

Abstract
Staphylococcus aureus (S. aureus) is an important pathogen of humans and livestock species, but an understanding of the clonal distribution of S. aureus causing different host-species infections in the same geographical environment and within the same period is lacking. By characterizing infections caused by S. aureus in bovine, pediatric, and adult patients in Shanghai, China, between 2012 and 2014, we identified methicillin-sensitive S. aureus (MSSA) ST188 as the major lineage causing infections in multiple host species. Whole-genome sequencing and phenotypic analyses demonstrated that ST188 might evolve from livestock, and there was no significant genomic or virulence difference between ST188 isolated from livestock and humans. The virulence of ST188 is related to its adhesion and nasal colonization ability. This result is in accord with the strong epithelial cell adhesion and biofilm formation properties of ST188. Furthermore, the adhesion- and biofilm-formation-related genes are present in multiple copies and exhibit significantly increased expression in ST188. In conclusion, S. aureus ST188 is the major lineage causing human and livestock infections in Shanghai, China. Due to its high expression of the factors associated with bacterial adhesion and biofilm formation, ST188 has the ability to colonize and infect different host species.
Introduction
S. aureus is a major human and economically important livestock species pathogen that is responsible for a variety of infectious diseases1,2. S. aureus can cause skin and soft tissue infections, pneumonia, bacteremia, and endocarditis in human beings1, and it is also considered one of the most relevant pathogens causing skin abscesses, mastitis, and skeletal infections in livestock3. Current typing methods, such as multi-locus sequence typing (MLST), have been applied to examine the population structure of S. aureus strains with respect to their specificity for infecting humans or animals. The majority of human and animal S. aureus isolates belong to a small number of host specificity clones4,5.
S. aureus mainly causes hospital-associated infections in predisposed individuals. In China, as in most Asian countries, the multiple-antibiotic-resistant strains ST239 and ST5 are of predominantly human hospital-associated methicillin-resistant S. aureus (HA-MRSA) lineages6,7. These two lineages account for nearly 90% of all HA-MRSA isolates in Chinese hospitals6. Recent studies have demonstrated that ST239 displays adaptive evolution with attenuated virulence for successful colonization in the environment with high antibiotics pressure. ST239 has also acquired specific molecular determinants to promote nasal colonization, immune evasion and virulence via horizontal gene transfer8. Neither ST239 nor ST5, which exhibit host specificity and restriction, have been isolated from livestock animals in China9. In contrast, emerging community-associated S. aureus (CA-SA) strains have collectively demonstrated increased virulence potential enabling the infection of otherwise healthy people outside of hospital settings10. The dominant CA-SA strain in the United States is the highly virulent USA300 (ST8), while the predominant Asian CA-SA lineage is ST5911. ST59 exhibits more pronounced virulence than the geographically matched HA-MRSA clones ST5 and ST23912. By high expression of core genome-encoded virulence determinants, CA-SA has acquired high-virulence phenotypes13.
Although nearly all S. aureus lineages are host-specific, there are strains that have a broader host tropism14. Livestock-associated S. aureus (LA-SA) ST398 was originally reported among pigs and pig farmers in the Netherlands in 2003, and it has been identified as the most pandemic LA-MRSA in Europe and North America15. However, in the Americas, ST398 is now also recognized as a causative agent of infections in humans living in animal-free environments, suggesting host-adaptive diversification of ST39816. Whole-genome sequencing (WGS) revealed this difference in its genetic composition, which was associated with significantly enhanced adhesion of human ST398 isolates and transmission among humans17. Although we have observed a considerably high rate of CA-SA infections caused by highly virulent ST398 in China18, the predominant LA-MRSA lineage in China is ST915, and most LA-ST398 infections in China have been reported to be MSSA infections9. This incongruent distribution implies that the clonal distribution of S. aureus causing different host species infections has considerable geographical variation.
To analyze the clonal distribution of S. aureus causing infections in different host species within the same geographical environment and within the same period, we investigated bovine, pediatric, and adult patient infections caused by S. aureus in Shanghai, China, between 2012 and 2014. WGS and in vivo infection models were used to clarify the characteristics of host-adaptable S. aureus.
Results
S. aureus ST188 is the major lineage causing infection in humans and livestock in Shanghai, China
The clonal distribution of S. aureus causing infections in different host species showed that many S. aureus lineages were host-specific (Fig. 1). The predominant lineages causing livestock-specific infection were ST97 (39.2%) and ST520 (16.0%) (Fig. 1a). The major lineages causing human-specific infections were ST5 (36.3%) and ST239 (18.0%) (Fig. 1c). However, several lineages showed the capacity to cause disease in multiple host species: ST188, ST398, ST7, ST59, and ST1 (Fig. 1a–c). Among them, ST188 was the major host-adapted lineage, causing 9.9% (21/212) of livestock-associated infections, 15.7% (25/159) of pediatric patient infections and 5.6% (44/791) of adult patient infections; moreover, t189 was of the epidemic spa type (>80%) in all types of host-associated ST188 (Supplementary Figure 1A). The Agr type was detected in all S. aureus ST188, and both human-associated (100%) and livestock-associated (100%) ST188 carried agr-group I.
Fig. 1. Clonal distribution of S. aureus causing infections of different host species in Shanghai, China, between 2012 and 2014.

a Clonal distribution of S. aureus causing bovine mastitis. b Clonal distribution of S. aureus causing infections in pediatric patients. c Clonal distribution of S. aureus causing infections in adult patients. Clones marked in gray: sequence types causing infections in both humans and livestock
Phylogenetic analysis of host-adapted ST188 isolates
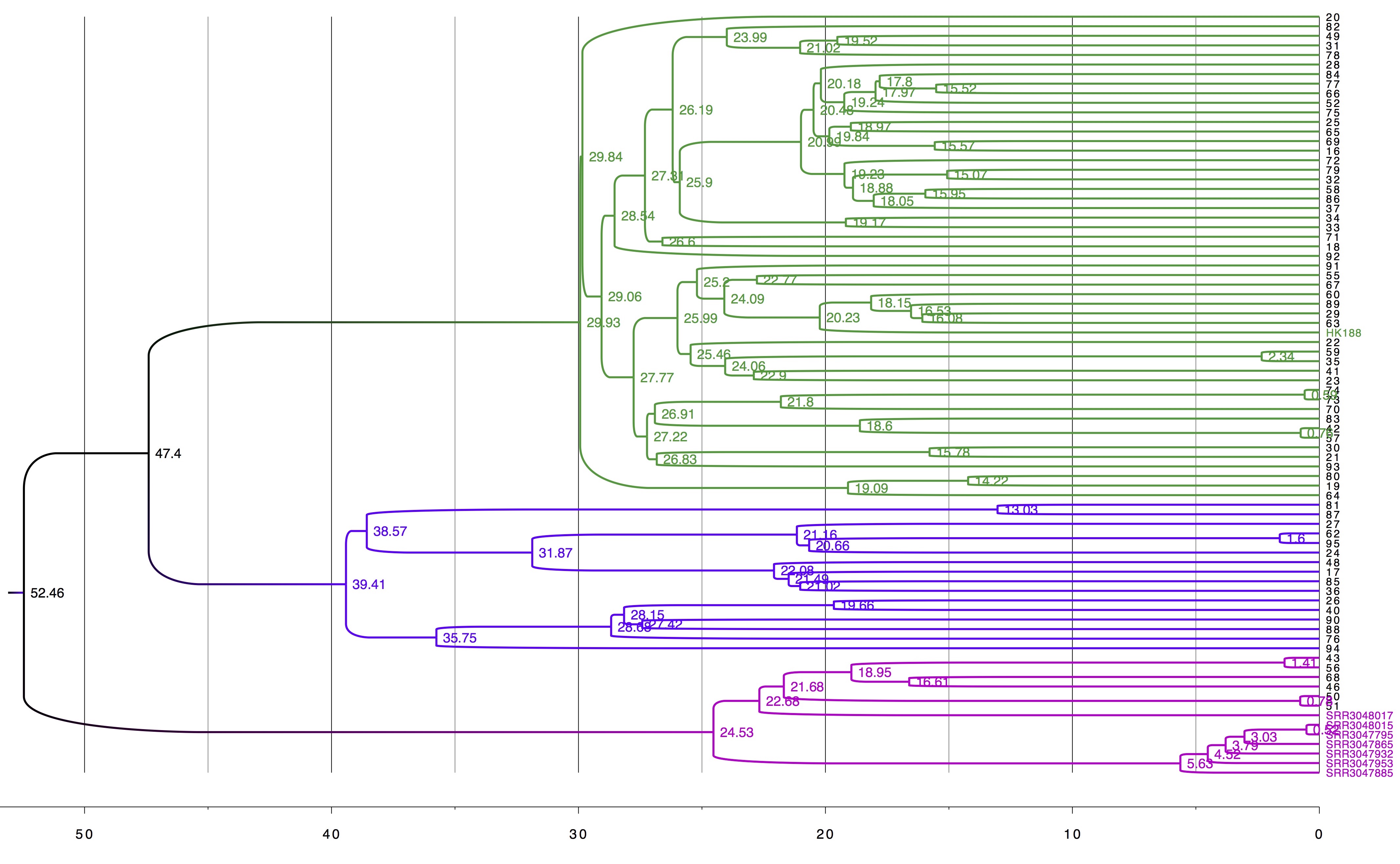
A total of 72 recoverable ST188 isolates were whole-genome sequenced. The core-genome SNPs were applied for phylogenetic tree reconstruction using maximum likelihood estimation (Fig. 2). All host-adapted ST188 isolates were divided into three major phylogenetic clades, with each supported by 100% bootstrapping. Most of the livestock-associated ST188 were in Clade 1, including four isolates of primate origin LA-MRSA ST18819. Human-adapted ST188 were mainly in Clades 2 and 3. Clade 3 accounted for 63.8% (51/80) of ST188 isolates sampled, including human-adapted MRSA CUHK_HK18820. Notably, these three clades were all mixtures of samples collected from both humans and livestock. Furthermore, we applied both a parsimony-based method and a Bayesian-based method to estimate the host state of the most common ancestor of ST188 isolates. Both analysis methods indicated a mixed state of the ancestor isolate (Supplementary Figure 2).
Fig. 2. Maximum-likelihood phylogenetic tree of ST188.

Seventy-two S. aureus ST188 isolates (39 from adult patients, 19 from pediatric patients and 14 from livestock) were used for phylogenetic reconstruction. Relationships are shown with respect to the draft genome of MRSA CUHK_HK188 and seven isolates of MRSA ST188 from North America. Branches are colored by geographic origin of isolates. Black branch: isolates in this study. Pink branch: isolated from North America. Green branch: isolated from Hong Kong. The outer dots indicate the host origin of isolates. Rose-red dots: human-adapted isolates. Blue dots: livestock-adapted isolates. Orange dots: environmental isolates
We further mapped antibiotic susceptibility information, source of infection, and infection type for each isolate to the phylogeny (Fig. 3). All ST188 isolates isolated in Shanghai, China, were MSSA and were susceptible to FOX, SXT, TEC, LZD, OX, and VA. Although the majority of MSSA ST188 in this study were resistant to penicillin (91.7%), they were still susceptible to most of the antibiotics tested. There was no significant antibiotic susceptibility differences between ST188 isolated from livestock and that isolated from humans. ST188 isolates isolated from pediatric and adult patients were well mixed in Clades 2 and 3. In Clade 2, isolates 62 and 95 only differed by 9 SNPs. Similarly, in Clade 3, the pairs 73/74, 42/57 and 59/35 only differed by 2, 4, and 14 SNPs, respectively. Importantly, the isolates in each pair were exclusively sampled from the same hospital, suggesting that these ST188 isolates in each pair were either recently infected from the hospital environment or resulted from patient-to-patient transmission (Fig. 3). Compared with the well-known epidemic host-adapted S. aureus ST398, which mainly leads to soft tissue infection21, human-adapted MSSA ST188 mainly caused respiratory infection (56.9%).
Fig. 3. Antibiotic resistance of ST188 isolates from different sources of infection.

Branches are colored by different clades and scaled with time (years). See Supplementary Figure 3 for further information about the time scale of the emergence of ST188. The splits represent the resistance (red) or sensitivity (blue) to antibiotics. Sample sources are denoted by blue stars
Time scale of the recent emergence of S. aureus ST188 isolates
To further estimate the time origin of S. aureus ST188 isolates, we applied Bayesian-based analysis to the isolates characterized as described above (see Supplementary Methods). Because no longitudinal isolates were obtained and no previous estimate of the mutation rate of ST188 isolates has been reported, we could not precisely calibrate the substitution rate of ST188 isolates. However, under the assumption that substitution rates of subtypes in a given species would mostly be similar, we applied a rough substitution rate that was previously calibrated in S. aureus ST398 isolates for Bayesian dating22. The results demonstrated that the median time of emergence of all ST188 isolates’ most recent common ancestor (MRCA) was approximately 52.46 years ago (95% HPD, 33.7–82.7), around 1960, pointing to a recent origin of ST188. This time-scaled phylogeny also indicates that ST188 isolates may have first arisen in livestock and were then transmitted to other species. However, the divergence of the three major clades in ST188 occurred more recently: within Clade 1, 24.53 years ago; within Clade 2, 39.41 years ago; and within Clade 3, 29.93 years ago. This pattern indicates that the expansion of ST188 was not simultaneous with its emergence but occurred at least 10–20 years later (Supplementary Figure 3).
ST188 isolates exhibit high nasal colonization and biofilm formation ability
The nasal cavity is the predominant location of S. aureus colonization in the host body23. Previous studies showed that ST188 could be detected from the nares of both human and animals24,25. We thus compared the nasal colonization ability in BALB/c mice of ST188 with that of the well-known highly virulent host-adapted S. aureus ST398, CA-MRSA USA300 (ST8), and low-virulence HA-MRSA ST239. The results indicated that there was no significant difference either between ST188 isolated from livestock or humans or between ST188 and HA-MRSA ST239. All host-adapted ST188 isolates showed a significantly higher nasal colonization ability than that of CA-MRSA USA300 and host-adapted S. aureus ST398 (P < 0.01) (Fig. 4a). The epithelial cell adhesion ability of ST188 was much stronger than that of CA-MRSA USA300 (P < 0.001) and host-adapted ST398 (P < 0.01) (Fig. 4b), further confirming the higher colonization of the ST188 lineage.
Fig. 4. Nasal colonization, cell adhesion, and biofilm formation ability of host-adapted S. aureus ST188.

a The nasal colonization ability of ST188 in mice was compared with that of CA-MRSA USA300 (ST8), HO-MRSA ST398 and HA-MRSA ST239 (4 isolates were randomly selected/lineage). Each mouse (one mouse/isolate) received 1 × 108 colony-forming units (CFUs) in the nares. Control animals (n = 4) received only sterile PBS. b Adhesion of S. aureus to human alveolar epithelial cells A549. Colony counts of adhesive and internalized bacteria on/in A549 epithelial cells after infection. Control group (n = 4) received only sterile PBS. c Semiquantitative biofilm assays demonstrating the biofilm formation ability of ST188 isolates (10 isolates were randomly selected). *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant (P ≥ 0.05). LO livestock-originated, HO human-originated
S. aureus biofilm formation is important for persistent infection, antibiotic resistance and immune evasion26. Compared with that of CA-MRSA USA300 and host-adapted ST398, the biofilm formation ability of ST188 was much stronger (P < 0.05). There was no significant difference either between ST188 isolated from livestock and humans or between ST188 and HA-MRSA ST239 (Fig. 4c).
Furthermore, we observed smaller abscesses caused by ST188 isolates compared with those caused by CA-MRSA USA300 (P < 0.001) and host-adapted ST398 (P < 0.001) using a mouse skin abscess model (Fig. 5a, b); this result is consistent with the lower expression of important virulence factors such as the Agr system and α-hemolysin in ST188 isolates (Fig. 5c, d). There was no significant difference between ST188 and HA-MRSA ST239.
Fig. 5. The ability of host-adapted S. aureus ST188 to cause invasive infection.

a Representative abscesses on day 2 after infection (4 isolates were randomly selected/lineage). Each mouse (one mouse/isolate) received 5 × 107 CFUs on the right dorsum by intradermal injection. Control animals (n = 4) received only sterile PBS. b Abscess areas (=π (length × width)/2) on day 2 after infection. c, d Quantitative RT-PCR analysis of RNAIII and hla gene expression in randomly selected ST188 (10 isolates were randomly selected), CA-MRSA USA300(ST8), HO-MRSA ST398, and HA-MRSA ST239 (4 isolates were randomly selected/lineage), from cells grown to the late logarithmic growth phase (4 h). **, P < 0.01; ***, P < 0.001; NS, not significant (P ≥ 0.05)
Carrying of virulence genes and phages in the genomes of ST188 isolates
The distribution of virulence genes differed among isolates. There were no significant differences in virulence genes carried between ST188 isolated from livestock and ST188 isolated from humans. Some genes were present in all of the isolates, including eta, hemolysin genes, and eight adhesion genes (cna, ebh, atl, spa, ebp, icaA, icaB, and sdrC) (Supplementary Figure 1B). More than 80% of the ST188 isolates carried the other four adhesion genes (eap, sdrD, sdrE, and icaC), but none of the isolates harbored tst, sasX, fnbA, fnbB, icaD, or pvl. Most enterotoxin genes (sea, sed, see, seg, seh, sei, sej, sek, sem, sen, seo, seq ser, and seu) could not be detected in ST188 isolates, but seb, sec, and sel genes showed lower detection rates in both livestock- and human-adapted ST188 (Gene list see supplementary methods).
A multiplex PCR assay was used to distinguish among the seven most prominent S. aureus prophages, Sa1int to Sa7int27. Sa3int has been considered to be a molecular marker for distinguishing livestock-adapted from humans-adapted S. aureus28. In the present study, 100% of human-adapted and 64.3% of livestock-adapted ST188 isolates carried Sa3int. Sa1int was harbored in 21.4% of livestock-adapted ST188 and 32.5% of human-adapted ST188. Sa5int only presented in 14.4% of human-adapted ST188 isolates, and no ST188 isolate harbored Sa2int, Sa4int, Sa6int, or Sa7int (Supplementary Figure 1C).
Adhesion- and biofilm-formation-related genes are present in multiple copies and are highly expressed in host-adapted ST188 isolates
Duplicate segments in a genome usually play important roles in bacterial phenotype and virulence. We found many genes related to adhesion and biofilm formation, such as icaB, icaC, ebh, cna, atl, spa, sdrE, and eap present in multiple copies in some of the ST188 isolates, based on WGS data (Fig. 6a). The expression levels of icaB, icaC, ebh, spa, cna, and sdrE were higher in ST188 isolates than in host-adapted ST398, as indicated by reverse-transcription polymerase chain reaction (RT-PCR) (Fig. 6b, c). These results are consistent with the robust epithelial cell adhesion and biofilm formation properties of ST188.
Fig. 6. The carrying and expression levels of genes related to adhesion and biofilm formation.

a The mapping depths of 19 adhesion and biofilm formation genes were detected in all ST188 isolates of this study. A gene with a two-fold higher mapping depth than the average genome sequencing depth was determined as a gene with multiple (≥2) copies (dark blue splits). Genes with one copy were indicated by light blue splits, and genes represented by white splits were determined as absent. b, c Quantitative RT-PCR analysis of genes with multiple (≥2) copies in host-adapted S. aureus ST188 compared with that of host-adapted S. aureus ST398 (10 isolates were randomly selected/lineage). *, P < 0.05; **, P < 0.01; NS, not significant (P ≥ 0.05)
Discussion
Although S. aureus ST188 has not yet been described as a global pandemic strain causing hospital-associated infections (HAIs) and community-associated infections (CAIs), it has been increasingly linked to HAIs and CAIs, particularly across the Asia-Pacific region29. The strain is more likely to cause HAI in adult patients and CAI in pediatric patients30,31. Most reported infections caused by ST188 have been associated with MSSA, but methicillin resistance in ST188 has been reported19. Recent epidemiological data show that this strain has a high colonization rate in the nasal cavity of healthy people and animals and could cause infections in various animals24,25, implying that S. aureus ST188 has the capacity to colonize and cause disease in multiple host species. By characterizing human and bovine S. aureus infections between 2012 and 2014, we identified MSSA ST188 as the major lineage causing infections in multiple host species in Shanghai, China. MRSA ST188 was not isolated between 2012 and 2014 in Shanghai, but in our follow-up monitoring, MRSA ST188 causing human infections could be detected, suggesting that ST188 is currently developing antibiotic resistance, thus necessitating continuous monitoring.
ST188 is a double locus variant (DLV) of ST1. However, genetic analysis has indicated large differences between ST188 and ST1, suggesting complex evolutionary processes underlying ST188. WGS is an effective way to analyze the evolution and spread of bacterial strains, but most studies have focused on the globally important host-adapted ST398 instead of ST188. Price et al. applied WGS to characterize 89 isolates of ST398 and suggested that livestock-adapted MRSA ST398 originated as MSSA in humans16. In this study, 72 isolates of host-adapted MSSA ST188 were whole-genome sequenced. Phylogenetic reconstruction of human and livestock-adapted ST188 suggested that ST188 isolates are readily transmitted among host species. Isolates from livestock did not differ from human-associated isolates and were embedded within them. Additionally, isolates from different hospitals were well-mixed in Clades 2 and 3, and no hospital-specific branch was found. This observation highlighted the potential role of common community commensals in HAI and indicated the recent transmission or continuous infection of ST188 from the same source. Furthermore, our data showed a mixed state of the ancestor isolate, suggesting that ST188 may have infected both humans and livestock before their common ancestor. We further reconstructed the clock-like evolution of ST188 isolates. The median time of clonal emergence was estimated to be ~52.46 years ago, indicating that ST188 isolates apparently first arose in livestock and were then transmitted to humans. However, due to the limited sequencing information of ST188 available worldwide, the evolutionary relationship must be further elucidated.
The predominant Asian CA-SA lineage ST59 can also be isolated from multiple host species9. In addition, the worldwide epidemic host-adapted ST398 is also an important strain causing CAI in Asian populations18, suggesting that the S. aureus lineage causing CAI in humans may be an important risk factor for livestock infection. Most CA-SA clones have gained increased virulence potential via high expression of important virulence components and by causing acute invasive infections, such as severe skin and soft tissue infections (SSTIs)16. However, in the present study, the host-adapted ST188 mainly caused HAI in immunocompromised adult inpatients, suggesting that the pathogenesis of ST188 was different from that of ST398. Human-adapted ST398 isolates harbored prophage 3, which contains the immune evasion complex (IEC) genes encoding the chemotaxis inhibitory protein (Chp), staphylococcal complement inhibitor (Scn), and staphylokinase (Sak), is highly human specific and is less prevalent in livestock-adapted ST39814. However, in the present study, 64.3% of livestock-adapted ST188 isolates carried prophage 3. We tested 44 virulence genes, but 20 of them (including enterotoxin, tst, eta, sasX, and pvl) were undetectable in ST188. In addition, no significant difference was found in virulence genes carried between ST188 isolated from livestock and ST188 isolated from humans. The molecular mechanism by which ST188 causes infections in multiple host species requires further clarification.
The agr operon encodes a global regulatory system in S. aureus, controlling the expression of the genes encoding extracellular virulence factors. Based on the major sequence variations of agr, the operon could be divided into four types32. Both highly virulent CA-SA USA300 (ST8) and the well-known highly virulent host-adapted S. aureus ST398 belong to agr type I33,34; in this study, all ST188 isolates were also agr type I. Agr consistently shows high expression levels in highly virulent S. aureus strains, but in our study, RNAIII (an mRNA with global regulatory properties) showed lower expression levels in ST188 than in USA300 and ST398. Similarly, the abscesses caused by ST188 were much smaller than those caused by USA300 and ST398 in a mice skin abscess model. An in vivo experiment on mice showed that the pathogenicity of ST188 was most likely related to nasal colonization. A robust nasal colonization ability enabled ST188 to successfully colonize and progress to a series of widespread infections. This result was consistent with the strong epithelial cell adhesion and biofilm formation properties of ST188, which was further confirmed by the presence of multiple copies and increased expression of genes related to adhesion and biofilm formation in ST188. The icaADBC operon encodes the enzymes necessary for polysaccharide intercellular adhesin (PIA) production, which is the major component of staphylococci biofilms35. Ebh is displayed on the surface of S. aureus and has been proposed to form a specialized surface structure involved in cellular adhesion. Ebh also plays roles in the determination of S. aureus cell size and complement resistance36. Cna has been proven to contribute to tissue colonization under various pathological conditions37. SPA promotes colonization and immune evasion of S. aureus38. SdrE is involved in fibrinogen-bridged S. aureus–platelet interactions and is required on the bacterial surface for stable adhesion to platelets39. Based on the above mentioned evidence, we suspect that ST188 obtained the ability to colonize different hosts by high expression of the factors associated with bacterial adhesion and biofilm formation.
In conclusion, our data provide important insight into the current epidemic status, pathogenicity, transmission, and phylogenetic relationship of host-adapted ST188. Our results reveal that ST188 is an epidemiologically important emerging host-adapted strain. The virulence of ST188 is strongly related to its adhesion and colonization ability, and we have provided evidence of the role of genes related to adhesion and biofilm formation in the colonization of ST188. Further studies are needed to elucidate the molecular mechanisms by which ST188 breaks the barrier of species and is transmitted.
Materials and methods
Bacterial isolates and growth conditions
We collected and analyzed 791 clinical isolates from adult patients of Renji Hospital affiliated with Shanghai Jiaotong University and Huashan Hospital affiliated with Fudan University and 158 clinical isolates from pediatric patients of Shanghai Children’s Medical Center affiliated with Shanghai Jiaotong University between 2012 and 2014. Two hundred twelve livestock-associated isolates were isolated from dairy cows with mastitis between 2012 and 2014 in farms near Shanghai, China. S. aureus identification was based on Gram staining and classical microbiological tests, and isolates were further characterized using the VITEK 2 Compact GP ID Card (bioMérieux, Marcy l’Etoile, France). S. aureus ATCC43300 was used as a quality-control organism. All S. aureus isolates were stored at −80 °C.
Molecular typing of S. aureus isolates
MLST of S. aureus isolates was performed by detection of seven housekeeping genes (arcC, aroE, glpF, gmk, pta, tpi, and yqiL)40. The sequences of housekeeping genes were submitted to the S. aureus MLST database (http://www.mlst.net). The sequence of the polymorphic X region of the spa gene was submitted to the S. aureus spa type database (http://www.spaserver.ridom.de)41. The agr type was detected by multiplex PCR as previously described32.
DNA extraction and WGS
Some isolates were contaminated and then discarded, or they died during long-term preservation or during transportation. Ultimately, 72 S. aureus ST188 isolates (39 from adult patients, 19 from pediatric patients and 14 from livestock) were used for WGS. WGS was carried out using the HiSeq 4000 sequencing platform (Illumina Inc., San Diego, CA) with a 2 × 150 bp read length. (Majorbio Bio-Pharm Technology, Shanghai, China). The fragment size for the pair-end libraries was 500 bp. The raw data were filtered before assembly, and clean reads were obtained after removing the adapter sequences and low-quality sequences (Q < 20). In addition, sequences containing more than 10% ambiguous N bases or sequences shorter than 30 bp in length were also removed. The Illumina sequences generated and used in this study available in the Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under study accession numbers SRR6227128 to SRR6227199.
SNP calling and phylogenetic analysis
The Sickle tool was used for WGS data trimming. The whole-genome sequence of the S. aureus MW2 strain (ST1, GenBank accession code: BA000033.2) was used as the reference template for read mapping. The genome of MRSA ST188 CUHK_HK188 (GenBank accession no.: JFFV00000000), isolated from a human in Hong Kong20, and seven isolates of MRSA ST188 (GenBank accession no.: SRR3048015 from the human nasal cavity; SRR3047795 and SRR3047865 from the environment; and SRR3047932, SRR3047953, RR3047885 and SRR3048017 from primates) isolated from North America were added for comparison19 (detailed information is provided in Supplementary Methods).
Detecting the presence of virulence-associated genes
We detected the presence of 44 virulence-associated genes. The DNA sequences of virulence genes were used as a genomic template for mapping the sequencing reads of each S. aureus ST188 isolate individually (details in Supplementary Methods). Furthermore, we checked the mapping depth of the 19 adhesion and biofilm formation genes. A gene with a two-fold higher mapping depth than the average genome sequencing depth was determined as a gene with multiple (≥2) copies.
Phage integrase multiplex PCRs
Prophages carried by ST188 isolates were detected by using a previously described method27. A multiplex PCR scheme was used to detect the seven most important S. aureus integrase families, Sa1int to Sa7int. Primers were designed according to the phage integrase genes, and multiplex PCR was performed using a multiplex PCR kit (Qiagen, Germany).
Mouse skin abscess and nasal colonization models
All animal experiments were performed following the guidelines for the Care and Use of Laboratory Animals of the Chinese Association for Laboratory Animal Sciences. The animal protocol was approved by the Committee on the Ethics of Animal Experiments of Renji Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, China. Outbred, immune-deficient hairless female mice (4–6 weeks old) were used for the abscess model. BALB/c female mice (4–6 weeks old) were used for the nasal colonization model (details in Supplementary Methods).
Adhesion of S. aureus to human alveolar epithelial cells A549
Human alveolar epithelial cells A549 were cultured in DMEM medium with fetal bovine serum (FBS, 10%) at 37 °C and 5% CO2. Bacteria were grown to the mid-logarithmic growth phase and washed twice with DMEM medium. A549 cells and bacteria were used at a 1:10 ratio (MOI = 10) and incubated for 2 h. Culture supernatants were discarded, and cells were washed three times with sterile PBS to remove non-adherent bacteria. A549 cells were lysed by the addition of 0.1% deoxysodium cholate solution. Bacterial CFU were enumerated by serial dilutions of epithelial cell lysates and plating onto trypticase soy agar (TSA) plates.
Quantitative reverse-transcription (RT) PCR
The expression of adhesion and biofilm formation genes in ST188 was detected by RT-PCR. We chose ST398 as the reference group because ST398 is an important host-species-adaptable S. aureus lineage with high virulence but low biofilm formation ability18,21. Complementary DNA was synthesized from total RNA using the QuantiTect reverse transcription system (Qiagen) according to the manufacturer’s instructions. Complementary DNA samples were amplified using the QuantiTect SYBR green PCR kit (Qiagen). Reactions were performed using a 7500 Sequence Detector (Applied Biosystems). We used purified chromosomal DNA at concentrations of 0.005–50 ng/ml to form a standard curve. All quantitative reverse-transcription polymerase chain reaction (qRT-PCR) experiments were performed in duplicate with gyrB as a control.
Statistical analysis
Unpaired two-tailed Student’s t-tests were performed to analyze statistical significance. All data were analyzed using GraphPad Prism, version 6.0, and P values <0.05 were deemed statistically significant.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81671975, 81601737, 81401735), the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (81421001), and Shanghai Health System Talents Training Program (2017BR001, 20152513).
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41426-018-0048-7).
References
- 1.Lowy FD. Staphylococcus aureus infections. New Engl. J. Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Leonard FC, Markey BK. Meticillin-resistant Staphylococcus aureus in animals: a review. Vet. J. 2008;175:27–36. doi: 10.1016/j.tvjl.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Bradley AJ, Leach KA, Breen JE, Green LE, Green MJ. Survey of the incidence and aetiology of mastitis on dairy farms in England and Wales. Vet. Rec. 2007;160:253–257. doi: 10.1136/vr.160.8.253. [DOI] [PubMed] [Google Scholar]
- 4.Hasman H, et al. Spa type distribution in Staphylococcus aureus originating from pigs, cattle and poultry. Vet. Microbiol. 2010;141:326–331. doi: 10.1016/j.vetmic.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 5.Woolhouse M, Gaunt E. Ecological origins of novel human pathogens. Crit. Rev. Microbiol. 2007;33:231–242. doi: 10.1080/10408410701647560. [DOI] [PubMed] [Google Scholar]
- 6.Li T, Song Y, Zhu Y, Du X, Li M. Current status of Staphylococcus aureus infection in a central teaching hospital in Shanghai, China. BMC Microbiol. 2013;13:153. doi: 10.1186/1471-2180-13-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ko KS, et al. Distribution of major genotypes among methicillin-resistant Staphylococcus aureus clones in Asian countries. J. Clin. Microbiol. 2005;43:421–426. doi: 10.1128/JCM.43.1.421-426.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li M, et al. MRSA epidemic linked to a quickly spreading colonization and virulence determinant. Nat. Med. 2012;18:816–819. doi: 10.1038/nm.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li T, et al. Molecular characteristics of Staphylococcus aureus causing bovine mastitis between 2014 and 2015. Front. Cell. Infect. Microbiol. 2017;7:127. doi: 10.3389/fcimb.2017.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.David MZ, et al. What is community-associated methicillin-resistant Staphylococcus aureus? J. Infect. Dis. 2008;197:1235–1243. doi: 10.1086/533502. [DOI] [PubMed] [Google Scholar]
- 11.Glasner C, et al. Staphylococcus aureus spa typet437: identification of the most dominant community-associated clone from Asia across Europe. Clin. Microbiol. Infect. 2015;21:163.e1–163.e8. doi: 10.1016/j.cmi.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 12.Li M, et al. Virulence determinants associated with the Asian community-associated methicillin-resistant Staphylococcus aureus lineage ST59. Sci. Rep. 2016;6:27899. doi: 10.1038/srep27899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, et al. Comparative analysis of virulence and toxin expression of global community-associated methicillin-resistant Staphylococcus aureus strains. J. Infect. Dis. 2010;202:1866–1876. doi: 10.1086/657419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uhlemann, A. C. et al. Identification of a highly transmissible animal-independent Staphylococcus aureus ST398 clone with distinct genomic and cell adhesion properties. MBio3, 10.1128/mBio.00027-12 (2012). [DOI] [PMC free article] [PubMed]
- 15.Chuang YY, Huang YC. Livestock-associated meticillin-resistant Staphylococcus aureus in Asia: an emerging issue? Int. J. Antimicrob. Agents. 2015;45:334–340. doi: 10.1016/j.ijantimicag.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 16.Price, L. B. et al. Staphylococcus aureus CC398: host adaptation and emergence of methicillin resistance in livestock. MBio3, 10.1128/mBio.00305-11 (2012). [DOI] [PMC free article] [PubMed]
- 17.Spoor, L. E. et al. Livestock origin for a human pandemic clone of community-associated methicillin-resistant Staphylococcus aureus. MBio4, 10.1128/mBio.00356-13 (2013). [DOI] [PMC free article] [PubMed]
- 18.Wang Y, et al. Role of the ESAT-6 secretion system in virulence of the emerging community-associated Staphylococcus aureus lineage ST398. Sci. Rep. 2016;6:25163. doi: 10.1038/srep25163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soge OO, et al. Transmission of MDR MRSA between primates, their environment and personnel at a United States primate centre. J. Antimicrob. Chemother. 2016;71:2798–2803. doi: 10.1093/jac/dkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ip, M., Wang, Z., Lam, W. Y., Zhou, H. & Tsui, S. Draft genome sequence of methicillin-resistant Staphylococcus aureus CUHK_188 (ST188), a Health Care-Associated Bacteremic Isolate from Hong Kong. Genome Announc.210.1128/genomeA.00255-14 (2014). [DOI] [PMC free article] [PubMed]
- 21.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. Lancet. 2010;375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uhlemann, A. C. et al. Evolutionary dynamics of pandemic methicillin-sensitive Staphylococcus aureus ST398 and its international spread via routes of human migration. MBio8, 10.1128/mBio.01375-16 (2017). [DOI] [PMC free article] [PubMed]
- 23.Wertheim HF, et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005;5:751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 24.Gharsa H, et al. Molecular characterization of Staphylococcus aureus from nasal samples of healthy farm animals and pets in Tunisia. Vector Borne Zoonotic Dis. 2015;15:109–115. doi: 10.1089/vbz.2014.1655. [DOI] [PubMed] [Google Scholar]
- 25.Chen BJ, et al. Factors associated with Staphylococcus aureus nasal carriage and molecular characteristics among the general population at a Medical College Campus in Guangzhou, South China. Ann. Clin. Microbiol. Antimicrob. 2017;16:28. doi: 10.1186/s12941-017-0206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Archer NK, et al. Staphylococcus aureus biofilms: properties, regulation, and roles in human disease. Virulence. 2011;2:445–459. doi: 10.4161/viru.2.5.17724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goerke C, et al. Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. Bacteriol. 2009;191:3462–3468. doi: 10.1128/JB.01804-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verkaik NJ, et al. Immune evasion cluster-positive bacteriophages are highly prevalent among human Staphylococcus aureus strains, but they are not essential in the first stages of nasal colonization. Clin. Microbiol. Infect. 2011;17:343–348. doi: 10.1111/j.1469-0691.2010.03227.x. [DOI] [PubMed] [Google Scholar]
- 29.Chen FJ, Siu LK, Lin JC, Wang CH, Lu PL. Molecular typing and characterization of nasal carriage and community-onset infection methicillin-susceptible Staphylococcus aureus isolates in two Taiwan medical centers. BMC Infect. Dis. 2012;12:343. doi: 10.1186/1471-2334-12-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu Y, et al. Dissemination and molecular characterization of Staphylococcus aureus at a Tertiary Referral Hospital in Xiamen City, China. BioMed. Res. Int. 2017;2017:1367179. doi: 10.1155/2017/1367179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiao Y, et al. Clinical and molecular characteristics of invasive community-acquired Staphylococcus aureus infections in Chinese children. BMC Infect. Dis. 2014;14:582. doi: 10.1186/s12879-014-0582-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lina G, et al. Bacterial competition for human nasal cavity colonization: role of Staphylococcal agr alleles. Appl. Environ. Microbiol. 2003;69:18–23. doi: 10.1128/AEM.69.1.18-23.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vindel A, et al. Molecular epidemiology of community-associated methicillin-resistant Staphylococcus aureus in Spain: 2004-12. J. Antimicrob. Chemother. 2014;69:2913–2919. doi: 10.1093/jac/dku232. [DOI] [PubMed] [Google Scholar]
- 34.Aspiroz C, et al. Molecular characterisation of methicillin resistant Staphylococcus aureus strains ST398 in patients with skin infections and their relatives. Enferm. Infecc. Microbiol. Clin. 2012;30:18–21. doi: 10.1016/j.eimc.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Vandecasteele SJ, Peetermans WE, Merckx R, Van Eldere J. Expression of biofilm-associated genes in Staphylococcus epidermidis during in vitro and in vivo foreign body infections. J. Infect. Dis. 2003;188:730–737. doi: 10.1086/377452. [DOI] [PubMed] [Google Scholar]
- 36.Cheng AG, Missiakas D, Schneewind O. The giant protein Ebh is a determinant of Staphylococcus aureus cell size and complement resistance. J. Bacteriol. 2014;196:971–981. doi: 10.1128/JB.01366-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhem MN, et al. The collagen-binding adhesin is a virulence factor in Staphylococcus aureus keratitis. Infect. Immun. 2000;68:3776–3779. doi: 10.1128/IAI.68.6.3776-3779.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garofalo A, et al. The length of the Staphylococcus aureus protein A polymorphic region regulates inflammation: impact on acute and chronic infection. J. Infect. Dis. 2012;206:81–90. doi: 10.1093/infdis/jis311. [DOI] [PubMed] [Google Scholar]
- 39.George NP, Wei Q, Shin PK, Konstantopoulos K, Ross JM. Staphylococcus aureus adhesion via Spa, ClfA, and SdrCDE to immobilized platelets demonstrates shear-dependent behavior. Arterioscler. Thromb. Vasc. Biol. 2006;26:2394–2400. doi: 10.1161/01.ATV.0000237606.90253.94. [DOI] [PubMed] [Google Scholar]
- 40.Maiden MC, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl Acad. Sci. USA. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koreen L, et al. spa typing method for discriminating among Staphylococcus aureus isolates: implications for use of a single marker to detect genetic micro- and macrovariation. J. Clin. Microbiol. 2004;42:792–799. doi: 10.1128/JCM.42.2.792-799.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.