

Abstract
The LARP7 gene encodes a chaperone protein of the noncoding RNA 75 K, and mutations in this gene have been identified in patients with Alazami syndrome. Herein, we report another Japanese patient with Alazami syndrome and novel compound heterozygous variants in LARP7 (i.e., c.370delG, p.Glu124fs*38 and c.641_667+25del involving the splice donor site of intron 8). These findings provide further evidence that biallelic LARP7 defects cause the phenotype of Alazami syndrome.
Alazami syndrome is an autosomal recessive developmental disorder that is characterized by severe intellectual disability, postnatal growth retardation and distinct dysmorphic facial features that include a protruding forehead, deep-set eyes, narrow palpebral fissures, a broad nose and malar hypoplasia (OMIM #615071). This syndrome was first identified in a large consanguineous Saudi Arabian family in 2012. A homozygous frameshift mutation in the La ribonucleoprotein domain family member 7 gene (LARP7), which encodes a chaperone protein of the noncoding RNA 75 K, was identified in all of the affected patients in the family.1
Because only four families with Alazami syndrome and LARP7 mutations have been reported thus far, the phenotypic spectrum of LARP7-related disease remains unclear.1–4 Herein, we report an additional patient with Alazami syndrome with novel compound heterozygous mutations in the LARP7 gene and review the clinical and genetic findings from patients with Alazami syndrome.
A male Japanese patient was born at 38 weeks of gestation after an uncomplicated pregnancy and delivery. At birth, his length was 46 cm (–1.3 standard deviation [SD]), and his weight was 2.44 kg (−1.1 SD). He was found to have subcoronal hypospadias and an inguinal hernia. He also exhibited distinct facial features that included a prominent forehead, narrow palpebral fissures, deep-set eyes, hypertelorism, a broad nose and malar hypoplasia (Figure 1a). He had severe hypotonia in infancy. His motor development was obviously delayed; he held up his head at 9 months, rolled over at 10 months and walked at 2 years. At the last examination at 2 years of age, he measured 80.6 cm (−1.14 SD) and weighed 9.45 kg (−1.28 SD). He could not speak any meaningful words and exhibited intellectual disability (total IQ, 67).
Figure 1.
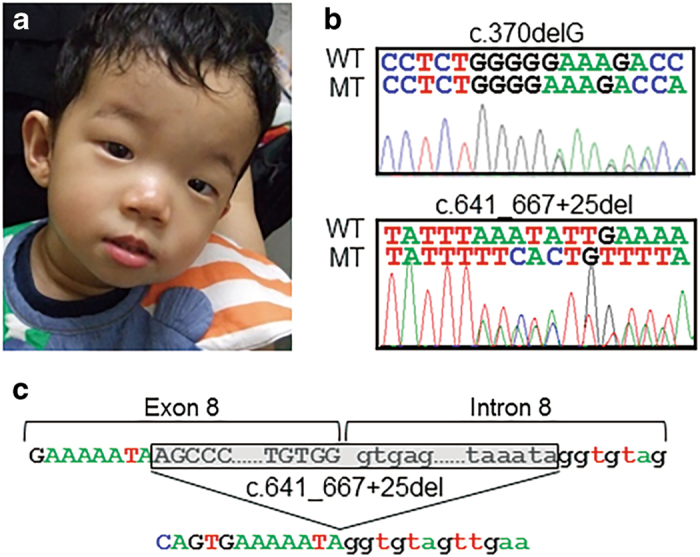
Clinical and genetic findings in the patient. (a) A front view of the patient at 2 years of age showing the distinct facial features of a prominent forehead, narrow palpebral fissures, deep-set eyes, hypertelorism, a broad nose and malar hypoplasia. (b) Electropherograms displaying the two LARP7 mutations in the patient. The upper (c.370delG) and lower (c.641_667+25del) sequences are the sense and antisense sequences, respectively. WT, wild-type allele; MT, mutant allele. (c) A schematic representation of the 52-bp deletion (c.641_667+25del) in LARP7. The exonic and intronic sequences are indicated by capital and lower-case letters, respectively. The deleted sequences are shaded in gray. The deletion involved the splice donor site of intron 8 and was predicted to cause an exon skipping of exon 8.
His non-consanguineous parents were clinically normal. His father and mother were 180 cm (+0.6 SD) and 152 cm (−1.3 SD) in height, respectively. There were no family histories of neurodevelopmental diseases or congenital malformations.
This study was approved by the Institutional Review Board of the Nagasaki University Graduate School of Biomedical Sciences. Because clinical assessment alone was unable to identify a conclusive diagnosis, we sought to identify disease-causing mutations with a trio whole-exome sequencing (WES) strategy using a SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA) on a HiSeq 2500 platform (Illumina, San Diego, CA, USA). Written informed consent was obtained from the parents. DNA samples were obtained from peripheral blood samples from the patient and the parents. The reads in the FASTQ files were aligned to the human reference genome using Novoalign version 3.0 (http://www.novocraft.com/). The trio-based genomic variation information was detected with the Genome Analysis Toolkit software version 3.4–46.5 Subsequently, de novo, homozygous, heterozygous and X-linked variations were extracted and annotated with the ANNOVAR software.6 This process excluded variants with allele frequencies >0.5% in any of the following databases: the Exome Aggregation Consortium (http://exac.broadinstitute.org/), the NHLBI GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), the Human Genetic Variation Database (http://www.hgvd.genome.med.kyoto-u.ac.jp) and the 1KJPN database of the Tohoku Medical Megabank (http://www.dist.megabank.tohoku.ac.jp). Heterozygous variations sharing the same GENCODE v19 genes were also extracted to detect compound heterozygous mutations. The mutations were confirmed via Sanger sequencing using a BigDye terminator and a 3130xl Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA).
Utilizing the aforementioned strategy, we identified several candidate variants. Of these variants, compound heterozygous mutations in LARP7 (c.370delG and c.641_667+25del involving exon 8 and intron 8; NM_001267039) were proposed as the best candidates based on mutual references to the WES data and the Online Mendelian Inheritance in Man database information of known diseases (www.omim.org) (Figure 1b). The father and mother of the proband were heterozygous for the c.370delG and c.641_667+25del variants, respectively. These two variants have not been recorded in the Human Genetic Variation Database (http://www.hgvd.genome.med.kyoto-u.ac.jp/) or the Exome Aggregation Consortium Database (http://exac.broadinstitute.org/). A 1-bp deletion in exon 6 (c.370delG) was predicted to cause a frameshift at codon 124 in LARP7 and a consequent termination at codon 161 (p.Glu124fs*38; NP_001253968). The deletion involving the splice donor site of intron 8 was predicted to cause an exon skipping of exon 8 during transcript processing, a frameshift mutation at codon 192 of LARP7 and a resultant termination at codon 204 (p.Phe192fs*13) (Figure 1c).
The LARP7 mutations identified in the present patient as well as the previously reported mutations are all frameshift mutations that are thought to cause early truncation of the LARP7 protein and thereby induce nonsense-mediated mRNA decay (Table 1).7 This supposition suggests that a severe loss-of-function of LARP7 is likely the main mechanism underlying the phenotype of Alazami syndrome.1–4
Table 1. Clinical and genetic features of patients with Alazami syndrome.
| Alazami et al. 1 | Ling et al. 2 | Holink et al. 3 | Holohan et al. 4 | Present case | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
||
| Age at the last examination (yrs) | 12 | 5 | 22 | 20 | 15 | 17 | 10 | 8 | 5 | 2.5 | 6 | 2.5 | 8 | 2 |
||
| Sex | Male | Female | Male | Male | Male | Female | Male | Female | Female | Female | Male | Male | Male | Male |
||
| | ||||||||||||||||
|
LARP7 mutations | ||||||||||||||||
| cDNA | c.1024_1030dup (homo) |
c.213_214dup c.651_655del | c.1091_1094del (homo) | c.1045_1051dup (homo) | c.756_757del (homo) | c.370del G c.641_667+25del |
||||||||||
| Protein | p.Thr344Lysfs* (homo) |
p.Ser72fs p.Lys219fs | p.Arg364fs (homo) | p.Thr351fs* (homo) | p.Arg253fs* (homo) | p.Glu124fs p.Phe192fs |
||||||||||
|
Birth measurements | ||||||||||||||||
| Gestational age | ND | ND | ND | ND | ND | ND | ND | ND | ND | 40 wks | 40 wks 3 days | ND | ND | 38 wks |
||
| Weight (SD) | ND | ND | ND | ND | ND | ND | ND | ND | ND | 2608 g (−1.35) | 2980 g | 2000 g | ND | 2444 g (−1.1) |
||
| Height (SD) | ND | ND | ND | ND | ND | ND | ND | ND | ND | 47.6 m (−0.91) | 45 cm | ND | ND | 46.3 cm (−1.3) |
||
| OFC (SD) | ND | ND | ND | ND | ND | ND | ND | ND | ND | 32.4 cm (−1.35) | 34 cm | ND | ND | 33 cm (−0.1) |
||
|
Present measurements | ||||||||||||||||
| Weight (SD) | 14.6 kg (−3.3 ) | 8 kg (−4.0) | 33 kg (−4.5) | 37 kg (−4.0) | 31 kg (−3.5) | 24 kg (−3.9) | 25 kg (−2.0) | 13 kg (−3.4) | 10 kg (−3.3) | 7.6 kg (−5.5) | 16.5 kg (−1.0) | 9.1 kg (−3.0) | ND | 9.75 kg (−1.1) |
||
| Height (SD) | 117 cm (−7.0) | 90 cm (−4.0) | 129 cm (−10.5) | 138 cm (−9.7) | 138 cm (−9.5) | 133 cm (−5.6) | 135 cm (−5.0) | 109 cm (−3.6) | 92 cm (−4.0) | 72.7 cm (−4.0) | 107.4 cm (−2.5) | 81 cm (−3.0) | (<−3.5) | 82 cm (−1.4) |
||
| OFC (SD) | 48 cm (−3.3) | 40 cm (−7.0) | 51 cm (−3.0) | 53 cm (−2.0) | 53 cm (−2.0) | 49 cm (−3.3) | 51 cm (−1.5) | 49.5 cm (−1.5) | 48 cm (−1.7) | 46 cm (−1.0) | 48.4 cm (−2.0) | 44.5 cm (−4.0) | ND | 45 cm (−1.5) |
||
|
Developmental status | ||||||||||||||||
| Motor developmental delay | 100% (14/14) | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
|
| Intellectual disability | 100% (14/14) | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
|
|
Clinical features | ||||||||||||||||
| Trianglar face | 85% (11/13) |
+ | + | + | + | + | + | + | + | + | + | − | + | ND | − | |
| Prominent forehead | 100% (14/14) |
+ | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Deep-seated eyes | 100% (13/13) |
+ | + | + | + | + | + | + | + | + | + | + | + | ND | + | |
| Narrow palpebral fissures | 69% (9/13) |
+ | + | + | + | + | − | + | − | + | − | − | + | ND | + | |
| Sparse eyebrows | 83% (10/12) |
+ | + | + | + | + | + | + | + | + | ND | − | + | ND | − | |
| Broad nose | 100% (14/14) |
+ | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Low-set ears | 54% (7/13) |
+ | + | − | + | + | − | + | − | + | + | − | − | ND | − | |
| Wide mouth | 85% (11/13) |
+ | + | + | + | + | + | + | + | + | + | − | + | ND | − | |
| Full lips | 69% (9/13) |
− | + | + | − | − | + | + | + | − | + | + | + | ND | + | |
| Widely spaced teeth | 77% (10/13) |
+ | + | + | + | + | − | + | + | − | + | + | + | ND | − | |
| Malar hypoplasia | 85% (11/13) |
+ | + | + | + | + | − | + | + | + | + | − | + | ND | + | |
| Strabismus | 33% (4/12) |
− | + | + | + | − | − | + | − | − | ND | − | − | ND | − | |
| Thickend skin over hands | 33% (4/12) |
+ | − | + | − | − | − | + | − | − | ND | + | − | ND | − | |
| Scoliosis | 25% (3/12) |
+ | + | − | − | − | − | − | − | − | ND | + | − | ND | − | |
| Other findings | ||||||||||||||||
| Short Achilles' tendon | Short Achilles' tendon | Blue/grey sclera, easily anxious and agitated | Clinodactyly of the big toes | Cleft palate, torticollis | Hypertelorism, hypotonia, small hands | Hypospadias, inguinal hernia, hypertelorism hypotonia | ||||||||||
Abbreviations: ND, not described; OFC, occipitofrontal circumference; SD, standard deviation; +, present; −, absent.
A comprehensive comparison of the clinical findings between the present patient and the previously reported cases revealed several important points (Table 1). First, severe intellectual disability, motor developmental delay and characteristic facial features, including deep-set eyes, a broad nose, narrow palpebral fissures, a prominent forehead and malar hypoplasia, are common characteristic findings in patients with biallelic LARP7 mutations. Second, although normal prenatal growth has been observed in some afflicted patients, severe postnatal growth disturbances that range from −2.5 to −10 SD have been found to be common among the previously reported patients. However, the phenotypic variety may be broader than initially presumed. Indeed, the present patient exhibited a relatively normal height up to 2 years of age. Third, the present patient has hypospadias, which has not been observed in any previously reported patients. Hypospadias is a common congenital malformation of the male external genitalia that is characterized by an aberrant opening of the urethra on the ventral side of the penis. While several genes, such as SRD5A2, AR, HOXA13 and MAMLD1, have been identified as causative genes in hypospadias, pathogenic mutations have thus far been identified in only a very small portion of patients with hypospadias.8 This finding is consistent with the notion of hypospadias as a highly heterogeneous condition that involves multiple genetic and environmental factors. Although whether this phenotype is a part of the spectrum of the manifestations of LARP7 mutations remains to be determined, the present study may broaden the genetic variability that is associated with then occurrence of hypospadias.
In conclusion, our study has provided further evidence that biallelic LARP7 defects cause Alazami syndrome. Further studies are needed to determine the clinical spectrum of patients with Alazami syndrome and the pathogenesis of LARP7 mutations.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
We wish to thank the patient and his family for their participation in this study. We also wish to thank Hiroyuki Mishima, Tatsuro Kondoh and Tadashi Matsumoto for their fruitful discussions about this article. This work was supported by a grant for the Initiative on Rare and Undiagnosed Diseases in Pediatrics (no. 17gk0110012h0101) from the Japan Agency for Medical Research and Development (AMED), Tokyo, Japan.
Footnotes
The authors declare no conflict of interest.
References
- Alazami AM, Al-Owain M, Alzahrani F, Shuaib T, Al-Shamrani H, Al-Falki YH et al. Loss of function mutation in LARP7, chaperone of 7SK ncRNA, causes a syndrome of facial dysmorphism, intellectual disability, and primordial dwarfism. Hum Mutat 2012; 33: 1429–1434. [DOI] [PubMed] [Google Scholar]
- Ling TT, Sorrentino S. Compound heterozygous variants in the LARP7 gene as a cause of Alazami syndrome in a Caucasian female with significant failure to thrive, short stature, and developmental disability. Am J Med Genet A 2016; 170A: 217–219. [DOI] [PubMed] [Google Scholar]
- Hollink IH, Alfadhel M, Al-Wakeel AS, Ababneh F, Pfundt R, de Man SA et al. Broadening the phenotypic spectrum of pathogenic LARP7 variants: two cases with intellectual disability, variable growth retardation and distinct facial features. J Hum Genet 2016; 61: 229–233. [DOI] [PubMed] [Google Scholar]
- Holohan B, Kim W, Lai TP, Hoshiyama H, Zhang N, Alazami AM et al. Impaired telomere maintenance in Alazami syndrome patients with LARP7 deficiency. BMC Genomics 2016; 17(Suppl 9): 749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet 2004; 36: 801–808. [DOI] [PubMed] [Google Scholar]
- Achermann JC, Hughes IA. Disorders of sex developmentIn:Melmed S, Polonsky KS, Larsen PR, Kronenberg HM (eds) Williams Textbook of Endocrinology12 edn. Saunders: Philadelphia, PA, USA, 2011, 914–915. [Google Scholar]
Data Citations
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.