

ABSTRACT
In 2016, Michigan experienced the largest outbreak of shigellosis, a type of bacillary dysentery caused by Shigella spp., since 1988. Following this outbreak, we isolated 16 novel Shigella-infecting bacteriophages (viruses that infect bacteria) from environmental water sources. Most well-known bacteriophages infect the common laboratory species Escherichia coli and Salmonella enterica, and these phages have built the foundation of molecular and bacteriophage biology. Until now, comparatively few bacteriophages were known to infect Shigella spp., which are close relatives of E. coli. We present a comprehensive analysis of these phages' host ranges, genomes, and structures, revealing genome sizes and capsid properties that are shared by very few previously described phages. After sequencing, a majority of the Shigella phages were found to have genomes of an uncommon size, shared by only 2% of all reported phage genomes. To investigate the structural implications of this unusual genome size, we used cryo-electron microscopy to resolve their capsid structures. We determined that these bacteriophage capsids have similarly uncommon geometry. Only two other viruses with this capsid structure have been described. Since most well-known bacteriophages infect Escherichia or Salmonella, our understanding of bacteriophages has been limited to a subset of well-described systems. Continuing to isolate phages using nontraditional strains of bacteria can fill gaps that currently exist in bacteriophage biology. In addition, the prevalence of Shigella phages during a shigellosis outbreak may suggest a potential impact of human health epidemics on local microbial communities.
IMPORTANCE Shigella spp. bacteria are causative agents of dysentery and affect more than 164 million people worldwide every year. Despite the need to combat antibiotic-resistant Shigella strains, relatively few Shigella-infecting bacteriophages have been described. By specifically looking for Shigella-infecting phages, this work has identified new isolates that (i) may be useful to combat Shigella infections and (ii) fill gaps in our knowledge of bacteriophage biology. The rare qualities of these new isolates emphasize the importance of isolating phages on “nontraditional” laboratory strains of bacteria to more fully understand both the basic biology and diversity of bacteriophages.
KEYWORDS: bacteriophage, cryo-electron microscopy, environmental microbiology, genomics, structural biology
INTRODUCTION
In 2016, Michigan experienced the largest outbreak of shigellosis in nearly 30 years (1). At least 180 cases were reported to the Michigan Department of Health and Human Services (MDHHS), with all cases occurring in Genessee and Saginaw Counties (2). A concurrent shigellosis outbreak occurred in Ingham County, where Michigan State University (MSU) is located, with 30 reported cases (3). Shigella spp. cause more than 164.7 million cases of shigellosis annually worldwide, leading to 1.1 million deaths every year, particularly among children younger than 5 years old (4). The genus Shigella is comprised of four species: S. boydii, S. dysenteriae, S. flexneri, and S. sonnei. The species most commonly isolated from shigellosis patients is S. flexneri, which is found predominantly in developing countries (5); however, shigellosis caused by S. sonnei, traditionally found in developed countries, is on the rise (6). Attempts to reduce the local and worldwide burdens of shigellosis are complicated by the low infectious dose (7) and increasing antibiotic resistance of Shigella (8). In February 2017, the World Health Organization designated Shigella a priority target for research and development of new drugs (9).
Bacteriophages (phages) are viruses that infect bacteria. With many species of bacteria becoming resistant to antibiotics, phages are reemerging as an attractive alternative and have already been used to combat a wide variety of bacterial infections (10). One of the first bacteriophages ever isolated was a Shigella phage, discovered by Felix d'Herelle in 1917 (11, 12), which was subsequently shown to cure children suffering from severe S. dysenteriae infection (13). However, relatively few studies on the isolation or characterization of Shigella phages have been performed since then. Information for only ∼35 Shigella phages has been deposited in public databases, and detailed studies of these phages are sparse. By contrast, over 400 Escherichia and Salmonella phages are readily available, some of which, such as ϕX174, P22, λ, and T4, have been used as model systems for decades.
With recent shigellosis outbreaks in Michigan, we isolated 16 new Shigella phages from our local environment, increasing the number of known Shigella phages from 35 to 51. Source materials were collected and screened for phages in October 2016 as part of a graduate school course. Surprisingly, significantly more Shigella phages were isolated than Escherichia and Salmonella phages. Information for hundreds of Escherichia coli and Salmonella phages exists in public databases, and many of these isolates came from environmental samples. Conversely, most previously isolated Shigella phages were isolated from wastewater or clinical samples, with very few being found in natural environments (14–16). Of these 16 new Shigella phages, seven have uncommon genome sizes and similarly uncommon T=9 capsid geometry. Given the information for all known phages, only ∼2% share similar genome sizes, and only two T=9 capsid structures have been seen previously (17, 18). Moreover, some isolates may be important for clinical diagnostics or treatment due to their host range. Therefore, the isolation and characterization of Shigella phages further our understanding of bacteriophage biology. In addition, continuing to isolate and study these phages may have implications for monitoring, diagnosing, and controlling shigellosis outbreaks worldwide.
RESULTS
Water is a rich source for isolating Shigella phages.
To test for the presence of Shigella-infecting bacteriophages in the environment and to increase open-ended experimentation in graduate education, we developed a short, 2-week module in a microbiology course at Michigan State University (MSU). Students devised hypotheses, targeted specific locations, and then collected material from those locations within the vicinity of MSU (see Materials and Methods). For all samples, native bacteria were removed by filtration, and then each filtrate was used to inoculate various strains of Enterobacteria in either solid or liquid media. The types of bacteria used in these initial tests were nonpathogenic laboratory strains, including Shigella flexneri (serotypes Y, 2a, and 5a), Escherichia coli (types B and K-12), and Salmonella enterica serovar Typhimurium (strain 14028s). At least one phage was isolated on each type of bacteria, producing a total of 18 distinct isolates, all of which originated from river samples (Table 1). An overwhelming majority of these were isolated on S. flexneri (n = 14), with comparatively few from E. coli (n = 2) or S. enterica (n = 2). This was surprising, as many previously described Shigella phages were induced from clinical isolates or found in untreated wastewater samples (see, e.g., references 19 to 23), with relatively few from the environment (14–16). Conversely, considerably more E. coli and Salmonella phages have been isolated, and from a variety of sources.
TABLE 1.
Sources and host ranges of the environmental isolatesa
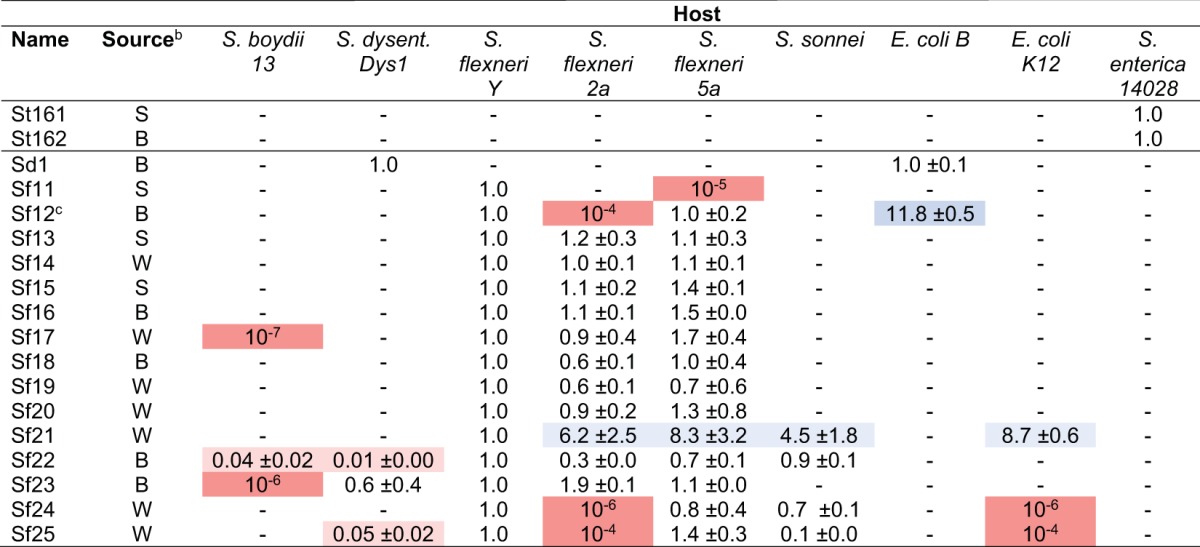
Efficiencies of plating (mean ± standard deviation) are shown. Colors indicate that the plating efficiency on the strain was significantly higher (blue) or lower (red) than that on S. flexneri Y for the majority of samples. S. dysenteriae Dys1 was used for normalization for Sd1, and S. enterica 14028s was used for normalization for phages St161 and St162.
b Sediment (S) was collected from the Red Cedar River downstream from a wastewater treatment plant, biofilm (B) was collected from a buoy in the middle of the Grand River, and water (W) was collected from the Red Cedar River.
c Temperature sensitive (forms plaques only below 37°C).
Six Shigella phages infect more than one species of Shigella.
After their initial isolation, all 18 phages (the total of 16 Shigella phages and 2 Salmonella phages) were purified and grown to high titer outside the classroom setting for subsequent, more complex analyses. The first of these was an analysis of host range that included pathogenic hosts. To initially determine host range qualitatively, phage was spotted directly onto agar plates seeded with test bacteria. The library of test strains included the initial strains used for screening plus S. boydii, S. dysenteriae, S. sonnei, and Citrobacter freundii. After overnight incubation at 37°C, a clearing of the spot resulted in a positive score, whereas a confluent lawn of growth resulted in a negative score. Phages were then named and numbered according to their host; e.g., “Sf” phages infected S. flexneri (starting at Sf11 due to previous characterization of SfX), “St” phages infected S. enterica serovar Typhimurium (starting at St161 due to previous characterization of ST160), etc.
Table 1 illustrates that 16 out of the 18 total isolates infected one or more species of Shigella. Of these, eight isolates infected exclusively S. flexneri. Five isolates could infect both Shigella and E. coli, and four isolates infected more than one species of Shigella but not E. coli. From the latter group, a total of six phages were found to infect more than one species of Shigella. We anticipated some overlap between Shigella and E. coli phages, since the host genera are phylogenetically the same (24–26). The remaining two non-Shigella phages infected exclusively S. enterica. Surprisingly, no phage could infect C. freundii, despite its documented prevalence in the environment and close relation to the other strains tested (26). These results indicate that a majority of the isolates are indeed Shigella phages, with a relatively even distribution between specialist and generalist lifestyles.
To determine whether Shigella spp. were present in the environment, 16S rRNA sequencing was performed for portions of the water and sediment collected from the Red Cedar River plus the biofilm from the Grand River. Eight distinct morphotypes were isolated from the Red Cedar River: six of these shared 16S rRNA sequence identity with Bacillus spp. and the remaining two with Pseudomonas spp. From the biofilm, genera most closely resembled Leclercia, Aeromonas, Pseudomonas, Enterobacter, Serratia, Citrobacter, and Pantoea. The biofilm also contained Plesiomonas shigelloides, which is a species of enterobacteria that shares a similar lipopolysaccharide O antigen with Shigella sonnei but is otherwise physiologically distinct (27). Despite purposefully screening for colonies with Shigella phenotypes, no Shigella spp. were identified. However, based on the host range specificity of most isolates, an alternative environmental host seems unlikely for these phages.
In 2017, no outbreaks of shigellosis were reported near MSU. Environmental samples were collected and screened as before, with students choosing locations and materials for screening. These samples included water and sediment from the Red Cedar River, which were collected both at and near the 2016 locations. The incorporation of C. freundii in the initial screen gave us eight bacterial strains to test, producing a total of 120 samples that were analyzed. Despite an additional 22 samples, only two phages were isolated: one on S. flexneri and one on C. freundii. These results suggest that during an outbreak, the prevalence of Shigella phages in the environment may be much greater than previously realized, particularly for aquatic environments. In the absence of an outbreak, these phages may still be present in surface water, albeit at much lower quantities.
Genome analyses suggest six distinct species of phage.
To characterize the original 2016 phages at a genetic level, we extracted and sequenced whole genomes from purified isolates. Once assembled, most genomes fell into one of three size groups: 40.0 to 50.0 kbp (n = 5), 85.0 to 95.0 kbp (n = 7), or 160.0 to 170.0 kbp (n = 6). The accession numbers for all genomes and their basic characteristics are listed in Table 2, with representative genome maps in Fig. 1. To investigate overall sequence similarities between these phages, dot plots were generated by comparing all genomes to each other. By this analysis, 17 of 18 isolates formed five distinct clusters, of which the Shigella phages formed four (Fig. 2A). These five clusters were further supported by phylogenetic analysis (Fig. 2B). Most phages belonged to the following established groups, as defined in reference 18: (i) FelixO1virus, (ii) T1virus, (iii) SETP3virus, (iv) T4virus, and (v) RB49virus. The remaining isolate, Shigella phage Sf11, appeared to be distinct from these groups. Sf11 instead belonged to the relatively small E1-like cluster, which is comprised of only four other phages. Two groups contained multiple isolates, with Sf13 through Sf19 belonging to group I and Sf21 through Sf25 belonging to group IV.
TABLE 2.
Genome properties of environmental isolates
| Isolate | Length (kbp) | No. of ORFs | No. of tRNAs | % GC | Accession no. | Taxonomic family | Taxonomic genusa | % ANI to reference genomeb |
|---|---|---|---|---|---|---|---|---|
| St161 | 42.7 | 62 | 0 | 51.2 | MF158036 | Siphoviridae | SETP3virus | 64.4 |
| St162 | 42.7 | 62 | 0 | 51.2 | MF158037 | Siphoviridae | SETP3virus | 62.3 |
| Sf11 | 46.5 | 80 | 2 | 46.0 | MF158038 | Siphoviridae | E1virus | 50.6 |
| Sf12 | 47.6 | 75 | 1 | 44.3 | MF158039 | Siphoviridae | T1virus | 48.4 |
| Sd1 | 48.2 | 73 | 2 | 44.5 | MF158042 | Siphoviridae | T1virus | 42.7 |
| Sf13 | 87.6 | 133 | 26 | 38.9 | MF158040 | Myoviridae | FelixO1virus | 86.5 |
| Sf14 | 87.6 | 131 | 26 | 39.0 | MF327003 | Myoviridae | FelixO1virus | 84.6 |
| Sf15 | 88.5 | 134 | 26 | 39.0 | MF158041 | Myoviridae | FelixO1virus | 84.6 |
| Sf16 | 88.6 | 134 | 26 | 39.0 | MF158043 | Myoviridae | FelixO1virus | 83.7 |
| Sf17 | 90.1 | 138 | 26 | 39.0 | MF327004 | Myoviridae | FelixO1virus | 84.6 |
| Sf18 | 90.3 | 137 | 26 | 39.0 | MF158044 | Myoviridae | FelixO1virus | 85.6 |
| Sf19 | 90.4 | 137 | 26 | 39.0 | MF327005 | Myoviridae | FelixO1virus | 83.7 |
| Sf20 | 164.0 | 272 | 0 | 40.6 | MF327006 | Myoviridae | RB49virus | 96.0 |
| Sf21 | 166.0 | 266 | 11 | 35.5 | MF327007 | Myoviridae | T4virus | 82.7 |
| Sf22 | 166.3 | 267 | 11 | 35.5 | MF158045 | Myoviridae | T4virus | 85.4 |
| Sf23 | 167.7 | 271 | 10 | 35.4 | MF158046 | Myoviridae | T4virus | 81.7 |
| Sf24 | 168.1 | 271 | 10 | 35.3 | MF327008 | Myoviridae | T4virus | 84.4 |
| Sf25 | 168.6 | 272 | 9 | 35.4 | MF327009 | Myoviridae | T4virus | 85.4 |
FIG 1.

Genome maps of representative isolates. Simplified representations of the Shigella phage genomes, using representative isolates from the groups defined in Fig. 2, are shown. The ruler is in kilobase pairs.
FIG 2.

Genomic and phylogenetic analysis of environmental isolates. (A) Dot plot of all phage genomes compared with each other, including reference genomes of related phages shown in red. Species are separated by blue (long) lines, with individual isolates separated by red (short) lines. (B) Bayesian inference phylogenetic tree of environmental isolates. The scale bar represents 0.4 nucleotide substitutions per site.
The genomes of Shigella phages Sf11, Sf12, and Sd1 were of similar size, ranging from 46.5 kbp to 48.2 kbp. The Sf12 and Sd1 genomes were generally organized with a similar gene order (Fig. 1, top panel) and were most closely related to the T1virus Escherichia phage phiEB49 (28). Morphologically, they also exhibited T1-like tail flexibility (Fig. 3, blue). Conversely, Sf11 appeared to be unique, as it did not group with any other phage isolated here in terms of either synteny or evolutionary history (Fig. 1, second panel, and 2B). Sf11 was also able to infect only two of the three S. flexneri serotypes tested and was the only S. flexneri-infecting phage to have this limited host range, suggesting additional differences. Genetically, the closest relative to Sf11 is IME207, which is a member of the diverse E1virus group (S. R. Casjens, personal communication). However, it is still significantly different from this phage, sharing only 51% average nucleotide identity (ANI), and may represent a new “subcluster E” within the E1virus cluster of phages.
FIG 3.

Morphology of all phage isolates. Representative micrographs as viewed by negative-stain TEM are shown. The scale bar is 50 nm. Colored bars below the micrographs correspond to phylogenetic groups.
Phages Sf20 through Sf25 closely resembled T4viruses in terms of both gene order and morphology, encoding 268 to 273 open reading frames (ORFs) and 9 to 11 tRNAs (Fig. 1, bottom panel). These “Sf21-like” phages were slightly more diverse, with 88 to 91% ANI between isolates. The exceptions in the Sf21 phage group were the isolates Sf24 and Sf25, which shared 96% sequence identity. However, the percent similarity between these genomes and the T4 genome was 85% or less, excluding the possibility that they are truly T4. These phages also exhibited a diversity of host ranges, infecting either multiple species of Shigella or two Shigella species plus E. coli K-12. The genomes of phages Sf21 through Sf25 had an average GC content of 35.4%, which is typical for the T4-like viruses. Conversely, phage Sf20 followed a gene order similar to those of Sf21 through Sf25, but it lacked any tRNAs and had a higher GC content of 40.6%. These qualities are also shared by RB49 viruses, which form a subcluster within the T4virus genus (29). When examined by negative-stain transmission electron microscopy (TEM), Sf20 through Sf25 exhibited T4-like morphology, with long tail fibers and prolate capsids (Fig. 3, yellow and green).
A majority of the isolates, Sf13 through Sf19, were members of the FelixO1virus genus. These all had genomes of 87.6 to 90.4 kbp in size and were closely related but not identical, sharing 90 to 97% average nucleotide identity between isolates (Fig. 1, third panel). Their GC content of approximately 39% corresponds well with subcluster C in the FelixO1virus cluster of phages (29), to which other Moogle viruses belong. Although our phages also share 90 to 94% sequence identity with the Moogle viruses, the host for the latter group is C. freundii, which our phages are unable to infect. When their morphology was determined by negative-stain TEM, the Shigella phages also displayed prototypical FelixO1-like morphology, having icosahedral heads, contractile tails, and short tail fibers (Fig. 3, red).
Uncommon genome and capsid properties of Sf13-like viruses.
Including FelixO1 and Moogle, only 48 out of 2,293 (∼2%) phage genomes in the NCBI RefSeq database fall within the size range of 85.0 to 95.0 kbp. The rarity of this genome size has been noted by several groups, though a biological explanation has been elusive (29–31). Perhaps because of their underrepresentation in databases, no structural analysis for any virus with this size genome has been conducted. As part of their life cycles, viruses package their genomes into capsids, which are composed of coat proteins that form an ordered lattice. Capsids are commonly organized into an icosahedron, with 12 vertices and 20 faces, and are classified according to the number of subunits per face (32). The simplest structure has 60 subunits, with a triangulation number (T) equal to 1. A wide range of capsid structures have been resolved, with T=7 and T=13 organizations being common among phages (18, 33). Phage capsids with T=7 geometry typically package double-stranded DNA (dsDNA) genomes of 40.0 to 50.0 kbp, whereas T=13 capsids typically package segmented dsRNA genomes or dsDNA genomes of ∼120 kbp. With dsDNA genomes of 85.0 to 91.0 kbp, our newly isolated phages did not obviously fit into either category. Thus, we hypothesized that the capsids of these phages may have similarly uncommon structures.
To determine the sizes and basic properties of these phage capsids, we performed cryo-electron microscopy and three-dimensional image reconstructions for all seven Moogle/Sf13-like isolates. As shown in a representative micrograph and three-dimensional reconstruction (Fig. 4A and B, EMD-8869), the pattern of the asymmetric unit is characteristic of a triangulation number equal to 9. Capsids with T=9 symmetry have been described for only two other viruses, the bacteriophages N4 and Basilisk (17, 34), which have genome sizes of 70.2 and 82.0 kbp, respectively. A second unusual characteristic of these new isolates' capsids was the presence of a decoration-like protein (arrow in Fig. 4C), giving the capsid a maximum diameter of 786 Å. Decoration proteins have been characterized for some bacteriophages, and most of these proteins contain at least one immunoglobulin (Ig)-like domain (34, 35), with the exception of the Dec protein of phage L, which is formed primarily by β-sheets (36). Using a tBLASTn sequence alignment against all known decoration proteins, phages Sf13 through Sf19 were found to contain gene products with significant similarity to T5 pb10 (35). This protein is composed of an α-helical capsid-binding domain at the N terminus and a single C-terminal Ig-like domain. Rather than a single Ig-like domain, the decoration proteins in phages Sf13 through Sf19 contain two Ig-like domains. These are similar to both the pb10 Ig-like domain and each other, which could represent a duplication of the Ig-like domain-coding region.
FIG 4.

Cryo-electron microscopy and three-dimensional image reconstruction of a T=9 Shigella phage. (A) Representative micrograph of vitrified Sf14 particles. The arrow indicates the tail fibers visible on a representative virion. (B) Cryo-EM reconstruction of the capsid at ∼15-Å resolution, colored according to radial distance. (C) Enlarged view of the region in panel B outlined by the white box, illustrating the two types of hexamers. The black arrow indicates the presence of a decoration protein in one type of hexamer.
Structurally, the location of this decoration protein on the capsid surface is also unlike those of previously described proteins. The decoration protein of N4 binds along the edges of each hexamer (34), whereas those of T4, RB49, and T5 bind at the center of each hexamer (35, 37, 38). The phage L decoration protein is more complex, binding preferentially to the quasi-3-fold but not the true 3-fold sites (36, 39). In phages Sf13 through Sf19, the decoration-like protein appears to be present on only one of the hexamers. Thus, while the structure of this decoration protein is most similar to that of pb10 of T5, its behavior is more similar to that of Dec of phage L. The duplication of the Ig-like region in the gene could have contributed to this protein's ability to bind preferentially to specific locations on the capsid.
DISCUSSION
Comprehensive isolation and characterization of Shigella phages.
In 2016, we isolated 18 bacteriophages from rivers around MSU. By testing multiple types of samples on a library of bacterial strains, we captured a significantly larger portion of the local phage community than if we had used a single host. One sample in particular yielded the most phages in terms of both overall abundance and diversity: the biofilm from a water buoy in the Grand River. Seven phages (38% of the total isolates) were isolated from this sample. These phages also had vastly different host ranges, genomes, and morphologies. Since mixed-species biofilms have been shown to “trap” virus particles (40), phages likely accumulated in the biofilm before sampling. This could explain the greater diversity from the biofilm than from the other samples, as phages suspended in river water are likely transient, and those from sediment could be easily buried by additional deposits.
Previously, information for only 35 Shigella phages had been deposited into public databases. The 16 Shigella phages described here represent a significant increase in the repertoire of known phages with this host genus. To advance our knowledge of this underrepresented group of phages, we employed multiple analyses to investigate fundamental aspects of their biology. Our results indicate that they are diverse in terms of host range, genetics, structure, and evolutionary history. Many well-described bacteriophages infect E. coli or Salmonella, and these have been crucial to laying the foundation of phage biology. However, some gaps still exist. For example, while T=9 capsids and genomes of 85.0 to 95.0 kbp are uncommon in E. coli and Salmonella phages, they may be more common in Shigella phages. Additional isolation and characterization of similar phages may indicate that some qualities are more common than previously thought.
Phage therapy, outbreaks, and local microbial populations.
All the phages infecting enterobacteria in this study were isolated from rivers, one of which receives effluent from a wastewater treatment plant in East Lansing, MI. Sampling for this study occurred in the first 2 weeks of October 2016, when shigellosis cases were unusually high for the area (3). The phage community during that time appeared to differ from previous compositions, and results from the 2017 survey suggest that they differ from current compositions as well.
In 2013, a metagenomic study of phages in wastewater influent and effluent was conducted using water from an East Lansing, MI, treatment plant (41). While the hosts of these phages could not be determined, that study indicated that most phages were tailed, with the long, non-contractile-tailed phages of the family Siphoviridae representing ∼43% of the total phage population. The long, contractile-tailed Myoviridae and the short-tailed Podoviridae were found in similar proportions of ∼30% and ∼23%, respectively. Here, most of our isolates belong to Myoviridae (72%), with comparatively few Siphoviridae (28%). In addition, we did not isolate any members of the family Podoviridae, despite our methods being optimized for the podoviruses P22, CUS-3, and Sf6, which infect Salmonella enterica, E. coli, and Shigella flexneri, respectively (42, 43). The greater proportion of myoviruses and siphoviruses found here may be due to temporal shifts in microbial populations (44, 45) across years or seasons. However, wastewater may be altered during an outbreak of bacillary dysentery, which could impact the composition of environmental phage populations. Additional studies investigating both microbial ecology and epidemiology over time would be necessary to investigate this relationship further.
The 2013 study also indicates that bacteriophages can still be detected after hypochlorite treatment in the East Lansing wastewater treatment plant (41), though their hosts are retained or destroyed in the treatment facility. Phages have been used as indicators for the presence of pathogenic bacteria and viruses in untreated or treated sewage (46), as well as surface water and groundwater (47). However, the prevalence and persistence of pathogen-associated bacteriophages in the absence of contamination has not been well explored. Here, we investigated the prevalence of pathogen-associated bacteriophages in the absence of contamination but in the presence of local outbreaks. Additional sampling in 2017 revealed a much lower abundance of Shigella-infecting phages, with one Shigella phage isolated from 120 samples compared with the 16 Shigella phages isolated from 98 samples in 2016. These differences in environmental phage populations could be linked to the prevalence of bacterial infections in the community.
While some of our isolates may be clinically relevant, understanding the link between human health and environmental phage populations is of critical importance if phage therapy replaces or supplements conventional antibiotics (48). Wastewater treatment generally removes most antibiotics, but their low level of persistence in wastewater effluent and reclaimed water (49, 50) may have contributed to the current antibiotic resistance problem we face today. If some bacteriophages used for phage therapy survive wastewater treatment and persist in the environment, we could encounter similar problems in the future. Thus, ensuring that we understand the full impact of releasing phages into the environment, whether intentionally or not, is a necessary part of developing and implementing treatments that use them.
Mutual benefit of phage hunting.
Providing students with research experience has been a long-standing goal in colleges and universities. The Science Education Alliance-Phage Hunters Advancing Genomics and Evolutionary Science (SEA-PHAGES) program has been at the forefront of student phage hunting, and this type of student-driven inquiry has been immensely successful (51, 52). The student response to the 2-week exercise described here was measured qualitatively by student evaluations. From these, 80% of the students found the phage hunting both helpful and meaningful, and 10% found the exercise moderately useful. Given the small sample sizes between 2016 and 2017, we will continue to monitor student evaluations in future years. Overall, however, the results of our study emphasize the mutual benefit of this type of research experience, not only for students but for researchers as well. The creativity and resourcefulness of students immensely benefitted our search for Shigella phages. While not all samples produced phage, several groups collected material that would have been difficult for us, or that we would not have thought to sample or visit on our own. Besides engaging students in science and research, student-based inquiry can bring new perspectives to established researchers and fields, facilitating significant advances across disciplines.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The following strains of bacteria used for host range tests and phage amplification have been described previously: S. flexneri serotype Y (PE577 [53]), S. flexneri serotype 2a (CSF100), S. flexneri serotype 5a (SD100), Escherichia coli K-12 (EV36 [54, 55]) and B (REL606 [56, 57]), and Salmonella enterica serovar Typhimurium (DB7136 [58]). S. flexneri serotypes 2a (CSF100) and 5a (SD100) are nonpathogenic derivatives of 2457T (59) and M90T Sm (60, 61), respectively. SD100 was cured of the virulence plasmid pWR100 by serial passaging at 42°C on Congo red agar. White colonies appeared after several passages, suggesting the functional loss of the type III secretion system (T3SS) encoded by pWR100 (62). The loss of T3SS function was confirmed by PCR, using primers specific for the virB and icsA/virG genes of the plasmid. CSF100 also forms white colonies on Congo red agar. S. boydii serotype 13 (ATCC 12032), S. dysenteriae serotype 1 (strain CDC-3823/69), and S. sonnei (strain 16372) were provided by Shannon Manning at the Shiga Toxin-Producing Escherichia coli (STEC) Center at Michigan State University.
Phage isolation and purification.
Since “phage hunting” has become a powerful teaching tool (51), we challenged Michigan State University graduate students to isolate enteric phages from local environmental sources. This was done during a short, 2-week module in a microbiology course, with 21 students enrolled in 2016 and 18 students enrolled in 2017. Students devised hypotheses, targeted specific locations, and then collected material from those locations within the vicinity of Michigan State University. The materials that students chose to collect in 2016 included soil from a horse farm (n = 1), soil from a field frequented by waterfowl (n = 1), river sediment downstream from a wastewater treatment plant (n = 1), river water (n = 2 for the same location), biofilm from a river buoy (n = 1), and a swab from an apple skin (n = 1). In 2017, samples included river or pond water (n = 3), river or lake sediment (n = 2), water left at the bottom of hand dryers in a university bathroom (n = 1), and soil from MSU gardens (n = 2). Approximately 15 to 25 g of soil or river sediment or 35 to 50 ml of river water was collected in sterile conical tubes. Luria broth (LB) was added to solid media to a final volume of 50 ml and then vortexed. Samples were then centrifuged for 10 min at 6 000 × g, and the supernatant was collected. If necessary, liquid samples were also spun to remove visible debris. For the apple skin, a sterile cotton swab was dipped in LB, used to swab the apple, dipped and swirled into 5 ml LB, and then processed as a liquid sample. Supernatant or liquid samples were then filtered through a 0.45-μm filter to remove bacteria and any particulates, and 250-μl aliquots of filtrate were added to LB plates with 0.3% top agar overlays seeded with test strains of bacteria. Plates were incubated overnight at 37°C and screened for plaque formation. In addition, 5-ml liquid enrichments were performed overnight. For each test strain, a 250-μl aliquot of sample was added to 5 ml LB seeded with a 1:100 dilution of an overnight bacterial culture and then incubated at 37°C. If the culture cleared, 250 μl of this lysate was then applied to a plate seeded with the same strain of bacteria and screened for plaque formation.
After overnight incubation, single plaques were picked and incubated with their respective permissive host in liquid LB until the culture cleared or for a maximum of 6 h. The culture was centrifuged for 20 min at 6,000 × g to remove cellular debris, and the supernatant was subjected to an additional round of centrifugation for 90 min at 26,000 × g to pellet the phage. Phage dilution buffer (10 mM Tris, 10 mM MgCl2) was then added, and the pellet was nutated at 4°C overnight. A final spin of 10 min at 6 000 × g removed any remaining cell debris, and the supernatant was retained. This high-titer phage suspension was used as a stock for further characterization.
To test for the presence of Shigella, an unfiltered sample was retained for bacterial isolation and subsequent 16S rRNA sequencing. Biofilm from the buoy, plus water and sediment from the Red Cedar River, was examined. To isolate single colonies, the sample was diluted into LB, and 100 μl of a 10−5 dilution was spread onto LB-Lennox or MacConkey agar plates. The plates were incubated at 37°C for 48 h, and colonies with diverse morphologies were chosen for further analysis. These were streaked with a sterile loop onto LB agar plates and incubated for at least 16 h, or for up to 3 days for small-colony-forming species. 16S rRNA sequences were then determined by PCR and Sanger sequencing, using the primers 5′-AGAGTTTGATCMTGGCTCAG-3′ (16S_27F) and 5′-ACGGCTACCTTGTTACGACTT-3′ (16S_1492R) based on a previous report (63).
Host range assays.
Initial host range assays were performed by dipping a sterile toothpick into a high-titer phage stock and stabbing the toothpick into LB agar plates overlaid with test bacteria. Plates were incubated at 37°C overnight. A clearing around the stab was indicative of a positive growth score, and quantitative plaque assays were conducted on these strains. Efficiencies of plating are reported with standard deviation for three replicate experiments.
Genomic methods and phylogenetic analysis.
Genomic DNA was extracted from 1 × 1010 to 1 × 1011 phage particles using phenol-chloroform (64) and sequenced at the Michigan State University Research Technology Support Facility (RTSF). The RTSF Genomics Core prepared genomes for sequencing using the Illumina TruSeq Nano DNA library kit on a Sciclone workstation (PerkinElmer) following the manufacturer's protocols. Completed libraries were quality controlled and quantified using a combination of Qubit dsDNA HS (Thermo Fisher) and Caliper LabChipGX HS DNA (PerkinElmer) assays. All libraries were pooled in equimolar quantities and then quantified using an Illumina library quantification kit (Kapa Biosystems). The pool was loaded on an Illumina MiSeq standard v2 flow cell and sequenced in a 2- by 250-bp paired-end format using a v2 500 cycle reagent cartridge. Bases were called by Illumina Real Time Analysis v1.18.54, the output of which was demultiplexed and converted to FastQ format with Illumina Bcl2fastq v2.18.0.
Raw reads were trimmed and assembled into contigs using the A5 Pipeline (65) (version 08-25-2016) and tested for quality using FastQC (66). Assembly coverages ranged from 925× to 2,819×, with Sf21 having the lowest coverage and St162 having the highest. The exception was Sf25, with a coverage of 504×, and this genome contained some regions with ambiguous sequence. To resolve this, additional PCR and Sanger sequencing confirmed the sequences of these regions. Genome termini were oriented according to reference sequences where possible. Open reading frames were identified using GeneMark.hmm S (67) and annotated using blast2GO (68). tRNAs were identified using tRNAscan-SE (69). Genome maps were generated by DNA Master (70) (version 5.23.1, 12 June 2017), and dot plot images were constructed using Gepard version 1.40 (71).
To build phylogenetic trees, all genomes were aligned to each other and to the reference sequences of related phages using ClustalW 2.0 (72). From this alignment, a Bayesian inference tree was constructed using MrBayes (73) (version 3.2.6) under a mixed model with haploid genome and gamma variation settings. Two replicates of tree construction were run concurrently until convergence. Trees were viewed using FigTree (74) (version 1.4.3).
Negative staining and cryo-electron microscopy.
General phage morphology was first determined by negative-stain transmission electron microscopy (TEM). Approximately 108 to 109 total purified phage particles were applied to a continuous carbon grid (Ted Pella, Inc.), which had been plasma cleaned for 20 s in a Fischione model 1020 plasma cleaner. A few drops of 1 to 2% uranyl acetate stain were subsequently added to the grid, with the excess removed by blotting.
To prepare samples for cryo-TEM, small (∼5-μl) aliquots of purified virus particles were vitrified using established procedures (75). Samples were applied to plasma-cleaned holey Quantifoil grids (R2/2) as described above and then vitrified using a pneumatic plunge freezing device into liquid ethane (76). Samples were transferred to a precooled Gatan 914 specimen holder, which maintained the specimen at liquid nitrogen temperature.
All virus particles were imaged in a JEOL JEM-2200FS TEM at 200 keV controlled by SerialEM (77) (version 3.5.0_beta) with the use of an in-column Omega energy filter with a slit width of 35 eV. Samples were viewed at a nominal magnification of ×25,000 (2.07 Å/pixel). Micrographs were recorded using a Direct Electron DE-20 camera (Direct Electron, LP, San Diego, CA, USA) cooled to −38°C, at a capture rate of 25 frames per second. Movie correction was performed on whole frames using the Direct Electron software package, version 2.7.1 (78).
Icosahedral cryo-reconstructions of phage virions.
Using AUTO3DEM (79), version 4.05.1, a subset of 7 to 276 particle images was used as input to the random-model computation procedure to generate an initial three-dimensional (3D) density map at ∼25-Å resolution (80). The initial maps were then used to initiate determination and refinement of particle orientations and origins for the complete set of images. For data sets that had fewer than 150 particles, all particles were used for the random-model computation. Particle counts ranged 7 to 817 for each phage type. Phases of the particle structure factor data were corrected to compensate for the effects caused by the microscope contrast-transfer function. The Fourier shell correlation criterion (FSC0.5) was used to estimate the resolution of each reconstruction using the “gold standard” approach, and resolution of maps ranged from ∼15 Å to 44 Å. Graphical representations were generated using the UCSF Chimera visualization software package (81).
Accession number(s).
The sequences determined in this study were submitted to GenBank under the accession numbers given in Table 2. A representative three-dimensional reconstruction was deposited in the EMDB (accession number EMD-8869).
ACKNOWLEDGMENTS
This work has been funded by the AAAS Marion Milligan Mason Award for Women in the Chemical Sciences to K.N.P. and is based in part upon work supported by the National Science Foundation under Cooperative Agreement no. DBI-0939454 to S.M.D.
The funders had no role in study design, data collection and interpretation, or the decision to submit this work for publication.
We thank the 2016 and 2017 classes of Integrative Microbial Biology for sampling the environment and performing initial phage screens. The 2016 class of Integrative Microbial Biology included Paulo Carneiro, Taylor Chicoine, Dan Clairborne, Shelby Dechow, Lucas Demey, Russell Fling, Josh Franklin, Ari Grode, Alicia Layer, Cole McCutcheon, Joel Rankin, Heather Selheimer, Yike Shen, Darian Smercina, Kameron Sugino, Matthew Swiatnicki, John Williams, Soo Yoon, and Ryan Yost. The 2017 class of Integrative Microbial Biology included Alex Aaring, Osama Alian, Ari Bintarti, Xavier Brandon, Kayla Conner, Kody Duhl, Nick Ether, Rachel Greenberg, Emily Greeson, Zoe Hansen, Keren Herrera, Brian Hsueh, Kristin Jacob, Jonathan Kaletka, Justin Lee, Reid Longley, Olivia Walser, and Kun Wang. We thank Madeline Hilton for subsequent sampling and phage isolation from the Red Cedar River. We are also grateful to Eric Lyons (University of Arizona) for helpful discussions and Sherwood Casjens (University of Utah) for critical reading of the manuscript.
S.M.D. purified and amplified phages and determined host ranges. J.A.D. extracted and sequenced genomes, which were assembled and annotated by S.M.D. J.R.S. and S.M.D. collected TEM images. J.R.S. and W.F.D. collected cryo-EM data and performed reconstructions. S.M.D. and K.N.P. designed experiments and wrote the manuscript.
We declare no competing interests.
REFERENCES
- 1.Lee LA, Ostroff SM, McGee HB, Johnson DR, Downes FP, Cameron DN, Bean NH, Griffin PM. 1991. An outbreak of shigellosis at an outdoor music festival. Am J Epidemiol 133:608–615. doi: 10.1093/oxfordjournals.aje.a115933. [DOI] [PubMed] [Google Scholar]
- 2.Michigan Department of Health and Human Services. 2016. 2016 Shigellosis outbreak—Genessee and Saginaw Counties. http://www.michigan.gov/mdhhs/0,5885,7-339-71550_5104_53072-397310-,00.html.
- 3.Ingham County Health Department. 17 October 2016. Increase in local cases of Shigella prompts health department to remind Ingham County residents to wash hands. http://www.hd.ingham.org/About/ArticlesPublications/tabid/3378/ctl/ArticleView/mid/4286/articleId/6474/Increase-in-local-cases-of-Shigella-prompts-health-department-to-remind-Ingham-County-residents-to-wash-hands.aspx.
- 4.Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, Levine MM. 1999. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Organ 77:651–666. [PMC free article] [PubMed] [Google Scholar]
- 5.Connor TR, Barker CR, Baker KS, Weill FX, Talukder KA, Smith AM, Baker S, Gouali M, Pham Thanh D, Jahan Azmi I, Dias da Silveira W, Semmler T, Wieler LH, Jenkins C, Cravioto A, Faruque SM, Parkhill J, Wook Kim D, Keddy KH, Thomson NR. 2015. Species-wide whole genome sequencing reveals historical global spread and recent local persistence in Shigella flexneri. Elife 4:e07335. doi: 10.7554/eLife.07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thompson CN, Duy PT, Baker S. 2015. The rising dominance of Shigella sonnei: an intercontinental shift in the etiology of bacillary dysentery. PLoS Negl Trop Dis 9:e0003708. doi: 10.1371/journal.pntd.0003708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DuPont HL, Levine MM, Hornick RB, Formal SB. 1989. Inoculum size in shigellosis and implications for expected mode of transmission. J Infect Dis 159:1126–1128. doi: 10.1093/infdis/159.6.1126. [DOI] [PubMed] [Google Scholar]
- 8.Gu B, Cao Y, Pan S, Zhuang L, Yu R, Peng Z, Qian H, Wei Y, Zhao L, Liu G, Tong M. 2012. Comparison of the prevalence and changing resistance to nalidixic acid and ciprofloxacin of Shigella between Europe-America and Asia-Africa from 1998 to 2009. Int J Antimicrob Agents 40:9–17. doi: 10.1016/j.ijantimicag.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 9.World Health Organization. 2017. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 10.Abedon ST, Kuhl SJ, Blasdel BG, Kutter EM. 2011. Phage treatment of human infections. Bacteriophage 1:66–85. doi: 10.4161/bact.1.2.15845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.d'Herelle F. 1917. An invisible antagonist microbe of dysentery bacillus. C R Hebd Seances Acad Sci 165:373–375. [Google Scholar]
- 12.Note by M. F. d'Herelle. 2011. On an invisible microbe antagonistic to dysentery bacilli. Note by M. F. d'Herelle, presented by M. Roux. Comptes Rendus Academie des Sciences 1917; 165:373–5. Bacteriophage 1:3–5. doi: 10.4161/bact.1.1.14941. [DOI] [Google Scholar]
- 13.d'Herelle F. 1931. Bacteriophage as a treatment in acute medical and surgical infections. Bull N Y Acad Med 7:329–348. [PMC free article] [PubMed] [Google Scholar]
- 14.Jun JW, Kim JH, Shin SP, Han JE, Chai JY, Park SC. 2013. Characterization and complete genome sequence of the Shigella bacteriophage pSf-1. Res Microbiol 164:979–986. doi: 10.1016/j.resmic.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Jun JW, Yun SK, Kim HJ, Chai JY, Park SC. 2014. Characterization and complete genome sequence of a novel N4-like bacteriophage, pSb-1 infecting Shigella boydii. Res Microbiol 165:671–678. doi: 10.1016/j.resmic.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 16.Jun JW, Giri SS, Kim HJ, Yun SK, Chi C, Chai JY, Lee BC, Park SC. 2016. Bacteriophage application to control the contaminated water with Shigella. Sci Rep 6:22636. doi: 10.1038/srep22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grose JH, Belnap DM, Jensen JD, Mathis AD, Prince JT, Merrill BD, Burnett SH, Breakwell DP. 2014. The genomes, proteomes, and structures of three novel phages that infect the Bacillus cereus group and carry putative virulence factors. J Virol 88:11846–11860. doi: 10.1128/JVI.01364-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Effantin G, Boulanger P, Neumann E, Letellier L, Conway JF. 2006. Bacteriophage T5 structure reveals similarities with HK97 and T4 suggesting evolutionary relationships. J Mol Biol 361:993–1002. doi: 10.1016/j.jmb.2006.06.081. [DOI] [PubMed] [Google Scholar]
- 19.Huan PT, Whittle BL, Bastin DA, Lindberg AA, Verma NK. 1997. Shigella flexneri type-specific antigen V: cloning, sequencing and characterization of the glucosyl transferase gene of temperate bacteriophage SfV. Gene 195:207–216. doi: 10.1016/S0378-1119(97)00144-3. [DOI] [PubMed] [Google Scholar]
- 20.Mavris M, Manning PA, Morona R. 1997. Mechanism of bacteriophage SfII-mediated serotype conversion in Shigella flexneri. Mol Microbiol 26:939–950. doi: 10.1046/j.1365-2958.1997.6301997.x. [DOI] [PubMed] [Google Scholar]
- 21.Sun Q, Lan R, Wang Y, Wang J, Wang Y, Li P, Du P, Xu J. 2013. Isolation and genomic characterization of SfI, a serotype-converting bacteriophage of Shigella flexneri. BMC Microbiol 13:39. doi: 10.1186/1471-2180-13-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jakhetia R, Marri A, Stahle J, Widmalm G, Verma NK. 2014. Serotype-conversion in Shigella flexneri: identification of a novel bacteriophage, Sf101, from a serotype 7a strain. BMC Genomics 15:742. doi: 10.1186/1471-2164-15-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schofield DA, Wray DJ, Molineux IJ. 2015. Isolation and development of bioluminescent reporter phages for bacterial dysentery. Eur J Clin Microbiol Infect Dis 34:395–403. doi: 10.1007/s10096-014-2246-0. [DOI] [PubMed] [Google Scholar]
- 24.Rolland K, Lambert-Zechovsky N, Picard B, Denamur E. 1998. Shigella and enteroinvasive Escherichia coli strains are derived from distinct ancestral strains of E. coli. Microbiology 144:2667–2672. doi: 10.1099/00221287-144-9-2667. [DOI] [PubMed] [Google Scholar]
- 25.Pupo GM, Lan R, Reeves PR. 2000. Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc Natl Acad Sci U S A 97:10567–10572. doi: 10.1073/pnas.180094797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alnajar S, Gupta RS. 2017. Phylogenomics and comparative genomic studies delineate six main clades within the family Enterobacteriaceae and support the reclassification of several polyphyletic members of the family. Infect Genet Evol 54:108–127. doi: 10.1016/j.meegid.2017.06.024. [DOI] [PubMed] [Google Scholar]
- 27.Janda JM, Abbott SL, McIver CJ. 2016. Plesiomonas shigelloides revisited. Clin Microbiol Rev 29:349–374. doi: 10.1128/CMR.00103-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Battaglioli EJ, Baisa GA, Weeks AE, Schroll RA, Hryckowian AJ, Welch RA. 2011. Isolation of generalized transducing bacteriophages for uropathogenic strains of Escherichia coli. Appl Environ Microbiol 77:6630–6635. doi: 10.1128/AEM.05307-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grose JH, Casjens SR. 2014. Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae. Virology 468-470:421–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hatfull GF. 2008. Bacteriophage genomics. Curr Opin Microbiol 11:447–453. doi: 10.1016/j.mib.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deschavanne P, DuBow MS, Regeard C. 2010. The use of genomic signature distance between bacteriophages and their hosts displays evolutionary relationships and phage growth cycle determination. Virol J 7:163. doi: 10.1186/1743-422X-7-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caspar DL, Klug A. 1962. Physical principles in the construction of regular viruses. Cold Spring Harbor Symp Quant Biol 27:1–24. doi: 10.1101/SQB.1962.027.001.005. [DOI] [PubMed] [Google Scholar]
- 33.Carrillo-Tripp M, Shepherd CM, Borelli IA, Venkataraman S, Lander G, Natarajan P, Johnson JE, Brooks CL III, Reddy VS. 2009. VIPERdb2: an enhanced and web API enabled relational database for structural virology. Nucleic Acids Res 37:D436–D442. doi: 10.1093/nar/gkn840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi KH, McPartland J, Kaganman I, Bowman VD, Rothman-Denes LB, Rossmann MG. 2008. Insight into DNA and protein transport in double-stranded DNA viruses: the structure of bacteriophage N4. J Mol Biol 378:726–736. doi: 10.1016/j.jmb.2008.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vernhes E, Renouard M, Gilquin B, Cuniasse P, Durand D, England P, Hoos S, Huet A, Conway JF, Glukhov A, Ksenzenko V, Jacquet E, Nhiri N, Zinn-Justin S, Boulanger P. 2017. High affinity anchoring of the decoration protein pb10 onto the bacteriophage T5 capsid. Sci Rep 7:41662. doi: 10.1038/srep41662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parent KN, Deedas CT, Egelman EH, Casjens SR, Baker TS, Teschke CM. 2012. Stepwise molecular display utilizing icosahedral and helical complexes of phage coat and decoration proteins in the development of robust nanoscale display vehicles. Biomaterials 33:5628–5637. doi: 10.1016/j.biomaterials.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sathaliyawala T, Islam MZ, Li Q, Fokine A, Rossmann MG, Rao VB. 2010. Functional analysis of the highly antigenic outer capsid protein, Hoc, a virus decoration protein from T4-like bacteriophages. Mol Microbiol 77:444–455. doi: 10.1111/j.1365-2958.2010.07219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fokine A, Islam MZ, Zhang Z, Bowman VD, Rao VB, Rossmann MG. 2011. Structure of the three N-terminal immunoglobulin domains of the highly immunogenic outer capsid protein from a T4-like bacteriophage. J Virol 85:8141–8148. doi: 10.1128/JVI.00847-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang L, Gilcrease EB, Casjens SR, Johnson JE. 2006. Highly discriminatory binding of capsid-cementing proteins in bacteriophage L. Structure 14:837–845. doi: 10.1016/j.str.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 40.Sutherland IW, Hughes KA, Skillman LC, Tait K. 2004. The interaction of phage and biofilms. FEMS Microbiol Lett 232:1–6. doi: 10.1016/S0378-1097(04)00041-2. [DOI] [PubMed] [Google Scholar]
- 41.O'Brien E, Munir M, Marsh T, Heran M, Lesage G, Tarabara VV, Xagoraraki I. 2017. Diversity of DNA viruses in effluents of membrane bioreactors in Traverse City, MI (USA) and La Grande Motte (France). Water Res 111:338–345. doi: 10.1016/j.watres.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 42.Parent KN, Tang J, Cardone G, Gilcrease EB, Janssen ME, Olson NH, Casjens SR, Baker TS. 2014. Three-dimensional reconstructions of the bacteriophage CUS-3 virion reveal a conserved coat protein I-domain but a distinct tailspike receptor-binding domain. Virology 464-465:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parent KN, Gilcrease EB, Casjens SR, Baker TS. 2012. Structural evolution of the P22-like phages: comparison of Sf6 and P22 procapsid and virion architectures. Virology 427:177–188. doi: 10.1016/j.virol.2012.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitman RL, Przybyla-Kelly K, Shively DA, Nevers MB, Byappanahalli MN. 2008. Sunlight, season, snowmelt, storm, and source affect E. coli populations in an artificially ponded stream. Sci Total Environ 390:448–455. doi: 10.1016/j.scitotenv.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 45.Lopez-Bueno A, Tamames J, Velazquez D, Moya A, Quesada A, Alcami A. 2009. High diversity of the viral community from an Antarctic lake. Science 326:858–861. doi: 10.1126/science.1179287. [DOI] [PubMed] [Google Scholar]
- 46.Mandilara GD, Smeti EM, Mavridou AT, Lambiri MP, Vatopoulos AC, Rigas FP. 2006. Correlation between bacterial indicators and bacteriophages in sewage and sludge. FEMS Microbiol Lett 263:119–126. doi: 10.1111/j.1574-6968.2006.00414.x. [DOI] [PubMed] [Google Scholar]
- 47.Espinosa AC, Arias CF, Sanchez-Colon S, Mazari-Hiriart M. 2009. Comparative study of enteric viruses, coliphages and indicator bacteria for evaluating water quality in a tropical high-altitude system. Environ Health 8:49. doi: 10.1186/1476-069X-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meaden S, Koskella B. 2013. Exploring the risks of phage application in the environment. Front Microbiol 4:358. doi: 10.3389/fmicb.2013.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie Y, Chen JZ, He JL, Miao XY, Xu M, Wu XW, Xu BY, Yu LY, Zhang WH. 2014. Antimicrobial resistance and prevalence of resistance genes of obligate anaerobes isolated from periodontal abscesses. J Periodontol 85:327–334. doi: 10.1902/jop.2013.130081. [DOI] [PubMed] [Google Scholar]
- 50.Kulkarni P, Olson ND, Raspanti GA, Goldstein RER, Gibbs SG, Sapkota A, Sapkota AR. 2017. Antibiotic concentrations decrease during wastewater treatment but persist at low levels in reclaimed water. Int J Environ Res Public Health 14:E668. doi: 10.3390/ijerph14060668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jordan TC, Burnett SH, Carson S, Caruso SM, Clase K, DeJong RJ, Dennehy JJ, Denver DR, Dunbar D, Elgin SC, Findley AM, Gissendanner CR, Golebiewska UP, Guild N, Hartzog GA, Grillo WH, Hollowell GP, Hughes LE, Johnson A, King RA, Lewis LO, Li W, Rosenzweig F, Rubin MR, Saha MS, Sandoz J, Shaffer CD, Taylor B, Temple L, Vazquez E, Ware VC, Barker LP, Bradley KW, Jacobs-Sera D, Pope WH, Russell DA, Cresawn SG, Lopatto D, Bailey CP, Hatfull GF. 2014. A broadly implementable research course in phage discovery and genomics for first-year undergraduate students. mBio 5:e01051-. doi: 10.1128/mBio.01051-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanauer DI, Graham MJ, Sea P, Betancur L, Bobrownicki A, Cresawn SG, Garlena RA, Jacobs-Sera D, Kaufmann N, Pope WH, Russell DA, Jacobs WR Jr, Sivanathan V, Asai DJ, Hatfull GF. 2017. An inclusive research education community (iREC): impact of the SEA-PHAGES program on research outcomes and student learning. Proc Natl Acad Sci U S A doi: 10.1073/pnas.1718188115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morona R, Mavris M, Fallarino A, Manning PA. 1994. Characterization of the rfc region of Shigella flexneri. J Bacteriol 176:733–747. doi: 10.1128/jb.176.3.733-747.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vimr ER, Troy FA. 1985. Regulation of sialic acid metabolism in Escherichia coli: role of N-acylneuraminate pyruvate-lyase. J Bacteriol 164:854–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vimr ER, Troy FA. 1985. Identification of an inducible catabolic system for sialic acids (nan) in Escherichia coli. J Bacteriol 164:845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daegelen P, Studier FW, Lenski RE, Cure S, Kim JF. 2009. Tracing ancestors and relatives of Escherichia coli B, and the derivation of B strains REL606 and BL21(DE3). J Mol Biol 394:634–643. doi: 10.1016/j.jmb.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 57.Jeong H, Barbe V, Lee CH, Vallenet D, Yu DS, Choi SH, Couloux A, Lee SW, Yoon SH, Cattolico L, Hur CG, Park HS, Segurens B, Kim SC, Oh TK, Lenski RE, Studier FW, Daegelen P, Kim JF. 2009. Genome sequences of Escherichia coli B strains REL606 and BL21(DE3). J Mol Biol 394:644–652. doi: 10.1016/j.jmb.2009.09.052. [DOI] [PubMed] [Google Scholar]
- 58.Winston F, Botstein D, Miller JH. 1979. Characterization of amber and ochre suppressors in Salmonella typhimurium. J Bacteriol 137:433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wei J, Goldberg MB, Burland V, Venkatesan MM, Deng W, Fournier G, Mayhew GF, Plunkett G 3rd, Rose DJ, Darling A, Mau B, Perna NT, Payne SM, Runyen-Janecky LJ, Zhou S, Schwartz DC, Blattner FR. 2003. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect Immun 71:2775–2786. doi: 10.1128/IAI.71.5.2775-2786.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sansonetti PJ, Kopecko DJ, Formal SB. 1982. Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect Immun 35:852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Onodera NT, Ryu J, Durbic T, Nislow C, Archibald JM, Rohde JR. 2012. Genome sequence of Shigella flexneri serotype 5a strain M90T Sm. J Bacteriol 194:3022. doi: 10.1128/JB.00393-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buchrieser C, Glaser P, Rusniok C, Nedjari H, D'Hauteville H, Kunst F, Sansonetti P, Parsot C. 2000. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol Microbiol 38:760–771. doi: 10.1046/j.1365-2958.2000.02179.x. [DOI] [PubMed] [Google Scholar]
- 63.Eden PA, Schmidt TM, Blakemore RP, Pace NR. 1991. Phylogenetic analysis of Aquaspirillum magnetotacticum using polymerase chain reaction-amplified 16S rRNA-specific DNA. Int J Syst Bacteriol 41:324–325. doi: 10.1099/00207713-41-2-324. [DOI] [PubMed] [Google Scholar]
- 64.Dover JA, Burmeister AR, Molineux IJ, Parent KN. 2016. Evolved populations of Shigella flexneri phage Sf6 acquire large deletions, altered genomic architecture, and faster life cycles. Genome Biol Evol 8:2827–2840. doi: 10.1093/gbe/evw177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tritt A, Eisen JA, Facciotti MT, Darling AE. 2012. An integrated pipeline for de novo assembly of microbial genomes. PLoS One 7:e42304. doi: 10.1371/journal.pone.0042304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. Babraham Institute, Cambridge, United Kingdom. [Google Scholar]
- 67.Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29:2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 69.Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 44:W54–W57. doi: 10.1093/nar/gkw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lawrence J. 1989. DNA master. http://cobamide2.bio.pitt.edu/computer.htm.
- 71.Krumsiek J, Arnold R, Rattei T. 2007. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23:1026–1028. doi: 10.1093/bioinformatics/btm039. [DOI] [PubMed] [Google Scholar]
- 72.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 73.Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 74.Rambaut A. 2012. FigTree v1.4: molecular evolution, phylogenetics and epidemiology. http://tree.bio.ed.ac.uk/software/figtree/.
- 75.Baker TS, Olson NH, Fuller SD. 1999. Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol Mol Biol Rev 63:862–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schrad JR, Young EJ, Abrahao JS, Cortines JR, Parent KN. 2017. Microscopic characterization of the Brazilian giant samba virus. Viruses 9:E30. doi: 10.3390/v9020030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mastronarde DN. 2005. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 78.Wang Z, Hryc CF, Bammes B, Afonine PV, Jakana J, Chen DH, Liu X, Baker ML, Kao C, Ludtke SJ, Schmid MF, Adams PD, Chiu W. 2014. An atomic model of brome mosaic virus using direct electron detection and real-space optimization. Nat Commun 5:4808. doi: 10.1038/ncomms5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yan X, Sinkovits RS, Baker TS. 2007. AUTO3DEM—an automated and high throughput program for image reconstruction of icosahedral particles. J Struct Biol 157:73–82. doi: 10.1016/j.jsb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yan X, Dryden KA, Tang J, Baker TS. 2007. Ab initio random model method facilitates 3D reconstruction of icosahedral particles. J Struct Biol 157:211–225. doi: 10.1016/j.jsb.2006.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goddard TD, Huang CC, Ferrin TE. 2007. Visualizing density maps with UCSF Chimera. J Struct Biol 157:281–287. doi: 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]