

Abstract
As the need for nasal, ocular, spinal, and articular therapeutic compounds increases, toxicology assessments of drugs administered via these routes play an important role in human safety. This symposium outlined the local and systemic evaluation to support safety during the development of these drugs in nonclinical models with some case studies. Discussions included selection of appropriate species for the intended route; conducting nonclinical studies that closely mimic the intended use with adequate duration; functional assessment, if deemed necessary; evaluation of local tissues with special histological staining procedure; evaluations of systemic toxicity and safety margin assessments based on local toxicity and systemic toxicity.
Keywords: Safety margins, local toxicity, safety evaluation, intravitreal, intracameral, intranasal, intra-articular, intrathecal, epidural, spinal cord, toxicity, granuloma, analgesics, anti-spasticity
Intranasal Drug Administration (Jeff Tepper)
Introduction
The nasal route has been shown to be an effective route of administration for both topical and systemic drugs. The nose functions as the primary portal of entry for inspired air, which can carry both intended and unintended particulates, vapors and gases. The nose has two important additional functions, olfaction and protection of the lung by heating, humidifying and filtering the incoming airstream. In this latter function, large and/or irritating particles or droplets can trigger reflex sneezing and thus be expelled avoiding lung deposition or systemic absorption. However, if properly formulated and delivered, drugs can be absorbed locally or via the nasal mucosa into the systemic circulation to therapeutic effect. Small particulates not absorbed in the nose can be inhaled into the lung where they may act locally, be absorbed into the systemic circulation or be cleared by mucociliary action and macrophage uptake. Bolus fluids delivered to the nose can also be absorbed in the nasal cavity or inhaled/aspirated (intended or unintended) to deliver drugs into the lung where absorption and clearance can also occur. Clearance of larger inhaled particles also occurs by the gastrointestinal (GI) tract. These particles and droplets adhere to mucus in the nose, are propelled toward the back of the nose by cilia (mucociliary clearance), drain into the nasopharynx and are eventually swallowed into the esophagus and stomach. Besides clearance of the drug from the nose, this mechanism can also result in drug exposure via the GI tract.1 Thus, intranasal administration of drugs can reach various local and systemic targets resulting in both intended pharmacological, as well as unintended toxic effects.
Advantages and Issues Associated with Intranasal (IN) Delivery
There are notable advantages to delivering drugs via the nose, which should be considered when determining the most appropriate route of administration for novel or repurposed pharmaceuticals.2 First, IN drug delivery is simple to administer and non-invasive, allowing self-medication in most patients including nauseous or vomiting patients. Besides local delivery to the nose and sinuses, it is underappreciated that the nose can be a route of delivery for multiple target sites. Currently, there are several marketed peptide and small molecule drugs in which the intended target of delivery is the systemic circulation, (e.g., salmon calcitonin and oxytocin), or the brain, (e.g., sumatriptan and zolmitriptan) and there are drugs in development targeting the lungs with IN administration. Because of the relatively large vascular surface area, lipophilic drugs with molecular weight < 1 kDa, are readily absorbed obtaining systemic absorption rates similar to the intravenous (IV) route.1,3,4 Indeed, some drugs delivered to the nose have better bioavailability and tolerability then the same drug delivered orally. Also, bypassing the GI tract has the advantage of avoiding hepatic first-pass metabolism. Additionally, IN delivery may be particularly beneficial for CNS acting drugs because of the proximity of the nasal mucosa to the brain. As discussed below, this juxtaposition may facilitate rapid achievement of therapeutic drug concentrations in the brain and/or cerebrospinal fluid.
As in any route, there are limitations to the type of drugs that can be administered via the nose. Large molecules, such as many proteins, peptides (> 1 kDa), and hydrophilic drugs tend to be poorly absorbed. Absorption is also limited by rapid nasal mucociliary clearance. As such, significant effort has been put into developing mucoadhesives and permeability enhancing formulations to allow such drugs to be more efficiently delivered by this route. Although the hepatic first pass effect may be eliminated, the nose is also a metabolically active organ, which may result in decreased delivery of the active drug. Another disadvantage is that IN administration can be highly variable due to rapid physiological changes. Such issues may limit or provide an obstacle for some compounds in which precise dosing must occur (eg. insulin).
Fortunately, both the listed advantages and issues associated with IN drug administration are also observed in common laboratory animals due to similar anatomy and physiology. Therefore, results from animal pharmacology and toxicology studies can be readily extrapolated to IN administration in humans. In humans and animals, by manipulation of formulation, particle size, ventilation, volume of administration and/or exposure position, various topical and systemic sites of delivery can be targeted using IN administration.
General Anatomy and Physiology
The nasal septum divides the two nasal cavities in all mammalian species. Air flows through the nasal cavity (nostrils) into the vestibule to reach the main anterior chamber of the nose. The vestibule has keratinized, stratified squamous epithelium whose function is both support and protection. The volume is small (~0.6 cm3, in humans) with low vascularization and poor permeability. After the vestuble, air flows into the atrium, which limits airflow rate but has similar function as the vestibule. It is lined with non-ciliated cuboidal/columnar epithelium. Once air passes through the atrium into the main chamber of the nose, it reaches an area of high vascularity, permeability and surface area (~130 cm2, in humans). The primary cell types in the respiratory region of the nose are goblet cells, ciliated pseudostratified cuboidal/columnar epithelium and basal cells. Goblet cells produce mucus, while the ciliated epithelial cells propel the mucus towards the distal end of the nasopharynx (mucociliary clearance). Basal cells serve as progenitor cells. These three cell types sit on convoluted folds, turbinates, which restrict airflow resulting in close contact between the inspired air and the respiratory epithelium. It is this area of the nose that is the primary portal of entry for most drugs into the blood. Finally, in the most dorsal posterior aspect of the nose, the olfactory area is found. This section of the nose contains the olfactory neurons, sustentacular and basal cells that function to allow olfaction, provide support and serve as progenitor cells, respectively.5
Other important anatomical features of the nose, important for drug delivery, include the vasculature, lymphatics, nerves and glands. The arterial supply originates from three sources and flows anteriorly, in the opposite direction to the inspired airflow. This countercurrent serves to warm and humidify inspired air, particularly in the region of Kiesselbach’s plexus. Here squamous epithelium transition to respiratory epithelium where most lipophilic drugs are absorbed. This highly vascular area contains valveless veins, fenestrated capillaries and a porous endothelial basement membrane that, in response to neural, mechanical, thermal, psychological or chemical stimuli, alters blood volume that can result in rapid changes in the rate of drug absorption.6 Similarly, secretions from serous and seromucous glands can secrete large amounts of fluid and mucus under parasympathetic control that can have profound effects on drug absorption. Lipid soluble drugs can also be absorbed via the lymphatics which drain into surrounding lymph nodes and, along with the nasal-associated lymphoid tissue, serve as an important site of recognition and elimination of pathogens but can also serve as the site of mucosal antibody development for nasally inhaled vaccines.
The nose is primarily innervated by branches of three cranial nerves (CN), namely the olfactory (CN I), trigeminal (CN V) and facial (CN VII). Efferent fibers of the facial nerve control the muscles in the region, but for drug delivery, its importance lies in the parasympathetic fibers stimulating nasal gland secretion and vasodilatation that can quickly and dramatically affect drug absorption. The trigeminal nerve is a predominant nerve in the head and face and is well represented in the nose. Afferent branches of the trigeminal nerve (maxillary nerve) are responsible for mechanical/touch pain and temperature sensation. These nerve endings are not covered with an epithelial layer which allows direct access of volatiles and small particulates to neuronal receptors. For drug delivery, this is important as these receptors are associated with the tolerability/irritancy of the drug and formulation. Finally, the first cranial nerve (olfactory nerve), enters the nose from the olfactory bulb through the cribiform plate at the most cranial aspect of the nose. These unmyelinated nerves have been targeted by drug companies as a means of delivering therapeutics directly to the brain via the nose.
Comparative Anatomy
Although the anatomy and physiology of the mammalian nose is basically similar across species, there are also significant differences. Such differences in nasal structure and function need to be accounted for when delivering drugs to the nasal cavity as well as interpreting results of IN toxicology studies. For instance, humans and non-human primates have simple turbinate structure while mice, rats, rabbit and dogs, of the common laboratory animals, have complex turbinate scrolls. The number and shape of the turbinates affects airflow patterns, which in turn affects deposition of drugs. The turbinate structure also can produce a larger surface area that affects drug absorbance. If interested, further detail on the comparative anatomy and epithelial cell biology of the nose has recently been reviewed.7
Table 1 presents the relative volume, surface area and turbinate complexity of the nose of various species. The table is important because these surface area values are used to normalize dose in IN toxicity studies, similar to the use of body weight or body surface area for systemically administered drugs.
Table 1.
| Species | Volume (mL) | Total Surface Area (cm2) | Turbinate Complexity |
|---|---|---|---|
| Human | 19 | 181 | Simple scroll |
| Dog | 20 | 220.7 | Very complex, membranous scroll |
| Monkey | 8 | 61.6 | Simple scroll |
| Rabbit | 6 | 61 | Complex, membranous scroll |
| Rat | 0.4 | 20 | Complex scroll |
| Mouse | 0.03 | 2.8 | Complex scroll |
Physiology
The anatomical features described above create the structure and environment for the important physiological functions of the nose. Of paramount importance is the nose’s function as a conduit for obtaining oxygen in the atmosphere that is warmed and humidified prior to entry into the lungs. Olfaction is of secondary importance in humans but a critical function in other species that have developed highly complex internal structures to facilitate improved olfaction, resulting in improved survival of the species. Another important physiologic function of the nose is protection of the lung from infection and injury. Foreign material is filtered and swept away by mucociliary clearance. Reflex alterations in breathing, produced by trigeminal irritation, also serve to protect the lung from upper and lower airway irritants. Other protective mechanisms associated with the nose are its immunological responsiveness and its high metabolic activity that includes, cytochrome P450s, glucuronyltransferases, carboxylesterases, glutathione S-transferase, aldehyde dehydrogenases, catalase and glutathione peroxidase among others.12–13
Drug Delivery and Devices for Toxicology Studies
Multiple factors including head position, delivery device and volume, the use of anesthesia and the method of administration contribute to where an IN dose deposits and how it is absorbed in both humans and animals.4 For small rodents (mice and rats), pipette application of bolus fluid is the most common method of IN administration in pharmacology and toxicology studies. Dose administration should be made to coincide with inspiration allowing capillary action to draw the solution into the nostril while the animal is held upright. Do not insert the tip into the nostril or too close to the animal as it may cause injury, especially during repeated dose studies. Using this procedure, one pipette tip can be used with each dose group, but should be changed if contaminated by an animal or for other reasons. Large doses can be delivered but the total delivered dose should be subdivided into smaller drops with a droplet size approximately as indicated in Table 2 for optimal systemic absorption. Each drop should be administered to coincide with inspiration in alternating nares. After delivery of each drop, an approximate 3–5 second pause prior to delivering the next drop helps prevent fluid accumulation and swallowing, thus helping to improve absorption of the drug. If the animal expels or sneezes the dose while being dosed, it should be documented but usually the animal is not re-dosed, especially in longer term studies. Following the completion of each IN dose administration, the head of the animal should be restrained in a tilted back position for approximately 5–10 seconds to prevent the loss of the drug from the nares. Intranasal administration is generally well tolerated in rodents but weight loss and nasal irritation from the procedure may be observed if dosing is more frequent than three times/day.
Table 2.
| Species | Device | Volume/Nostril (μL) | Nasal Volume (μL) | Dose Volume/Nasal Volume | Maximum Doses/Day |
|---|---|---|---|---|---|
| Mice | Micropipette | 5 | 30 | 17% | 3 |
| Rats | Micropipette | 10 | 400 | 3% | 3 |
| Monkeys | 1 mL Syringe Clinical Device |
100 | 8,000 | 1% | 3 |
| Dogs | 1 mL Syringe Clinical Device |
100 | 20,000 | 0.5% | 6 |
Similar methods can be used in nonrodents for bolus fluid administration with a syringe, but in addition, a clinical device, such as a liquid pump or dry powder insulfflator can be used. Note that use of a clinical device is not required for regulatory animal studies but can provide additional useful information. Dogs and primates are generally hand held by one technician while the second administers the dose. The procedure is well tolerated in dogs. Primates can be used, if justified, but are more difficult to dose as the procedure is considered stressful in this species, thus the recommendation to limit the volume and number of IN administrations per day (Table 2).
Drug deposition and absorption can be affected by direct contact with the device, concentrated solutions, extreme pH, cold fluids and rapid impaction. These chemical and mechanical issues may cause irritation, secretions, tearing, itching, sneezing, bleeding, and pain, all of which can result in decrease absorption of the drug.
If nasal sprayers for liquid and powder insulfflators are employed to reduce the particle size, which may aide nasal absorption, increased lung deposition may also occur. In animals, larger IN volumes can be aspirated into the lungs and be systemically absorbed by the pulmonary vasculature. This observation is frequently exploited in pharmacology and drug development studies to allow a somewhat non-invasive means of exposing the lung. In mice, five minutes after IN instillation of 99technicium-sulfur colloid (99Tc), radioactivity was detected in the lung (T. Sweeney, personal communication). The amount of 99Tc deposited in the lung was volume-dependent and reached a maximum of 45% in anesthetized mice administered a 75 μL bolus. Similar results have been obtained in mice using intranasal administration of a luminescent live bacterial vaccine.14 The authors reported that 10 μL volumes were deposited primarily in the upper respiratory tract (mostly nose) while volumes ≥ 50 μL resulted in approximately 50% deposition in the lower respiratory tract. Visweswaraiah et al. 2002, demonstrated that a significant portion of an IN instilled volume (10–50 μL) is swallowed if the mouse was unanesthetized and that nasal absorption is enhanced when repeated small volumes are administered.15
The brain can also be targeted with IN drugs. Currently there are several drugs on the market exploiting this route of administration.4, 16 With IN administration, drugs that enter or drain into the sinus cavernosa are absorbed in the venous sinus, which comes in direct contact with the walls of carotid artery. Drugs that can penetrate the blood brain barrier (e.g., midazolam), can be rapidly transported into the brain by this counter-current transfer. Lymph pathways, perivascular spaces along the olfactory and trigeminal nerve, may act to transport molecules to the brain. As the olfactory nerves enter the nose through the cribriform plate, the nerves project into the olfactory cleft. By targeting this most dorsal posterior aspect of the nose, using small particle sized aerosols, these nerves can be directly accessed by drugs, as well as potential toxic agents, as has been recently suggested for some nanomaterials.17
Formulation
To target these diverse anatomical sites (systemic circulation, lung and brain), effort has gone into improving IN formulations to enhance persistence and absorption of drugs in the nose.3, 5, 18 In general, aqueous solution and dry powder formulations have been currently used in marketed products, but novel polymeric microspheres and nanoparticles are being evaluated to improve exposure. To improve durability and penetration of aqueous solutions, synthetic surfactants, bile salts, phospholipids and cyclodextrins have been used while the use of micelles, liposomes and various emulsions have been explored for similar purpose. For dry powders, bioadhesives and other formulations that reduce drug clearance have been tried and currently are being tested in clinical trials.
Intranasal Toxicity Study Designs and Assessment
In general, the design of IN regulatory toxicology studies does not differ from written guidance.19–21 Use of the same route of administration, drug concentration and dosing regimen (duration and frequency) similar to or greater than the intended clinical dosing schedule is generally expected. However, for IN drugs, achieving sufficient dose multiples and/or similar frequency of dosing relative to those used in clinical trials may be difficult to achieve due to animal welfare concerns. The concentration of the drug solution should at least be equivalent to that expected to be used in clinical studies. Usually sufficient multiples (10–20x) can be achieved based on differences in relative surface area between species (Table 1), unless the solubility of the drug is particularly limited. In some circumstances, increasing the frequency of dosing can be used to increase total daily dose.
All common laboratory animals used in toxicology studies can be used in studies in which drug administration is by the intranasal route. A rodent and nonrodent species is generally expected for GLP toxicology studies, but it is typically acceptable to perform intranasal chronic studies in the most appropriate single species when the drug is a reformulation of an already approved product. Most commonly, rats and dogs are the rodent and nonrodent species of choice; however, mice and primates can be successfully used. In particular, there is a wealth of information regarding adverse histological findings in these species as well as species-specific findings which appear to have little relevance to humans. As in any toxicology study, the rodent and/or nonrodent species should be sufficiently justified.
As for animal number, in general there are no special considerations beyond what is commonly used for dose-range finding, repeat dose and chronic studies. As is typical, satellite animals are used for toxicokinetics. Usually a vehicle control is sufficient but if there are novel or previously unused excipients in the IN formulation, a buffer control, such as phosphate buffered saline, is advisable. If the intent is to deliver drug via the IN route to obtain lung deposition, as is frequently done in pharmacology studies, extra animals may be desired as deep anesthesia used to facilitate lung deposition and drug aspiration may result in excess mortality. However, for IN drug development, if lung deposition is intended or if the particle size of the formulation is less than 5 μm, nose-only aerosol inhalation studies are typically requested by regulatory agencies in order to characterize the effects of the drug on the pulmonary tissues.
In addition to all standard measurements obtained in regulated toxicology studies (body weights, food consumption, clinical observations, clinical pathology, ophthalmology, necropsy, organ weights, histopathology, etc.), specific assessments should, or may be needed for drugs administered by the IN route. Typically, clinical observations, with particular attention to the site of administration, body weight and food consumption are sufficient for clinically evaluating the health of animals in IN toxicology studies. However, functional effects on olfaction, including anosmia, have been reported following intranasal exposure to compounds, including some pharmaceutical preparations, which may require investigation.
Although beyond the scope of many contract research organizations and typically not validated at this time, there are animal models that could be used to assess aspects of olfactory function. A simple test of olfaction, the buried cookie test, can be used to evaluate odor perception, discrimination and habituation.22–23 More sophisticated and sensitive operant conditioning techniques can also be used. Negative electrical potentials produced by the olfactory epithelium can be measured in an electro-olfactogram analogous to an electroretinogram for the eye. These sophisticated tests can be performed in humans and animals (in-vivo/ex-vivo) and correlate directly with olfaction, demonstrating signal changes with olfactory tissue damage.24
As discussed above, pain receptors in the nose are not covered by squamous epithelium, which gives chemical stimuli almost direct access to the free nerve endings that may result in upper airway irritation. If IN administration appears to be causing irritant effects, the irritant potency can be assessed. Trigeminal irritation causes characteristic changes in the breathing pattern of mice that can be assessed in head-out plethysmographs and is the subject of an American Standard Test Method (ASTM 981-04, 2012).25 Good correlation between the depression in breathing rate in mice and irritancy reported by humans exposed to numerous chemicals and pollutants has been observed.26–27
Of particular importance in IN administration toxicology studies is the histopathology assessment of the nasal cavity. Typically four, or as many as six, nasal sections are taken after decalcifying the nasal bones.28,11 Note that this procedure typically adds a week or two to the pathology processing time. Similarly, additional assessment of the olfactory regions of the brain should be undertaken, especially if the drug is centrally active. In addition to the standard three sections normally taken for histopathology, it is suggested that 2–4 more sections are obtained.29 As IN administration also can cause exposure to the oropharynx, larynx, trachea and lung, as well as the esophagus and stomach, particular attention should be paid to these structures in the pathology assessment. Additionally, submandibular, tracheobronchial and mediastinal draining lymph nodes should be evaluated, if enlarged.
The nasal cavity and brain, as well as other tissues mentioned above, are typically stained with hematoxylin and eosin. Special stains may be recommended for evaluation of olfactory epithelium and the olfactory bulb, if issues arise. If mucus hyperplasia is present, alcian blue (pH 2.4) and periodic acid Schiff staining can highlight the mucus producing cells. Olfactory marker protein (OMP), an olfactory receptor neuronal marker and adenylyl cyclase 3 (AC3), found in olfactory cilia, may also be useful. Several less olfactory specific neuronal markers may also be used for example, β-tubulin, fluoro-jade, silver stain, and caspase 3.
Common, non-neoplastic, test article-related or background findings in nasal mucosae include: shortening (resorption) or loss of cilia, epithelial cell dedifferentiation, hyperplasia/metaplasia, mucous (goblet cell) hyperplasia/metaplasia, eosinophilic inclusions/droplets, inflammation, mucosal edema, degeneration, atrophy, necrosis, and ulceration and hyperostosis of the turbinates.28, 30 Of note, mucous cell (goblet cell) hyperplasia and eosinophil inclusions/droplets are common findings in rodent toxicology studies that may have little relevance to human pathology.
Establishing Safety Margins for Clinical Trials
For first-in-human (FIH) dose calculation, no-observable-adverse-effect-level (NOAEL) margins over clinical (after applying the appropriate safety factor) should be calculated three ways to account for both systemic as well as local tissue safety. The primary calculation of local tolerability is made by taking the ratio of the NOAEL animal dose normalized by the species-specific nasal surface area relative to the clinical dose normalized by the surface area of the human nose (Table 1). Unlike systemic administration, the concentration of the solution at the animal NOAEL is divided by the highest solution concentration intended for use in the clinical study. That ratio should be 1 or greater (no safety factor is applied to this calculation). Finally, the ratio of systemic exposure (area-under-the-curve, AUC) at the animal NOAEL divided by the human AUC can be calculated if known. As this is not generally known for FIH studies, the ratio of the body surface area (BSA) adjusted dose of the animal NOAEL is compared to the highest intended human dose, as per FDA guidance.31
Summary/Conclusions
For some drugs, administration via the nose can be a non-invasive method to deliver appropriate levels of drug to patients at various topical sites within the nose and lung as well as targeting the systemic circulation. To accomplish targeting such diverse sites, variables such as the IN device, drug volume and form (liquid, gas, vapor, powder), particle size, chemical properties and formulation can be manipulated to influence the site of drug deposition and absorption. Species differences in nasal architecture, surface area and physiology may be important in understanding and interpreting pharmacology and toxicology data as toxicity can be manifest locally and/or systemically altering both structure and function. Because of the potential proximity of drug to the olfactory epithelium and olfactory bulb, neurotoxicity may also be observed. Functional olfactory assessments in animals may be needed on a case by case basis. Histopathological changes are generally the most sensitive predictors of toxicity and usually are the basis of the NOAEL determination. For first-in-human studies, safety margins should be calculated three ways to demonstrate an appropriate starting dose. Overall though, animal toxicology studies have proved useful in the development of IN drugs despite anatomical and physiological differences..
Think Global, Act Local: Toxicological Evaluation of Intraocular Drugs (Brian Short)
Intraocular Routes of Delivery and Delivery Systems
Intraocular drug delivery is the method of choice to deliver therapeutics for posterior eye diseases and the method is being evaluated as an alternative to topical ocular drugs for chronic anterior eye diseases. Intraocular drugs are delivered by several routes and this presentation focused on intravitreal injection into the posterior chamber and intracameral injection into the anterior chamber (Figure 1). Intravitreal and intracameral drug products represent a wide variety of formulations, including solutions, suspensions, and sustained release ocular drug delivery systems composed of nano- or micro-particles, gels or biodegradable or nonbiodegradable implants.32–37 Intravitreal drug products are categorized as new molecular entities (NME), combination drug products, and reformulations. A NME is further categorized as chemically synthesized small molecule, referred to as a new chemical entity (NCE) or a biotechnology derived protein, referred to as a new biological entity (NBE). Reformulations are drug products of previously approved drug substances for the same or another ocular route or a non-ocular route. Intracameral drug products are usually reformulations of previous approved drug substances for the topical route although examples exist from other routes.32 Intraocular drug products that are drug-drug combinations are uncommon and beyond the scope of this summary, although the same principles apply.
Figure 1.
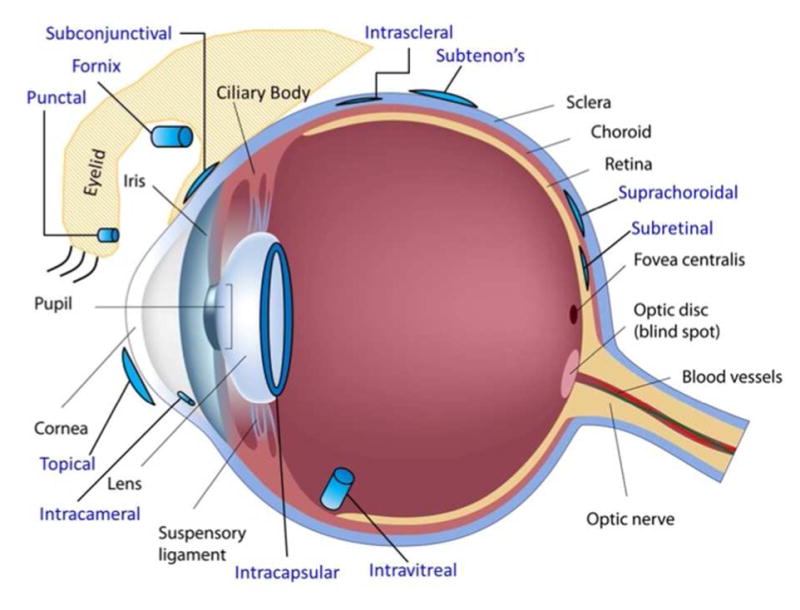
Ocular delivery routes (©Copyright 2016 DrugDel Consulting, LLC. Used with permission).
Species Anatomy, Selection and Number for Intraocular Toxicity Studies
Non-rodent species such as rabbits, dogs, monkeys, and minipigs are used in intraocular toxicity studies since rodent eyes are too small to inject solutions and suspensions due to small vitreal and anterior chamber volume and the proportionally larger size of the lens that can be damaged during injection and too small to properly fit drug delivery systems used in clinical trials. 38–42
The selection of one or two species to be used for pivotal intravitreal toxicity studies depends on several factors. For a NME, two species are generally conducted unless justification can be made for a single species, which may be the case for a NBE with pharmacological relevance in only one species, such as monkey, and recent evidence of the limited interpretive value of toxicity studies of ocular toxicity studies in rabbits.40–44 For a NCE, two species are generally conducted and relevance can be based in part on metabolism.40 For a reformulation, if a drug substance has not been previously administered by the proposed ocular route, then intravitreal toxicity studies in two species with complete ocular and systemic evaluation should be carried out with the new formulation. 45 However, justification of one species may be made based on 1) lack of pharmacological relevance of a biologic in a second species, 2) the compound belongs to a chemical class known to have a favorable safety profile. A study in a single most appropriate species is usually adequate if the drug substance has been previously administered by the intravitreal route.44
Pigmented (Dutch-Belted, New Zealand Red) or non-pigmented (New Zealand White; NZW) rabbits, beagle dogs, cynomolgus monkeys, and minipigs are usually used for intravitreal toxicity studies. All species have adequate vitreal volume to inject up to 50 mcL of solution or suspension or similar volume of drug delivery systems (Table 3).46. 47. Non-pigmented NZW are the most commonly used species, especially for small molecule intravitreal formulations. NZW rabbits have a large historical data base and rabbits in general are considered more sensitive than humans to ocular irritation and toxicity but are not always the most sensitive species.39 Rabbit retinal vascular architecture is merangiotic, meaning that blood vessels extend along the surface of the retina whereas most other laboratory species and humans have holangiotic architecture where blood vessels are present within the inner retina. Rabbits are useful for assessing pharmacokinetics and toxicity of novel excipients or impurities of small molecule and biologic intravitreal formulations and toxicity of small molecule intravitreal formulations. Unfortunately, ocular biologics may cause a profound ocular immune response during single or repeat dose toxicity studies in rabbits which may lead to severe ocular inflammation and secondary retinal injury, which limits their usefulness with ocular biologic drug candidates.42 Pigmented rabbit strains, including Dutch-belted and New Zealand Red, have been used instead of NZW rabbits if a drug binds to melanin, which may or may not be predictive of toxicity.38, 39 However, use of NZW rabbits for intravitreal GLP studies is acceptable since intravitreal studies in a second nonpigmented species such as monkeys, dogs, and minipigs is adequate to assess the role of pigmentation. Furthermore, the use of NZW rabbits as the sole species and strain of an intravitreal reformulation is justified if there were no previous toxicological issues in pigmented species that related to melanin binding during development of the original ocular drug product.
Table 3.
Intraocular Species and Volumes
| Species | Intravitreal Injection Volume (uL) | Vitreous Volume (mL) | HED ratio based on Vitreous Volume | Anterior Chamber Volume (mL)a | Iridocorneal Angle (degrees) |
|---|---|---|---|---|---|
| Rabbit | 50 | 1.5 a | 2.7 | 0.287 | ND |
| Monkey | 50 | 2.0 a,b | 2 | 0.123 | 34 ± 2c |
| Dog | 50 | 3.0 a | 1.3 | 0.77 | 42.4 ± 4c |
| Minipig | 50 | 3.0 a | 1.3 | NA | 40 ± 3c |
| Human | 50–100 | 4.0 a | -- | 0.31 | 35.8 ± 12.2d |
Cynomolgus monkeys are the most common species used for pivotal intravitreal toxicity studies for biologics due to pharmacologic relevance which determines species specificity and their similarities to the human eye. Recent evidence that the determination of safety based on monkeys data was more relevant than rabbit data for translation to humans suggests that the monkey is likely the single and most relevant species for the safety testing of ocular biologics.42 Rabbits may have value for evaluating tolerability of intravitreal biologics that may contain novel excipients or undesirable high levels of host cell proteins or endotoxin before use in monkey studies. Dogs have been used successfully for intravitreal toxicity studies, however, they have a tapetum lucidum, lack a macula/fovea and are least like humans for pharmacokinetics of intravitreal formulations due to rapid distribution from the vitreous humor to aqueous humor. Gottingen or Yucatan minipigs offer an alternative species since they have a large vitreal volume, like to dogs, and holangiotic retinal vascular architecture and innervation similarities to humans, monkeys and dogs. They have no tapetum lucidum, which may bind drug, but lack a macula/fovea.
Species selection for intracameral toxicity studies depends on the type of formulation. For an intracameral drug delivery system it is important to consider the comparative size of the anterior chamber angle and corresponding angle opening distance and angle recess area across species. Anterior chamber angle dimensions are larger in dogs and minipigs and more like humans compared to rabbits and monkeys, although there is much variability among humans (Table 3).48–50 Intracameral implants migrate inferiorly into the anterior chamber angle and ideally should fit deep into the limbus to avoid contact with the corneal endothelium. If implant dimensions are larger than the anterior chamber angle dimensions, they will not fit in the limbus may cause mechanical damage to the corneal endothelium and iris. Dogs have anterior chamber angle size that is like humans (Table 3). 49, 50 Cynomolgus monkeys and rabbits have a smaller anterior angle size and therefore are more prone to endothelial damage elicited by chronic physical contact between the implant and cornea, which may not translate to risk of injury to humans. Therefore, dogs are the preferred species for intracameral toxicity studies of intracameral implants. In some cases, rabbits have been used for intracameral gels and implants and no tolerability issues were reported.35–37 There are no reports of intracameral injection in minipigs, but anterior chamber angle, angle opening distance, and angle recess area in minipigs is smaller than in dogs.48 Since all species vary in anterior segment angle size, use of anterior segment optical coherent topography is recommended to quantitate angle size for intracameral implant fit and screen animals before implant injection.
Intraocular Toxicity Study Designs and Assessment
Optimal intraocular toxicity studies can only be designed if pharmacodynamic and pharmacokinetic parameters of the formulation are understood and clinical trial design has defined the target product profile. This includes the number and frequency of injections, the maximal anticipated human dose, and if applicable, the amount of the drug delivery system material, including number of implants. Generally, the duration of an intraocular toxicity study needs to be as long or longer than the human study, which is comparable to that outlined in the ICH M3(R2) guidance document. 51 Pivotal studies of an intraocular drug delivery system to support an investigational new drug (IND) application may be 6 to 9 months or longer based on the biodegradation profile of the drug delivery system. The number of injections should be equal or greater than the number planned for clinical trials and the frequency of administration should be at least equal to that intended for clinical usage. 40
Pharmacokinetic or pharmacodynamic parameters such as Tmax and half-life or duration of pharmacological response should guide timing of sacrifice intervals for an intraocular solution or suspension and may be helpful in evaluating in-life parameters, including safety pharmacology assessments. In vitro and/or in vivo drug release profiles and anticipated biodegradation profile should guide timing of sacrifice intervals for an intraocular drug delivery system to allow evaluation at or slightly before all drug has been released and when most of the drug delivery system has biodegraded. Unilateral or bilateral intraocular injection is acceptable in toxicity studies. Although some experts and laboratories advocate bilateral dosing for ocular drugs 41, unilateral treatment allows a valuable comparison between the treated and untreated eye and is generally acceptable to clinicians and regulatory agencies. Placebo drug delivery systems may differ in amount of material between doses within a study and high and low dose placebo drug delivery formulations can be evaluated in one eye each in the control group. Sponsors should provide appropriate background information and relevant data for an early development meeting to discuss their nonclinical plan including specifics of pivotal intraocular study designs, study parameters and frequency of evaluation.
The maximal intravitreal dose for solutions or suspensions is limited by 1) the maximum volume that can safety be injected intravitreally in animals without causing a spike in intraocular pressure (~50 microliters) 39 and 2) the maximum concentration based on limit of solubility of a small molecule or feasibility of manufacturing a biologic at concentrations that are usually only slightly higher than the clinical concentration and 3) the tolerability of the intraocular drug product, which includes a variety of excipients and in the case of biologics, host cell proteins, and endotoxin. Intravitreal or intracameral high dose selection for drug delivery systems is limited by the size and number of particles or implants that can be injected without causing mechanical trauma or a profound inflammatory foreign body reaction with secondary tissue injury. It is often the case that no more than a 2 or 3 fold multiple of the maximal anticipated human dose is achieved in intraocular toxicity studies. The number of animals euthanized for the main study phase(s) is usually a minimum of 3 animals/sex/group for monkeys, dogs, and minipigs and 4–5/sex/group for rabbits and the number of animals used for the recovery phase is usually 2 animals/sex/group for control and high dose groups.
Intraocular toxicity in-life parameters for ocular assessment usually include ocular gross observations, slit lamp biomicroscopy, fundoscopy (direct or indirect), and tonometry. 52 Emerging technologies are also used, including photographic imaging to follow biodegradation of drugs delivery systems and spectral-domain optical coherence tomography (SD-OCT) for providing cross-sectional or 3-dimensional images of the posterior segment for intravitreal studies or the anterior chamber for intracameral studies 39,53 Intravitreal toxicity studies evaluate retinal function by electroretinography (ERG), and may include fluorescein angiography if retinal vessels changes are expected secondary to leakage or inflammation and confocal scanning laser opththalmology of the retina if for fundus imaging and dynamic retinal angiography.39,53 Intracameral toxicity studies evaluate corneal endothelium with specular microscopy, measure anterior chamber angle characteristics and track location and size of drug delivery systems with gonioscopy, and measure corneal thickness with pachymetry.39,53 Intraocular toxicity studies conduct histopathology of up to 5 sections of the globe, optic nerve, and ocular adnexa. The decision to evaluate systemic parameters including clinical pathology, organ weights, and a full list of systemic tissues for histopathology is based on the classification of the drug substance as a NME or reformulation and the detection of systemic exposure above the EC50 and/or clinical signs following intraocular injection. Usually a NME or a reformulation that has not been given by the proposed route or that contains a novel excipient will need evaluation of systemic parameters in ocular toxicity studies. 41,45 If the drug substance has been given by the proposed ocular route and systemic exposure is neglible or the drug substance has a favorable safety profile, then systemic evaluation of the reformulation may not needed.
Correlation of Ophthalmology and Pathology and Establishing Adversity
Integration of ophthalmology and pathology findings in intraocular studies needs close coordination between the ophthalmologist, pathologist, and histology technicians.39 Ophthalmology findings and images help guide histologists in tissue trimming and guide the pathologist in evaluating the eye and correlating their findings with the ophthalmologist’s findings. Pathology and ophthalmology reports should define and justify which findings are adverse or nonadverse to help the study team to justify a NOAEL in the toxicology report. 54 Adverse ophthalmological findings for intraocular studies represent clinically impaired visual function and may include persistent, high intraocular pressure (>21 mm Hg) and profound ERG or SD-OCT changes. Adverse ophthalmological findings for intravitreal toxicity studies can includes persistent, moderate to severe inflammation, vitreal opacity, aqueous flare, aqueous or vitreal cells, or perivascular sheathing that prevents continued administration. Adverse pathological findings for intravitreal toxicity studies includes moderate to severe retinal, vitreal and/or uveal inflammation, retinal or vascular necrosis, epiretinal membranes (Figure 2), or retinal detachment.39,55 Nonadverse pathological findings in intravitreal toxicity studies include minimal to mild retinal, vitreal and/or uveal inflammation, perivascular cuffing, implant casts or remnants, and scleral fibrosis secondary to the injection procedure.
Figure 2.
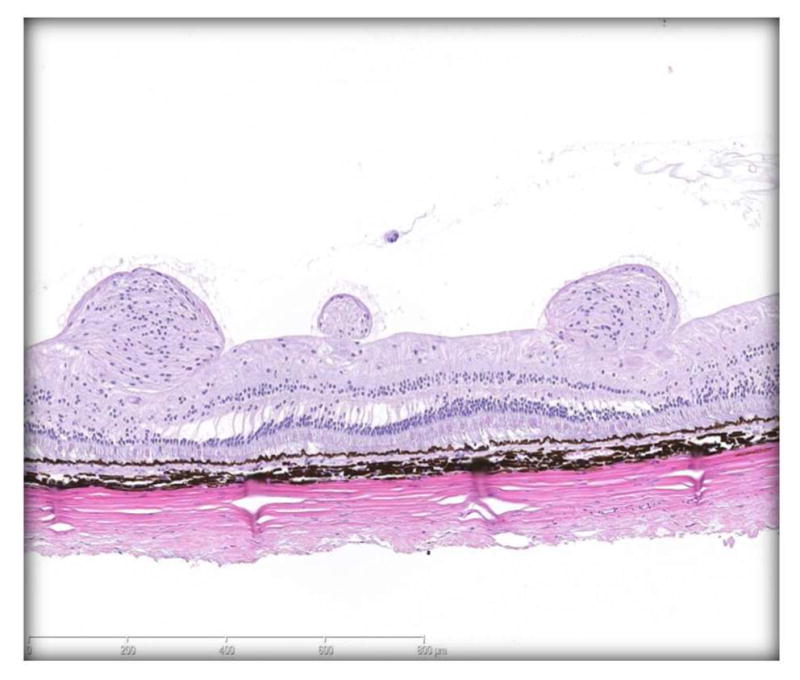
Adverse finding of epiretinal membrane in retinal of monkey eye injected intraviteally with implants. Note thick epiretinal membrane with nodular and linear arrays of cells on inner retinal layer. Underlying separation of retinal nuclear layers.
Adverse ophthalmological findings for intracameral studies includes moderate to severe decreased corneal endothelial cell density, corneal swelling and haze, increased corneal thickness, or corneal opacity.39 Adverse pathological findings in intracameral toxicity studies are secondary to mechanical contact of the implant with the corneal endothelium and include moderate or severe corneal endothelial attenuation, corneal edema, corneal neovascularization and iris adhesion. Nonadverse pathological findings in intracameral studies include minimal to mild corneal endothelial attenuation, corneal endothelial or Schwalbe’s line cell hyperplasia (Figure 3), and implant casts. 56
Figure 3.
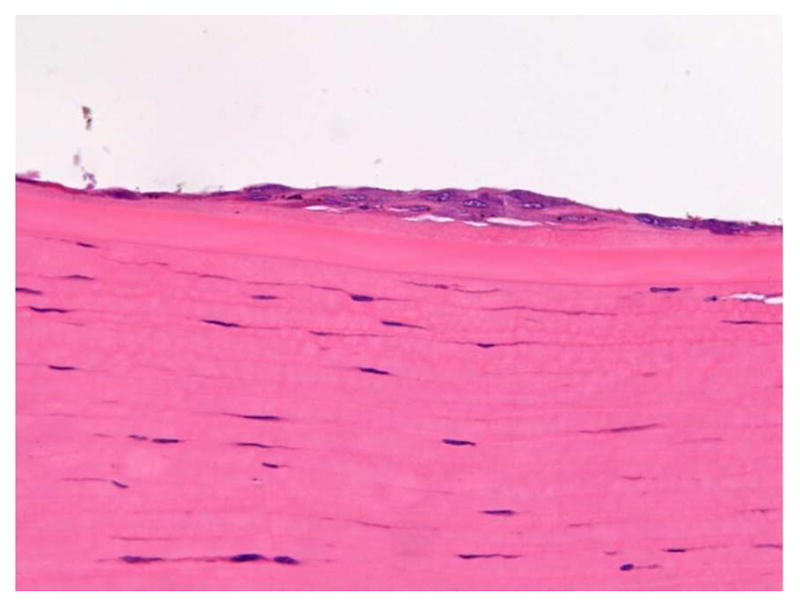
Nonadverse finding in cornea near limbus of dog injected with intracameral implants. Note small focal area of minimal endothelial hyperplasia in response to mechanical contact with implants.
Intraocular Safety Margins and Translation to the Clinic
Vitreous volume differences exist among species and since vitreous fluid is not usually sampled in animal or human studies, animal to human vitreous volume ratios are used to calculate a human equivalent dose (HED) and animal to human intravitreal safety margins. Vitreous volumes of 1.5 mL in rabbits, 2.0 mL in monkeys, 3.0 mL in dogs and minipigs, and 4.0 mL in humans can be used to calculate intravitreal safety margins (Table 1). 46, 47 For example, an intravitreal dose of 1 mg/eye in rabbits, monkeys, and dogs or minipigs represents a HED of 2.7, 2.0, and 1.3 mg/eye, respectively. Publications that cited vitreal volume of monkeys of up to 3.2 or 4.0 mL38, 39 are overestimates based on vitreal distribution volume. 57 rather than direct vitreous measurement. 46, 47 Safety of intraocular drug delivery systems is often related to the biodegradation profile and secondary mechanical trauma or foreign body effects of the drug delivery system and not the drug since the daily release rate of a drug delivery system is much lower than the drug dose from the approved ocular drug product. Nonadverse findings with 2-to 3-fold more implants or other drug delivery system material per injection interval in animals than the maximal human dose is generally considered a safe margin for an intravitreal drug delivery system. For intracameral drug delivery systems, the diameter and fit of the implant in the anterior chamber angle is the most important factor for safety prediction of the drug delivery system and there may be no or a slim safety margin multiple for nonadverse findings. Since the dog anterior chamber angle dimensions are like humans despite a larger anterior chamber volume in dogs, safety of an intracameral implant in dogs will support studies in patients with similar anterior angle dimensions as in dog studies. Therefore, no drug margin of safety is typically calculated for intraocular drug delivery systems other than comparing daily release rate to the daily dose of the approved ocular drug product.
Development of Therapeutics for Spinal Delivery (Tony L. Yaksh)
Abstract
Delivery of drugs into the epidural or intrathecal space has particular relevance in the use of therapeutics that have a spinal target. This “topical” application of molecules to the spinal cord has unique properties, including the use of small volumes and high concentrations to deliver the dose into the small volumes and the fact that the intrathecal cerebrospinal fluid is poorly mixed. These variables emphasize the potential for local toxicity. The present commentary reviews issues pertinent to the preclinical development of drugs for spinal delivery and the robust assessment of their safety.
The rationale for the development of drugs for spinal delivery is based on i) a spinal target of drug action, ii) limited neuraxial bioavailability after systemic delivery and/or, iii) adequate neuraxial systemic bioavailability but drug effects observed at doses leading to non-spinal side effects. Spinal therapeutic targets have included pain and spasticity, neurodegenerative disorders and spinal pathologies, including neuraxial cancers and bacterial and fungal infections. The rapid advances in spinal biology has led to the development of novel targets and drug delivery approaches.58 In the following sections we will consider issues pertinent to the development of spinally delivered drugs.
Spinal Cord
Spinal anatomy
The spinal cord extends extracranially from the caudal medulla to the sacral cord and terminal nerve roots. It lies within the lumens of the vertebral bodies (spinal canal) and is covered by the meninges: the pia (a single cell layer lying tightly on the spinal cord), the arachnoid (a cellular layer) along with the contiguous dura mater (fibroblast collagen matrix). Between the arachnoid and the pia is the subarachnoid space (SAS) forming a moderately compliant fluid sac filled with cerebrospinal fluid (CSF). The spinal space has a complex arterial supply from the vertebral and radicular arteries. Extradurally, the cord is surrounded by a large venous plexus draining into the azygous vein. Sensory and motor nerve roots exit the spinal cord and shed their dural covering as they pass laterally through the root foramena.59
Cerebrospinal fluid
The CSF is a relatively acellular, low-protein, glucose-rich media that resembles a plasma ultrafiltrate. However, it has an electrolyte composition that differs from plasma, and an exclusive chronobiologic cycle for sodium.60 Historically, CSF was thought to arise exclusively from the intracranial choroid plexus. However, evidence of increases in CSF pressure in the lumen of the neural tube during the first month of embryonic life, before the formation of the choroid plexus, suggests that CSF may arise from other structures like cerebral capillaries and ependymal epithelium. Still, in the adult, approximately seventy percent of the CSF production takes place in the choroid plexuses of the lateral ventricles and in the tela choroidea of the third and fourth ventricles. CSF secretion occurs by a combination of a passive plasma filtration that follows a pressure gradient and an active, ATP-dependent enzymatic process that generates an osmotic gradient that moves water (supported by aquaporins) into the subarachnoid space.61,62 Intracranial CSF flows along the ventricular system to the cisterna where it proceeds over the cerebral convexities to be absorbed into the venous outflow by the subarachnoid granules of the sagittal sinus. It also serves to fill the extracranial (spinal) SAS. The dynamics of spinal CSF movement are complex. Though, historically, CSF was believed to undergo movement (flow) from the cisterna to the caudal cord and back, there is in fact little if any large-scale rostrocaudal movement of spinal CSF. Although only modest rostrocaudal net movement of spinal CSF is observed, it is subject to pulsations correlating with the cardiac and respiratory cycles secondary to the oscillatory filling and emptying of the intracranial and spinal arterial tree, and the subsequent compression resulting from filling of the cranial vault.60,63 The respiratory cycle results in increased intrathoracic pressure-evoked movement of venous blood into the large-capacitance, valveless epidural venous plexus.7 The effects of these arterial and venous oscillatory pressure pulses on CSF in the compliant spinal neuraxis serve to move the intradural fluid contents in a rostrocaudal oscillatory fashion leading to increased local mixing of the CSF.
Delivery of Drugs to the Spinal Cord
Drugs may be delivered to the spinal cord by injection into the epidural space or directly into the SAS by percutaneously placed needles and/or catheters placed through those needles, typically at the lumbar level. Epidural drugs must pass through the meninges to access the SAS. Once in the SAS, the agents with a spinal target must then diffuse from the SAS to the parenchyma. Importantly, it can be appreciated that these components represent specific barriers that must be crossed for the drug to access the target site. The dura represents a molecular filter allowing ready passage of small molecules. The arachnoid presents a diffusion barrier that is more readily traversed by small lipophilic molecules. The pia presents a modest barrier for small molecules but impedes movement into the parenchyma of larger solutes such as very large molecules and adenoviruses.65–67
Distributional Properties of Spinally Delivered Molecules and Injection Parameters
Rostrocaudal movement
The intrathecal fluid space is limited and poorly mixed. Accordingly, solutes delivered into the intrathecal space tend to remain proximal to the injection site. Rostrocaudal distribution is enhanced by increases in volumes, and rate of infusion. 68,69 through volumes that are given, are restricted by the limited intrathecal space.
Solute clearance
CSF is cleared by reabsorption in subarachnoid granules in the sagittal sinus and to some degree in the root sleeves, where lymphatic drainage may occur.70 Solutes delivered as a bolus intrathecal injection show an initial dilution in the local CSF followed by a second phase clearance from the CSF. This second phase decline in concentration reflects i) the local dilution of solute, ii) actual clearance from the CSF by movement into the adjacent meninges (where lipophilic compounds bind in the lipid rich environment of the arachnoid layer and epidural fat) and iii) into the spinal parenchyma, and iv) where, aside from uptake into cells, the molecule moves into the vasculature for clearance into the systemic circulation.71 Polar molecules (morphine), large molecules (ziconotide, albumin) or particles (adenoviruses) following a bolus show a slower clearance from the local CSF than small molecules of moderate lipid solubility (e.g. fentanyl), a property reflected by the rapid appearance of a lipid soluble solute in the blood.71
Intrathecal Drug Formulations
Neuraxial formulations are water-based and osmolarity is in the range of 300 mOsM with a pH between 5 and 7. Adjuvants such as surfactants, antioxidants or antimicrobials are avoided (see:72). Surfactant/detergents (Tween, polyethylene glycol) or solubilizing agents such as dimethylformamide or dimethylsulfoxide cannot be routinely considered as inherently compatible with spinal delivery.72 The safety of a spinal therapeutic with such adjuvants must consider direct tissue effects of the formulation. Further, some solvents (DMSO) can interact with plastic-based hardware employed for drug delivery. Cyclodextrins have been widely employed in preclinical formulations producing increased solubility and altered distribution of otherwise water insoluble agents.73 Other formulation strategies may employ several approaches such as liposomes, microspheres and nanoparticles, allowing large doses to be delivered with reduced peak concentrations and increased duration of drug action, though persistence of the matrix in the SAS must be considered.74
Neuraxial Drug Toxicity
Examples of neuraxial toxicity
From the earliest days of intrathecal drug delivery, issues of tissue toxicity were noted including CSF pleocytosis and changes in DRG Nissl staining after local anesthetics.75, 76 Here, we will note two examples of spinal drug-related toxicity: i) local anesthetic root injury and ii) morphine-induced granulomas.
Local anesthetics. Since the 1990s, symptoms characterized by perineal sensory loss, lower limb weakness and bladder and bowel dysfunction were reported after continuous spinal anesthesia (cauda equina syndrome) through small bore catheters over hours to days. This was taken to reflect damage to spinal nerve roots.77–79 Preclinical studies showed that bolus delivery of several anesthetics produced a concentration-dependent incidence of pathology, characterized by axonal degeneration, grey matter vacuolization, infiltration of macrophages and degeneration of Schwann cell sheaths. 80,81 Comparable results were observed in rabbits.82,83 (but see84) and dogs 85 (but see86). Proposed mechanisms of the concentration- and drug-dependent nerve injury include: i) increased release of glutamate,87 ii) direct effects on DRG cells increasing free calcium, 88 and iii) formation of micelles, yielding detergent-like effects upon lipid membranes.89
Opioids. Intrathecal infusion of morphine in the guinea pig, 90 dog, 91 sheep, 92 and human93 has no effect on spinal parenchyma, but yields a space-occupying mass in the intrathecal space arising from the adjacent dura-arachnoid. In preclinical models, the mass is produced by continuous (but not bolus) delivery94 of several (morphine, hydromorphone) but not all (fentanyl) opioids over 2–3 weeks, in an opioid antagonist-independent fashion.95,96 Histopathology shows the mass is composed largely of fibroblasts and a collagen matrix. This origin of the mass is not certain, but is diminished by mast cell stabilizers. This has led to the hypothesis that the granuloma reflects an opioid receptor-independent degranulation of mast cells, release of fibroblast activating products and the formation of a fibroblast-collagen-based reaction proximal to the catheter, e.g., a meningeal scar.90,96
Pre-clinical assessment of drug safety
Issues of adequate safety evaluations of neuraxial drugs have been extensively discussed.97–100. Common factors of the two examples of pathology noted above are that their toxicity is uniformly dependent upon route of delivery, local concentration, and time of spinal drug exposure. Several organizing principles will be briefly noted here.
At a minimum, drugs for neuraxial delivery must undergo preclinical safety assessments by the appropriate route (epidural/intrathecal) with the delivery motif proposed (e.g., single bolus, repeated bolus vs. continuous infusion) and the time frame over which the intended clinical use is to be performed: transient pain (e.g., acute postoperative) vs. a chronic pain state (e.g. cancer, chronic back pain). Parenchymal and root drug exposure is greatest for a given dose after intrathecal delivery. Typically, epidural dosing requires large volumes and doses. Hence, drugs to be employed epidurally should have a parallel consideration by an intrathecal route in the event that an epidural dose is inadvertently given intrathecally.
Duration of assessment. From a practical perspective, the duration of drug delivery is limited by the exposure achievable in the model. In general, with current technology and catheter characteristics this may be up to 1 month in a rodent and 3 months in a large (non rodent) animal model with the limiting factor being the tissue reactions being driven by the catheter alone. (see103). Agents such as single injection toxicants producing enduring effects require survival intervals that cover the possibility of evolving adverse effects. (see, for example, 101).
While small animal models are decidedly useful for mechanistic and pharmacological studies, the use of large animals reflects upon the issue of scaling of intrathecal volumes and varying kinetics across spinal canals of different sizes and thus the local drug tissue exposure profile (see Table 4).
As noted above, a primary variable in local drug toxicity appears to be concentration vs. dose. This leads to several considerations. First, the study drug formulation must be delivered in concentrations that meet or, preferably, exceed those to be developed for human use. Second, it is possible to appreciate that we might estimate relative local exposure after bolus delivery by dividing the dose delivered in the species by estimated volume of CSF, as presented in Table 4. Thus, a 20 mg/mL drug given in 0.5 mL in the dog and the dose divided by the estimated dog CSF volume (16 mL) would give a concentration of 0.625 mg/mL, which, when compared to the same calculation carried out for a human (20 mg/mL x 1/130 = 0.15 mg/mL), gives a relative concentration of 4.2x in the dog than in the humans, suggesting that, dose for dose, the dog spinal cord will see a 4.2x greater exposure than the human cord. While useful to provide for planning and initial estimates of how robustly the model is defining toxicity in the animal model relative to the human exposure, the numbers must be considered circumspectly, as the numerical treatment assumes a redistribution in the entire CSF volume (e.g. 16 mL and 130 mL, respectively). These volumes reflect the total neuraxis and not just the spinal cord, where estimates would be approximated by volumes that are intuitively at most 1/3 of the total intrathecal space. Moreover, as noted, the spinal space is poorly mixed and the majority of injectate distributes asymmetrically as a declining gradient with the peak being proximal to the catheter tip. Accordingly, the concentrations to which the local cord is exposed are higher than those defined by calculations based on total CSF volume. This problem is accentuated when the drug is delivered in a small volume or at a low rate where intrathecal distribution is limited.45 The important variable is what is the concentration to which the local tissue is exposed?
If demonstration of target engagement is possible, e.g., analgesia, muscle flaccidity, reduction in a target molecule, etc., maximum tolerated dosing in the animals can be expressed in terms of multiples of therapeutic doses. This can assist in defining an upper drug dose/concentration to be studied (see100).
Study groups must involve the minimum of a control and a study concentration of test article. Study drug must be prepared in formulations to be employed in the patient, e.g., the GMP product and the same vehicle. Note that in the event that any vehicle other than normal saline is to be employed, consideration should be made to employ a normal saline group to assist in determining if any response can be attributed to the vehicle used in formulation. Note that in the event that any vehicle other than normal saline is to be employed, consideration should be made to employ a normal saline group to assist in determining if any response can be attributed to the vehicle used in formulation.
Generally speaking, to assess drug-related spinal toxicity, “dose response curves” should be generated by changing the concentration vs. changing the volume delivered. Increasing volume results in a greater drug spread, but does not necessarily alter the concentrations to which the local neuraxial tissue is exposed.
Systemic safety may also be required for drugs administered spinally. Elimination of spinally-administered drugs eventually occurs through the systemic circulation, thus systemic effects and plasma concentrations should be measured to demonstrate systemic safety.
The principal characteristic separating a pharmacological investigation from one defining safety is the implementation of systematic histopathology of the target tissues. Issues related to the selection of histopathological analyses and the timing for intrathecal drugs has been discussed elsewhere (see103).
Adult vs. neonate. Neuraxial anesthesia and pain management therapies are relevant to the neonatal and pediatric population. The current concerns over the effects of general anesthetics on neural development in the young population has increased the focus on the neuraxial route. Yet, there has been a paucity of data to indicate the relative safety of neuraxial anesthetics in the same population (see 100). Given issues of pathway development and synaptic connectivity, assessment of the effects of neuraxial agents on concurrent and future spinal function must be considered in further application of analgesics and analgesic agents to this space. 100
TABLE 4.
Spinal Delivery and CSF Formation Rates and Volumes by Species
| SPECIES | ROUTE | ACUTE (bolus) | CATH | CHRONIC (infusion) | Qcsf (mL/hr)a | CSF Vol (mL)a,b | IT Bolus (mL) c |
|---|---|---|---|---|---|---|---|
| Mouse | IT | Yes | No | No | 0.020 | 0.04 | 0.005 |
| Rat | EP/IT | Yes | Yes | Yes | 0.13–0.32 | 0.25 | 0.01 |
| Guinea Pig | EP/IT | Yes | Yes | Yes | 0.21 | 0.30 | 0.01 |
| Rabbit | EP-IT | Yes | Yes | Yes | 0.36–0.6 | 1.4–2.3 (2) | 0.2 |
| Dog | EP/IT | Yes | Yes | Yes | 1.9–4.0 | 7.8–24 (16) | 0.5 |
| Pig | EP/IT | Yes | Yes | Yes | 7.8 | 50 | 1.0 |
| Goat | EP/IT | Yes | Yes | Yes | 9.8 | 25–30 (23) | 2.0 |
| Primate | EP/IT | Yes | Yes | Yes | 1,7–2.5 | 15 | 0.5 |
| Human | EP/IT | Yes | Yes | Yes | 21–24 | 100–160 (130) | 2.0 |
Values typically determined by ventriculocisternal perfusion (Adapted from Artru 1999 (107)).
These estimated volumes are typically reflective of the total neuraxial (spinal and supraspinal) CSF volumes (see text). Numbers in parentheses are means for calculation.
Nominal volumes. Significant variations can be found in the published literature
IT: Intrathecal; EP: Epidural.
In summary, an important implication of the above commentary is that changes in drug delivery profile (bolus vs. infusion), volume or rate of drug delivery, formulation (pH/osmolarity/ionic or additive content) and concentration must be considered as defining variables for the characterization of potential toxicity. Experience with alterations in these variables for intrathecally delivered drugs (e.g., opioids and local anesthetics) emphasizes that such changes have a clear impact upon the assertion of safety of the formulation.
Important Things to Know About Intra-Articular Testing of Drugs in the Animals (Alison M. Bendele)
Advantages and Disadvantages of Intra-articular Therapy
Intra-articular (IA) administration of treatments for various arthropathies (such as osteoarthritis, physical trauma, rheumatoid and gouty arthritis or pseudogout) has the advantage of direct targeting of agents to the joint for a variety of outcomes including the potential for anabolic effects (cell based or growth factor repair strategies), anti-inflammatory, analgesic, lubricant or anti-degenerative activities. In so doing, IA administration has the potential to maximize efficacy and minimize toxicity. The disadvantage of this route of dosing (in various species) is the necessity for administration by an experienced person with sufficient proficiency in the technique to not only avoid iatrogenic injury, hemorrhage or infection, but also deliver the appropriate dose to the correct location.
General Anatomy
Despite some range of motion and minor structural differences, comparative anatomy of joints is remarkably similar across animal species and humans.108–110 For purposes of this discussion the focus will be on the knee joint as it is the largest synovial joint, the easiest to aspirate clinically, can be afflicted by any of the major arthropathies, and is the most commonly treated; thus positioning itself as a primary joint of interest for any drug candidate. 111 However, virtually any joint can be injected. The knee joint (Figure 4) has a considerable amount of space in which injected materials can distribute. Synovial surfaces and recesses extend high up the sides of the femur and down the sides of the tibia and there are often areas in the joint (microcavities and irregularities) at the cruciate insertion where cyst-like indentions in the bone accumulate synovial fluid and injected materials in a reservoir (of sorts) that freely communicates with the femorotibial and femoropatellar joint spaces. 112 In the normal animal, synovial fluid lightly coats these surfaces with a thin layer, but does not fill the total joint space. When arthritis is present, synovial fluid volumes increase and can fill most of the space, often to the point of distention. 111 Therefore knee volume and synovial fluid volume would differ in the normal knee but would likely be similar in the diseased knee in which inflammation/synovial effusion is present. Various methods have been used to estimate cartilage and synovial surface areas, as well as, evaluating circulating biomarkers in an attempt to determine these volumes (Table 5).113–117 Biomechanics of knee joint movement helps distribute materials (especially cells or particulates) to certain specific locations within the knee depending on the species and its tendency to preferentially load bear more on the medial or lateral side of the joint.118 For example, particulates injected into the rabbit knee (a lateral load bearing animal) will often lodge in the posterior lateral recesses of the tibial synovium. This becomes important in gross and microscopic toxicity evaluation since synovium in these areas may be more affected than synovium in other areas (unpublished observation, A Bendele).
Figure 4.
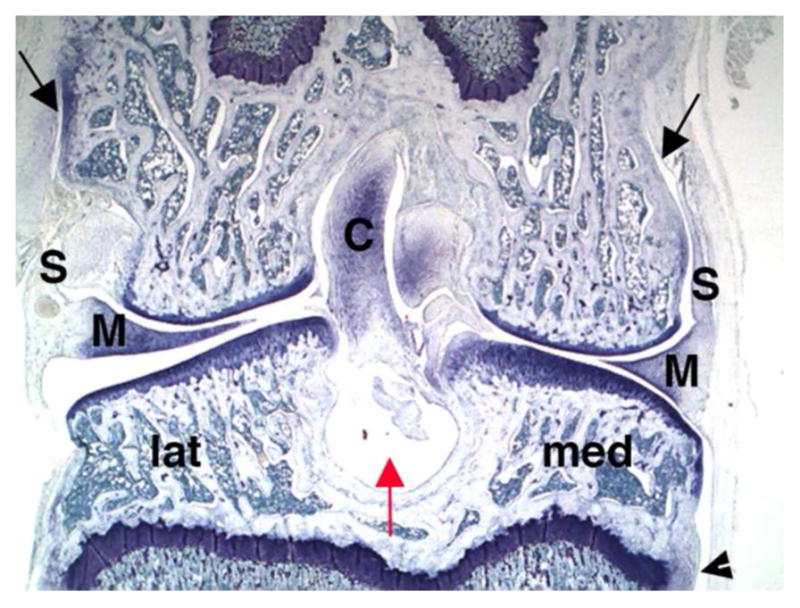
Frontal section of normal rat knee shows shape of medial (med) and lateral (lat) sides with synovium (S), menisci (M) and cruciate (C) ligaments. Black arrows show extent of synovial space extending up the sides of the femur and arrow head shows the same on tibia. The red arrow indicates a cystic area of bone in which synovial fluid and any injected material might be present. (Toluidine blue, 16X).
Table 5.
Drug Delivery: Knee Injection Volume Estimates/Scaling for Dose
| Species (Ratio inj vol/synovial fluid volume in norm joint (ref values col 2,3) | Estimated# synovial fluid volume (mL) w/reference | Body weight and injection volumes (BBP* normal animal experience) | Maximal lavage volumes (BBP), w/o distention to estimate joint volume in normal animals |
|---|---|---|---|
| Mouse | No data available | 35g Dose: 5–10 mcL | 20 mcL |
| Rat (300g) (0.03/0/035=0.86) | 0.035# Wehr, 2007113 |
175–250g Dose: 10–20 mcL 300–350g Dose: 30–50 mcL |
50 mcL 100 mcL |
| Guinea Pig | No data available | 0.7–1kg Dose: 50–100 mcL | 200 mcL |
| Rabbit (3.5kg) (0.35/0.4=0.88) | 0.4 (norm)−0.68 (disease) Matsuzaka, 2002114 |
3–4kg Dose: 0.3–0.5 mL | 1 mL, can direct draw for synovial fluid |
| Dog (25kg) (1.0/1.2=0.83) | 1.2# Wehr, 2007113 |
8–12kg Dose: 0.5–1 mL 20–30kg Dose: 1–2 mL |
2 mL, can direct draw for synovial fluid (50–100 mcL 80% of the time) |
| Monkey (Cyno) (0.5/0.58=0.86) | 0.58# Athanasious, 1991115 |
5–8kg Dose: 0.5–1 mL | No data available |
| Human (2/7=0.28) | 7 (normal) – 14 (disease) Heilman, 1996116 |
70kg (approximate) Dose: 2 mL (HA etc) |
Direct draw of ~500mcL Courtney et al112 |
BBP-Bolder BioPATH, Inc, Boulder Colorado, in house data
Calculations were made using cartilage surface area measures (human:animal species) dog:6-fold lower; rat 200 fold lower; rabbit:17 fold lower: monkey 12 fold lower
Species Selection Rationale for Toxicology Studies
Species selection for IA toxicity testing usually occurs with a background knowledge of some efficacy testing in the various animal models which are commonly used for preclinical development of drugs and treatments for arthritis. Since most of these animal efficacy models have histopathology evaluation as an endpoint (exception being analgesics where load bearing improvement is monitored), there should be some information available about IA responses of the joint to administration of the test article in laboratory animals to help with species selection for the definitive IA toxicology studies. However, there are some definite pros and cons beyond local reaction characteristics to using the various laboratory animal species.
The mouse is commonly used for efficacy testing due to the abundance of both standard and athymic, transgenic and knock-out strains. 119,120 Osteoarthritis models include the meniscal destabilization (DMM), anterior cruciate ligament transection (ACLT) and to a lesser extent the medial meniscal tear (MMT). Chemical agents, including monoiodoacetate (MIA), mono-sodium urate crystals (MSU), enzymes, as well as, other antigens have been explored to produce an arthropathic condition. While more delicate than the larger species, agents can be administered by the IA route in mice. Potential issues associated with using mice for efficacy and toxicity testing include spontaneous medial cartilage degeneration (due to preferential load bearing of the medial side) which increases in severity as mice age, and tendency for cruciate ligament damage from joint injections which can cause instability and result in lesions similar to those seen in the ACLT model. 121,122 Joint injection volumes must be very small to avoid joint distention and clinical evaluation of joint swelling and pain are more challenging to monitor in mice than in larger species.
The rat is commonly used for arthritis efficacy testing due to its ease of use and number of available models. The rat is the most commonly utilized model for efficacy testing for osteoarthritis treatments. 123 The medial meniscal tear (MMT) which can be used to evaluate anabolics for repair or anti-degenerative therapies and to a lesser extent analgesic therapies, is widely used. Other OA model variations include, partial medial meniscectomy (PMM), ACLT and the ACLT with PMM. Like the mouse, chemically induced models like monoiodoacetate (MIA), purified peptidoglycan-polysaccharide polymerscollagenase (PGPS), adjuvants, enzymes, and other antigens, which are used mainly for evaluation of anti-inflammatory properties and pain amelioration, have been used to evaluate potential treatments. Furthermore, with the growth of gene therapies, IA injections in immune response models, like collagen induced arthritis (CIA), have also gained favor. 124 The rat is a near perfect laboratory animal species for efficacy and toxicity evaluation because it has very little spontaneous knee cartilage degeneration with age and it is one of the more balanced load bearing animals. Meaning, the rat does not preferentially load the medial side of the knee joint, as is the case with mice and guinea pigs or lateral as is the case with rabbits.123, 128 Cartilage cysts can occur in most rat strains, especially on the lateral side of the joint, but knowledge of this in conjunction with frontal sectioning of the joint allows for easy identification of this spontaneous change. Clinical toxicity evaluation (knee swelling and gait abnormalities) post IA injection are easily performed in the rat.
Like the mouse and rat, the guinea pig can be used in a variety of surgical and chemically induced efficacy models of arthropathy. However, one relatively unique feature of this species is the development of bilateral naturally occurring osteoarthritis (OA) primarily on the medial side of the knee (due to preferential medial load bearing) starting at 3 months of age with progression to severe OA by 12 to 18 months. 125 Therefore this laboratory animal offers the opportunity to evaluate the toxicity profile of agents in a natural disease setting should that be needed but the presence of spontaneous lesions can complicate the interpretation of toxicity findings if a background OA lesion is not desired. 126
Rabbit models of arthritis include surgically and chemically induced models of OA and inflammation, as well as, mechanical models (such as impact, repetitive loading and immobilization models).127 A notable difference for models involving rabbits, like the meniscectomy or ACLT, is that the rabbit preferentially load-bears on the lateral side of the knee joint.128 For this reason, the lateral side of the joint should be the focus of efficacy and/or toxicity evaluation. Rabbits have very little spontaneous cartilage degeneration to complicate histopathology in the efficacy or toxicity evaluation process.129 Clinical monitoring of pain and swelling post IA injection can be challenging due to the sedentary nature and unusual gait (hopping) of rabbits. However, rabbits do offer an opportunity to study effects of an IA agent in a larger volume joint.
The Beagle dog is an excellent animal for use for both efficacy and toxicity testing of intra-articular agents.130 The most commonly used animal model of OA in the Beagle is the partial medial meniscectomy (PMM). The ACLT can be performed in dogs but requires the use of large hounds (Walker hounds) since beagles do not reliably develop OA after ACLT. 131 Use of beagles for both efficacy and toxicity testing allows for determination of a therapeutic index (dose of a therapeutic agent that causes the therapeutic effect as compared to the dose that causes toxicity).132 Clinical swelling and gait abnormalities associated with treatment are easily monitored and beagles have very little spontaneous cartilage degeneration to complicate histopathology evaluation in either efficacy or toxicity studies.
Monkeys (Cynomolgus) are regularly used as non-rodent species in preclinical toxicology studies, due to their close phylogenic relationship to humans and the comprehensive understanding of underlying spontaneous pathology.133 Monkey models of arthropathy include MMT, CIA and some chemically induced models, so efficacy data may be available for comparison to toxicity data. One should be aware that cynomolgus monkeys have fibrocartilage tibial plateaus rather than hyaline as is the case in humans and all other species. Cartilage cysts are common along with some spontaneous medial tibial cartilage degeneration, which may complicate interpretation of efficacy and toxicity evaluation.121
In summary, with respect to species selection, the rat and the dog are most likely to generate efficacy data to help with dose selection and general response of the joint to treatment with the test article. These two species are relatively easy to inject IA, have reasonable joint volumes, and very little spontaneous cartilage degeneration to complicate histopathology interpretation of toxicity findings. Both species are commonly used for all types of toxicology testing, so handling procedures are routine in most laboratories and they are easy to monitor clinically for swelling and gait abnormalities post IA injection (in a toxicity setting).
Intra-articular Drug Delivery
IA drug delivery in animals should be done using the smallest gauge and shortest needle possible to avoid trauma to the joint. The volume should be tailored to the size of the joint so that distension does not occur as this will result in irritation/inflammation and more rapid clearance of the material from the space (Table 5). Care should be taken to not insert the needle too far back (posterior) in the joint, as it is possible to hit the popliteal artery in the back of the knee and cause hemorrhage. Insertions placed too far from the central area of the patellar tendon have the potential to damage the articular cartilage, especially the femoral surfaces, so this should be avoided. Animals must be anesthetized for the injection to insure proper needle placement with delivery of the material into the joint space and to avoid iatrogenic injury to the cartilage as a result of unexpected movement.
Intra-articular Toxicity Assessment-Clinical Evaluation
IA toxicity assessment should include body weight measure as well as visual evaluation for joint swelling 24 hours post injection with later times added as needed depending on the acute response. Knee caliper measures can easily be done in the rat. In the dog, a cord (rope) can be placed around the knee and then the length measured to document absence or degree of swelling. Standing pain can be measured using force plates and moving pain by gait analysis can be done for both rats and dogs, if swelling is observed, and knowledge of the functional significance of this is desired.
Intra-articular Toxicity Assessment-Histopathology
The most important parameter for IA toxicity testing is histopathology evaluation with particular emphasis on the presence (or absence) of irreversible changes. These would include chondrocyte death and collagen matrix loss usually subsequent to the loss of matrix producing cells. If significant chondrocyte death occurs, joint destruction will always follow (Figure 5). Chondrocyte death occurs as a result of certain types of inflammation (mainly neutrophil, persistent and aggressive) or direct toxicity to the chondrocytes. If chondrocyte death is observed in toxicity studies it is important to try to determine whether it is a result of inflammation or direct toxicity to chondrocytes since inflammation induced chondrocyte loss may be eliminated by changing vehicles or other manipulations whereas direct toxicity to the chondrocyte is generally not something that can be eliminated (Table 6). Reversible changes such as mild acute or chronic synovitis, cartilage matrix proteoglycan loss, minor subchondral bone resorption may be observed in the short-term interval post injection but can recover completely after a period of time. Almost anything, including saline, injected into the joint will result in some minor acute synovitis and minor cartilage proteoglycan loss (unpublished observation, A Bendele).
Figure 5.
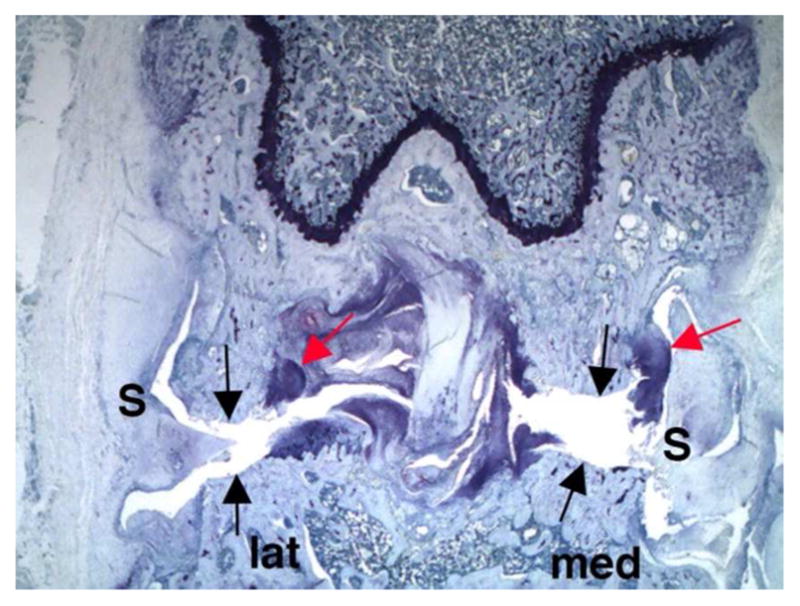
Frontal section of rat knee injected with a toxic material shows synovium (S) that is thickened as a result of fibrosis and inflammation. Black arrows indicate tibial (lower) and femoral (upper) surfaces completely denuded of hyaline cartilage as a result of chondrocyte death. Red arrows indicate cartilage proliferation in the marginal zones, a common response to load bearing cartilage loss. Bone sclerosis is present on both medial (med) and lateral (lat) sides. (Toluidine blue, 16X).
Table 6.
Features of Irreversible Joint Lesions-Chondrocyte Death Leading to Matrix Loss
| Direct Toxicity to Chondrocytes | Indirect Toxicity to Chondrocytes Via Inflammation |
|---|---|
| 1. Generally Affects all surfaces to some degree. Usually kills the chondrocytes within a few days of exposure | 1. Generally affects marginal zone chondrocytes 1st, ±pannus with slower spreading to other areas |
| 3. May spare marginal zone chondrocytes, later stage see large osteophytes | 2. Does not spare marginal zone chondrocytes, ±osteophytes |
| 3. Acute inflammation (PMN’s and edema) may be present, but subsides within a week to MNIC predominantly | 3. Persistent with neutrophils, edema, some MNIC, may become chronic active |
| 4. Late stage (takes > 2 weeks) see severe matrix loss and bone erosion (often diffuse) with recontouring of surfaces | 4. Femurs often more severely affected than tibia but in late stage may be diffuse |
Particulate agents that release drug over long periods of time often lodge in the synovium and cause some chronic inflammation which resolves as the agents are processed/removed by macrophages.
Intra-articular Toxicology Studies-General Comments
When designing IA toxicology studies, reasonable volumes for the species (Table 5) should be used and time should be allowed for clearance of viscous material (like hyaluronic acid) before superimposing more injections. Accumulation of highly viscous materials can result in significant joint inflammation and chondrocyte death/matrix destruction. Simply allowing these materials to be largely cleared out of the joint prior to the next injection will avoid this problem. Residence time for various agents (aqueous vs polymers) may be minutes to months and this should be taken into consideration when designing studies. Growth factor and other repair strategies may show better toxicity (and efficacy) profiles when dosed in a cyclical manner rather than attempting to have the agents in continuous residence. This is because some remodeling of the repair tissue may need to occur before the next round of repair stimulation happens. Using this approach both in toxicity and efficacy studies may help to avoid excessive marginal zone proliferation (pleuripotential cells that form osteophytes at the edge of the joint) or degenerative changes in repair tissue in the hyaline cartilage.
In all intra-articular toxicology studies, it is important to use animals that have a closed articular/epiphyseal growth plate, as cartilage responses to injury in immature animals are different from those seen in more mature animals with a well-formed tidemark. This occurs at about 11 weeks in the rat and 7 months in the dog, ages that will be more representative of what might occur in mature humans. It is recommended to always use frontal (Figures 4 and 5) histologic sectioning (not sagittal) for histopathology evaluation. Load-bearing differences between medial and lateral sides can influence lesion development and progression (both sides are not equal) and some spontaneous changes tend to occur on one side or the other. Frontal sectioning allows for accurate identification of medial and lateral areas of the joint for a detailed assessment of both induced and spontaneous lesions. For example, more load-bearing on the medial side of the joint might result in greater chondrocyte death (medially) associated with inflammatory changes that were diffuse and load-bearing differences often result in increased subchondral bone on the preferentially loaded side. Designed studies should always be of sufficient duration to determine if chondrocyte death will occur as a result of single or multiple treatments. Cationic dye stains such as toluidine blue are actually much more useful than the standard hematoxylin and eosin (H&E) stains for joint microscopy. The cationic dye binds to the negatively charged proteoglycans in the cartilage matrix and thus helps identify areas of decreased proteoglycan. This in turn helps the pathologist identify areas of early chondrocyte death since proteoglycan is lost with time (mild at first then permanently and severely) when the chondrocytes are dead. Inflammatory cell infiltrates, synovial fibrosis, subchondral bone resorption and most other changes that are important in the evaluation of joint pathology can easily be observed using the toluidine blue stain.
The primary purpose of toxicology studies should be to determine that treatments do not result in irreversible damage (chondrocyte death) to the joint especially with single or repeated administration. Safety evaluation and determination of margins can be done using synovial fluid concentrations of drug if this information is available or by using increasing concentrations of an agent in a fixed (and appropriate) volume for the joint being injected. However, if solubility or other factors dictate that a fixed concentration of the agent must be evaluated, then increasing the injection volume relative to the total volume of the joint space may allow confidence that margins are achieved (Table 5). Having some animal efficacy data in the species used for toxicity testing allows calculation of a therapeutic index and thus provides some increased confidence in the safely margins.
Regulatory insights regarding nonclinical studies needed to support development of drugs intended for less common routes of administration (Armaghan Emami)
The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidances M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals and S6 Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals, describe the nonclinical safety studies necessary to support the clinical development of small molecule and biotechnology-derived pharmaceutical drug products, respectively. These guidances state that is preferable to evaluate the local tolerance of a drug as part of the general toxicity studies rather than standalone studies; however, they do not address specific considerations for alternate routes of administration. The U.S. Food and Drug Administration (FDA; the Agency) guidance for industry on Nonclinical Safety Evaluation of Reformulated Drug Products and Products Intended for Administration by an Alternate Route (herein referred to as the FDA reformulation guidance) represents the Agency’s current thinking regarding the nonclinical evaluations needed for the development of compounds intended for various routes of administration. The FDA reformulation guidance provides considerations for all routes of administration of a reformulated drug product and many route-specific considerations which can be applied to the development of novel therapeutics. Notably, the guidance states that for all drug product reformulations and for all drug products with new routes of administration, acute and/or repeat-dose toxicity studies with complete histological evaluation should be conducted using the clinical route of administration. If systemic exposure by the new route is equivalent to or less than that of an approved route, and there are no novel excipient concerns, histological evaluation may be limited to locally exposed tissues. The FDA guidance specifically addresses route-specific recommendations for the oral, dermal, intravenous, ocular, otic, inhalation, intranasal, vaginal, rectal, intraoral, intracavernosal, intraurethral, intravesicular, extended release injected/implanted formulations, intrathecal, epidural, and subcutaneous, and intramuscular routes. The FDA reformulation guidance does not specifically address the intra-articular route of administration or how safety margins should be derived based on local toxicity findings to support human dosing.
General design considerations for local toxicity evaluation
In general, the design of the nonclinical studies should reproduce as closely as possible the intended clinical dosing regimen, taking into consideration the drug concentration, the volume to be administered, and route-specific unique tissue considerations such as the size/volume of the targeted space (e.g., intranasal, intravitreal, intrathecal, intra-articular space) of the animal. The study designs should include groups that meet and exceed the dose and concentration levels intended for human study in order to establish potential margins for safety and characterize the potential for local toxicity. In addition to systemic safety for the drug, the studies must establish a safety margin for the concentration of the drug product in the local space (such as olfactory tissues, intravitreal, intrathecal, or synovial joint fluid). Appropriate design of nonclinical toxicology studies intended to characterize the local tissue effects of a drug to support human safety should be carefully considered sponsors should provide justification why the concentrations of the drug tested in the nonclinical species adequately mimic and exaggerate the anticipated concentrations of the drug in the patients.
Local safety assessment
In nonclinical toxicology studies intended to support clinical studies for unique routes of administration, local safety must be considered since the local tissue concentrations following local administration usually far exceed the concentrations in that tissue compared to when the drug was administered via a previously approved systemic route of administration. In addition, the local injection sites frequently contain uniquely vulnerable tissues (e.g., cartilage, neuronal tissues, ocular tissue, olfactory nerves), some with limited or reduced capacity to recover from insult. For reformulations of previously approved drug products, the systemic safety is frequently already adequately characterized and development programs usually only have to focus on local safety. From a systemic safety perspective, the gold standard for comparisons across species is to directly compare key pharmacokinetic parameters such as Cmax and AUC. When these data are not yet available, such as prior to any human dosing, allometric scaling based on body surface area is used to compare dosing in animals to humans. However, plasma levels and body surface area comparisons may not reflect the local tissue concentrations of the drug and are not generally appropriate for establishing a safety margins for local toxicity.
When assessing local tissue toxicity, the concentration of the drug product being tested is a critical factor because for any drug that is administered into a relatively enclosed space. The greatest potential for local toxicity is at the point of administration where the drug concentration is the highest. In these circumstances, a direct comparison of the concentrations tested in the animals to the proposed human drug product concentration provides an appropriate determination of the exposure margin for adverse effects in the immediate local tissue environment. However, once administered, the drug will diffuse from the injection site over time at a rate generally consistent with the movement of extracellular fluids within the tissues and the physicochemical properties of the molecule.
However, as noted previously, many of the administration sites, such as intra-articular spaces, intrathecal spaces, and even intra-ocular spaces can be viewed as virtual “fluid” filled chambers into which the drug product is introduced. With time, the drug product diffuses throughout the “virtual space”, reaches a steady-state concentration within the tissue, and is eventually cleared from the area. However, clearance from these spaces varies in different tissues and prior to complete clearance, the drug may reside for some time in the tissues at a virtual “steady state” concentration. This concentration at “steady state” is another way to compare exposures across species without having to attempt to extract fluids or tissues to assess drug concentrations in the local environment. Frequently, the safety assessment is actually based on three different exposure margin assessments: 1) comparison of plasma levels or body surface area comparisons for systemic safety, 2) local concentration at the injection site, and 3) concentration at virtual “steady state” to ensure appropriate scaling to ensure the adequacy of the toxicology study design to inform human risk assessment. As noted in Figure 6, a local tissue toxicology assessment must adequately compare the local tissue concentration at the injection site (Panel A) and the local tissue concentration in the virtual space (Panel B) once the drug equilibrates within the tissue. For the purposes of this illustration the virtual space could be ocular tissue volume, intra-articular spaces in joints, intrathecal spaces (CSF), or even intra-otic fluid volume.
Figure 6.

Need for Safety Margins Based on both Local Injection Concentration and Local “Steady-state” Concentrations in a Virtual Space.
The differences in the local environment into which the drug is administered across species must be taken into consideration when designing and interpreting toxicology studies employing a novel route of administration. Therefore, adequate toxicology studies must test the concentration to be tested in humans in an appropriate animal model. In order to assure that the toxicology study is designed adequately, sponsors should attempt to estimate the size of the virtual space in the test species, assure that the study tests the clinical concentration and scale the volume to be administered to the virtual space into which the drug will be injected, and ensure that the concentration at steady state is at least 1 x, as illustrated in Figure 7.
Figure 7.

Hypothetical Example of Scaling Nonclinical Dosing to Mimic Human Dosing for “Virtual Space” injections.
Estimating virtual spaces across species is not always a simple task, as various estimates for a variety of tissues have been proposed in the published literature using a variety of different approaches, sponsors should provide the justification for the values they employ to estimate these parameters across species and present their safety margins for both systemic and local effects.
Every toxicology study should be designed to not only test the predicted human exposures but also exaggerate exposure to the test article in order to fully characterize the toxicological potential of the drug product. For local tissues, this can be completed by increasing the concentration of the drug product administered, increasing the volume of the drug product solution, or increasing the frequency of the injections. Ideally, the studies should mimic the way a drug could be overused in a clinical setting. As such, it is logical to test larger doses of a fixed concentration solution via changes in volume. However, changes in volume can confound the study design for many local tissue assessments. For example, injection of too large a volume into a relatively enclosed space, such as a knee joint, can result in adverse effects related to the volume administered rather than the drug product administered. Sponsors should clearly delineate why they have selected the doses in their toxicology studies, taking into consideration the limitations of the local tissue sites. The evaluation of the safety of a “virtual space” local injection should be done via analysis of all exposure margins which can be clearly depicted in tabular format, such as the example provided in Table 7.
Table 7.
Local SM calculation based on local concentration at the injection site and Virtual space volume.
| Dose (mg) | Concentration* (mg/mL) | Virtual Space Volume** (mL) | Virtual Space Conc. (mg/mL) | Margins | ||
|---|---|---|---|---|---|---|
| By Local Conc. | By Virtual Space Conc. | |||||
| Human proposed study | A | C | E | A/E | ||
| Toxicology study NOAEL |
B | D | F | B/F | D/C | (B/F)/(A/E) |
Concentration of the drug product administered
Note that references should be provided to support the virtual space volumes for specific species
If there are insufficient data or a safety concern about any of the excipients in the formulation, the inclusion of a saline, water, or untreated control in the safety studies greatly facilitates interpretation of the study results and can have an impact on decisions pertaining to viable formulation development.
When designing nonclinical toxicology studies, sponsors should also consider including study arms that test drug product material at or near the end of the expected shelf-life or material that contains adequate levels of drug product degradants in order for these studies to be useful to qualify drug product degradants from both a systemic and local perspective. The safety justification for any compounds identified as leachables in the long-term stability samples may also be tested in this study which may obviate the need for a second toxicology study to support degradants and leachables prior to the new drug application (NDA) submission. Careful consideration of the entire development program can reduce animal use and ultimately cost of the development program.
The intra-articular route is not discussed in the FDA guidance document. As always, companies are encouraged to contact the review division for specific recommendations and consider unique aspects of the local environment.
Summary
Nonclinical toxicology studies are necessary to adequately inform the risk:benefit for experimental dosage forms for the proposed patient population and any subjects enrolled in clinical trials during development. For orally administered drugs, this safety assessment can be accomplished via standard oral toxicology development programs. However for many indications, it is desirable to obtain adequate local tissue concentrations of a drug and minimize systemic adverse effects. As such novel routes of administration have been employed which result in greater concentrations of drugs in local tissues than can be obtained following oral administration. As such, additional local tissue toxicity studies are necessary. These studies should characterize the impact on all tissues in the local environment that were not adequately characterized by existing toxicology studies. That local tissue assessment requires an understanding of not only the tissues in the local environment, but also how the drug is likely to distribute through and be cleared from the local tissues. One way to do this is to consider the “virtual space” in which the drug distributes to ensure that the toxicology studies adequately mimic the clinical setting. The FDA has provided some recommendations to address specific concerns regarding unique routes of administrations; however, guidances rarely provide specific details on how the studies are ultimately reviewed. As always, investigators who wish to pursue such development programs are encouraged to discuss their programs with regulatory agencies to ensure that studies are designed adequately to meet the needs of the program and minimize the use of animals.
Acknowledgments
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article with the exception of the work by Tony Yaksh, whose work was supported in part by the NIH grant R01 DA15353 (TLY) and the Mallinckrodt Pharmaceuticals Research Fellowship Program (AFC).
Footnotes
Disclaimer: This publication reflects the views of the authors and should not be construed to represent FDA’s views or policies.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Illum L. Nasal clearance in health and disease. J Aerosol Med. 2006 Spring;19(1):92–9. doi: 10.1089/jam.2006.19.92. [DOI] [PubMed] [Google Scholar]
- 2.Grassin-Delyle S, Buenestado A, Naline E, Faisy C, Blouquit-Laye S, Couderc LJ, Le Guen M, Fischler M, Devillier P. Intranasal drug delivery: an efficient and non-invasive route for systemic administration: focus on opioids. Pharmacol Ther. 2012 Jun;134(3):366–79. doi: 10.1016/j.pharmthera.2012.03.003. Reed (1993) [DOI] [PubMed] [Google Scholar]
- 3.Fortuna A, Alves G, Serralheiro A, Sousa J, Falcão A. Intranasal delivery of systemic-acting drugs: small-molecules and biomacromolecules. Eur J Pharm Biopharm. 2014 Sep;88(1):8–27. doi: 10.1016/j.ejpb.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Mittal D, Ali A, Md S, Baboota S, Sahni JK, Ali J. Insights into direct nose to brain delivery: current status and future perspective. Drug Deliv. 2014 Mar;21(2):75–86. doi: 10.3109/10717544.2013.838713. [DOI] [PubMed] [Google Scholar]
- 5.Pires A, Fortuna A, Alves G, Falcão A. Intranasal drug delivery: how, why and what for? J Pharm Pharm Sci. 2009;12(3):288–311. doi: 10.18433/j3nc79. Review. [DOI] [PubMed] [Google Scholar]
- 6.Gizurarson S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr Drug Deliv. 2012 Nov;9(6):566–82. doi: 10.2174/156720112803529828. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harkema JR. Comparative Biology of the Normal Lung. In: Parent RA, editor. Comparative Anatomy and Epithelial Cell Biology of the Nose. 2. Chapter 2. San Francisco, CA: Elsevier; 2015. pp. 7–18. [Google Scholar]
- 8.Gizurarson S. Animal models for intranasal drug delivery studies. A review article. Acta Pharm Nord. 1990;2(2):105–22. Review. [PubMed] [Google Scholar]
- 9.Ménache MG, Hanna LM, Gross EA, Lou SR, Zinreich SJ, Leopold DA, Jarabek AM, Miller FJ. Upper respiratory tract surface areas and volumes of laboratory animals and humans: considerations for dosimetry models. J Toxicol Environ Health. 1997 Apr 11;50(5):475–506. doi: 10.1080/00984109708984003. [DOI] [PubMed] [Google Scholar]
- 10.Gross E, Morgan K. Architecture of the nasal passages and larynx. In: Parent RA, editor. Comparative Biology of the Normal Lung: A Treatise on Pulmonary Toxicology. Vol. 1. Chapter 2. Telford Press; Caldwell, NJ: 1992. pp. 7–25. [Google Scholar]
- 11.Morgan KT. Approaches to the Identification and Recording of Nasal Lesions in Toxicology Studies. Toxicologic Pathology. 1991;19(4):337–351. doi: 10.1177/0192623391019004-103. [DOI] [PubMed] [Google Scholar]
- 12.Toskala E. AAOA allergy primer: immunology. Int Forum Allergy Rhinol. 2014;4:S21–S27. doi: 10.1002/alr.21380. [DOI] [PubMed] [Google Scholar]
- 13.Reed CJ. Drug metabolism in the nasal cavity: relevance to toxicology. Drug Metab Rev. 1993;25(1–2):173–205. doi: 10.3109/03602539308993975. Review. [DOI] [PubMed] [Google Scholar]
- 14.Miller MA, Stabenow JM, Parvathareddy J, Wodowski AJ, Fabrizio TP, Bina XR, Zalduondo L, Bina JE. Visualization of murine intranasal dosing efficiency using luminescent Francisella tularensis: effect of instillation volume and form of anesthesia. PLoS One. 2012;7(2):e31359. doi: 10.1371/journal.pone.0031359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visweswaraiah A, Novotny LA, Hjemdahl-Monsen EJ, Bakaletz LO, Thanavala Y. Tracking the tissue distribution of marker dye following intranasal delivery in mice and chinchillas: a multifactorial analysis of parameters affecting nasal retention. Vaccine. 2002;20:3209–3220. doi: 10.1016/s0264-410x(02)00247-5. [DOI] [PubMed] [Google Scholar]
- 16.Djupesland PG. Nasal drug delivery devices: characteristics and performance in a clinical perspective-a review. Drug Deliv Transl Res. 2013 Feb;3(1):42–62. doi: 10.1007/s13346-012-0108-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bakand S, Hayes A. Toxicological Considerations, Toxicity Assessment, and Risk Management of Inhaled Nanoparticles. Int J Mol Sci. 2016 Jun 14;17(6) doi: 10.3390/ijms17060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brayden DJ, Oudot EJ, Baird AW. Drug delivery systems in domestic animal species. Handb Exp Pharmacol. 2010;(199):79–112. doi: 10.1007/978-3-642-10324-7_4. [DOI] [PubMed] [Google Scholar]
- 19.ICH M3(R2), Guidance for Industry: Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. U.S. Department of Health and Human Services, Food and Drug Administration Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); Jun, 2009. [Google Scholar]
- 20.ICH S6(R1), Guidance for Industry: S6 Addendum to Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. U.S. Department of Health and Human Services, Food and Drug Administration. Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); May, 2012. [Google Scholar]
- 21.U.S. Food and Drug Administration. Guidance for Industry and Review Staff: Nonclinical Safety Evaluation of Reformulated Drug Products and Products Intended for Administration by an Alternate Route 2015 [Google Scholar]
- 22.Rattazzi L, Cariboni A, Poojara R, Shoenfeld Y, D’Acquisto F. Impaired sense of smell and altered olfactory system in RAG-1(−/−) immunodeficient mice. Front Neurosci. 2015 Sep 9;9:318. doi: 10.3389/fnins.2015.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang M, Crawley JN. Simple behavioral assessment of mouse olfaction. Curr Protoc Neurosci. 2009;48:8.24.1–8.24.12. doi: 10.1002/0471142301.ns0824s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lapid H, Hummel T. Recording odor-evoked response potentials at the human olfactory epithelium. Chem Senses. 2013 Jan;38(1):3–17. doi: 10.1093/chemse/bjs073. [DOI] [PubMed] [Google Scholar]
- 25.American Society for Testing and Materials (ASTM) ASTM Designation E 981084. ASTM; 2012. Standard Test Method for Estimating Sensory Irritancy of Airborne Chemicals. [Google Scholar]
- 26.Kane LE, Barrow CS, Alarie Y. A short-term test to predict acceptable levels of exposure to airborne sensory irritants. Am Ind Hyg Assoc J. 1979 Mar;40(3):207–29. doi: 10.1080/15298667991429516. [DOI] [PubMed] [Google Scholar]
- 27.Kuwabara Y, Alexeeff GV, Broadwin R, Salmon AG. Evaluation and application of the RD50 for determining acceptable exposure levels of airborne sensory irritants for the general public. Environ Health Perspect. 2007;115:1609–16. doi: 10.1289/ehp.9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harkema JR, Carey SA, Wagner JG. The nose revisited: a brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol Pathol. 2006;34(3):252–69. doi: 10.1080/01926230600713475. [DOI] [PubMed] [Google Scholar]
- 29.Rao DB, Little PB, Malarkey DE, Herbert RA, Sills RC. Histopathological evaluation of the nervous system in National Toxicology Program rodent studies: a modified approach. Toxicol Pathol. 2011 Apr;39(3):463–70. doi: 10.1177/0192623311401044. [DOI] [PubMed] [Google Scholar]
- 30.Tepper JS, Kuehl PJ, Cracknell S, Nikula KJ, Pei L, Blanchard JD. Symposium Summary: “Breathe In, Breathe Out, Its Easy: What You Need to Know About Developing Inhaled Drugs”. Int J Toxicol. 2016 Feb 7; doi: 10.1177/1091581815624080. [DOI] [PubMed] [Google Scholar]
- 31.U.S. Food and Drug Administration. Draft guidance for industry and reviewers estimating the safe starting dose in clinical trials for therapeutics in adult healthy volunteers. 2002 http://www.fda.gov/cber/gdlns/dose.htm.
- 32.Kim Y, Chiang B, Wu X, Prausnitz M. Ocular delivery of macromolecules. J Control Release. 2014;0:172–181. doi: 10.1016/j.jconrel.2014.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bansal P, Garg S, Sharma Y, Vernkatesh P. Posterior segment drug delivery devices: Current and novel therapies in development. 2016;32:135–144. doi: 10.1089/jop.2015.0133. [DOI] [PubMed] [Google Scholar]
- 34.Barar J, Aghanejad A, Fathi M, Omidi Y. Advanced drug delivery and targeting technologies for the ocular diseases. Bioimpacts. 2016;6:49–67. doi: 10.15171/bi.2016.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Kudisch M, Mudhumba S, et al. Biocompatibility and pharmacokinetic analysis of an intracameral polycaprolactone drug delivery implant for glaucoma. Invest Ophthalmol Vis Sci. 2016;57:4341–4346. doi: 10.1167/iovs.16-19585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chou S, Luo L, Lai J. In vivo pharmacological evaluations of pilocarpine-loaded antioxidant-functionalized biodegradable thermogels in glaucomatous rabbits. Scientific Reports. 2017;7:42344. doi: 10.1038/srep42344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaefer E, Smith S, Salmon J, et al. Evaluation of intracameral pentablock copolymer thermosensitive gel for sustained drug delivery to the anterior chamber of the eye. J Ocul Pharmacol Ther. 2017 Mar 16; doi: 10.1089/jop.2016.0181. [DOI] [PubMed] [Google Scholar]
- 38.Short B. Safety evaluation of ocular drug delivery formulations: Techniques and practical considerations. Toxicol Pathol. 2008;36:49–62. doi: 10.1177/0192623307310955. [DOI] [PubMed] [Google Scholar]
- 39.Ferrell Ramos M, Attar M, Stern M, Brassard J, Kim A, Matsumoto S, Vangi C. Safety evaluation of ocular drugs. In: Faqi AS, editor. A Comprehensive Guide to Toxicology in Preclinical Drug Development. 2. Elsevier Inc; 2017. pp. 758–812. [Google Scholar]
- 40.Wier A, Wilson S. Nonclinical regulatory aspects for ophthalmic drugs. In: Weir A, Collins M, editors. Assessing Ocular Toxicology in Laboratory Animals. Springer Science+Business Media; 2013. pp. 259–294. [Google Scholar]
- 41.Novack G, Moyer E. How much nonclinical safety data are required for a clinical study in ophthalmology? J Ocul Pharmacol and Ther. 2016;32:5–10. doi: 10.1089/jop.2015.0120. [DOI] [PubMed] [Google Scholar]
- 42.de Zafra C, Sasseville V, Matsumoto S, et al. Inflammation and immunogenicity limit the utility of the rabbit as a nonclinical species for ocular biologic therapeutics. Regul Toxicol Pharmacol. 2017;86:221–230. doi: 10.1016/j.yrtph.2017.03.013. [DOI] [PubMed] [Google Scholar]
- 43.ICH S6(R1) Pre-Clinical Safety Evaluation of Biotechnology-derived Pharmaceuticals. 2011 www.ich.org/fileadmin/Public_Web_Site/ICH.../S6_R1/.../S6_R1_Guideline.pdf.
- 44.ICH S6(R1) S6 Addendum to Pre-Clinical Safety Evaluation of Biotechnology-derived Pharmaceuticals. 2011 https://www.fda.gov/downloads/Drugs/.../Guidances/UCM194490.pdf.
- 45.FDA Guidance for Industry, Nonclinical Safety Evaluation of Reformulated Drug Products and Products Intended for Administration by an Alternate Route. 2015 https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079245.pdf.
- 46.Vezina M. Comparative ocular anatomy in commonly used laboratory animals. In: Weir A, Collins M, editors. Assessing Ocular Toxicology in Laboratory Animals. Springer Science+Business Media; 2013. pp. 1–21. [Google Scholar]
- 47.Struble C, Howard S, Relph J. Comparison of ocular tissue weights (volumes) and tissue collection techniques in common preclinical animal species. European Assoc for Vision and Eye Research Conference; October 1–4, 2014; Abstract S005. [Google Scholar]
- 48.Samuelson D. A reevaluation of the comparative anatomy of the eutherian irideocorneal angle and associated ciliary body musculature. Vet Comp Ophthalmol. 1996;6:153–172. [Google Scholar]
- 49.Almazan A, Tsai S, Miller P, et al. Iridocorneal angle measurements in mammalian species: normative data by optical coherence tomography. Vet Ophthalmol. 2013;16:163–166. doi: 10.1111/j.1463-5224.2012.01030.x. [DOI] [PubMed] [Google Scholar]
- 50.Fernandez-Vigo J, Garcia-Feijoo J, Martinez-de-la-casa J, et al. Fourier domain optical coherence tomography to assess the iridocorneal angle and correlation study in a large Caucasian population. BMC Ophthalmol. 2016;16:42–49. doi: 10.1186/s12886-016-0219-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.ICH M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals Questions and Answers(R2) 2013 https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292340.pdf. [PubMed]
- 52.Munger R, Collins M. Assessment of ocular toxicity potential: Basic theory and techniques. In: Weir A, Collins M, editors. Assessing Ocular Toxicology in Laboratory Animals. Springer Science+Business Media; 2013. pp. 23–52. [Google Scholar]
- 53.Nork T, Rasmussen C, Christian B, Croft M, Murphy C. Emerging imaging technologies for assessing ocular toxicity in laboratory animals. In: Weir A, Collins M, editors. Assessing Ocular Toxicology in Laboratory Animals. Springer Science+Business Media; 2013. pp. 53–121. [Google Scholar]
- 54.Kerlin R, Bolon B, Burkhardt J, et al. Scientific and regulatory policy committee: Recommended (“best”) practices for determining, communicating, and using adverse effect data from nonclinical studies. Toxicol Pathol. 2016;44:147–162. doi: 10.1177/0192623315623265. [DOI] [PubMed] [Google Scholar]
- 55.Schafer K, Render J. Toxicologic pathology of the eye: Alterations of the lens and posterior segment. In: Weir A, Collins M, editors. Assessing Ocular Toxicology in Laboratory Animals. Springer Science+Business Media; 2013. pp. 219–257. [Google Scholar]
- 56.Schafer K, Render J. Toxicologic pathology of the eye: Histologic preparation and alterations of the anterior segment. In: Weir A, Collins M, editors. Assessing Ocular Toxicology in Laboratory Animals. Springer Science+Business Media; 2013. pp. 159–217. [Google Scholar]
- 57.Pearson P, Jaffe G, Martin D, et al. Evaluation of a delivery system providing long-term release of cyclosporine. Arch Ophthalmol. 1996;114:311–317. doi: 10.1001/archopht.1996.01100130307014. [DOI] [PubMed] [Google Scholar]
- 58.Yaksh TL, Fisher C, Hockman T, Wiese A. Current and Future Issues in the development of spinal agents for the management of pain. Curr Neuropharmacol. 2016 doi: 10.2174/1570159X14666160307145542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pollay M. The function and structure of the cerebrospinal fluid outflow system. Cerebrospinal Fluid Res. 2010;7:9. doi: 10.1186/1743-8454-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haughton V, Mardal KA. Spinal fluid biomechanics and imaging: an update for neuroradiologists. AJNR Am J Neuroradiol. 2014;35(10):1864–1869. doi: 10.3174/ajnr.A4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sakka L, Coll G, Chazal J. Anatomy and physiology of cerebrospinal fluid. Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128(6):309–316. doi: 10.1016/j.anorl.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 62.Hladky SB, Barrand MA. Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS. 2014;11(1):26. doi: 10.1186/2045-8118-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhadelia RA, Madan N, Zhao Y, et al. Physiology-Based MR Imaging Assessment of CSF Flow at the Foramen Magnum with a Valsalva Maneuver. Am J Neuroradiol. 2013;34(9):1857–1862. doi: 10.3174/ajnr.A3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Batson OV. The Function of the Vertebral Veins and their Role in the Spread of Metastases. Ann Surg. 1940;112(1):138–149. doi: 10.1097/00000658-194007000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bernards CM, Hill HF. Morphine and Alfentanil Permeability through the Spinal Dura, Arachnoid, and Pia Mater of Dogs and Monkeys. Anesthesiology. 1990;73(6):1214–1219. doi: 10.1097/00000542-199012000-00020. [DOI] [PubMed] [Google Scholar]
- 66.Miyanohara A, Kamizato K, Juhas S, et al. Potent spinal parenchymal AAV9-mediated gene delivery by subpial injection in adult rats and pigs. Mol Ther Methods Clin Dev. 2016;3:16046. doi: 10.1038/mtm.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brochner CB, Holst CB, Mollgard K. Outer brain barriers in rat and human development. Front Neurosci. 2015;9:75. doi: 10.3389/fnins.2015.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Greene NM. Distribution of Local-Anesthetic Solutions within the Subarachnoid Space. Anesth Analg. 1985;64(7):715–730. [PubMed] [Google Scholar]
- 69.Casati A, Fanelli G, Cappelleri G, et al. Does speed of intrathecal injection affect the distribution of 0.5% hyperbaric bupivacaine? (vol 81, pg 355, 1998) Brit J Anaesth. 1998;81(6):1001–1001. doi: 10.1093/bja/81.3.355. [DOI] [PubMed] [Google Scholar]
- 70.Brinker T, Stopa E, Morrison J, Klinge P. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS. 2014;11:10. doi: 10.1186/2045-8118-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bernards CM. Recent insights into the pharmacokinetics of spinal opioids and the relevance to opioid selection. Curr Opin Anaesthesiol. 2004;17(5):441–447. doi: 10.1097/00001503-200410000-00015. [DOI] [PubMed] [Google Scholar]
- 72.Grouls RJE, Korsten EHM, Yaksh TL. General considerations for the formulation of drugs for spinal delivery. In: Yaksh TL, editor. Spinal Drug Delivery. Amsterdam: Elsevier Science B.V; 1999. pp. 371–393. [Google Scholar]
- 73.Yaksh TL, Jang JD, Nishiuchi Y, Braun KP, Ro SG, Goodman M. The utility of 2-hydroxypropyl-beta-cyclodextrin as a vehicle for the intracerebral and intrathecal administration of drugs. Life Sci. 1991;48(7):623–633. doi: 10.1016/0024-3205(91)90537-l. [DOI] [PubMed] [Google Scholar]
- 74.Lagarce F, Benoit JP. Sustained release formulations for spinal drug delivery. J Drug Deliv Sci Tec. 2004;14(5):331–343. [Google Scholar]
- 75.Barker A. Elimination of stovaine after spinal analgesia. Br Med J. 1904;2:789–791. [Google Scholar]
- 76.Wossidlo E. Experimentelle untersuchungen uber veranderungen Nissl’schen granulma beider lumbalanasthesie. Arch f Klin Chir. 1908;86:1017–1053. [Google Scholar]
- 77.Rigler ML, Drasner K, Krejcie TC, et al. Cauda equina syndrome after continuous spinal anesthesia. Anesth Analg. 1991;72(3):275–281. doi: 10.1213/00000539-199103000-00001. [DOI] [PubMed] [Google Scholar]
- 78.Schell RM, Brauer FS, Cole DJ, Applegate RL., 2nd Persistent sacral nerve root deficits after continuous spinal anaesthesia. Can J Anaesth. 1991;38(7):908–911. doi: 10.1007/BF03036972. [DOI] [PubMed] [Google Scholar]
- 79.Snyder R, Hui G, Flugstad P, Viarengo C. More cases of possible neurologic toxicity associated with single subarachnoid injections of 5% hyperbaric lidocaine. Anesth Analg. 1994;78(2):411. doi: 10.1213/00000539-199402000-00046. [DOI] [PubMed] [Google Scholar]
- 80.Drasner K, Sakura S, Chan VW, Bollen AW, Ciriales R. Persistent sacral sensory deficit induced by intrathecal local anesthetic infusion in the rat. Anesthesiology. 1994;80(4):847–852. doi: 10.1097/00000542-199404000-00018. [DOI] [PubMed] [Google Scholar]
- 81.Sakura S, Kirihara Y, Muguruma T, Kishimoto T, Saito Y. The comparative neurotoxicity of intrathecal lidocaine and bupivacaine in rats. Anesth Analg. 2005;101(2):541–547. doi: 10.1213/01.ANE.0000155960.61157.12. table of contents. [DOI] [PubMed] [Google Scholar]
- 82.Ready LB, Plumer MH, Haschke RH, Austin E, Sumi SM. Neurotoxicity of intrathecal local anesthetics in rabbits. Anesthesiology. 1985;63(4):364–370. [PubMed] [Google Scholar]
- 83.Yamashita A, Matsumoto M, Matsumoto S, Itoh M, Kawai K, Sakabe T. A comparison of the neurotoxic effects on the spinal cord of tetracaine, lidocaine, bupivacaine, and ropivacaine administered intrathecally in rabbits. Anesth Analg. 2003;97(2):512–519. doi: 10.1213/01.ANE.0000068885.78816.5B. [DOI] [PubMed] [Google Scholar]
- 84.Malinovsky JM, Charles F, Baudrimont M, et al. Intrathecal ropivacaine in rabbits: pharmacodynamic and neurotoxicologic study. Anesthesiology. 2002;97(2):429–435. doi: 10.1097/00000542-200208000-00021. [DOI] [PubMed] [Google Scholar]
- 85.Ganem EM, Vianna PT, Marques M, Castiglia YM, Vane LA. Neurotoxicity of subarachnoid hyperbaric bupivacaine in dogs. Reg Anesth. 1996;21(3):234–238. [PubMed] [Google Scholar]
- 86.Kroin JS, McCarthy RJ, Penn RD, Kerns JM, Ivankovich AD. The effect of chronic subarachnoid bupivacaine infusion in dogs. Anesthesiology. 1987;66(6):737–742. doi: 10.1097/00000542-198706000-00005. [DOI] [PubMed] [Google Scholar]
- 87.Koizumi Y, Matsumoto M, Yamashita A, Tsuruta S, Ohtake T, Sakabe T. The effects of an AMPA receptor antagonist on the neurotoxicity of tetracaine intrathecally administered in rabbits. Anesth Analg. 2006;102(3):930–936. doi: 10.1213/01.ane.0000195550.67356.6a. [DOI] [PubMed] [Google Scholar]
- 88.Gold MS, Reichling DB, Hampl KF, Drasner K, Levine JD. Lidocaine toxicity in primary afferent neurons from the rat. J Pharmacol Exp Ther. 1998;285(2):413–421. [PubMed] [Google Scholar]
- 89.Kitagawa N, Oda M, Totoki T. Possible mechanism of irreversible nerve injury caused by local anesthetics: detergent properties of local anesthetics and membrane disruption. Anesthesiology. 2004;100(4):962–967. doi: 10.1097/00000542-200404000-00029. [DOI] [PubMed] [Google Scholar]
- 90.Eddinger KA, Rondon ES, Shubayev VI, et al. Intrathecal Catheterization and Drug Delivery in Guinea Pigs: A Small-animal Model for Morphine-evoked Granuloma Formation. Anesthesiology. 2016;125(2):378–394. doi: 10.1097/ALN.0000000000001166. [DOI] [PubMed] [Google Scholar]
- 91.Yaksh TL, Horais KA, Tozier NA, et al. Chronically infused intrathecal morphine in dogs. Anesthesiology. 2003;99(1):174–187. doi: 10.1097/00000542-200307000-00028. [DOI] [PubMed] [Google Scholar]
- 92.Gradert TL, Baze WB, Satterfield WC, Hildebrand KR, Johansen MJ, Hassenbusch SJ. Safety of chronic intrathecal morphine infusion in a sheep model. Anesthesiology. 2003;99(1):188–198. doi: 10.1097/00000542-200307000-00029. [DOI] [PubMed] [Google Scholar]
- 93.Deer TR, Pope JE, Hayek SM, et al. The Polyanalgesic Consensus Conference (PACC): Recommendations for Intrathecal Drug Delivery: Guidance for Improving Safety and Mitigating Risks. Neuromodulation. 2017;20(2):155–176. doi: 10.1111/ner.12579. [DOI] [PubMed] [Google Scholar]
- 94.Allen JW, Horais KA, Tozier NA, et al. Time course and role of morphine dose and concentration in intrathecal granuloma formation in dogs: a combined magnetic resonance imaging and histopathology investigation. Anesthesiology. 2006;105(3):581–589. doi: 10.1097/00000542-200609000-00024. [DOI] [PubMed] [Google Scholar]
- 95.Allen JW, Horais KA, Tozier NA, Yaksh TL. Opiate pharmacology of intrathecal granulomas. Anesthesiology. 2006;105(3):590–598. doi: 10.1097/00000542-200609000-00025. [DOI] [PubMed] [Google Scholar]
- 96.Yaksh TL, Allen JW, Veesart SL, et al. Role of meningeal mast cells in intrathecal morphine-evoked granuloma formation. Anesthesiology. 2013;118(3):664–678. doi: 10.1097/ALN.0b013e31828351aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yaksh TL, Allen JW. The use of intrathecal midazolam in humans: A case study of process. Anesth Analg. 2004;98(6):1536–1545. doi: 10.1213/01.ANE.0000122638.41130.BF. [DOI] [PubMed] [Google Scholar]
- 98.Yaksh TL, Allen JW. Preclinical insights into the implementation of intrathecal midazolam: A cautionary tale. Anesth Analg. 2004;98(6):1509–1511. doi: 10.1213/01.ANE.0000121768.79904.7F. [DOI] [PubMed] [Google Scholar]
- 99.Yaksh TL. Spinal delivery and assessment of drug safety. In: Bolon B, Butt MT, editors. Fundamental Neuropathology for Pathologists and Toxicologists: Principles and Techniques. 1. John Wiley & Sons, Inc; 2011. pp. 449–462. [Google Scholar]
- 100.Walker SM, Yaksh TL. Neuraxial analgesia in neonates and infants: a review of clinical and preclinical strategies for the development of safety and efficacy data. Anesth Analg. 2012;115(3):638–662. doi: 10.1213/ANE.0b013e31826253f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wiese AJ, Rathbun M, Butt MT, et al. Intrathecal substance P-saporin in the dog: distribution, safety, and spinal neurokinin-1 receptor ablation. Anesthesiology. 2013;119(5):1163–1177. doi: 10.1097/ALN.0b013e3182a95164. [DOI] [PubMed] [Google Scholar]
- 102.Flack SH, Bernards CM. Cerebrospinal fluid and spinal cord distribution of hyperbaric bupivacaine and baclofen during slow intrathecal infusion in pigs. Anesthesiology. 2010;112(1):165–173. doi: 10.1097/ALN.0b013e3181c38da5. [DOI] [PubMed] [Google Scholar]
- 103.Butt MT. Evaluation of the Adult Nervous System in Preclinical Studies. In: Butt MT, editor. Fundamental Neuropathology for Pathologists and Toxicologists: Principles and Techniques. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2011. pp. 321–338. [Google Scholar]
- 104.Artru A. CSF dynamics, cerebral edema and intracranial pressure. In: Albin MS, editor. Textbook of Neuroanesthesia. New York, NY: McGraw Hill; 1997. pp. 61–115. [Google Scholar]
- 105.Davison H, Segal MD, editors. Physiology of the CSF and Blood brain Barriers. Boca Raton, FL: CRC Press; 1996. [Google Scholar]
- 106.Himwich WA. Cerebral Circulation, Blood Brain Barrier and Cerebrospinal Fluid. In: Swenson MJ, editor. Dukes’ Physiology of Domestic Animals. London: Cornell University Press; 1977. pp. 157–174. [Google Scholar]
- 107.Artru A. Spinal Cerebrospinal Fluid Chemistry and Physiology. In: Yaksh TL, editor. Spinal Drug Delivery. Amsterdam: Elsevier, Inc; 1999. pp. 177–237. [Google Scholar]
- 108.Proffen BL, McElfresh M, Fleming BC, Murray MM. A Comparative A anatomical study of the human and six animal species. The Knee. 2012;19(4):493–499. doi: 10.1016/j.knee.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tantisricharoenkul G, Linde-Rosen M, Araujo P, et al. Anterior cruciate ligament: an anatomical exploration in humans and in a selection of animal species. Knee Surg Sports Traumatol Arthrosc. 2014;22:961. doi: 10.1007/s00167-013-2463-6. [DOI] [PubMed] [Google Scholar]
- 110.Ingham SJM, de Carvalho RT, Martins CAQ, et al. Anterolateral ligament anatomy: a comparative anatomical study. Knee Surg Sports Traumatol Arthrosc. 2015 doi: 10.1007/s00167-015-3956-2. [DOI] [PubMed] [Google Scholar]
- 111.Edwards Jo., editor. Notes on Rheumatology. University College London; 2000. Normal Joint Structure. Archived from the original on 19 Nov 2012. Retrieved 5 April 2013. [Google Scholar]
- 112.Courtney Philip, et al. Joint aspiration and injection and synovial fluid analysis Best Practice & Research Clinical Rheumatology. 2013;23(2):161–192. doi: 10.1016/j.berh.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 113.Wehr B, Grams A, Hudelmaier M, Kotyk J, Wachsmuth L, Eckstein F. Tibial cartilage surface area, thickness and volume in various animal species and in humans. Osteoarthritis Cartilage. 2007;15:C54–C55. [Google Scholar]
- 114.Matsuzaka S, Sato S, Miyauchi S. Estimation of joint fluid volume in the knee joint of rabbits by measuring the endogenous calcium concentration. Clinical Experimental Rheumatology. 2002;20:531–534. [PubMed] [Google Scholar]
- 115.Athanasiou KA, Rosenwasser MP, Buckwalter JA, Malinin TI, Mow VC. Interspecies comparisons of in situ intrinsic mechanical properties of distal femoral cartilage. Journal Orthopaedic Research. 1991;9:330–340. doi: 10.1002/jor.1100090304. [DOI] [PubMed] [Google Scholar]
- 116.Heilmann HH, Lindenhayn K, Walther HUU. Synovial volume of healthy and arthrotic human knee joints. Zeitschrift für Orthopädie und ihre Grenzgebiete. 1996;134:144–148. doi: 10.1055/s-2008-1039786. [DOI] [PubMed] [Google Scholar]
- 117.Kraus VB, Stabler TV, Kong J, Varju G, McDaniel G. Measurement of synovial fluid volume using urea. Osteoarthritis Cartilage. 2007;15:1217–1220. doi: 10.1016/j.joca.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ateshian GA, Mow VC. Friction, lubrication, and wear of articular cartilage and diarthrodial joints. In: Mow VC, Huiskes R, editors. Basic Orthopaedic Biomechanics and Mechano-Biology. Vol. 3. Philadelphia: Lippincott Williams & Wilkins; 2005. pp. 447–494. [Google Scholar]
- 119.Chu CR, Szczodry M, Bruno S. Animal Models for Cartilage Regeneration and Repair. Tissue Engineering Part B, Reviews. 2010;16(1):105–115. doi: 10.1089/ten.teb.2009.0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Little CB, Zaki S. What constitutes an “animal model of osteoarthritis” – the need for consensus? Osteoarthritis and Cartilage. 2012 Apr;20(4):261–267. doi: 10.1016/j.joca.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 121.Bendele AM. Animal Models. In: Bronner F, Farach-Carson MC, editors. Bone and Osteoarthritis. London: Springer-Verlag; 2007. pp. 149–163. [Google Scholar]
- 122.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative-recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010;18:S17–23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 123.Gerwin N, Bendele A, Glasson S, Carlson CS. The OARSI histopathology initiative-recommendations for histological assessments of osteoarthritis in the rat. Osteoarthritis Cartilage. 2010;18:S24–34. doi: 10.1016/j.joca.2010.05.030. [DOI] [PubMed] [Google Scholar]
- 124.Jou I-M, Shiau A-L, Chen S-Y, Wang C-R, Shieh D-B, Tsai C-S, Wu C-L. Thrombospondin 1 as an effective gene therapeutic strategy in collagen-induced arthritis. Arthritis & Rheumatism. 2005;52:339–344. doi: 10.1002/art.20746. [DOI] [PubMed] [Google Scholar]
- 125.Bendele AM, Hulman JF. Spontaneous cartilage degeneration in guinea pigs. Arthritis Rheum. 1988;31:561–565. doi: 10.1002/art.1780310416. [DOI] [PubMed] [Google Scholar]
- 126.Kraus V, Huebner J, DeGroot J, Bendele A. The OARSI histopathology initiative-recommendations for histological assessment of osteoarthritis in the guinea pig. Osteoarthritis Cartilage. 2010;18:S35–52. doi: 10.1016/j.joca.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Colombo C, Butler M, O’Byrne E, Hickman L, Swartzendruber D, Selwyn M, Steinetz B. A new model of osteoarthritis in rabbits. I. Development of knee joint pathology following lateral meniscectomy and section of the fibular collateral and sesamoid ligaments. Arthritis Rheum. 1983;26(7):875–86. doi: 10.1002/art.1780260709. [DOI] [PubMed] [Google Scholar]
- 128.Gushue David L, Houck Jeff, Lerner Amy L. Rabbit knee joint biomechanics: Motion analysis and modeling of forces during hopping. Journal of Orthopaedic Research. 2005 Jul;23(4):735–742. doi: 10.1016/j.orthres.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 129.Laverty S, Girard CA, Williams JM, Hunziker EB, Pritzker KPH. The OARSI histopathology initiative-recommendations for histological scoring of osteoarthritis in the rabbit. Osteoarthritis Cartilage. 2010;18:S53–65. doi: 10.1016/j.joca.2010.05.029. [DOI] [PubMed] [Google Scholar]
- 130.Connor JR, Lepage C, Swift BA, Yamashita D, Bendele AM, Maul D, Kumar S. Protective effects of a cathepsin K inhibitor, SB-553484, in the canine medial meniscectomy model of osteoarthritis. Osteoarthritis Cartilage. 2009;17(9):1236–43. doi: 10.1016/j.joca.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 131.Brandt KD, Braunstein EM, Visco DM, O’Connor B, Heck D, Albrecht M. Anterior (cranial) cruciate ligament transection in the dog: a bona fide model of osteoarthritis, not merely of cartilage injury and repair. J Rheumatology. 1991;18:436–446. [PubMed] [Google Scholar]
- 132.Trevor A, Katzung B, Masters S, Knuidering-Hall M. Pharmacology Examination & Board Review. 10. New York: McGraw-Hill Medical; 2013. Chapter 2: Pharmacodynamics; p. 17. [Google Scholar]
- 133.Chamanza Ronnie, Marxfeld Heike A, Blanco Ana I, Naylor Stuart W, Bradley Alys E. Incidences and Range of Spontaneous Findings in Control Cynomolgus Monkeys (Macaca fascicularis) Used in Toxicity Studies. Toxicologic Pathology. 38(4):642–657. doi: 10.1177/0192623310368981. First published date: May-06-2010. [DOI] [PubMed] [Google Scholar]