

Abstract
We present six novel patients affected by lipid storage myopathy (LSM) presenting mutations in the ETFDH gene. Although the diagnosis of multiple acyl-coenzyme-A dehydrogenase deficiency (MADD) in adult life is difficult, it is rewarding because of the possibility of treating patients with carnitine or riboflavin, leading to a full recovery. In our patients, a combination of precipitating risk factors including previous anorexia, alcoholism, poor nutrition, and pregnancy contributed to a metabolic critical condition that precipitated the catabolic state.
In the present series of cases, five novel mutations have been identified in the ETFDH gene. We propose clinical guidelines to screen patients with LSM due to different defects, in order to obtain a fast diagnosis and offer appropriate treatment. In such patients, early diagnosis and treatment as well as avoiding risk factors are part of clinical management.
Specific biochemical studies are indicated to identify the type of LSM, such as level of free carnitine and acyl-carnitines and studies or organic acidemia. Indeed, when a patient is biochemically diagnosed with secondary carnitine deficiency, a follow-up with appropriate clinical-molecular protocol and genetic analysis is important to establish the final diagnosis, since riboflavin can be supplemented with benefit if riboflavin-responsive MADD is present. In muscle biopsies, increased lipophagy associated with p62-positive aggregates was observed. The clinical improvement can be attributed to the removal of an autophagic block, which appears to be reversible in this LSM.
Introduction
Lipid Storage Myopathies (LSMs) are a heterogeneous group of disorders of lipid metabolism characterized by impaired oxidation of fatty acids (FAs). LSM can arise from different defects in lipid metabolism (carnitine transport, beta-oxidation, and endogenous triglyceride catabolism); almost all associated with dys-metabolism of FAs and their derivatives (Liang and Nishino 2011).
Primary Systemic Carnitine Deficiency (SCD, MIM# 212140) is due to a defect in the plasma membrane high-affinity carnitine transporter (OCTN2, encoded by SLC22A5 gene), causing systemic carnitine depletion.
In Multiple Acyl-coenzyme-A Dehydrogenase Deficiency (MADD, MIM# 231680), symptoms and age at onset are highly variable and characterized by recurrent episodes of lethargy, vomiting, hypoglycaemia, metabolic acidosis, and hepatomegaly, often preceded by a metabolic stress. Muscle involvement, myalgia, weakness, and LSM may occur. MADD is also known as “glutaric aciduria type II”, because it results in large excretion of glutaric, lactic, ethyl-malonic, butyric, isobutyric, 2-methyl-butyric, and isovaleric acids. The organic aciduria in late-onset MADD patients is often intermittent and evident during illness or catabolic stress.
A clinical improvement of MADD myopathy following riboflavin supplementation has been observed in patients affected by either the infantile-severe, the adult, or the late-onset myopathic form. Most late-onset cases are characterized by a secondary carnitine-deficient LSM. MADD can be caused by mutations in three different genes (ETFA, ETFB, and ETFDH) which are involved in the electron transfer in the mitochondrial respiratory chain. In most MADD patients, the disease is caused by mutations in the ETFDH gene (MIM # 231675), encoding the ETF dehydrogenase enzyme protein.
We present six patients affected by a carnitine-deficient LSM, with novel mutations in the ETFDH gene.
Methods
Patients
We investigated six patients from four families affected with a carnitine/riboflavin-responsive form of LSM. The diagnosis was based on characteristic muscle pathology features, low levels of plasma/muscle carnitine, high levels of acyl-carnitines, and glutaric acidemia.
Clinical re-evaluation was done during follow-up, including neuromuscular examinations, muscle MRI or CT imaging, and blood sample collection for genetic analysis.
Muscle Biopsy
Informed consent was obtained from all patients for undergoing muscle biopsy as part of the diagnostic procedure. Muscle biopsies were used for histopathological evaluation, following a panel of routine histochemical and histoenzymatic stains, and for ultrastructural analysis.
Biochemical Analysis
Carnitine and its fractions (acyl-carnitines) were extracted from muscle and plasma and their levels were determined using a standardized radiochemical method. Organic acids profile was investigated by mass spectrometry. Mitochondrial respiratory chain enzymes activity (OX-PHOS) was measured by a standard spectrophotometric method.
Genetic Analysis
Genomic DNA was extracted from blood sample using a standard method, and used for sequencing analysis of all exons and their flanking regions of the ETFDH gene.
Case Reports
Patient 1
This woman was hospitalized at age 36 years for psychiatric disturbances, alcoholism, and poor nutrition. She referred muscle pain in the upper limbs and weakness in the lower limbs with difficulty walking. She was unable to rise from the floor without assistance, had lost body weight over previous months, had difficulty walking and rising from a chair, and was unable to climb stairs. She was inadequately nourished, and her weakness had progressed steadily. She presented temporal lobe epilepsy, CK = 868 U/L, myopathic EMG, and had a subacute onset of carnitine-deficient LSM. Plasma acyl-carnitines and urinary organic acids profiles indicated an increased acyl-carnitine level. There was glutaric aciduria type II (ethyl-malonic acid = 70.5 nMol/mol creatinine, normal values 0.1–17.9). Muscle biopsy revealed LSM and low muscle carnitine levels (11% of controls). Mitochondrial enzyme activities were decreased in muscle after normalization to citrate synthase: NADH-dehydrogenase = 286.2 (normal values 391–663 nmol/min non-collagen protein), NADH-Cytochrome C reductase = 17.5 (n.v. 21.3–86.9), and Succinate Cytochrome C reductase = 4.35 (n.v.5.8–19.1). Treatment with a low-fat, high-protein diet and 4 g/day l-carnitine (because of secondary muscle carnitine deficiency with elevated acyl-carnitines) produced some improvement; however, only riboflavin supplementation (200 mg/day) produced marked improvement.
At age 45 years, she presented a crisis after an ab-ingestis pneumonia with septic shock, metabolic acidosis, dysphagia, and respiratory insufficiency, which required tracheostomy and assisted ventilation. On neurological examination, she had waddling gait, diffuse hypotonia, and weakness in lower limbs and hand grip. She slowly improved with carnitine, riboflavin, and carbamazepine.
Patient 2
This woman at age 38 years had a subacute onset of weakness of upper and lower girdle muscles, neck flexors, and respiratory muscles and became virtually quadriplegic and respirator-dependent. CK was 2,277 U/L, and a polymyositis was suspected. A steroid therapy was therefore tried for a short period of time while the patient was in intensive care unit. EMG was myopathic. A first muscle biopsy showed carnitine-deficient LSM with abnormal palmitate oxidation, which demonstrates a block of beta-oxidation. Treatment with a low-fat, high-protein diet, medium-chain triglyceride (MCT) oil supplementation, and 4 g/day of l-carnitine (because of muscle carnitine deficiency with increased acyl-carnitines and low palmitate beta-oxidation) produced some improvement and normalization of carnitine in plasma and muscle in a post-treatment muscle biopsy, which showed a decreased amount of lipid droplets but atrophic fibres. Riboflavin supplements (200 mg/day) produced improvement, preventing further metabolic crises. A muscle CT scan was recently repeated and showed only a mild muscle atrophy in both legs.
Patient 3
This 23-year-old woman presented extremely thin and weak muscles of undetermined cause. She complained of progressive arm, trunk, and lower limb weakness and exercise intolerance, developed myalgias, and had difficulty riding a bike or raising arms. One year later she had a thrombosis of inferior cava vein, recurrent episodes of hypoglycaemia (31 mg/dL), vomiting, conspicuous (10 kg) body weight loss, high CK (15,000 U/L), and diffuse myalgias. She became virtually quadriplegic due to nutritional deficiency (body weight was 48 kg) and had to be fed by nasogastric tube. On neurological examination, she presented a waddling gait, was able to go from a lying to sitting position only with the help of hands, could neither raise her arms in horizontal position nor raise legs from bed, and had a proximal muscle hypotrophy, winging scapulae, and distal leg hypotrophy. Manual muscle test by the MRC scale showed severe weakness of head flexors, deltoid, biceps, triceps, and arms extra-rotators, and of lower girdle muscles, especially of iliopsoas, tibialis anterior, peroneus, and EDL. On spirometry, a moderate restrictive deficiency was found. Brain MRI was normal, and muscle MRI revealed oedema and marked atrophy of upper girdle, thigh, and leg muscles (Fig. 1).
Fig. 1.
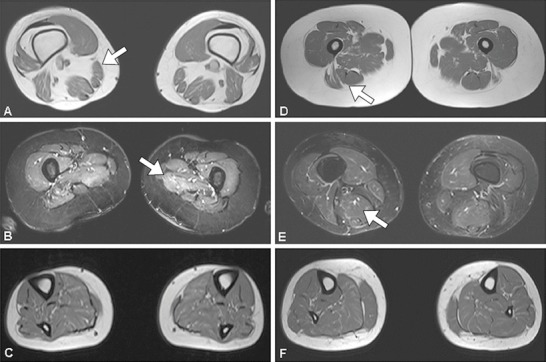
Muscle MRI imaging in patient 3 (a, b, c) and patient 4 (d, e, f) using T1 sequences (a, d, c, f) and STIR sequences (b, e). Note atrophy of the thigh muscles (a, b) and in the leg muscles (c) in patient 3, while thigh muscles (d, e) and leg muscles (f) in patient 4 are not atrophic. Note hyper-intense signal in STIR sequences at the thigh level (b, e) due to myo-oedema
Muscle biopsy showed the features of LSM (Fig. 2). We found a reduction of total muscle carnitine (4.34, n.v. 10.5–29.5), total plasma carnitine (6.77 mMol/L, n.v. 36.2–72.9), and free plasma carnitine (3.34 nMol/mL, n.v. 27.6–61.9) (Table 1). Plasma acyl-carnitines profile by mass spectroscopy was normal. Because of carnitine deficiency in muscle and plasma, she was treated simultaneously with riboflavin (200 mg/day) and l-carnitine (2 g/day) both orally and by infusion; with this treatment she could have her nasogastric tube removed and then, when she was able to walk, only by oral therapy. She gained body weight and muscle strength.
Fig. 2.
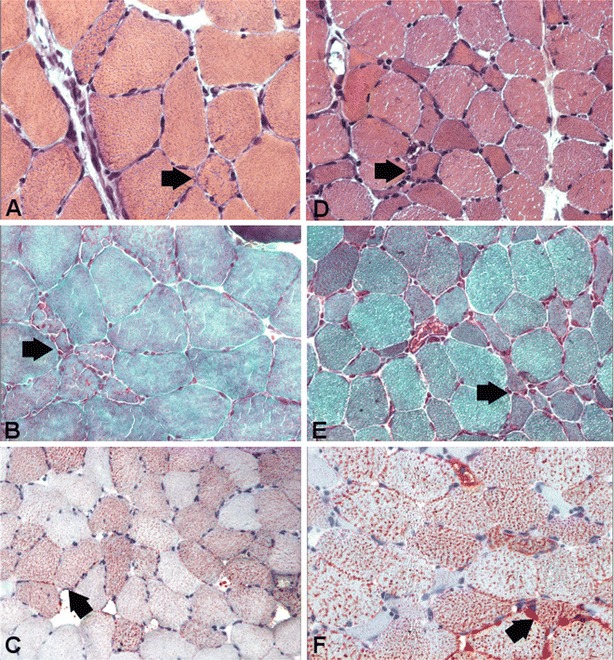
Muscle biopsy morphology in patient 3 (a, b, c) and patient 4 (d, e, f) stained for hematoxylin-eosin (a, d), Gomori trichrome (b, e), and Oil-Red-O (c, f). Note that in patient 3 there is lipid storage and fibre size variability, whereas in patient 4 there are many atrophic fibres (d, e) and lipid storage (f). The atrophy is likely due to a prolonged steroid treatment
Table 1.
Clinical data, laboratory and genetic findings, and treatment
| Patient no. | Gender | Age at onset (years) | Age at biopsy (years) | Causes of metabolic stress | Muscle carnitine levels (% of controls) | ETFDH gene mutations | Treatment |
|---|---|---|---|---|---|---|---|
| 1 | F | 35 | 36 | Poor nutrition and alcoholism | 11 | c.412C>T, p.L138F, exon 4; c.1531G>A, p.D511N, exon 12 | Riboflavin (200 mg/day), carnitine (4 g/day), and carbamazepine |
| 2 | F | 38 | 41 | Poor nutrition | 17 | c.560C>T, p.A187V, exon 5; c.1027T>C, p.W343R, exon 11 | Carnitine (2 g/day), MCT oil, riboflavin (200 mg/day), and steroids |
| 3 | Fa | 23 | 23 | Hypoglycaemia, venous thrombosis, and weight loss | 30 |
c.451A>G (p.T151A), exon 4 c.1649T>G, p.L550P, exon 12, |
Heparin (5,000 U/L), 10% glucose i.v., riboflavin (200 mg/day), and carnitine (2 g/day) |
| 4 | Fa | 24 | 28 | Sepsis and immunosuppressive treatment | 12 | Steroids, riboflavin, and methotrexate/cell-cept | |
| 5 | Fb | 16 | 16 | Pregnancy and delivery | 10 | c.152G>A (p.R51Q), exon 2 c.606+5insT, exon 5 |
Carnitine (2 g/day) and steroids |
| 6 | Mb | 33 | 33 | Cold exposure | 20 | Riboflavin (200 mg/day) |
Novel mutations are indicated in bold
a,bPairs of siblings
Patient 4
This is the older sister of patient 3. She was a 28-year-old, overweight woman, had experienced progressive lower limb weakness and myalgias for 4 years, and exercise intolerance, intermittent hypoglycaemic episodes, and increased CK (11,000 U/L). She was misdiagnosed with polymyositis, and treated with IVIg, steroids (caused a cushingoid syndrome), methotrexate, and mycophenolate mofetil (Cell-cept) without benefit. On neurological examination, she had slow gait, kyphoscoliosis, slight weakness of head flexors, deltoid, arm extra-rotators, and iliopsoas muscles. CK was 188 U/L. Spirometry was normal. On abdominal ultrasound, a slight hepatomegaly was found. Muscle biopsy showed an LSM (Fig. 2). Muscle carnitine level was reduced (1.77, n.v. 10.5–29.5). Plasma acyl-carnitine profile was normal. She was started treatment with riboflavin (200 mg/day) and l-carnitine (2 g/day) and regained muscle strength.
Patient 5
This 16-year-old girl presented an acute post-partum carnitine deficiency syndrome, previously described (Angelini et al. 1978). She had generalized muscle weakness after the delivery, and, 2 months later had neck and limb muscle weakness, and myalgia and was unable to chew or lift arms and was quadriplegic. On muscle biopsy, there was an LSM, with low plasma and muscle carnitine levels (10%). She slowly recovered following a low-fat diet, with carnitine and MCT oral supplementation. Treatment with riboflavin was not done because after delivery, she joined her family to Germany and was lost to follow-up. We learned about her death from her brother, reporting that she was hospitalized for a respiratory infection, followed by a Reye-like syndrome. Steroid treatment was tried, but she went into coma and died at age 17 years.
Patient 6
This is the 33-year-old brother of patient 5. After being exposed to cold, he presented painful myalgia in the calves that progressed in the following days and extended to the thigh muscles. He could only walk 100 m because his lower limbs were swollen and stiff. CK was 3670 U/L and a first muscle biopsy showed LSM. Mitochondrial enzymes activities were reduced, after normalization to citrate synthase: NADH-dehydrogenase = 226.8 (normal values 391–663 nmol/min non-collagen protein), Succinate-dehydrogenase = 2.1 (n.v. 6.5–20.9), and Cytochrome oxidase = 14.71 (n.v. 24.5–57.5). A second muscle biopsy was performed, and mitochondria were isolated to conduct beta-oxidation studies, which showed a low oxidation of radiolabelled palmitate. Mitochondrial enzyme activities returned to be in the range of controls. On exercise tests with VO2max, he arrived only to 60 W. He was then started on riboflavin treatment at 200 mg/day, which resulted in a remarkable clinical improvement. A few months later, a second exercise test was normal.
Results
Patients
The clinical features of patients are summarized in Table 1. All patients had a juvenile/adult onset form with generalized muscle weakness, low muscle carnitine, and lipid storage in muscle, mostly localized in type 1 fibres. A variable decrease of OX-PHOS complexes documented mitochondrial involvement. Muscle ultrastructural analysis showed a massive increase of intra-cytoplasmic lipid droplets, which were usually localized nearby mitochondria and were found decreased after treatment.
Muscle Imaging
Muscle mass was investigated by imaging in three cases. By CT scan, an asymmetric atrophy of the legs was found in patient 2, whereas by muscle MRI, there was atrophy of both in upper and lower girdle muscles and in leg muscles in patient 3, and irregular muscle mass and some fatty replacement in patient 4 (Fig. 1).
Genetic Analysis
Eight different mutations in the ETFDH gene have been identified (Table 1): four missense mutations are novel (p.L138F, p.T151A, p.W343R, and p.L550P), and three missense (p.R51Q [rs534388496], p.A187V [rs369912835], and p.D511N) have already been reported (Wen et al. 2010, 2013; Sugai et al. 2012). Moreover, a new splice site mutation has been detected in patient 6 (c.606+5insT). It is likely that his sister (patient 5), with a similar clinical and pathological features, might have carried the same mutations. Novel mutations have been confirmed by restriction enzyme analysis and not found in about 100 control chromosomes.
Discussion
Most patients with MADD have a good response to carnitine and riboflavin (Wen et al. 2010). Olsen et al. (2007) noted that riboflavin-responsive MADD may result from a defect in ETFDH, which might be associated with a generalized mitochondrial dysfunction. In support to this notion, all our patients had mutations in the ETFDH gene, and variable decrease of OX-PHOS complexes.
Table 2 summarizes the clinical differences between the different defects of FAs metabolism, as a guideline for subsequent studies. In RR-MADD, there is a characteristic profile of acyl-carnitines in blood (Wen et al. 2010) and concomitant low free carnitine in plasma and muscle.
Table 2.
Clinical guidelines for screening defects of fatty acid metabolism
| Enzyme/cofactor | Phenotype | |||
|---|---|---|---|---|
| Myopathy, hypotonia | Cardiomyopathy | Myoglobinuria | Hypoglycaemia, hypoketonaemia | |
| Fatty acid transport | ||||
| Carnitine (OCTN2 gene mutations) | ++ | ++ | − | ++ |
| Carnitine palmitoyl transferase | ± | + | ++ | + |
| Beta-oxidation enzymes | ||||
| Long-chain acyl-CoA dehydrogenase | ++ | + | + | ++ |
| Medium-chain acyl-CoA dehydrogenase | ± | ± | − | ++ |
| Short-chain acyl-CoA dehydrogenase | + | ± | − | + |
| 3-Hydroxy acyl-CoA dehydrogenase | + | ++ | − | ++ |
| Neutral lipid storage (NLS) diseases | ||||
| NLSD-M | ++ | + | − | − |
| NLSD-I | + | − | − | − |
| Transferring flavoproteins | ||||
| Electron transfer flavoprotein (ETF) | ± | ± | − | ++ |
| ETF coenzyme Q reductase (ETF-QO) | ± | − | − | ++ |
| Riboflavin-responsive forms (RR-MADD) | + | − | − | ++ |
++ present, + sometimes present, ± rarely observed, − absent
Using the information from our cases, we developed a possible flow chart to make a diagnosis, since when secondary carnitine deficiency is suspected, l-carnitine supplementation brings a normal carnitine profile. Genetic analysis is important for the final diagnosis, since riboflavin can be added. Indeed, if clinical manifestations respond, it is worth undertaking ETFDH analysis, a common cause of MADD (Olsen et al. 2007).
It is noteworthy that an increased autophagic activity was found in our patients’ muscle biopsies, which we previously found by immunoblot experiments (Angelini et al. 2016). In two of our patients, we previously demonstrated a co-localized expression of TFEB and p62/SQSTM1 (marker of protein aggregates) in atrophic fibres. Immunoblot analysis of p62/SQSTM1, LC3, and TFEB showed a marked increased expression during the acute phase of the disease. The appearance of a lipidated LC3-II band implies the occurrence of autophagosome proliferation, while the concurrent increased p62/SQSTM1 expression was suggestive of a block of the autophagic flux (Angelini et al. 2016).
Other conditions such as neutral LSM (NLSD, due to PNPLA2 or ABDH5 gene mutations) do not recover, due to their lipolysis block. Unsurprisingly, NLSD patients present a variety of clinical manifestations, such as myopathy, hepatomegaly, and neuropathy, and respond poorly to treatment (Missaglia et al. 2015).
The data presented suggest that disturbances of mitochondrial protein integrity are a secondary consequence of RR-MADD. It is possible that the primary ETFDH deficiency may cause secondary impairment of mitochondrial OX-PHOS complexes due to accumulation of toxic metabolites and increased oxidative stress. Cornelius et al. (2012) observed a prolonged association of mutant ETFDH proteins in fibroblasts from such patients with the Hsp60 chaperon in mitochondrial matrix determining decreased heat stability. Risk factors to be considered in such patients are feverish state, bacterial or viral infections, physiological stresses like pregnancy, and restricted diet. Mitochondrial OX-PHOS status in macrophages during such metabolic crises is turned to an activated state resulting in a different metabolic anaerobic profile and increased ROS production (Angelini 2017; Mills et al. 2016).
An acute metabolic decompensation was fatal in patient 5, who had low muscle and plasma levels of carnitine, and LSM presumably for decreased oxidation of FAs and died of sepsis. Her brother presented a similar syndrome, but we were able to detect early his LSM with a low oxidation of radiolabelled palmitate in muscle and were able to treat his catabolic state.
A subgroup of patients with MADD showed a significant response to supplementary treatment with riboflavin, resulting in near-normalized biochemical and clinical parameters after high doses of oral supplementation: they are commonly referred as riboflavin-responsive MADD. This clinical entity has probably various aetiologies, that can be clinically differentiated from the primary SCD since patients present weakness of upper and lower girdle muscles, but no cardiomyopathy. The cause of the acute myopathy seems to be due to activation of lipophagy and block of metabolism, and lack of coenzyme FAD might result in instability of dehydrogenases, as observed in fibroblast cultures (Indo et al. 1991; Freneaux et al. 1992; Cornelius et al. 2012).
Synopsis
Here, six new cases of LSM with various phenotypes and precipitating factors are presented, with mutations of the ETFDH gene.
Conflict of Interest
Corrado Angelini declares that he has no conflict of interest. Daniela Tavian declares that she has no conflict of interest. Sara Missaglia declares that she has no conflict of interest.
Informed Consent
Informed consent was obtained from all patients for being included in the study.
Animal Rights
This article does not contain any studies with animals performed by any of the Authors.
Details of the Contribution of Individual Authors
Dr. Angelini collected and analysed the data, planned the study and wrote the manuscript. Drs. Tavian and Missaglia collected and analysed the data, and revised the manuscript.
Contributor Information
Corrado Angelini, Email: corrado.angelini@unipd.it.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Angelini C (2017) Metabolites-mitochondria-macrophages (MMM): new therapeutic avenues for inflammation and muscle atrophy. Transl Cancer Res 5(4):S8–S11 doi: 10.21037/tcr.2017.01.37
- Angelini C, Govoni E, Bragaglia MM, Vergani L. Carnitine deficiency: acute postpartum crisis. Ann Neurol. 1978;4:558–561. doi: 10.1002/ana.410040616. [DOI] [PubMed] [Google Scholar]
- Angelini C, Nascimbeni AC, Cenacchi G, Tasca E. Lipolysis and lipophagy in lipid storage myopathies. Biochim Biophys Acta. 2016;1862:1367–1373. doi: 10.1016/j.bbadis.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius N, Frerman FE, Corydon TJ, et al. Molecular mechanism of riboflavin-responsiveness in patients with ETF-QO variations and multiple acyl-CoA dehydrogenation deficiency. Hum Mol Genet. 2012;21:3435–3448. doi: 10.1093/hmg/dds175. [DOI] [PubMed] [Google Scholar]
- Freneaux E, Sheffield VC, Molin L, Shires A, Rhead WJ. Glutaric acidemia type II: heterogeneity in beta-oxidation flux, polypeptide synthesis, and complementary DNA mutations in the alpha-subunit of electron transfer flavoprotein in eight patients. J Clin Invest. 1992;90:1679–1686. doi: 10.1172/JCI116040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indo Y, Glassberg R, Yokota I, Tanaka K. Molecular characterization of variant alpha-subunit of electron transfer flavoprotein in three patients with glutaric acidemia type II and identification of glycine substitution for valine 157 in the sequence of the precursor, producing an unstable mature protein in a patient. Am J Hum Genet. 1991;49:575–580. [PMC free article] [PubMed] [Google Scholar]
- Liang WC, Nishino I. Lipid storage myopathy. Curr Neurol Neurosci Rep. 2011;11:97–103. doi: 10.1007/s11910-010-0154-y. [DOI] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167:457–470. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missaglia S, Tasca E, Angelini C, Moro L, Tavian D. Novel missense mutations in PNPLA2 causing late onset and clinical heterogeneity of neutral lipid storage disease with myopathy in three siblings. Mol Genet Metab. 2015;115:110–117. doi: 10.1016/j.ymgme.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Olsen RK, Olpin SE, Andersen BS, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Brain. 2007;130:2045–2054. doi: 10.1093/brain/awm135. [DOI] [PubMed] [Google Scholar]
- Sugai F, Baba K, Toyooka K, et al. Adult-onset multiple acyl CoA dehydrogenation deficiency associated with an abnormal isoenzyme pattern of serum lactate dehydrogenase. Neuromuscul Disord. 2012;22:159–161. doi: 10.1016/j.nmd.2011.08.004. [DOI] [PubMed] [Google Scholar]
- Wen B, Dai T, Li W, et al. Riboflavin-responsive lipid storage myopathy caused by ETFDH gene mutations. J Neurol Neurosurg Psychiatry. 2010;81:231–236. doi: 10.1136/jnnp.2009.176404. [DOI] [PubMed] [Google Scholar]
- Wen B, Li D, Shan J, et al. Increased muscle coenzyme Q10 in riboflavin responsive MADD with ETFDH gene mutations due to secondary mitochondrial proliferation. Mol Genet Metab. 2013;109:154–160. doi: 10.1016/j.ymgme.2013.04.007. [DOI] [PubMed] [Google Scholar]