

ABSTRACT
Positive-strand RNA viruses, including picornaviruses, utilize cellular machinery for genome replication. Previously, we reported that each of the 2B, 2BC, 2C, 3A, and 3AB proteins of Aichi virus (AiV), a picornavirus, forms a complex with the Golgi apparatus protein ACBD3 and phosphatidylinositol 4-kinase IIIβ (PI4KB) at viral RNA replication sites (replication organelles [ROs]), enhancing PI4KB-dependent phosphatidylinositol 4-phosphate (PI4P) production. Here, we demonstrate AiV hijacking of the cellular cholesterol transport system involving oxysterol-binding protein (OSBP), a PI4P-binding cholesterol transfer protein. AiV RNA replication was inhibited by silencing cellular proteins known to be components of this pathway, OSBP, the ER membrane proteins VAPA and VAPB (VAP-A/B), the PI4P-phosphatase SAC1, and PI-transfer protein β. OSBP, VAP-A/B, and SAC1 were present at RNA replication sites. We also found various previously unknown interactions among the AiV proteins (2B, 2BC, 2C, 3A, and 3AB), ACBD3, OSBP, VAP-A/B, and SAC1, and the interactions were suggested to be involved in recruiting the component proteins to AiV ROs. Importantly, the OSBP-2B interaction enabled PI4P-independent recruitment of OSBP to AiV ROs, indicating preferential recruitment of OSBP among PI4P-binding proteins. Protein-protein interaction-based OSBP recruitment has not been reported for other picornaviruses. Cholesterol was accumulated at AiV ROs, and inhibition of OSBP-mediated cholesterol transfer impaired cholesterol accumulation and AiV RNA replication. Electron microscopy showed that AiV-induced vesicle-like structures were close to ER membranes. Altogether, we conclude that AiV directly recruits the cholesterol transport machinery through protein-protein interactions, resulting in formation of membrane contact sites between the ER and AiV ROs and cholesterol supply to the ROs.
IMPORTANCE Positive-strand RNA viruses utilize host pathways to modulate the lipid composition of viral RNA replication sites for replication. Previously, we demonstrated that Aichi virus (AiV), a picornavirus, forms a complex comprising certain proteins of AiV, the Golgi apparatus protein ACBD3, and the lipid kinase PI4KB to synthesize PI4P lipid at the sites for AiV RNA replication. Here, we confirmed cholesterol accumulation at the AiV RNA replication sites, which are established by hijacking the host cholesterol transfer machinery mediated by a PI4P-binding cholesterol transfer protein, OSBP. We showed that the component proteins of the machinery, OSBP, VAP, SAC1, and PITPNB, are all essential host factors for AiV replication. Importantly, the machinery is directly recruited to the RNA replication sites through previously unknown interactions of VAP/OSBP/SAC1 with the AiV proteins and with ACBD3. Consequently, we propose a specific strategy employed by AiV to efficiently accumulate cholesterol at the RNA replication sites via protein-protein interactions.
KEYWORDS: ACBD3, Aichi virus, OSBP, PI4KB, PI4P, SAC1, VAP, cholesterol, picornavirus
INTRODUCTION
Positive-strand RNA viruses include various important pathogens in humans, such as poliovirus (PV), hepatitis C virus (HCV), dengue virus, and severe acute respiratory syndrome coronavirus. All known positive-strand RNA viruses rearrange the membranes of intracellular organelles, including the endoplasmic reticulum (ER), the Golgi apparatus, the ER-Golgi apparatus intermediate compartment, endosomes, and mitochondria, to generate specific compartments for viral RNA replication, called viral replication complexes, replication organelles (ROs), or membranous webs. This process involves viral and host proteins, as well as cellular lipids (1–3).
The family Picornaviridae is a group of nonenveloped positive-strand RNA viruses including PV, enterovirus 71 (EV71), rhinoviruses, and hepatitis A virus. The genome encodes a single polyprotein, which is cleaved into 11 or 12 proteins by virus-encoded proteases. For some picornaviruses, some or all of the proteins 2B, 2C, and 3A and cleavage intermediates 2BC and 3AB, which are membrane-associated nonstructural proteins, are involved in membrane rearrangement (4–12).
Studies of some picornaviruses and the flavivirus HCV have revealed that a lipid, phosphatidylinositol 4-phosphate (PI4P), is essential for viral RNA replication. In mammalian cells, PI4P is synthesized from phosphatidylinositol (PI) by any of the four PI 4-kinases, PI4KIIα, PI4KIIβ, PI4KIIIα (also known as PI4KA), and PI4KIIIβ (also known as PI4KB). PI4P is involved in membrane trafficking and lipid transport through the recruitment of certain effector proteins to membranes (13–15). HCV and some picornaviruses generate a PI4P-enriched environment at the site of viral RNA replication using ER-resident PI4KA (16–21) or Golgi apparatus-resident PI4KB (22–25). For PV and coxsackievirus B3 (CVB3), i.e., picornaviruses belonging to the genus Enterovirus, a model is proposed in which the viral protein 3A binds to Golgi apparatus-specific brefeldin A resistance guanine nucleotide exchange factor 1 (GBF1)/ADP-ribosylation factor 1 (Arf1) to recruit PI4KB, which is known as an Arf1 effector, to the Golgi/trans-Golgi network, resulting in PI4P accumulation at viral RNA replication sites (22). PI4P binds to viral RNA polymerase 3D to initiate and facilitate viral RNA synthesis. However, a recent study reported that the recruitment of PI4KB to CVB3 ROs is GBF1/Arf1 independent (26). It was also reported for EV71 that GBF1 and Arf1 play minor roles in RNA replication (27).
Aichi virus (AiV), a picornavirus belonging to the genus Kobuvirus, uses another strategy to recruit PI4KB (23, 24). AiV, which is associated with acute gastroenteritis in humans, was first isolated in 1989 from a patient in Japan (28) and was subsequently detected in various countries throughout the world. We have reported previously that the AiV membrane proteins 2B, 2BC, 2C, 3A, and 3AB bind to the Golgi apparatus protein acyl-coenzyme A binding domain containing 3 (ACBD3), which interacts with PI4KB to form a viral protein/ACBD3/PI4KB complex that produces PI4P at the sites of RNA replication (23). The level of PI4P produced is enhanced in AiV-replicating cells or 2B-, 2BC-, 2C-, 3A-, or 3AB-expressing cells (29). In addition, ACBD3 is indicated to be required for the recruitment of PI4KB to the RNA replication site (29). ACBD3 is not necessary for CVB3 and rhinovirus RNA replication (26, 30), whereas it facilitates enterovirus 71 replication (27). For the necessity of ACBD3 for PV replication, conflicting results have been reported (24, 31).
Recently, oxysterol-binding protein (OSBP) was demonstrated to be involved in RNA replication of some picornaviruses and HCV. In uninfected cells, OSBP binds to PI4P on the Golgi apparatus through its pleckstrin homology (PH) domain (32) and to vesicle-associated membrane protein-associated protein isoforms A and B (VAPA and VAPB [VAP-A/B]) on the ER through its FFAT (two phenylalanines in an acidic tract) motif (33). OSBP transfers cholesterol from the ER to the Golgi apparatus and transfers PI4P back at the membrane contact sites (MCSs) (34, 35). For picornaviruses and HCV, OSBP plays a critical role in the formation of the viral replication complex and in cholesterol accumulation at the sites of viral RNA replication (21, 25, 36–38). SAC1 and PI transfer protein β (PITPNB) are also involved in the cholesterol transfer system. SAC1 is a phosphatase that catalyzes dephosphorylation of PI4P on the ER to produce PI (35). PITPNB binds to the PI and transfers it from the ER to the Golgi apparatus (39). SAC1 and PITPNB have been reported to be involved in rhinovirus infection (25).
Here, we report that AiV hijacks the OSBP-mediated cholesterol transport system, which works at the MCS between the ER and the AiV ROs, for its RNA replication. We confirmed novel protein-protein interactions between various AiV proteins (2B, 2BC, 2C, 3A, and 3AB), ACBD3, and the components of this pathway, including OSBP, VAP-A/B, and SAC1. The hijacking strategy in which these interactions play important roles has not been reported in picornaviruses other than AiV. Notably, the OSBP-2B interaction has been indicated to result in the PI4P-independent recruitment of OSBP, suggesting preferential recruitment of OSBP over other PI4P-binding proteins. In addition, we discuss the possibility that ACBD3 is a novel component of the cholesterol transport pathway, based on the finding of interactions between ACBD3 and the component proteins.
RESULTS
Knockdown of OSBP, VAP-A/B, SAC1, and PITPNB, all of which are known to be components of the cholesterol transport pathway, inhibits AiV replication.
To determine the involvement of the OSBP-mediated cholesterol transport pathway in AiV RNA replication, we first examined whether AiV RNA replication is affected by the depletion of certain host proteins involved in the pathway, OSBP, VAP-A/B, and SAC1. Vero cells were transfected with small interfering RNA (siRNA) and, after 48 h, further transfected with AV-FL-Luc-5′rzm harboring a firefly luciferase gene, which enabled us to monitor AiV RNA replication by a luciferase assay. The lysates of the cells were prepared at the indicated times until 9 to 10 h after transfection of replicon RNA, when AiV replication has progressed enough to determine the difference in replication efficiency. The depletion of OSBP decreased AiV RNA replication to 18% of control levels at 9 h after transfection (Fig. 1A). Depletion of VAPA and VAPB decreased AiV RNA replication to 54 and 90%, respectively, of control levels at 9 h after transfection, but silencing of VAP-A/B decreased replication to 7% (Fig. 1B). Knockdown of SAC1 also decreased AiV RNA replication, albeit less effectively than that of OSBP or VAP-A/B. Cell viability was also determined, and a statistically significant but slight (3.3%) decrease was detected only when VAP-A/B were silenced (Fig. 1B). PITPNB has also been shown to be involved in rhinovirus replication as a component of this cholesterol transport pathway (25). Knockdown of PITPNB also decreased AiV RNA replication to 49% at 10 h after transfection, although its knockdown was inefficient (Fig. 1D). These results suggest the involvement of the OSBP-mediated cholesterol transport pathway in AiV RNA replication.
FIG 1.

Knockdown of VAP-A/B, SAC1, and OSBP, known to be components of the cholesterol transport pathway, inhibits AiV replication. (A to D, left) Vero cells were transfected with nontargeting control siRNA and OSBP siRNA (A); VAPA, VAPB, or VAP-A/B siRNA (B); SAC1 siRNA (C); or PITPNB siRNA (D). After 72 h, the cells were transfected with replicon RNA, and luciferase activity was measured at the indicated times after transfection. The maximum value obtained for nontargeting control siRNA-treated cells was taken as 100%. The experiment was repeated at least three times. The error bars indicate standard deviations (SD). *, P < 0.05; **, P < 0.01; ***, P < 0.005; ****, P < 0.001. (A to D, middle) Western blots for the levels of OSBP (A), VAP-A/B (B), SAC1 (C), or PITPNB (D) and α-tubulin in Vero cells treated with the indicated siRNAs for 72 h; for detection, rabbit antibodies against OSBP, VAP-A/B, SAC1, and PITPNB and mouse antibody against α-tubulin were used. Values obtained by densitometry and normalized to α-tubulin are indicated. (A to D, right) Viability of Vero cells treated with nontargeting control siRNA and OSBP siRNA (A); VAPA, VAPB, or VAP-A/B siRNA (B); SAC1 siRNA (C); or PITPNB siRNA (D). The data are means and SD for at least three independent experiments. ***, P < 0.005. NS, not significant.
VAP-A/B, SAC1, and OSBP are present at AiV RNA replication sites.
Next, to determine whether VAP-A/B, SAC1, and OSBP are actually present at the sites of AiV RNA replication, we examined their localization in replicon RNA-transfected cells. In uninfected cells, VAPA and VAPB were present mainly in the ER (Fig. 2A and B), as shown by colocalization with calnexin and calregulin (Fig. 3). SAC1 was distributed to the ER with some Golgi apparatus localization (Fig. 2D), as reported previously (40). OSBP was present predominantly in the Golgi apparatus, which was known (Fig. 2E). In AiV replicon-transfected cells, OSBP, VAP-A/B, and SAC1 were generally colocalized with the viral proteins 2B, 2C, and 3A (Fig. 2A, B, D, and E). For OSBP and VAP-A/B, we confirmed that the proteins were colocalized with double-stranded RNA (dsRNA) (Fig. 2C and E). Moreover, VAP-A/B were colocalized with ACBD3, which is a crucial host factor for AiV RNA replication (Fig. 2C), and were no longer colocalized with calnexin and calregulin (Fig. 3A and B). These results suggest that OSBP, VAP-A/B, and SAC1 are recruited to the sites of AiV RNA replication.
FIG 2.

VAP-A/B, SAC1, and OSBP are present at AiV RNA replication sites. (A to E) Vero cells were mock electroporated or electroporated with the replicon RNA, AV-FL-Luc-5′rzm. After 4 h, the cells were fixed and then immunostained with the indicated antibodies. Bars, 20 μm.
FIG 3.

The ER proteins calnexin (A) and calregulin (B) are not recruited to viral RNA replication sites. Vero cells were mock electroporated or electroporated with AiV replicon RNA, and 4 h later, the cells were immunostained with the indicated antibodies. Bars, 20 μm.
Interactions of VAP-A/B, SAC1, and OSBP with 2B, 2BC, 2C, 3A, and 3AB.
Based on the above-mentioned results, we tested whether OSBP, VAP-A/B, and SAC1 interact with the AiV proteins 2B, 2BC, 2C, 3A, and 3AB, which are present at the sites of AiV RNA replication (23). Mammalian two-hybrid (M2H) analyses revealed that both VAPA and VAPB could interact with 2B, 2BC, 2C, 3A, and 3AB (13- to 47-fold increases in luciferase activity), but not with L, an AiV nonstructural protein that is not localized at RNA replication sites (23) (Fig. 4A). Reciprocal coimmunoprecipitation analysis using FLAG-tagged VAP-A/B and hemagglutinin (HA)-tagged viral proteins confirmed that 2B, 2BC, 2C, 3A, and 3AB, but not L, were associated with VAP-A/B (Fig. 4D and 5A).
FIG 4.

Interactions of VAP-A/B, SAC1, and OSBP with 2B, 2BC, 2C, 3A, and 3AB. (A to C) Mammalian two-hybrid assay. Vero cells were transfected with the indicated combinations of pACT and pBIND constructs expressing VAP-A/B, SAC1, OSBP, or AiV protein together with pG5luc encoding firefly luciferase (Fluc). After 48 h, the Fluc activity was assayed, and relative Fluc activity was normalized to the negative control. (D to F) Coimmunoprecipitation of VAP-A/B, SAC1, or OSBP with L, 2B, 2BC, 2C, 3A, and 3AB. 293T cells were transfected with FLAG-tagged VAPA, VAPB, SAC1, or OSBP and HA-tagged L, 2B, 2BC, 2C, 3A, or 3AB, as indicated. Proteins were immunoprecipitated (IP) with anti-FLAG antibody or control IgG, and the resulting immunoprecipitates and whole-cell lysates were analyzed by immunoblotting (IB) with anti-FLAG and anti-HA antibodies.
FIG 5.

Immunoprecipitation using anti-HA antibody. (A and B) Transfections were performed as described for Fig. 4D to F. Immunoprecipitations were conducted with anti-HA antibody or control IgG, and the resulting immunoprecipitates and whole-cell lysates were detected by anti-HA and anti-FLAG antibodies, as indicated.
SAC1 was also revealed to interact with the viral proteins by M2H analysis (24- to 31-fold increases in luciferase activity) and coimmunoprecipitation analyses (Fig. 4B and E).
The interaction of OSBP with 2B, 2BC, or 2C caused >11-fold increase in luciferase activity, whereas the interaction of OSBP with 3A and 3AB resulted in 2.7- and 5.3-fold increases, respectively (Fig. 4C) in an M2H analysis. OSBP-2B and -2BC interactions were further determined by reciprocal coimmunoprecipitation (Fig. 4F and 5B). On the other hand, in analyses of OSBP-2C, -3A, and -3AB interactions, coimmunoprecipitation was detected when the proteins were immunoprecipitated with anti-FLAG antibody, but not with anti-HA antibody (Fig. 4F and 5B). These results suggest that 2B and 2BC interact with OSBP, whereas interaction of OSBP with 2C, 3A, and 3AB is not definite, or may be weak, since reciprocal coimmunoprecipitation was not detected.
VAP-A/B, SAC1, and OSBP interact with ACBD3, which is an essential host factor for AiV RNA replication.
Previous studies have demonstrated that the AiV proteins 2B, 2BC, 2C, 3A, and 3AB bind to ACBD3. In addition, as seen in Fig. 2, ACBD3 colocalized with VAP-A/B at the AiV RNA replication sites, and OSBP and SAC1 were also present at the replication sites. We therefore examined whether OSBP, VAP-A/B, and SAC1 interact with ACBD3. M2H analyses indicated that the interactions of OSBP, VAPA, VAPB, and SAC1 with ACBD3 caused 30-, 277-, 241-, and 171-fold increases, respectively, in luciferase activity (Fig. 6A). In addition, OSBP, VAP-A/B, and SAC1 were coimmunoprecipitated with ACBD3 (Fig. 6B). These results suggest interactions of OSBP, VAP-A/B, and SAC1 with ACBD3.
FIG 6.

VAP-A/B, SAC1, and OSBP interact with ACBD3, which is an essential host factor for AiV RNA replication. (A) A mammalian two-hybrid assay was performed to examine interaction of L, VAP-A/B, SAC1, and OSBP with ACBD3. The results are represented as described for Fig. 4A to C. The experiment was repeated at least three times. The error bars indicate standard deviations. (B) Coimmunoprecipitation of VAP-A/B, SAC1, or OSBP with ACBD3. 293T cells were cotransfected with plasmids encoding HA-ACBD3 and FLAG-VAPA, VAPB, SAC1, or OSBP. The cell lysates were analyzed as described for Fig. 4D to F. (C) ACBD3 induces Golgi apparatus localization of VAP-A/B or SAC1. (Top) FLAG-VAPA, FLAG-VAPB, FLAG-SAC1, and HA-ACBD3 were expressed individually in Vero cells and immunostained with anti-FLAG or anti-HA antibodies. (Bottom) Vero cells coexpressing HA-ACBD3 with FLAG-VAPA, FLAG-VAPB, or FLAG-SAC1 were immunostained with anti-FLAG, anti-HA, or anti-TNG46 antibodies. Bars, 20 μm.
We further investigated whether the interaction of VAP-A/B or SAC1 with ACBD3 observed in M2H and coimmunoprecipitation assays affects intracellular localization of ER-located VAP-A/B and SAC1. When expressed alone in Vero cells, FLAG-tagged VAP-A/B and SAC1 were localized in an ER-like network, and HA-ACBD3 was localized at the Golgi apparatus (Fig. 6C, top). The localizations of tagged VAP-A/B and ACBD3 were reminiscent of those of endogenously expressed proteins (Fig. 2). It is known that SAC1 is associated with the ER when expressed as a fusion with an epitope tag or green fluorescent protein (GFP) (40, 41). When coexpressed with HA-ACBD3, FLAG-VAPA, FLAG-VAPB, or FLAG-SAC1 was still localized in an ER-like network but concentrated on the Golgi apparatus, where each of them colocalized with HA-ACBD3 and the trans-Golgi marker TGN46 (Fig. 6C, bottom.). These results suggest that ACBD3 can recruit VAP-A/B and SAC1 to the Golgi apparatus and, thus, that interaction of VAP-A/B or SAC1 with ACBD3 can affect their intracellular localization.
OSBP can be recruited to AiV RNA replication sites independently of PI4P.
At AiV RNA replication sites, a viral protein/ACBD3/PI4KB complex is formed to produce PI4P. OSBP is known as a PI4P effector protein. To determine whether PI4KB-generated PI4P is required for the recruitment of OSBP to the sites of AiV RNA replication, we examined the effect of T00127-HEV1, a PI4KB-specific inhibitor (42), on the intracellular localization of OSBP. T00127-HEV1 inhibited AiV replication severely (23), resulting in insufficient production of the viral proteins in cells. Then, to solve this problem, the AiV polyprotein was expressed by the cytomegalovirus (CMV) promoter. Our previous study showed that CMV promoter-driven AiV polyprotein expression in Vero cells induced the formation of clusters containing 2B, 2C, 3A, ACBD3, PI4KB, and PI4P, which are similar to those formed in AiV replicon-replicating cells (29). Here, it was shown that OSBP also colocalized with 2B, 2C, 3A, and PI4P in cells expressing the polyprotein (Fig. 7A). When the polyprotein was expressed in T00127-HEV1-treated cells, each viral protein (2B, 2C, or 3A) and PI4P, which is probably synthesized by PI4Ks other than PI4KB, formed small clusters but did not colocalize with each other (Fig. 7B), as described previously (29); however, OSBP still colocalized with the viral proteins. Thus, colocalization of OSBP with the viral proteins is independent of PI4KB-produced PI4P, indicating that OSBP can be recruited to AiV RNA replication sites in a PI4P-independent manner.
FIG 7.
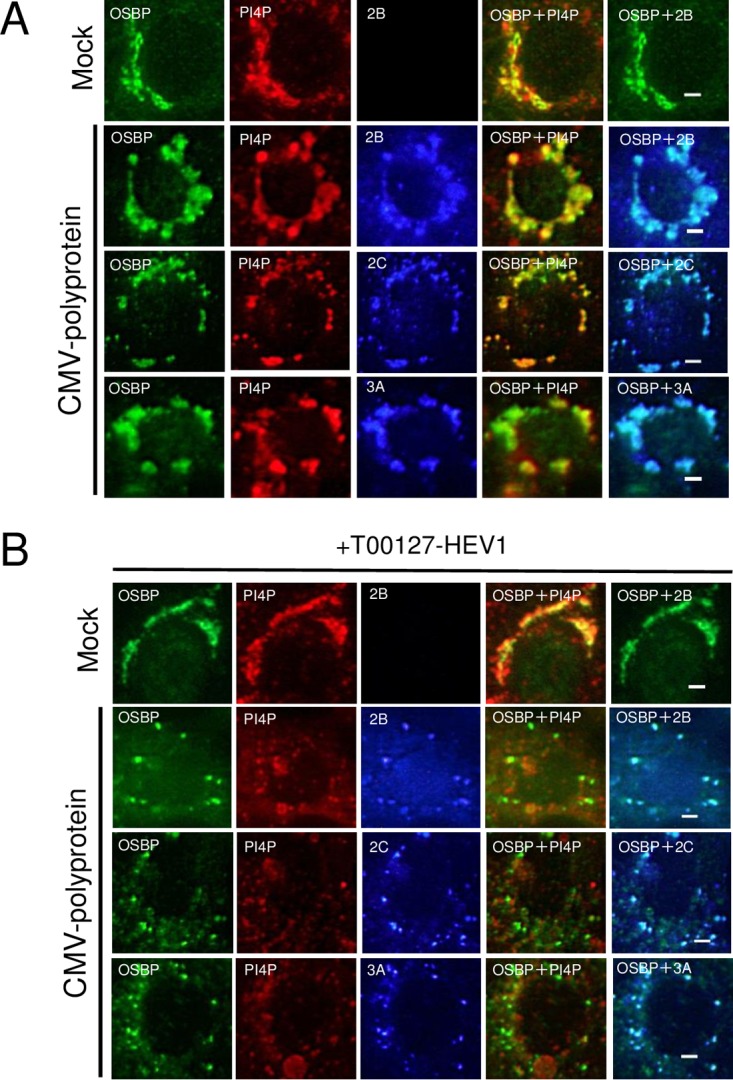
OSBP can be recruited to AiV RNA replication sites independently of PI4P. (A and B) Vero cells were mock transfected or transfected with pCMV-polyprotein and then cultured in medium with or without 5 μM T00127-HEV1, a PI4KB-specific inhibitor. At 24 h after transfection, the cells were fixed and stained with the indicated antibodies. Bars, 20 μm.
To examine whether the interactions of OSBP with the viral proteins and ACBD3 were involved in PI4P-independent recruitment of OSBP, FLAG-tagged OSBP, HA-tagged ACBD3, and 2B, 2C, or 3A were coexpressed in Vero cells, and immunostained. When expressed alone, FLAG-OSBP was localized throughout the cytoplasm and also at the Golgi apparatus (Fig. 8, top). Coexpression of FLAG-OSBP, HA-ACBD3, and 2B resulted in the colocalization of the proteins in the perinuclear region (Fig. 8, bottom). Conversely, in cells coexpressing 2C or 3A with FLAG-OSBP and HA-ACBD3, 2C, and 3A colocalized with HA-ACBD3, they did not relocate FLAG-OSBP to the perinuclear region; FLAG-OSBP was present throughout the cytoplasm (Fig. 8, bottom). These results showed that 2B can affect the intracellular localization of OSBP, but 2C, 3A, and ACBD3 cannot. Thus, they suggest that PI4P-independent recruitment of OSBP to AiV RNA replication sites is accomplished by the 2B-OSBP interaction.
FIG 8.

Effect of 2B, 2C, 3A, or ACBD3 on the localization of OSBP in coexpressing cells. (Top) FLAG-OSBP, HA-ACBD3, 2B, and 2C were expressed in Vero cells individually and then immunostained with anti-FLAG, anti-HA, anti-2B, and anti-2C antibodies, respectively. (Bottom) Cells coexpressing FLAG-OSBP with HA-ACBD3 and 2B, 2C, or 3A were triple stained with anti-FLAG, anti-HA, and anti-2B, anti-2C, or anti-3A antibodies. Bars, 20 μm.
To verify the specific recruitment of OSBP to AiV RNA replication sites, a similar analysis was performed on CERT, which also binds to VAP and PI4P and transfers ceramide from the ER to the Golgi apparatus. For analyses of interactions of CERT with the viral proteins and ACBD3, these interactions resulted in 2.9- to 4.9-fold increases in luciferase activities in M2H (Fig. 9A). In coimmunoprecipitation assays, only faint bands were detected in coimmunoprecipitations with anti-FLAG antibody, and no bands were observed with anti-HA antibody (Fig. 9B). Thus, no definite interaction of CERT with any of the viral proteins tested or with ACBD3 was confirmed. Coexpression of FLAG-CERT with HA-ACBD3 and 2B or 3A did not affect the intracellular localization of FLAG-CERT, although 2B and 3A colocalized with HA-ACBD3 (Fig. 9C). Finally, in AiV replicon-transfected cells, CERT was dispersed throughout the cell, and it did not colocalize with dsRNA and VAP-A/B, which localize at AiV RNA replication sites (Fig. 9D). Thus, CERT, which lacked definite ability to bind to the viral proteins and ACBD3, was not recruited to the AiV RNA replication sites, suggesting preferential recruitment of OSBP over other PI4P-binding proteins.
FIG 9.

CERT, which did not interact with the viral proteins and ACBD3, is not recruited to the AiV RNA replication sites. (A) Mammalian two-hybrid analysis was performed to determine interactions between CERT and 2B, 2BC, 2C, 3A, 3AB, or ACBD3, and the results are represented as described for Fig. 4A to C. The error bars indicate standard deviations. (B) Coimmunoprecipitation analysis of CERT with viral proteins. HA-tagged L, 2B, 2BC, 2C, 3A, and 3AB were coexpressed with FLAG-tagged CERT in 293T cells and immunoprecipitated with anti-FLAG (top) or anti-HA (bottom) antibodies or control IgG. The resulting immunoprecipitates and whole-cell lysates were detected by anti-HA and anti-FLAG antibodies as indicated. (C) (Left) FLAG-CERT was expressed and immunostained with anti-FLAG antibody. (Right) Cells coexpressing FLAG-CERT with HA-ACBD3 and 2B or 3A were triple stained with anti-FLAG, anti-HA, and anti-2B or anti-3A antibodies. Bars, 20 μm. (D) Vero cells were mock electroporated or electroporated with the replicon RNA, and after 4 h, the cells were fixed and stained with the indicated antibodies. Bars, 20 μm.
OSBP-mediated cholesterol trafficking to AiV RNA replication sites is critical for viral RNA replication.
OSBP transfers cholesterol from the ER to the Golgi apparatus (35). To examine whether cholesterol is accumulated at the AiV RNA replication sites, replicon-transfected cells were stained with filipin, a fluorescent cholesterol-binding polyene antibiotic, for cholesterol and with various antibodies. Filipin colocalized with 2B, 2C, 3A, and dsRNA (Fig. 10A). In addition, filipin could clearly form foci with ACBD3 and OSBP in AiV RNA-replicating cells, which were distinguished by expression of the viral nonstructural protein L (Fig. 10B). Similar observations were made with VAPA and VAPB, both of which colocalized with dsRNA (Fig. 10C). Thus, these results demonstrate accumulation of cholesterol at the AiV RNA replication sites associated with ACBD3, OSBP, and VAP-A/B.
FIG 10.

OSBP-mediated cholesterol trafficking to AiV RNA replication sites is critical for viral RNA replication. (A to C) Cholesterol accumulation at AiV RNA replication sites. Vero cells grown on medium containing lipid-free fetal bovine serum (FBS) were mock electroporated or electroporated with the replicon RNA. At 4 h after electroporation, the cells were fixed and stained with filipin III and the indicated antibodies. Bars, 20 μm. (D) Effect of 25-HC on AiV RNA replication. Vero cells were electroporated with AiV replicon RNA and then incubated with or without 25-HC. At 1 to 6 h after electroporation, the cells were assayed for luciferase activity. The luciferase activity of cells without 25-HC at 6 h after transfection was taken as 100%. The error bars indicate standard deviations. (E and F) Effect of 25-HC on intracellular localization of cholesterol in cells expressing the AiV polyprotein. Vero cells cultured in lipid-free FBS-supplemented medium with (F) or without (E) 25-HC were mock transfected or transfected with pCMV-polyprotein. After 24 h, the cells were fixed and stained with filipin III and the indicated antibodies. Bars, 20 μm.
To confirm whether transport of cholesterol by OSBP is required for AiV replication, the culture medium was treated with 25-HC, a high-affinity ligand of OSBP that inhibits cholesterol transfer by OSBP (35). The addition of 25-HC significantly reduced the replication of the AiV replicon by 92% at 6 h after transfection (Fig. 10D, left) without affecting cell viability (Fig. 10D, right).
To further confirm whether transport of cholesterol by OSBP contributes to the accumulation of cholesterol at the AiV RNA replication sites, we examined the effect of 25-HC on filipin localization in cells expressing the AiV polyprotein. In Vero cells expressing the polyprotein, filipin colocalized with 2B, 2C, and 3A (Fig. 10E), as observed in replicon-replicating cells (Fig. 10A). When the polyprotein was expressed in the presence of 25-HC, 2B, 2C, and 3A formed small clusters. Filipin was present throughout the cells and did not colocalize with the clusters containing 2B, 2C, or 3A (Fig. 10F), illustrating that 25-HC inhibits cholesterol accumulation at the viral protein-positive membrane structures. Taken together, these results suggest that OSBP-mediated cholesterol transport is important for AiV RNA replication and results in accumulation of cholesterol at the replication sites.
VAP-A/B and the viral proteins localize to distinct membranes in AiV RNA-replicating cells.
To characterize the membrane in which VAP-A/B or the AiV proteins are present in AiV-replicating cells, we performed a membrane flotation analysis of replicon-transfected cells in the presence of Nonidet P-40 (NP-40) (Fig. 11). Cholesterol- and glycosphingolipid-rich membrane domains, such as lipid rafts or caveolae, are resistant to treatment with nonionic detergents, including NP-40 (66, 67). In the analysis of the untreated homogenate of replicon-transfected cells, 2B, 2BC, 2C, 3A, and 3AB, as well as VAP-A/B and the ER marker calnexin, were associated with membranes. For the NP-40-treated homogenate, VAP-A and VAP-B were mostly shifted to the soluble fraction, like calnexin; however, a substantial portion of 2B, 2BC, 2C, or 3A and a small portion of 3AB remained in the insoluble fraction, indicating association of the viral proteins with detergent-resistant membranes. These results suggest that in AiV-replicating cells, VAP-A and VAP-B are localized in the ER membrane, similar to calnexin, whereas the viral proteins 2B, 2BC, 2C, and 3A (and a portion of 3AB) are associated with the detergent-insoluble membrane, whose nature is different from that of the ER membrane.
FIG 11.
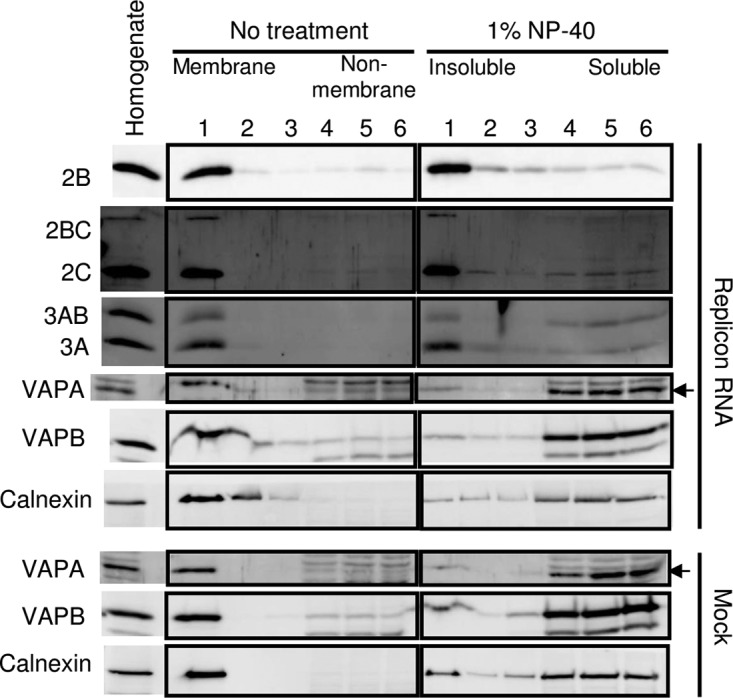
VAP-A/B and the viral proteins localize to distinct membranes in AiV RNA-replicating cells. The homogenate of mock-transfected or AiV replicon RNA-transfected Vero cells was treated or not with 1% NP-40 for 1 h on ice and then fractionated on a sucrose gradient. The fractions were subjected to immunoblotting to analyze the indicated proteins.
AiV-induced membrane structures are located in the vicinity of the ER membrane.
To investigate membrane alteration at the ultrastructure level in AiV RNA-replicating cells, we performed electron microscopy analysis. Figure 12A shows electron micrographs of mock- or AiV replicon-transfected cells at 6 h after transfection. The typical Golgi apparatus cisternae were not found in replicon-transfected cells at 6 h (data not shown). We observed two types of structures in the cytoplasm in AiV RNA-transfected cells, both of which were not found in mock-transfected cells. The first type of AiV-induced structure was single-membrane vesicle-like structures 200 to 400 nm in diameter (Fig. 12A, right, box 1, and B), and the second was 150- to 200-nm vesicle-like structures with unclear shapes (Fig. 12A, right, boxes 2, 3, and 4, and C). The former was observed at both 4 h (data not shown) and 6 h (Fig. 12B) after transfection. On the other hand, the latter was hardly observed at 4 h after transfection (data not shown), and some of the structures appeared to lack a membrane (Fig. 12C). These results suggest that the larger single-membrane vesicle-like structures convert into the smaller structures with unclear shapes, and some of the latter structures may start to collapse at 6 h after transfection. In common with clusters of the larger and the smaller structures, the membranes of the outermost structures were frequently observed in the vicinity of the ER membrane, which could be identified by association with ribosomes (Fig. 12B and C). These results demonstrated that AiV-induced vesicle-like structures were present in the vicinity of the ER membrane and therefore support the possibility that the OSBP-mediated cholesterol transport pathway operates between the ER and AiV ROs.
FIG 12.
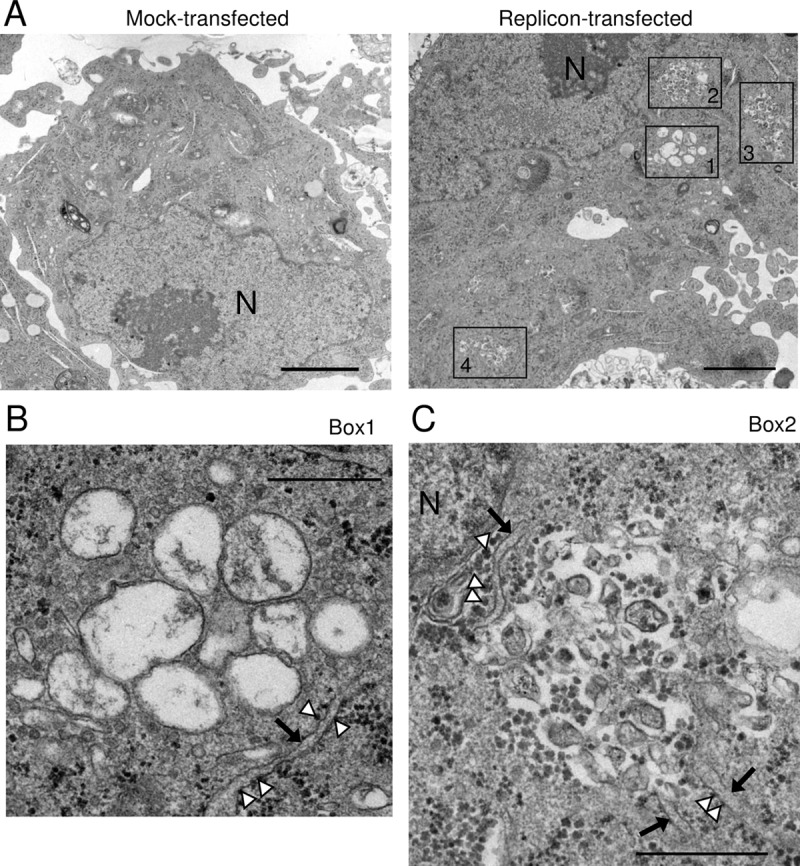
AiV-induced membrane structures are located in the vicinity of the ER membrane. (A) Electron micrographs of mock- or AiV replicon-transfected cells at 6 h after transfection. Clusters of AiV-induced structures are boxed. Higher-magnification images of the areas in box 1 and box 2 are shown in panels B and C, respectively. N, nucleus. Bars, 2 μm. (B and C) Electron micrographs of clusters of the larger single-membrane vesicle-like structures (B) and the smaller vesicle-like structures (C). The arrows and arrowheads indicate the ER membrane and ribosomes, respectively. Bars, 500 nm.
DISCUSSION
We previously demonstrated that the AiV protein/ACBD3/PI4KB complex is formed to produce PI4P at the sites of AiV RNA replication (23). PI4P production at the viral RNA replication sites has also been observed in enteroviruses and cardioviruses, other members of the family Picornaviridae, and in HCV (16–21). All these viruses have been shown to exploit the OSBP-mediated cholesterol transport pathway for viral RNA replication (21, 25, 36–38). In this study, we examined the involvement of the OSBP-mediated cholesterol transport pathway in AiV RNA replication. OSBP and some components of this pathway, VAP-A/B, SAC1, and PITPNB, were required for efficient AiV RNA replication. OSBP, VAP-A/B, and SAC1 were present at AiV RNA replication sites. We found for the first time that all or some of the viral proteins 2B, 2BC, 2C, 3A, and 3AB and a host protein, ACBD3, interact with the component proteins OSBP, VAP-A/B, and SAC1. The OSBP-2B interaction was suggested to enable the PI4P-independent recruitment of OSBP to AiV RNA replication sties. Cholesterol was accumulated at AiV RNA replication sites, and inhibition of OSBP-catalyzed cholesterol transfer by 25-HC inhibited cholesterol accumulation at the 2B-, 2C-, and 3A-containing membrane clusters in polyprotein-expressing cells and also inhibited AiV RNA replication. Membrane flotation analysis and electron microscopy suggested that the cholesterol transport pathway operates at the MCS between the AiV RO and the ER. These results suggest that the OSBP-mediated cholesterol transport pathway, which works at the RO-ER MCS, is essential for AiV RNA replication.
It is known for picornaviruses and HCV that cholesterol is accumulated at viral RNA replication sites by hijacking the OSBP-mediated cholesterol transport pathway. These viruses utilize the same hijacking strategy, where OSBP is indirectly recruited through binding to PI4P accumulated at viral RNA replication sites (21, 25, 36–38). On the other hand, this study showed that AiV also uses the OSBP-mediated cholesterol transport system to supply cholesterol to the ROs but suggested a different strategy to recruit the system: the newly found protein-protein interactions recruit not only OSBP to PI4P on the RO, but also the components of the system, VAP-A/B and SAC1. For other picornaviruses, such interactions have not been reported. VAP-A/B are reported to be involved in HCV replication (43–46), and HCV NS5A interacts with VAP-A/B; however, the involvement of VAP-A/B in OSBP-mediated cholesterol transport has not been shown for HCV. Direct recruitment of the cholesterol transport system has also been reported for a tombusvirus, tomato bushy stunt virus (TBSV), whose replication protein p33 interacts with OSBP-related proteins and Scs2p VAP protein (47), although involvement of PI4P in viral RNA replication has not been described (47).
The first notable finding concerning protein-protein interactions is that the 2B-OSBP interaction enables selective recruitment of OSBP, among PI4P-binding proteins, to AiV replication sites. This was indicated by the following results. First, coexpression of OSBP, ACBD3, and 2B resulted in the redistribution of OSBP to the perinuclear region, where ACBD3 and 2B were localized (Fig. 8, bottom). In contrast, similar experiments uncovered the fact that 2C and 3A, for which reciprocal coimmunoprecipitation with OSBP was not detected, could not recruit OSBP to the perinuclear region. Furthermore, another PI4P- and VAP-binding protein, CERT, which did not interact with the viral proteins and ACBD3, was not recruited to the perinuclear region in coexpression with ACBD3 and 2B or 3A (Fig. 9C). Indeed, CERT was not recruited to the AiV ROs (Fig. 9D). In addition, in the AiV polyprotein-expressing cells treated with a PI4KB-specfic inhibitor, OSBP maintained colocalization with the viral proteins (Fig. 7B). Although involvement of PI4P in recruiting OSBP is not excluded, PI4P would be essential as the scaffold of OSBP and as the resource for exchange with cholesterol. PI4P-independent and protein-protein interaction-based recruitment of OSBP to viral RNA replication sites will promote the selective recruitment of OSBP, among PI4P-binding proteins, to PI4P on ROs to facilitate cholesterol accumulation. Protein-protein interaction-based OSBP recruitment has not been reported for other picornaviruses and HCV. In those viruses, the recruitment of OSBP to viral RNA replication sites is dependent on PI4K activity or PI4P (25, 37, 38, 48). The processes for hijacking this cholesterol transport system may be diverse, even among closely related viruses.
Another important finding involving protein-protein interactions is that ACBD3 interacts with the components of the cholesterol transport pathway, OSBP, VAP-A/B, and SAC1. Interactions among OSBP, VAP-A/B, and SAC1 have previously been shown (33, 49). In addition to these known interactions, the newly found interactions involving ACBD3 would also contribute to recruiting the component proteins to AiV RNA replication sites and to stabilizing the complexes of the cholesterol transport pathway. Furthermore, in uninfected cells, ACBD3 forms a giantin/ACBD3/PI4KB complex at the Golgi apparatus (23, 50), and ACBD3 plays a critical role in the Golgi apparatus recruitment of PI4KB (23, 29), suggesting that ACBD3 is present at the site where PI4P is produced prior to recruitment of OSBP, VAP-A/B, and SAC1. Thus, we speculate that, in infected and uninfected cells, ACBD3 plays a role in setting up the cholesterol transfer system by recruiting PI4KB first and then facilitating recruitment of OSBP, VAP-A/B, and SAC1 to the RO or Golgi apparatus. Actually, ACBD3 was capable of relocalizing VAP-A/B and SAC1 (Fig. 6C), although we could not confirm the ability of ACBD3 to recruit OSBP, since coexpression of OSBP, ACBD3, and 2C or 3A did not relocalize OSBP (Fig. 8, bottom). Moreover, ACBD3 is reported to be involved in the regulation of ceramide trafficking between the ER and the Golgi apparatus by CERT (51, 52), which also binds to PI4P and VAP-A/B. ACBD3 may also have an important role in the ceramide transfer pathway involved in sphingolipid metabolism. More research is required to understand the role of ACBD3 in lipid trafficking between the ER and the Golgi apparatus.
The previous study of rhinovirus demonstrated involvement of VAP-B in viral RNA replication by analysis using siRNA (25). The present study is the first one exploring in detail the involvement of VAP-A/B in picornavirus RNA replication, leading to a new finding that they interact with the viral proteins and ACBD3. In addition, in uninfected cells, VAP/A-B colocalized with calnexin and calregulin (Fig. 3). In contrast, in AiV-replicating cells, VAP-A/B were recruited to AiV RNA replication sites and no longer colocalized with calnexin and calregulin (Fig. 2 and 3). On the other hand, the membrane flotation analysis of replicon-transfected cells showed that 2B, 2BC, 2C, and 3A reside in NP-40-resistant membranes, whereas VAP-A/B and calnexin are found exclusively in the NP-40 detergent-soluble fractions (Fig. 11). Electron microscopy showed that AiV-induced membrane structures are frequently located in the vicinity of the ER membrane (Fig. 12). These results suggest that, while these viral proteins are present on the cholesterol-enriched ROs, VAP-A/B are present on the ER membrane in AiV-replicating cells. OSBP-, VAP-A/B-, and PI4P-mediated cholesterol transport would occur at the MCS between the ER and the ROs, where OSBP binds to VAP-A/B on the ER and to PI4P on ROs to transfer cholesterol from the ER to ROs.
Taking the data together, we propose a model for cholesterol supply to the sites of AiV RNA replication (Fig. 13). Each of the AiV proteins 2B, 2BC, 2C, 3A, and 3AB forms a complex with ACBD3 and PI4KB to produce PI4P on viral ROs that probably originated from the Golgi apparatus (23). PI4P may recruit some PI4P-binding proteins, but 2B (and possibly 2BC) enables the PI4P-independent recruitment of OSBP, resulting in the selective recruitment of OSBP, among the PI4P-binding proteins, to PI4P on ROs. Then, the viral proteins and ACBD3 facilitate the recruitment of VAP-A/B to OSBP. At this time, VAP-A/B are likely to be present on the ER membranes. OSBP transfers cholesterol from the ER to ROs and transfers PI4P back (34, 35). SAC1 is recruited to the site of cholesterol/PI4P traffic through interactions with the AiV proteins and ACBD3 (Fig. 13B), as well as with VAP-A/B and OSBP (47), and dephosphorylates PI4P to PI on the ER, to which PITPNB binds. Then, PI is transferred to the RO by PITPNB, and the PI on the RO is phosphorylated by PI4KB, which had been recruited by the AiV proteins with the mediation of ACBD3. Thus, the cholesterol/PI4P traffic cycle operates at the MCS between the AiV RO and the ER. As a result, the ROs are enriched with cholesterol. Inhibition of the PI4KB activity or cholesterol transport by OSBP alters viral protein-positive membrane structures (Fig. 7 and 10E and F) (29). Therefore, the cholesterol/PI4P traffic and subsequent cholesterol accumulation seem to be important for the morphogenesis of AiV ROs.
FIG 13.
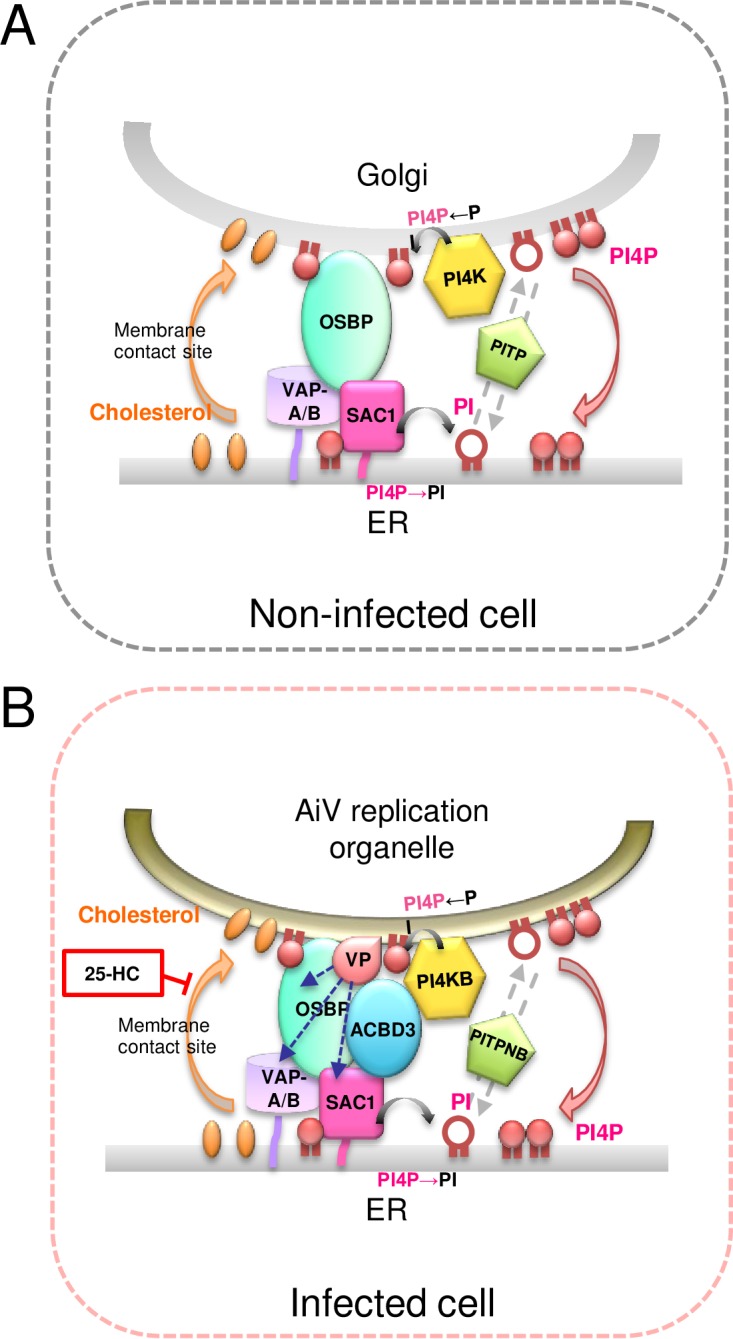
Model for cholesterol trafficking at membrane contact sites between the ER and AiV replication organelles. (A) In noninfected cells, OSBP bridges the ER, containing VAP-A/B and SAC1, and the Golgi apparatus, containing PI4P, and exchanges cholesterol trafficking from the ER to the Golgi apparatus for PI4P trafficking in the opposite direction. PI4P production by PI4K at the Golgi apparatus, PI4P dephosphorylation by SAC1 at the ER, and PI transport by PITP provide the energy driving the OSBP-mediated cholesterol transfer. (B) In infected cells, each of the viral proteins (VP) 2B, 2BC, 2C, 3A, and 3AB forms a complex with ACBD3 and PI4KB to synthesize PI4P on AiV replication organelles. Both VP and ACBD3 can directly interact with OSBP, VAP-A/B, and SAC1, resulting in the formation of membrane contact sites between the ER and AiV replication organelles. The AiV-controlled OSBP-mediated cholesterol transport pathway, where ACBD3 participates in tethering between the two organelles, allows the accumulation of cholesterol at AiV replication organelles.
Electron microscopy showed formation of the two types of AiV-induced vesicle-like structures in the cytoplasm in AiV RNA-transfected cells (Fig. 12). The larger vesicles, but not the smaller structures, were found 4 h after transfection with replicon RNA, suggesting that the larger clear vesicles convert into the smaller vesicle-like structures with unclear shapes. For enteroviruses, single-membrane structures were formed at the early stage and transformed into double-membrane structures as infection progressed (53, 54). The AiV-induced larger vesicles were single-membrane structures. On the other hand, the smaller structures were irregularly shaped, and some of them appeared to lack membranes. We could not find the structure surrounded by a clear double membrane that was observed in enterovirus-infected cells (53, 54). Further research is required to clarify the morphological alteration of intracellular membranes that occurred in AiV-replicating cells. The local lipid composition generates membrane curvature (55). Accumulation of cholesterol and PI4P may be involved in membrane curvature, resulting in the formation of vesicle-like structures that serve as platforms for RNA replication.
In conclusion, this study indicates that AiV hijacks the OSBP-mediated cholesterol transport pathway to maintain the lipid homeostasis of AiV ROs through protein-protein interactions that include the newly found ones involving ACBD3. Enteroviruses and HCV also hijack the same pathway, but they accomplish this by producing PI4P. Previously, our studies and others revealed different requirements for ACBD3 in viral RNA replication among AiV, enteroviruses, and HCV (23, 26, 27, 30, 56). Also, AiV and enteroviruses are known to use a PI-4 kinase different from that used by HCV and cardioviruses for PI4P production on their ROs (16–25). This study has provided new evidence demonstrating the commonality and diversity of the strategies of positive-strand RNA viruses for cholesterol accumulation at the ROs.
MATERIALS AND METHODS
Plasmids for expressing AiV proteins.
The plasmid pCMV-polyprotein expresses the AiV polyprotein under the control of the CMV promoter in mammalian cells (29). The plasmids pCI-HA-L, -2B, -2C, -2BC, -3A, and -3AB were described previously (23). Plasmids for expression of 2B, 2C, and 3A without the HA tag sequence were described previously (29).
Clones of ACBD3 and PI4KB.
The plasmid pCI-HA-ACBD3 was constructed by replacing the FLAG tag sequence inserted into the NheI/MluI sites of pCI-FLAG-ACBD3 (23) with the HA tag sequence, which was obtained by annealing the oligonucleotides NheI-kozak-HA-MluI Fw (5′-CTAGCCACCATGGAGTACCCATACGACGTACCAGATTACGCTA-3′) and NheI-kozak-HA-MluI-Rv (5′-CTAGCCACCATGGAGTACCCATACGACGTACCAGATTACGCTA-3′). pACT- and pBIND-ACBD3 and pACT- and pBIND-PI4KB were described previously (23).
pCI-FLAG-VAPA and pCI-FLAG-VAPB.
The VAPA- and VAPB-coding regions were amplified by reverse transcription (RT)-PCR from HeLa cell total RNA using the following primer pairs containing MluI/XbaI restriction sites: Mlu-VAPA-Fw1 (5′-CGACGCGTATGGCGTCCGCCTCAGG-3′) and Xba-VAPA-Rv (5′-GCTCTAGACTACAAGATGAATTTCCCTAGAAAG-3′) for VAPA and Mlu-VAPB-Fw1 (5′-CGACGCGTATGGCGAAGGTGGAGCAGG-3′) and Xba-VAPB-Rv (5′-GCTCTAGACTACAAGGCAATCTTCCCAATAAT-3′) for VAPB. The PCR products were digested by MluI and XbaI and cloned into the MluI/XbaI sites of pCI-FLAG-ACBD3 (23), generating pCI-FLAG-VAPA and pCI-FLAG-VAPB. The coding regions of VAPA and VAPB cloned were 249 and 243 amino acids (aa), respectively, in length.
pACT- and pBIND-VAPA and pACT- and pBIND-VAPB.
The VAPA- and VAPB-coding sequences were obtained by PCR using pCI-FLAG-VAPA and pCI-FLAG-VAPB, respectively, as templates and the following primer pairs: Mlu-VAPA-Fw2 (5′-CGACGCGTGGATGGCGTCCGCCTCAGGGGC-3′) and EcoRV-VAPA-Rv (5′-ATCCTACAAGATGAATTTCCCTAGAAAG-3′) for VAPA and Mlu-VAPB-Fw2 (5′-CGACGCGTGGATGGCGAAGGTGGAGCAGGT-3′) and EcoRV-VAPB-Rv (5′-ATCCTACAAGGCAATCTTCCCAATAATTAC-3′) for VAPB. The PCR fragments were digested with MluI and inserted into MluI/EcoRV-digested pACT and pBIND, generating pACT- and pBIND-VAPA or pACT- and pBIND-VAPB, respectively.
pCI-FLAG-OSBP.
The OSBP-coding sequence was amplified from pLEGFP-N1-hOSBP (36), which was kindly provided by M. Arita (National Institute of Infectious Diseases, Tokyo, Japan) using the primers MluI-OSBP-F (5′-AAACGCGTATGGCGGCGACGGAGCTGAGA-3′) and Sal-OSBP-Rv (5′-AAGTCGACTCAGAAAATGTCCGGGCATGAG-3′). The amplicon was digested with MluI and SalI and then cloned into the pCI vector containing a FLAG tag sequence, generating pCI-FLAG-OSBP.
pACT- and pBIND-OSBP.
The 3′ half of the OSBP-coding sequence (the XbaI-BamHI [blunt ended] fragment of pLEGFP-N1) was cloned into the XbaI-NotI (blunt ended) sites of pACT and pBIND. Then, the 5′ half of the OSBP-coding sequence (the SacI [blunt ended]-XbaI fragment of pLEGFP-N1-hOSBP) was cloned to the EcoRV-XbaI sites of pACT and pBIND, which contain the 3′ half of the OSBP sequence, generating pACT- and pBIND-OSBP.
pACT- and pBIND-CERT.
The plasmid, pEGFP-hCERT (57), which was kindly provided by K. Hanada (National Institute of Infectious Diseases, Tokyo, Japan), was digested with HindIII (blunt ended) and KpnI, and the resultant fragment containing the human CERT (hCERT)-coding sequence was cloned between the BamHI (blunt ended) and KpnI sites of pACT and pBIND, generating pACT- and pBIND-CERT.
pCI-FLAG-CERT.
The hCERT-coding sequence was amplified from pBS-hCERT (58), which was kindly provided by K. Hanada (National Institute of Infectious Diseases, Tokyo, Japan), using the primers XbaI-CERT-F (5′-TTTCTAGAATGTCGGATAATCAGAGCTGG-3′) and CERT-M (5′-CTAGAACAAATAGGCTTTCCTG-3′). The amplicon was digested with XbaI and cloned into the XbaI-SmaI sites of the pCI mammalian expression vector (Promega) containing a FLAG tag sequence between the NheI and MluI sites, generating pCI-FLAG-CERT.
pACT- and pBIND-SAC1.
The coding sequence of SAC1 was amplified by PCR using the following primer pair: Mlu-SAC1-Fw (5′-CGACGCGTGGATGGCGACGGCGGCCTAC-3′) and EcoRV-SAC1-Rv (5′-ATCTCAGTCTATCTTTTCTTTCTGGAC-3′) from a cDNA clone (Kazusa Genome Technologies Inc.; pFN21ASDA0851). The PCR products were digested with MluI and cloned into MluI/EcoRV-digested pACT and pBIND, yielding pACT- and pBIND-SAC1.
pCI-FLAG-SAC1.
pCI-FLAG-ACBD3 (23) was digested with MluI and NotI and then inserted into the cassette that was obtained by annealing the oligonucleotides pCI-BindAct MluI-NotI cassette Fw (5′-CGCGGGACGCGTTGATATCATCTAGAGC-3′) and pCI-BindAct MluI-NotI cassette Rv (5′-GGCCGCTCTAGATGATATCAACGCGTCC-3′), resulting in pCI-FLAG-cassette. pCI-FLAG-cassette was digested with MluI and NotI and cloned into the corresponding sites in pACT-SAC1, generating pCI-FLAG-SAC1.
Antibodies and reagents.
Rabbit or guinea pig polyclonal anti-2B, -2C, -3A, and -L antibodies; rabbit polyclonal anti-FLAG antibody; mouse monoclonal anti-HA antibody; rabbit polyclonal anti-ACBD3 antibody; mouse monoclonal anti-dsRNA antibody; and mouse monoclonal IgM anti-PI4P were described previously (23, 29). Rabbit polyclonal anti-OSBP, anti-VAPA, and anti-VAPB antibodies were purchased from Atlas Antibodies; rabbit polyclonal anti-SAC1 antibody (ab102983) was purchased from Abcam; rabbit polyclonal anti-PITPNB antibody was purchased from Sigma; and rabbit polyclonal anti-CERT antibody was purchased from Atlas Antibodies and Bethyl Laboratories. Rabbit polyclonal anti-calnexin and anti-calregulin antibodies were obtained from Santa Cruz Biotechnology. Secondary anti-mouse, anti-rabbit, and anti-guinea pig antibodies coupled to Alexa Fluor 488, 594, or 350 were purchased from Molecular Probes, and AMCA-conjugated anti-guinea pig was acquired from Millipore. Guinea pig antibodies that recognized either VAPA or VAPB were prepared (Medical & Biological Laboratories Co., Ltd. [MBL], Nagoya, Japan) by using the variable central domain of VAPA and VAPB as antigens, according to the method of Teuling et al. (59). The VAPA nucleotide sequence encoding aa 139 to 232 and the VAPB nucleotide sequence encoding aa 132 to 225 were amplified from pACT-VAPA using the primers Bam-VAPA-415Fw (5′-AAGGATCCTTGAATGATATGGAACCTAGC-3′) and Sal-VAPA-696Rv (5′-AAAGTCGACTTAAAGAAGTGAAGGAAGAGG-3′) and from pACT-VAPB using the primers Bam-VAPB-394Fw (5′-AAGGATCCCCACATGATGTAGAAATAAATA-3′) and Sal-VAPB-675Rv (5′-AAAGTCGACTTACAAGAGCCGGGTGCTAAG-3′) and cloned into pGEX-6P3 (Amersham Pharmacia Biotech). The glutathione S-transferase (GST) fusion proteins were expressed in Escherichia coli cells as described in the manufacturer's protocol and purified as described previously (23), and antibodies against these proteins were raised in guinea pigs (MBL, Nagoya, Japan). Filipin III was purchased from Cayman Chemical. 25-HC was obtained from Sigma and was added to the culture medium at a concentration of 3.1 μM. T00127-HEV1 (42) was kindly provided by M. Arita (National Institute of Infectious Diseases, Tokyo, Japan) and added to the culture medium at a concentration of 5 μM.
AiV replicon RNA.
pAV-FL-Luc-5′rzm carries full-length AiV cDNA in which the capsid-coding region is replaced by a firefly luciferase gene (60, 61). In vitro transcription and luciferase assays for examining viral RNA replication were described previously (23).
RNA and DNA transfection.
The replicon RNA was electroporated into Vero cells for analysis of the effect of 25-HC treatment and immunostaining, as described previously (23). To transfect siRNA-treated cells with the replicon RNA, Lipofectin reagent (Invitrogen) was used. DNA transfection was performed as described previously (23, 29).
Gene silencing with siRNA.
Control siRNA (On-Target Plus nontargeting siRNA 1) and siRNAs (On-Target Plus Smart Pool siRNA) were purchased from Dharmacon. Transfection of Vero cells with siRNA was performed as described previously (23).
M2H assays.
M2H assays were performed as described previously using a Checkmate mammalian two-hybrid system (Promega) (62).
Coimmunoprecipitation and immunoblotting.
Coimmunoprecipitation assays and immunoblotting were performed as described previously (23). Densitometry was performed using ImageJ software.
Immunofluorescence and lipid staining.
Immunostaining of Vero cells were performed as described previously (23, 29). For filipin III staining, Vero cells were cultured in medium containing 5% lipid-free fetal bovine serum for 24 h, followed by electroporation with replicon RNA or transfection with pCMV-polyprotein. The cells were fixed with 4% paraformaldehyde for 45 min and then permeabilized with phosphate-buffered saline (PBS) containing 0.2% saponin and 3% bovine serum albumin (BSA) for 30 min (except for dsRNA staining, in which the permeabilization step was omitted). The cells were incubated with appropriate primary and secondary antibodies diluted in PBS-3% BSA supplemented with 0.05 mg/ml filipin III and 0.05% saponin or Can Get Signal immunostain solution A (Toyobo). For 25-HC treatment, cells were grown in medium with 3.1 μM 25-HC for 24 h prior to and after transfection with pCMV-polyprotein. T00127-HEV1 treatment was performed as described previously (29). All images were acquired using a fluorescence microscope (Keyence BZ-8000).
Cell viability.
The viability of cells treated with siRNA or 25-HC was determined using the CellTiter-Glo luminescent cell viability assay (Promega).
Membrane flotation analysis.
Membrane flotation analysis was performed according to the method of Shi et al. (63) and Shih et al. (64) with modifications. Vero cells were mock transfected or transfected with AV-FL-Luc-5′rzm RNA by electroporation, and 6 h after transfection, the cells were scraped from the culture dishes. The cells were resuspended in 1 ml of hypotonic buffer (10 mM Tris-HCl [pH 7.5], 10 mM KCl, 5 mM MgCl2), incubated for 15 min on ice, and then passed through a 26-gauge needle 15 times. The homogenate was centrifuged at 1,000 × g for 5 min at 4°C to precipitate the nuclei and unbroken cells, and the supernatant was divided into two aliquots, one of which was treated with 1% NP-40 for 1 h on ice. Each aliquot (0.45 ml) was mixed with 1.8 ml of 72% sucrose in low-salt buffer (LSB) (50 mM Tris-HCl [pH 7.5], 25 mM KCl, 5 mM MgCl2) and overlaid with 2.25 ml of 55% sucrose in LSB and 0.5 ml of 10% sucrose in LSB. The sucrose gradient was centrifuged at 37,000 rpm for 14 h at 4°C in a Beckman SW55Ti rotor, and six fractions (0.83 ml) were collected from the top of the gradient. Each fraction was concentrated using a 10-kDa-cutoff filter (Amicon ultra-0.5 ml) and then subjected to immunoblot analysis.
Electron microscopy.
Vero cells were mock electroporated or electroporated with AiV replicon RNA and harvested 4 or 6 h after electroporation. Sample preparation and electron microscopy were performed as described previously (65).
ACKNOWLEDGMENTS
We thank Minetaro Arita for kindly providing T00127-HEV1 and pLEGFP-N1-hOSBP, Kentaro Hanada for his generous gifts of pBS-hCERT and pEGFP-hCERT, Tom Kouki for critical comments regarding EM, and Anna Nomura for excellent technical assistance.
This study was supported in part by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science; a Grant-in-Aid for Strategic Research Base Development Program for Private Universities from the Ministry of Education, Culture, Sports, Science and Technology, Japan; a Grant-in-Aid from the Japan Agency for Medical Research and Development; and a research grant from the Naito Foundation.
REFERENCES
- 1.Miller S, Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 6:363–374. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.den Boon J, Diaz A, Ahlquist P. 2010. Cytoplasmic viral replication complexes. Cell Host Microbe 8:77–85. doi: 10.1016/j.chom.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagy PD, Pogany J. 2011. The dependence of viral RNA replication on co-opted host factors. Nat Rev Microbiol 10:137–149. doi: 10.1038/nrmicro2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aldabe R, Barco A, Carrasco L. 1996. Membrane permeabilization by poliovirus proteins 2B and 2BC. J Biol Chem 271:23134–23137. doi: 10.1074/jbc.271.38.23134. [DOI] [PubMed] [Google Scholar]
- 5.Cho MW, Teterina N, Egger D, Bienz K, Ehrenfeld Z. 1994. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 202:129–145. doi: 10.1006/viro.1994.1329. [DOI] [PubMed] [Google Scholar]
- 6.Egger D, Teterina N, Ehrenfeld E, Bienz K. 2000. Formation of the poliovirus replication complex requires coupled viral translation, vesicle production, and viral RNA synthesis. J Virol 74:6570–6580. doi: 10.1128/JVI.74.14.6570-6580.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knox C, Moffat K, Ali S, Ryan M, Wileman T. 2005. Foot-and-mouth disease virus replication sites form next to the nucleus and close to the Golgi apparatus, but exclude marker proteins associated with host membrane compartments. J Gen Virol 86:687–696. doi: 10.1099/vir.0.80208-0. [DOI] [PubMed] [Google Scholar]
- 8.Krogerus C, Samuilova O, Pöyry T, Jokitalo E, Hyypiä T. 2007. Intracellular localization and effects of individually expressed human parechovirus 1 non-structural proteins. J Gen Virol 88:831–841. doi: 10.1099/vir.0.82201-0. [DOI] [PubMed] [Google Scholar]
- 9.Moffat K, Howell G, Knox C, Belsham GJ, Monaghan P, Ryan MD, Wileman T. 2005. Effects of foot-and-mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum-to-Golgi transport. J Virol 79:4382–4395. doi: 10.1128/JVI.79.7.4382-4395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suhy DA, Giddings TH, Kirkegaard K. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol 74:8953–8965. doi: 10.1128/JVI.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teterina NL, Gorbalenya AE, Egger D, Bienz K, Ehrenfeld E. 1997. Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J Virol 71:8962–8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Towner JS, Ho TV, Semler BL. 1996. Determinants of membrane association for poliovirus protein 3AB. J Biol Chem 271:26810–26818. doi: 10.1074/jbc.271.43.26810. [DOI] [PubMed] [Google Scholar]
- 13.Balla A, Balla T. 2006. Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol 16:351–361. doi: 10.1016/j.tcb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 14.D'Angelo G, Vicinanza M, Di Campli A, De Matteis MA. 2008. The multiple roles of PtdIns(4)P—not just the precursor of PtdIns(4,5)P2. J Cell Sci 121:1955–1963. doi: 10.1242/jcs.023630. [DOI] [PubMed] [Google Scholar]
- 15.Graham TR, Burd CG. 2011. Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol 21:113–121. doi: 10.1016/j.tcb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. 2009. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci U S A 106:7577–7582. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger KL, Kelly SM, Jordan TX, Tartell MA, Randall G. 2011. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase IIIalpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J Virol 85:8870–8883. doi: 10.1128/JVI.00059-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim YS, Hwang SB. 2011. Hepatitis C virus NS5A protein interacts with phosphatidylinositol 4-kinase type IIIα and regulates viral propagation. J Biol Chem 286:11290–11298. doi: 10.1074/jbc.M110.194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tai AW, Salloum S. 2011. The role of the phosphatidylinositol 4-kinase PI4KA in hepatitis C virus-induced host membrane rearrangement. PLoS One 6:e26300. doi: 10.1371/journal.pone.0026300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorobantu CM, Albulescu L, Harak C, Feng Q, van Kampen M, Strating JR, Gorbalenya AE, Lohmann V, van der Schaar HM, van Kuppeveld FJ. 2015. Modulation of the host lipid landscape to promote RNA virus replication: the picornavirus encephalomyocarditis virus converges on the pathway used by hepatitis C virus. PLoS Pathog 11:e1005185. doi: 10.1371/journal.ppat.1005185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sasaki J, Ishikawa K, Arita M, Taniguchi K. 2012. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J 31:754–766. doi: 10.1038/emboj.2011.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greninger AL, Knudsen GM, Betegon M, Burlingame AL, DeRisi JL. 2012. The 3A protein from multiple picornaviruses utilizes the Golgi adaptor protein ACBD3 to recruit PI4KIIIβ. J Virol 86:3605–3616. doi: 10.1128/JVI.06778-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roulin PS, Lötzerich M, Torta F, Tanner LB, van Kuppeveld FJ, Wenk MR, Greber UF. 2014. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe 16:677–690. doi: 10.1016/j.chom.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Dorobantu CM, van der Schaar HM, Ford LA, Strating JR, Ulferts R, Fang Y, Belov G. 2014. Recruitment of PI4KIIIβ to coxsackievirus B3 replication organelles is independent of ACBD3, GBF1, and Arf1. J Virol 88:2725–2736. doi: 10.1128/JVI.03650-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei X, Xiao X, Zhang Z, Ma Y, Qi J, Wu C, Xiao Y, Zhou Z, He B, Wang J. 2017. The Golgi protein ACBD3 facilitates enterovirus 71 replication by interacting with 3A. Sci Rep 7:44592. doi: 10.1038/srep44592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamashita T, Kobayashi S, Sakac K, Nakata S, Chiba S, Ishihara Y, Isomura S. 1991. Isolation of cytopathic small round viruses with BS-C-1 cells from patients with gastroenteritis. J Infect Dis 164:954–957. doi: 10.1093/infdis/164.5.954. [DOI] [PubMed] [Google Scholar]
- 29.Ishikawa-Sasaki K, Sasaki J, Taniguchi K. 2014. A complex comprising phosphatidylinositol 4-kinase IIIβ, ACBD3, and Aichi virus proteins enhances phosphatidylinositol 4-phosphate synthesis and is critical for formation of the viral replication complex. J Virol 88:6586–6598. doi: 10.1128/JVI.00208-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dorobantu CM, Ford-Siltz LA, Sittig SP, Lanke KH, Belov GA, van Kuppeveld FJ, van der Schaar HM. 2015. GBF1- and ACBD3-independent recruitment of PI4KIIIβ to replication sites by rhinovirus 3A proteins. J Virol 89:1913–1918. doi: 10.1128/JVI.02830-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Téoulé F, Brisac C, Pelletier I, Vidalain PO, Jégouic S, Mirabelli C, Bessaud M, Combelas N, Autret A, Tangy F, Delpeyroux F. 2013. The Golgi protein ACBD3, an interactor for poliovirus protein 3A, modulates poliovirus replication. J Virol 87:11031–11046. doi: 10.1128/JVI.00304-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levine TP, Munro S. 2002. Targeting of Golgi-specific pleckstrin homology domains involves both PtdIns 4-kinase-dependent and -independent components. Curr Biol 12:695–704. [DOI] [PubMed] [Google Scholar]
- 33.Wyles JP, McMaster CR, Ridgway ND. 2002. Vesicle-associated membrane protein-associated protein-A (VAP-A) interacts with the oxysterol-binding protein to modify export from the endoplasmic reticulum. J Biol Chem 277:29908–29918. doi: 10.1074/jbc.M201191200. [DOI] [PubMed] [Google Scholar]
- 34.de Saint-Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B, Drin G. 2011. Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J Cell Biol 195:965–978. doi: 10.1083/jcb.201104062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mesmin B, Bigay J, von Filseck JM, Lacas-Gervais S, Drin G, Antonny B. 2013. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155:830–843. doi: 10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 36.Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H. 2013. Oxysterol-binding protein (OSBP) family I is the target of minor enviroxime-like compounds. J Virol 87:4252–4260. doi: 10.1128/JVI.03546-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arita M. 2014. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol Immunol 58:239–256. doi: 10.1111/1348-0421.12144. [DOI] [PubMed] [Google Scholar]
- 38.Wang H, Perry JW, Lauring AS, Neddermann P, De Francesco R, Tai AW. 2014. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 146:1373–1385. doi: 10.1053/j.gastro.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carvou N, Holic R, Li M, Futter C, Skippen A, Cockcroft S. 2010. Phosphatidylinositol- and phosphatidylcholine-transfer activity of PITPβ is essential for COPI-mediated retrograde transport from the Golgi to the endoplasmic reticulum. J Cell Sci 123:1262–1273. doi: 10.1242/jcs.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohde HM, Cheong FY, Konrad G, Paiha K, Mayinger P, Boehmelt G. 2003. The human phosphatidylinositol phosphatase SAC1 interacts with the coatomer I complex. J Biol Chem 278:52689–52699. doi: 10.1074/jbc.M307983200. [DOI] [PubMed] [Google Scholar]
- 41.Nemoto Y, Kearns BG, Wenk MR, Chen H, Mori K, Alb JG Jr, De Camilli P, Bankaitis VA. 2000. Functional characterization of a mammalian Sac1 and mutants exhibiting substrate-specific defects in phosphoinositide phosphatase activity. J Biol Chem 275:34293–34305. doi: 10.1074/jbc.M003923200. [DOI] [PubMed] [Google Scholar]
- 42.Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H. 2011. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J Virol 85:2364–2372. doi: 10.1128/JVI.02249-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao L, Aizaki H, He J-W, Lai MM. 2004. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J Virol 78:3480–3488. doi: 10.1128/JVI.78.7.3480-3488.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evans MJ, Rice CM, Goff SP. 2004. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc Natl Acad Sci U S A 101:13038–13043. doi: 10.1073/pnas.0405152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Yamada O, Sakamoto T, Yoshida H, Iwai T, Matsushita Y, Shimamura H, Araki H, Shimotohno K. 2004. Down-regulation of viral replication by adenoviral-mediated expression of siRNA against cellular cofactors for hepatitis C virus. Virology 320:135–143. doi: 10.1016/j.virol.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 46.Hamamoto I, Nishimura Y, Okamoto T, Aizaki H, Liu M, Mori Y, Abe T, Suzuki T, Lai MM, Miyamura T, Moriishi K. 2005. Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J Virol 79:13473–13482. doi: 10.1128/JVI.79.21.13473-13482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barajas D, Xu K, de Castro Martín IF, Sasvari Z, Brandizzi F, Risco C, Nagy PD. 2014. Co-opted oxysterol-binding ORP and VAP proteins channel sterols to RNA virus replication sites via membrane contact sites. PLoS Pathog 10:e1004388. doi: 10.1371/journal.ppat.1004388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strating JR, van der Linden L, Albulescu L, Bigay J, Arita M, Delang L, Leyssen P, van der Schaar HM, Lanke KH, Thibaut HJ, Ulferts R. 2015. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep 10:600–615. doi: 10.1016/j.celrep.2014.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wakana Y, Kotake R, Oyama N, Murate M, Kobayashi T, Arasaki K, Inoue H, Tagaya M. 2015. CARTS biogenesis requires VAP-lipid transfer protein complexes functioning at the endoplasmic reticulum-Golgi interface. Mol Biol Cell 26:4686–4699. doi: 10.1091/mbc.E15-08-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sohda M, Misumi Y, Yamamoto A, Yano A, Nakamura N, Ikehara Y. 2001. Identification and characterization of a novel Golgi protein, GCP60, that interacts with the integral membrane protein giantin. J Biol Chem 276:45298–45306. doi: 10.1074/jbc.M108961200. [DOI] [PubMed] [Google Scholar]
- 51.Saito S, Matsui H, Kawano M, Kumagai K, Tomishige N, Hanada K, Echigo S, Tamura S, Kobayashi T. 2008. Protein phosphatase 2Cε is an endoplasmic reticulum integral membrane protein that dephosphorylates the ceramide transport protein CERT to enhance its association with organelle membranes. J Biol Chem 283:6584–6593. doi: 10.1074/jbc.M707691200. [DOI] [PubMed] [Google Scholar]
- 52.Shinoda Y, Fujita K, Saito S, Matsui H, Kanto Y, Nagaura Y, Fukunaga K, Tamura S, Kobayashi T. 2012. Acyl-coA binding domain containing 3 (ACBD3) recruits the protein phosphatase PPM1L to ER-Golgi membrane contact sites. FEBS Lett 586:3024–3029. doi: 10.1016/j.febslet.2012.06.050. [DOI] [PubMed] [Google Scholar]
- 53.Limpens RW, van der Schaar HM, Kumar D, Koster AJ, Snijder EJ, van Kuppeveld FJ, Bárcena M. 2011. The transformation of enterovirus replication structures: a three-dimensional study of single- and double-membrane compartments. mBio 2:e00166-11. doi: 10.1128/mBio.00166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. 2012. Complex dynamic development of poliovirus membranous replication complexes. J Virol 86:302–312. doi: 10.1128/JVI.05937-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jarsch IK, Daste F, Gallop JL. 2016. Membrane curvature in cell biology: an integration of molecular mechanisms. J Cell Biol 214:375–387. doi: 10.1083/jcb.201604003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hong Z, Yang X, Yang G, Zhang L. 2014. Hepatitis C virus NS5A competes with PI4KB for binding to ACBD3 in a genotype-dependent manner. Antiviral Res 107:50–55. doi: 10.1016/j.antiviral.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 57.Kawano M, Kumagai K, Nishijima M, Hanada K. 2006. Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J Biol Chem 281:30279–30288. doi: 10.1074/jbc.M605032200. [DOI] [PubMed] [Google Scholar]
- 58.Hanada K, Kumagai K, Yasuda S, Miura Y. 2003. Molecular machinery for non-vesicular trafficking of ceramide. Nature 426:803–809. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
- 59.Teuling E, Ahmed S, Haasdijk E, Demmers J, Steinmetz MO, Akhmanova A, Jaarsma D, Hoogenraad CC. 2007. Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J Neurosci 27:9801–9815. doi: 10.1523/JNEUROSCI.2661-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagashima S, Sasaki J, Taniguchi K. 2003. Functional analysis of the stem-loop structures at the 5′ end of the Aichi virus genome. Virology 313:56–65. doi: 10.1016/S0042-6822(03)00346-5. [DOI] [PubMed] [Google Scholar]
- 61.Nagashima S, Sasaki J, Taniguchi K. 2005. The 5′-terminal region of the Aichi virus genome encodes cis-acting replication elements required for positive- and negative-strand RNA synthesis. J Virol 79:6918–6931. doi: 10.1128/JVI.79.11.6918-6931.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ishikawa K, Sasaki J, Taniguchi K. 2010. Overall linkage map of the nonstructural proteins of Aichi virus. Virus Res 147:77–84. doi: 10.1016/j.virusres.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 63.Shi ST, Polyak SJ, Tu H, Taylor DR, Gretch DR, Lai MM. 2002. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology 292:198–210. doi: 10.1006/viro.2001.1225. [DOI] [PubMed] [Google Scholar]
- 64.Shih WL, Kuo ML, Chuang SE, Cheng AL, Doong SL. 2003. Hepatitis B virus X protein activates a survival signaling by linking Src to phosphatidylinositol 3-kinase. J Biol Chem 278:31807–31813. doi: 10.1074/jbc.M302580200. [DOI] [PubMed] [Google Scholar]
- 65.Nagashima S, Jirintai S, Takahashi M, Kobayashi T, Nishizawa T, Kouki T, Yashiro T, Okamoto H. 2014. Hepatitis E virus egress depends on the exosomal pathway, with secretory exosomes derived from multivesicular bodies. J Gen Virol 95:2166–2175. doi: 10.1099/vir.0.066910-0. [DOI] [PubMed] [Google Scholar]
- 66.Anderson RG. 1998. The caveolae membrane system. Annu Rev Biochem 67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- 67.Pike LJ. 2003. Lipid rafts: bringing order to chaos. J Lipid Res 44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]