi: 10.1016/j.cell.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ticau S, Friedman LJ, Ivica NA, Gelles J, Bell SP. Cell. 2015;161:513. doi: 10.1016/j.cell.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yardimci H, Loveland AB, Habuchi S, van Oijen AM, Walter JC. Mol Cell. 2010;40:834. doi: 10.1016/j.molcel.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Devbhandari S, Jiang J, Kumar C, Whitehouse I, Remus D. Mol Cell. 2017;65:131. doi: 10.1016/j.molcel.2016.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Looke M, Maloney MF, Bell SP. Genes Dev. 2017;31:291. doi: 10.1101/gad.291336.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burgers PMJ, Kunkel TA. Annu Rev Biochem. 2017;86:417. doi: 10.1146/annurev-biochem-061516-044709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao NY, O’Donnell ME. In: Encyclopedia of Cell Biology. Bradshaw RA, Stahl PD, editors. Academic Press; 2015. p. 396. [Google Scholar]
- 37.Langston LD, Zhang D, Yurieva O, Georgescu RE, Finkelstein J, Yao NY, Indiani C, O’Donnell ME. Proc Natl Acad Sci U S A. 2014;111:15390. doi: 10.1073/pnas.1418334111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simon AC, Zhou JC, Perera RL, van Deursen F, Evrin C, Ivanova ME, Kilkenny ML, Renault L, Kjaer S, Matak-Vinkovic D, Labib K, Costa A, Pellegrini L. Nature. 2014;510:293. doi: 10.1038/nature13234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun J, Shi Y, Georgescu RE, Yuan Z, Chait BT, Li H, O’Donnell ME. Nat Struct Mol Biol. 2015;22:976. doi: 10.1038/nsmb.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kunkel TA, Burgers PMJ. Bioessays. 2017:39. doi: 10.1002/bies.201700070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clausen AR, Lujan SA, Burkholder AB, Orebaugh CD, Williams JS, Clausen MF, Malc EP, Mieczkowski PA, Fargo DC, Smith DJ, Kunkel TA. Nat Struct Mol Biol. 2015;22:185. doi: 10.1038/nsmb.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kunkel TA, Burgers PM. Nat Struct Mol Biol. 2014;21:649. doi: 10.1038/nsmb.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyabe I, Kunkel TA, Carr AM. PLoS Genet. 2011;7:e1002407. doi: 10.1371/journal.pgen.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Mol Cell. 2008;30:137. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Science. 2007;317:127. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shcherbakova PV, Pavlov YI. Genetics. 1996;142:717. doi: 10.1093/genetics/142.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu C, Gan H, Han J, Zhou ZX, Jia S, Chabes A, Farrugia G, Ordog T, Zhang Z. Mol Cell. 2014;56:551. doi: 10.1016/j.molcel.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Georgescu RE, Schauer GD, Yao NY, Langston LD, Yurieva O, Zhang D, Finkelstein J, O’Donnell ME. Elife. 2015;4:e04988. doi: 10.7554/eLife.04988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schauer GD, O’Donnell ME. Proc Natl Acad Sci U S A. 2017;114:675. doi: 10.1073/pnas.1619748114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson RE, Klassen R, Prakash L, Prakash S. Mol Cell. 2015;59:163. doi: 10.1016/j.molcel.2015.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stillman B. Mol Cell. 2015;59:139. doi: 10.1016/j.molcel.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yeeles JT, Janska A, Early A, Diffley JF. Mol Cell. 2017;65:105. doi: 10.1016/j.molcel.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kurat CF, Yeeles JT, Patel H, Early A, Diffley JF. Mol Cell. 2017;65:117. doi: 10.1016/j.molcel.2016.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langston LD, Mayle R, Schauer GD, Yurieva O, Zhang D, Yao NY, Georgescu RE, O’Donnell ME. Elife. 2017;6:e29118. doi: 10.7554/eLife.29118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Georgescu R, Yuan Z, Bai L, de Luna Almeida Santos R, Sun J, Zhang D, Yurieva O, Li H, O’Donnell ME. Proc Natl Acad Sci U S A. 2017;114:E697. doi: 10.1073/pnas.1620500114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Asturias FJ, Cheung IK, Sabouri N, Chilkova O, Wepplo D, Johansson E. Nat Struct Mol Biol. 2006;13:35. doi: 10.1038/nsmb1040. [DOI] [PubMed] [Google Scholar]
- 57.Zhou JC, Janska A, Goswami P, Renault L, Abid Ali F, Kotecha A, Diffley JF, Costa A. Proc Natl Acad Sci U S A. 2017;114:4141. doi: 10.1073/pnas.1700530114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Costa A, Ilves I, Tamberg N, Petojevic T, Nogales E, Botchan MR, Berger JM. Nat Struct Mol Biol. 2011;18:471. doi: 10.1038/nsmb.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lyubimov AY, Costa A, Bleichert F, Botchan MR, Berger JM. Proc Natl Acad Sci U S A. 2012;109:11999. doi: 10.1073/pnas.1209406109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhai Y, Cheng E, Wu H, Li N, Yung PY, Gao N, Tye BK. Nat Struct Mol Biol. 2017;24:300. doi: 10.1038/nsmb.3374. [DOI] [PubMed] [Google Scholar]
- 61.Bochman ML, Bell SP, Schwacha A. Mol Cell Biol. 2008;28:5865. doi: 10.1128/MCB.00161-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coster G, Diffley JFX. Science. 2017;357:314. doi: 10.1126/science.aan0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Samel SA, Fernandez-Cid A, Sun J, Riera A, Tognetti S, Herrera MC, Li H, Speck C. Genes Dev. 2014;28:1653. doi: 10.1101/gad.242404.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun J, Evrin C, Samel SA, Fernandez-Cid A, Riera A, Kawakami H, Stillman B, Speck C, Li H. Nat Struct Mol Biol. 2013;20:944. doi: 10.1038/nsmb.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yuan Z, Riera A, Bai L, Sun J, Nandi S, Spanos C, Chen ZA, Barbon M, Rappsilber J, Stillman B, Speck C, Li H. Nat Struct Mol Biol. 2017;24:316. doi: 10.1038/nsmb.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li N, Zhai Y, Zhang Y, Li W, Yang M, Lei J, Tye BK, Gao N. Nature. 2015;524:186. doi: 10.1038/nature14685. [DOI] [PubMed] [Google Scholar]
- 67.Noguchi Y, Yuan Z, Bai L, Schneider S, Zhao G, Stillman B, Speck C, Li H. Proc Natl Acad Sci U S A. 2017;114:E9529. doi: 10.1073/pnas.1712537114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petojevic T, Pesavento JJ, Costa A, Liang J, Wang Z, Berger JM, Botchan MR. Proc Natl Acad Sci U S A. 2015;112:E249. doi: 10.1073/pnas.1422003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuan Z, Bai L, Sun J, Georgescu R, Liu J, O’Donnell ME, Li H. Nat Struct Mol Biol. 2016;23:217. doi: 10.1038/nsmb.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abid Ali F, Renault L, Gannon J, Gahlon HL, Kotecha A, Zhou JC, Rueda D, Costa A. Nat Commun. 2016;7:10708. doi: 10.1038/ncomms10708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barry ER, McGeoch AT, Kelman Z, Bell SD. Nucleic Acids Res. 2007;35:988. doi: 10.1093/nar/gkl1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu Y, Gristwood T, Hodgson B, Trinidad JC, Albers SV, Bell SD. Proc Natl Acad Sci U S A. 2016;113:13390. doi: 10.1073/pnas.1613825113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Froelich CA, Kang S, Epling LB, Bell SP, Enemark EJ. Elife. 2014;3:e01993. doi: 10.7554/eLife.01993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miller JM, Enemark EJ. Archaea. 2015;2015:305497. doi: 10.1155/2015/305497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Enemark EJ, Joshua-Tor L. Nature. 2006;442:270. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 76.Itsathitphaisarn O, Wing RA, Eliason WK, Wang J, Steitz TA. Cell. 2012;151:267. doi: 10.1016/j.cell.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Langston L, O’Donnell M. Elife. 2017;6:e23449. doi: 10.7554/eLife.23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Donmez I, Patel SS. Nucleic Acids Res. 2006;34:4216. doi: 10.1093/nar/gkl508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hacker KJ, Johnson KA. Biochemistry. 1997;36:14080. doi: 10.1021/bi971644v. [DOI] [PubMed] [Google Scholar]
- 80.Kaplan DL, Davey MJ, O’Donnell M. J Biol Chem. 2003;278:49171. doi: 10.1074/jbc.M308074200. [DOI] [PubMed] [Google Scholar]
- 81.McGeoch AT, Trakselis MA, Laskey RA, Bell SD. Nat Struct Mol Biol. 2005;12:756. doi: 10.1038/nsmb974. [DOI] [PubMed] [Google Scholar]
- 82.Patel SS, Hingorani MM, Ng WM. Biochemistry. 1994;33:7857. doi: 10.1021/bi00191a013. [DOI] [PubMed] [Google Scholar]
- 83.Trakselis MA. F1000Res. 2016:5. doi: 10.12688/f1000research.7509.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee SJ, Syed S, Enemark EJ, Schuck S, Stenlund A, Ha T, Joshua-Tor L. Proc Natl Acad Sci U S A. 2014;111:E827. doi: 10.1073/pnas.1322254111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Donmez I, Rajagopal V, Jeong YJ, Patel SS. J Biol Chem. 2007;282:21116. doi: 10.1074/jbc.M702136200. [DOI] [PubMed] [Google Scholar]
- 86.Manosas M, Xi XG, Bensimon D, Croquette V. Nucleic Acids Res. 2010;38:5518. doi: 10.1093/nar/gkq273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kanke M, Kodama Y, Takahashi TS, Nakagawa T, Masukata H. EMBO J. 2012;31:2182. doi: 10.1038/emboj.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Deursen F, Sengupta S, De Piccoli G, Sanchez-Diaz A, Labib K. EMBO J. 2012;31:2195. doi: 10.1038/emboj.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Watase G, Takisawa H, Kanemaki MT. Curr Biol. 2012;22:343. doi: 10.1016/j.cub.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 90.Fang L, Davey MJ, O’Donnell M. Mol Cell. 1999;4:541. doi: 10.1016/s1097-2765(00)80205-1. [DOI] [PubMed] [Google Scholar]
- 91.Kelman Z, Lee JK, Hurwitz J. Proc Natl Acad Sci U S A. 1999;96:14783. doi: 10.1073/pnas.96.26.14783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lewis JS, Spenkelink LM, Schauer GD, Hill FR, Georgescu RE, O’Donnell ME, van Oijen AM. Proc Natl Acad Sci U S A. 2017;114:10630. doi: 10.1073/pnas.1711291114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hizume K, Kominami H, Kobayashi K, Yamada H, Araki H. Biochemistry. 2017;56:2435. doi: 10.1021/acs.biochem.6b00922. [DOI] [PubMed] [Google Scholar]