

ABSTRACT
Host cell surface receptors are required for attachment, binding, entry, and infection by nonenveloped viruses. Receptor binding can induce conformational changes in the viral capsid and/or the receptor that couple binding with downstream events in the virus life cycle (intracellular signaling, endocytosis and trafficking, and membrane penetration). Virus-receptor interactions also influence viral spread and pathogenicity. The interaction between feline calicivirus (FCV) and its receptor, feline junctional adhesion molecule A (fJAM-A), on host cells is required for infection and induces irreversible, inactivating conformational changes in the capsid of some viral strains. Cryoelectron microscopy (cryo-EM) structures of FCV bound to fJAM-A showed several possible virus-receptor interactions. However, the specific residues on the viral capsid required for binding are not known. Capsid residues that may be involved in postbinding events have been implicated by isolation of soluble receptor-resistant (srr) mutants in which changes in the capsid protein sequence change the capacity of such srr mutants to be inactivated upon incubation with soluble fJAM-A. To clarify which residues on the surface of FCV are required for its interaction with fJAM-A and to potentially identify residues required for postreceptor binding events, we used the existing atomic-resolution structures of FCV and the FCV-fJAM-A cryo-EM structures to select 14 capsid residues for mutation and preparation of recombinant viral capsids. Using this approach, we identified residues on the FCV capsid that are required for fJAM-A binding and other residues that are not required for binding but are required for infection that are likely important for subsequent postbinding events.
IMPORTANCE Feline calicivirus (FCV) is a common cause of mild upper respiratory disease in cats. Some FCV isolates can cause virulent systemic disease. The genetic determinants of virulence for FCV are unknown. We previously found that virulent FCV isolates have faster in vitro growth kinetics than less virulent isolates. Differences in viral growth in vitro may correlate with differences in virulence. Here, we investigated the roles of specific FCV capsid residues on the receptor-virus interaction and viral growth in vitro. We show that the capsid protein genes of the virulent FCV-5 isolate determine its faster in vitro growth kinetics compared to those of the nonvirulent FCV-Urbana infectious clone. We also identified residues on the capsid VP1 protein that are important for receptor binding or for steps subsequent to receptor binding. Our data provide further insight into the specific molecular interactions between fJAM-A and the FCV capsid that regulate binding and infectious entry.
KEYWORDS: feline calicivirus, capsid protein, surface residue, feline junctional adhesion molecule A
INTRODUCTION
Feline calicivirus (FCV) is a member of the Vesivirus genus in the Caliciviridae family. FCV infection may be asymptomatic or cause mild upper respiratory signs, oral ulceration, and fever (1, 2). Like other members of the Caliciviridae, FCV is highly contagious, and morbidity can approach 100% in situations where animals are housed together. Although FCV infection is not normally life threatening, rare isolates of FCV can be highly virulent and cause outbreaks leading to high mortality (40 to 60%) (3–5). Because of its contagiousness, such FCV outbreaks can be devastating when they occur within veterinary hospitals or animal shelters (4, 6).
The functional cell surface receptor for FCV is feline junctional adhesion molecule A (fJAM-A), and introduction of fJAM-A into nonpermissive CHO K1 cells confers susceptibility to FCV (7, 8). JAM-A is a type I transmembrane glycoprotein that consists of two extracellular immunoglobulin (Ig)-like domains, D1 and D2, a transmembrane domain, and a short cytoplasmic tail (9, 10). It is found at tight junctions of endothelial and epithelial cells, where it regulates the integrity and permeability of these junctions. It is also expressed on the surfaces of platelets, leukocytes, and erythrocytes and is involved in leukocyte diapedesis and platelet aggregation (11–14).
The FCV genome is a single strand of positive-sense RNA (∼7.6 kb) that contains three open reading frames (ORFs). The 5′-most ORF, ORF1, encodes a polyprotein that contains the viral protease, the RNA-dependent RNA polymerase, and other nonstructural proteins required for replication and transcription. Two additional ORFs (2 and 3) are located in a subgenomic viral mRNA. These encode the viral structural proteins VP1 and VP2, respectively (15). Ninety dimers of the VP1 protein assemble into a T=3 icosahedral capsid (16). Each VP1 monomer has three structural domains: an N-terminal arm (NTA), a shell (S) domain, and a protruding (P) domain that consists of the P1 and P2 subdomains. The P2 subdomain contains the residues predicted to interact with fJAM-A and is the most variable. FCV VLPs (virus-like particles) self-assemble when the VP1 and VP2 structural proteins are expressed in insect or mammalian cells (17, 18).
Virus-receptor interactions are important determinants of host range, tissue and cell tropism, and disease pathogenesis. Binding of virus to receptor at the cell surface can trigger downstream events important for viral infection, including cell signaling, endocytic uptake and trafficking, and membrane penetration. In some cases, binding of the receptor(s) induces reversible or irreversible conformational changes in the viral capsid (19–22). We have shown that incubation of some (but not all) FCV isolates with purified fJAM-A ectodomain at 37°C leads to loss of infectivity, and that these interactions induce a conformational change in the viral capsid associated with an increase in hydrophobicity (16). FCV isolates that are inactivated upon incubation with the fJAM-A ectodomain were derived from animals with virulent systemic (VS) disease (e.g., FCV-5) or were isolated from kittens with severe pneumonia. In contrast, the F9 vaccine strain, a laboratory isolate (FCV-Urbana), or field isolates associated with mild or inapparent disease were resistant to receptor-mediated inactivation (23). In addition, more virulent FCV isolates have different tropisms in vivo and replicate to higher titers (10- to 100-fold higher yields at 4 h postinfection [p.i.]) in cell culture compared to those of less virulent field isolates of FCV (23, 24). While a virulent isolate (FCV-5) can be inactivated by incubation with the fJAM-A ectodomain, we identified soluble receptor-resistant (srr) mutants of FCV-5 that are resistant to inactivation and mapped the amino acid changes in the VP1 capsid protein that conferred resistance to surface and buried residues (16). Cryo-EM structures of FCV (F9 strain) bound to soluble fJAM-A showed that the D1 domain of fJAM-A bound the P domain capsid and led to substantial changes in the conformation of the capsid, with a 15° counterclockwise rotation of the P domain of the AB dimer and tilting of the P domain of the CC dimer away from the 2-fold axis of symmetry. In these studies, residues on the surface of the P domain dimer that potentially interacted with fJAM-A were proposed, but the low resolution of the structures prevented accurate identification of the interacting interfaces (25, 26). Taken together, these findings suggested a link between virulence, how FCV interacts with fJAM-A, and postreceptor binding conformational changes. We hypothesized that the enhanced virulence and pathogenicity of some FCV isolates is associated with an increase in infection efficiency mediated by changes in the interactions of the capsid with fJAM-A.
To better understand the molecular interactions between FCV and its receptor, fJAM-A, and how this interaction regulates the outcome of infection, we prepared recombinant chimeric viruses that contained the nonstructural ORF1 gene from FCV-Urbana as a backbone, combined with the genes encoding the structural genes encoding VP1 and VP2 from the virulent FCV-5 strain. Under both single- and multiple-cycle growth conditions, the recombinant FCV-5–Urbana chimeric virus (rFCV-5–Urbana) infected cells more efficiently than the recombinant FCV-Urbana (rFCV-Urbana), showing that the capsid genes of FCV are determinants of virus growth kinetics in tissue culture. We further performed sequence alignment of 273 VP1 sequences and identified a set of 14 surface-exposed residues in the P2 domain for mutation. We generated mutations in the background of the recombinant FCV-5–Urbana chimera and tested the capacity of rescued recombinant viruses to replicate and bind to fJAM-A. The residues selected for mutation were either highly conserved or differed between FCV-5 and FCV-Urbana. Three mutants were viable and had growth kinetics similar to those of the wild-type (WT) chimera. For the remaining 11 nonviable mutants, we assessed their capacity to assemble VLPs by expressing their capsid proteins, VP1 and VP2, in insect cells using a baculovirus expression system. Two nonviable mutants (P431A and G433A) did not form detectable VLPs. The remaining 9 mutants assembled VLPs. Of these, 7 (I437A, K479A, V481I, T484A, K493E, D513A, and H515A) bound to fJAM-A expressed on CHO cells and 2 (N459A and N462A) did not. These data suggest that certain surface residues were critical for VLP assembly or stability, that two residues were required for fJAM-A binding, and that seven surface residues are important for a postbinding step that, when blocked, prevents infectious virus recovery.
RESULTS
Recombinant chimeric FCV-5–Urbana grows to significantly higher levels than recombinant FCV-Urbana.
To test the hypothesis that the capsid proteins of FCV contain determinants important for efficient virus entry and spread in tissue culture, we first prepared a recombinant chimeric virus (rFCV-5–Urbana) that contained the FCV-Urbana nonstructural genes (ORF1) fused to the genes encoding the VP1 and VP2 capsid proteins (ORFs 2 and 3) derived from the virulent FCV-5 strain in the backbone of the rFCV-Urbana (pQ14-FCV-Urbana) infectious clone (Fig. 1). We then compared the single-cycle and multiple-cycle growth kinetics of rFCV-5–Urbana with those of rFCV-Urbana and the field isolate FCV-5.
FIG 1.
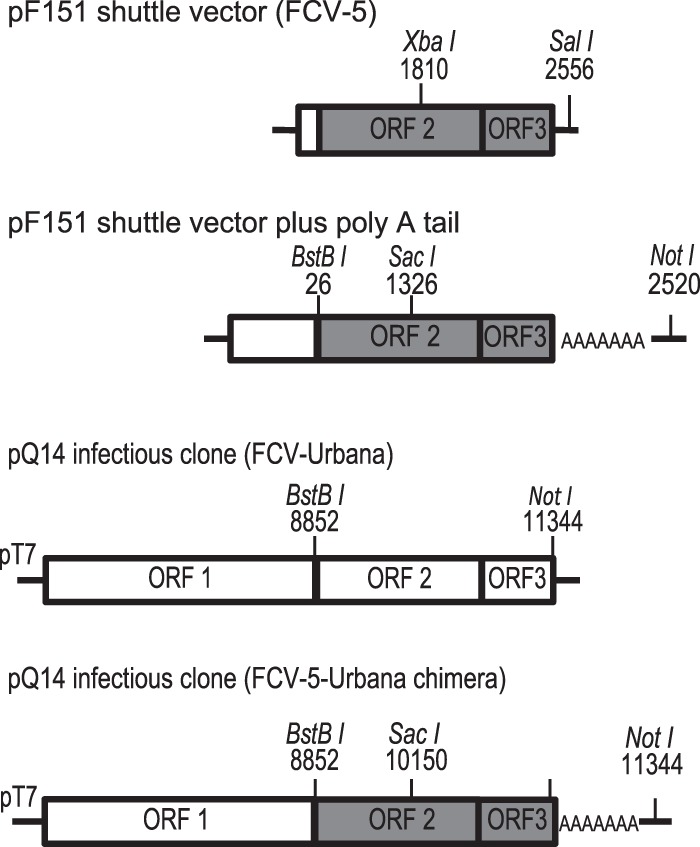
Schematic presentation of pF151 shuttle vector and the parental and chimeric cDNA clones used for preparing recombinant viruses. The restriction sites for cloning are depicted. ORFs 2 and 3 from the virulent FCV-5 strain are shaded gray.
Under single-cycle growth conditions (multiplicity of infection [MOI] of 5), all three viruses grew rapidly during a single round of virus replication with a lag phase of ∼2.5 h, followed by exponential growth that lasted ∼4 h (Fig. 2A). Peak titer was reached at 12 h p.i., corresponding to the end of a single cycle of replication. At 2.5 h p.i., there was no difference in titer; however, for the subsequent time points (from 4 h p.i. to 12 h p.i.), rFCV-5–Urbana had approximately 10-fold higher yield (P < 0.001) than rFCV-Urbana. The yields of the field isolate FCV-5 were between those of rFCV-5–Urbana and rFCV-Urbana and were approximately 5-fold higher than those of rFCV-Urbana at 4 to 12 h p.i.
FIG 2.
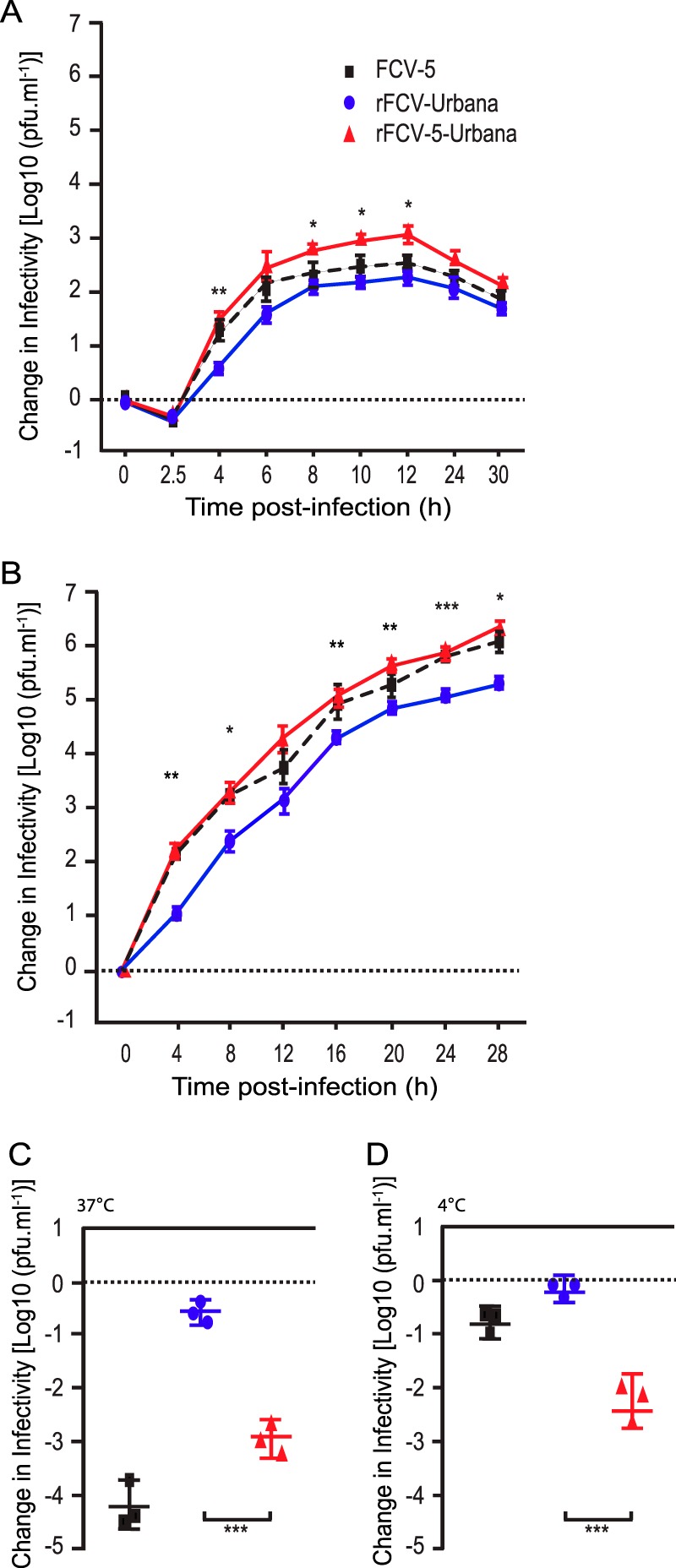
Single- and multiple-cycle growth kinetics of recombinant FCV and their inactivation by soluble GST-fJAM-A. CRFK monolayers were infected with viruses at an MOI of 5 (A) or 0.01 (B), and change in virus titer was determined by plaque assay. The mean log10 titer (log10 titer at each time point minus log10 titer at time zero) from three replicates is shown. Error bars represent the standard deviations from three independent experiments. Statistically significant differences in titer between recombinant FCV-Urbana and FCV-5–Urbana chimera as determined by two-way ANOVA are indicated by asterisks. For virus inactivation, 1 × 105 PFU of each virus was incubated with or without soluble GST-fJAM-A (27.5 mM) at 37°C (C) or 4°C (D) for 30 min. The remaining infectivity of each sample was assayed by plaque titration. The change in log titer was calculated by subtracting the titer of samples incubated with receptor from that of samples incubated with GST alone. The mean change in log10 titer ± SD from three replicates of a representative experiment is shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Under multiple-cycle growth conditions (MOI of 0.01), the growth kinetics of rFCV-5–Urbana were faster than those of rFCV-Urbana virus at all tested time points (Fig. 2B). By 4 h p.i., the yield of rFCV-5–Urbana was equivalent to that for FCV-5 and 10-fold higher than that for rFCV-Urbana (P < 0.001). Based on these results, we concluded that the capsid protein genes derived from FCV-5 contained determinants that led to the faster growth kinetics of FCV-5 than rFCV-Urbana.
Recombinant FCV-5–Urbana differs from rFCV-Urbana and FCV-5 in its inactivation profile by soluble fJAM-A at 37°C and 4°C.
We showed previously that in vitro incubation of purified FCV-5 virions with soluble glutathione S-transferase (GST)-tagged fJAM-A ectodomain (GST-fJAM-A) at 37°C, but not at 4°C, led to a 3- to 4-log decrease in infectivity (16). To assess if rFCV-5–Urbana was also inactivated by incubation with soluble fJAM-A, we incubated 27.5 μM GST-fJAM-A or GST with 106 PFU of rFCV-5–Urbana, rFCV-Urbana, or FCV-5 at either 4°C or 37°C for 30 min and then compared the remaining infectivity. As we had found previously, FCV-5 lost 4 logs of infectivity when incubated with GST-fJAM-A at 37°C but showed minimal change in infectivity when incubated at 4°C (Fig. 2C and D). Recombinant FCV-Urbana showed minimal change in infectivity when incubated with GST-fJAM-A at 37°C or 4°C (Fig. 2C and D). Recombinant FCV-5–Urbana virus lost 3 logs of infectivity when incubated with GST-fJAM-A at 37°C and, unexpectedly, 2 logs of infectivity at 4°C. The changes in infectivity at 37°C suggest that the FCV-5 capsid-fJAM-A interaction causes a conformational change in the capsid upon receptor engagement. The 2-log loss of infectivity of rFCV-5–Urbana at 4°C differed from results for parental FCV-5, which showed minimal infectivity loss at 4°C. This finding suggests that the genome packaged within the capsid also affects capsid stability.
Identification of specific surface residues within the FCV capsid that interact with fJAM-A.
Based on our finding that the capsid of FCV plays important roles in determining viral growth, we further hypothesized that specific interactions between the capsid and fJAM-A regulate the efficiency of cell infection. To test this hypothesis, we aligned 273 published VP1 sequences of FCV to identify conserved residues and mapped the degree of conservation of each residue on the surface of the P2 domain of FCV-5 (Fig. 3A). Highly conserved residues (colored blue) within VP1 align along the dimer interface of the P2 domain of the capsid and within a central pocket; less conserved residues (colored red) lie on the margins of the P2 domain. Residues predicted to interact with fJAM-A based on the cryo-EM structure of the F9 FCV strain bound to the ectodomain of fJAM-A are colored yellow (25) (Fig. 3B). We selected 14 residues to mutagenize in rFCV-5–Urbana on the P2 surface that were either predicted to contact fJAM-A or lay in conserved regions of the capsid (Table 1). Four residues (N478D, V481I, K493E, and R518D) differ between FCV-5 and FCV-Urbana and were mutated accordingly (Fig. 3C). The remaining nine residues (P431, G433, I437, N459, N462, K479, T484, D513, and H515) were highly conserved or were adjacent to highly conserved residues and were mutated to alanine. Conserved residue 454 is an alanine, so we mutated this residue to a serine.
FIG 3.

Surface residues of the P2 domain of FCV-5 VP1 were selected for site-directed mutagenesis in the recombinant FCV-5–Urbana clone. (A) Surface view of the P2 dimer of FCV-5 VP1 protein. Residues targeted for mutagenesis in the recombinant Urbana-FCV-5 clone are labeled with circles. Degree of conservation of surface-exposed residues on the P2 subdomain as predicted by Consurf 3.0. The solid yellow line indicates the dimer interface. (B) fJAM-A contact residues on the surface of the VP1 dimer predicted from the FCV-F9-fJAM-A cryo-EM structure (25) are highlighted in yellow. (C) An alignment of the amino acid sequences within the P2 domain of FCV-5 and FCV-Urbana. Residues 475 to 521 are shown. Nonidentical residues selected for mutagenesis are indicated.
TABLE 1.
Residues on the surface of the P2 subdomain of VP1 chosen for mutagenesis
| Residue in FCV-5 | Mutation | Conserved or variable | fJAM-A binding sites predicted by cryo-EMa |
|---|---|---|---|
| Residues differing between FCV-5 and FCV-Urbana | |||
| N478 | Db | Conserved | No |
| V481 | Ib | Variable | No |
| K493 | Eb | Variable | Yes |
| R518 | Db | Variable | No |
| Conserved residues | |||
| P431 | A | Conserved | No |
| G433 | A | Conserved | Yes |
| I437 | A | Conserved | Yes |
| A454 | S | Conserved | Yes |
| N459 | A | Conserved | No |
| N462 | A | Conserved | Yes |
| K479 | A | Conserved | Yes |
| T484 | A | Conserved | No |
| D513 | A | Conserved | No |
| H515 | A | Conserved | Yes |
Contact residues of VP1 were identified following fitting experiments of vaccine F9 strain with fJAM-A by cryo-EM (25).
These residues correspond to amino acids in FCV-Urbana at that position.
After three independent attempts to rescue mutant rFCV-5–Urbana, we recovered three recombinant mutant viruses (rFCV-5–Urbana-A454S, rFCV-5–Urbana-N478D, and rFCV-5–Urbana-R518D). Two of the viable mutants (N478D and R518D) contained residues that differed between FCV-5 and FCV-Urbana. Two mutants where the FCV-5 residue was mutated to that of FCV-Urbana were not recovered (V481I and K493E).
To investigate if the three viable mutants differed from rFCV-5–Urbana in their in vitro growth, we examined their single- and multiple-cycle growth kinetics. All three viable mutants (A454S, N478D, and R518D) had single- and multiple-cycle growth kinetics similar to those of rFCV-5–Urbana (Fig. 4A and B).
FIG 4.
Single- and multiple-cycle growth kinetics of viable FCV recombinant chimeric mutant viruses and inactivation profiles of these viruses by soluble fJAM-A. CRFK monolayers were infected with viruses at an MOI of 5 (A) or 0.01 (B), and the change in virus titer was determined by plaque assay. The mean change in log10 titer (log10 titer at each time point minus log10 titer at time 0) from three replicates is shown. Error bars represent the standard deviations from three independent experiments. Statistically significant differences in titer between rFCV-Urbana and rFCV-5–Urbana as determined by two-way ANOVA are indicated by asterisks. For virus inactivation, 1 × 105 PFU of virus was incubated in the presence or absence of soluble GST-fJAM-A (27.5 mM) at 37°C (C) or 4°C (D) for 30 min. The infectivity of each sample was assayed by plaque titration. The change in log titer was calculated by subtracting the titer of samples incubated with receptor from that of samples incubated without receptor. The mean change in log10 titer ± SD from three replicates of a representative experiment is shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We then assayed the capacity of soluble receptor to inactivate the viable mutant viruses. Like rFCV-5–Urbana, the mutants rFCV-5–Urbana-A454S and rFCV-5–Urbana-N478D lost about ∼3 logs in titer after incubation with soluble GST-fJAM-A. rFCV-5–Urbana-R518D lost 2 logs of titer at 37°C (Fig. 4C) and had no loss of titer when incubated with soluble fJAM-A at 4°C. Both rFCV-5–Urbana and rFCV-5–Urbana-A454S lost 2 logs of titer when incubated at 4°C, whereas rFCV-5–Urbana-N478D lost ∼1.5 logs of infectivity (Fig. 4D). These data suggest that the viable mutants have similar but not identical inactivation profiles when incubated with soluble fJAM-A.
Capacity of nonviable mutant virus capsid proteins to form VLPs.
Of the 11 nonviable mutants, two had mutations at residues that differed between FCV-5 and FCV-Urbana (V481I and K493E). The remaining 9 mutants had mutations at highly conserved sites on the surface of the P2 domain of the capsid. To determine if any of these mutations affected capsid assembly, we expressed the full-length precursor capsid gene (VP1 and VP2) using a baculovirus expression system and determined their capacity to assemble VLPs. The mutant capsid proteins were all expressed efficiently, with some differences in their level of expression relative to each other and WT FCV-5 capsid proteins (Fig. 5A and B). By negative-stain electron microscopy, purified FCV-5 capsids have a characteristic round appearance (∼35 nm in diameter) with cup-shaped depressions (Fig. 5C). VLPs with shape and size similar to those of capsids of WT FCV-5 virions formed when the full-length VP1 and VP2 capsid proteins of FCV-5 were expressed (Fig. 5C). We were unable to detect VLP formation for 2 (P431A and G433A) of the 11 nonviable mutants. The remaining 9 mutant capsid proteins assembled VLPs (Fig. 5C). Based on these findings, we conclude that mutants P431A and G433A were not recovered as viable viruses because of a failure to assemble capsids or instability of their capsids.
FIG 5.

Expression of recombinant FCV VP1-VP2 capsid proteins in cell lysates (A), after purification (B), andafter formation of VLPs (C), confirmed by negative-stain electron microscopy. VLPs were expressed using a baculovirus system. (A) Equal numbers of Sf9 cells were infected with VLP baculovirus stocks at an MOI of 1, and cell lysates were collected at 3 days p.i. The produced VLPs were purified following ultracentrifugation as described in Materials and Methods. (B) Equal volumes of purified VLPs were loaded per well. VP1 proteins were detected by immunoblotting with anti-capsid antibody 8D1A. Electron microscopy images of baculovirus-expressed FCV-5 VLPs (shown by arrows) were prepared as described in the text and negatively stained with 1% phosphotungstic acid. Mutant VLPs have morphologies similar to those of WT FCV-5 VLPs. Scale bar, 100 nm.
Binding of mutant VLPs to fJAM-A expressed on CHO cells.
To determine if the mutant VLPs were capable of binding fJAM-A, we assessed binding of the mutant VLPs to CHO cells transiently expressing fJAM-A. We initially tried to compare binding levels by flow cytometry, but poor signal to noise prevented discrimination of differences in binding between the VLPs other than binding or not binding. We therefore used immunofluorescence microscopy and scored VLPs as binders or nonbinders (Fig. 6). Of the nine mutant VLPs, two were nonbinders (N459A and N462A). The remaining 7 mutant VLPs bound to fJAM-A (Fig. 6). Binding was specific, as WT and mutant VLPs were unable to bind CHO cells expressing an empty vector. However, differences in the level of binding were not assessed. We conclude from these data that changes at positions 459 and 462 disrupted capsid-fJAM-A interactions and thus likely prevented recovery of viable virions. For the remaining 7 mutants, which were capable of binding fJAM-A but from which we were unable to recover viable viruses, we infer that viable mutant virions were not recovered either because of failure of a postreceptor binding event or because differences in binding that we were unable to assay led to inefficient infection.
FIG 6.

Capacity of VLP capsids to bind fJAM-A expressed on CHO-S cells. Binding was determined by immunofluorescent assay (IFA). CHO-S cells transiently expressing fJAM-A were bound with equal amounts of purified recombinant FCV VLPs at 4°C for 30 min. Unbound capsids were removed by washing, and the expression of fJAM-A and bound FCV capsids was assessed by immunofluorescence microscopy. Expressed fJAM-A (green) and bound (red) VLPs are shown in greyscale and merged images.
DISCUSSION
Differences in how viruses interact with their receptors determine tissue tropism (27), pathogenesis of disease (28, 29), and virus host range (30, 31). Thus, understanding the details of FCV-receptor interactions will provide insight into these processes. Previously, we have shown that differences in virulence between FCV isolates correlates with in vitro growth kinetics (23). In this study, our chimeric virus growth kinetics data showed that the capsid proteins derived from a virulent FCV-5 isolate conferred faster spreading in tissue culture than the parental nonvirulent infectious FCV-Urbana clone. Swapping the viral capsid protein also significantly affected the susceptibility of the recombinant chimeric virus to inactivation upon incubation with soluble GST-fJAM-A at 37°C; rFCV-5–Urbana lost 3 logs of infectious titer, while FCV-Urbana was not affected by incubation with GST-fJAM-A. Previously we showed that most, but not all, srr mutants had significantly lower rates of spread in tissue culture that were similar to those found for the F9 vaccine strain (16). Taken together, these data provide further support for a link between increased virulence of FCV isolates and faster kinetics of infection in vitro. Moreover, they suggest that differences in the interaction of FCV isolates with fJAM-A or downstream events triggered by that binding also correlate with viral growth kinetics and virulence.
The current available low-resolution (9-Å) cryo-EM structure of the FCV-F9 capsid in complex with fJAM-A indicates that fJAM-A binds FCV at the dimer interface of the P2 subdomains (25). Seven FCV-5 srr mutants also map to the surface of the P2 subdomain, close to the dimer interface (16). Therefore, we used this structure together with capsid amino acid residue conservation to guide our choice of 14 mutants. Surprisingly, 2 mutants (V481I and K493E) whose mutations only changed the capsid residue of FCV-5 to that of FCV-Urbana in the rFCV-5–Urbana background were nonviable. These two nonviable mutants assembled VLPs and bound to fJAM-A. These findings suggest that postreceptor binding steps are critical to infectivity but also indicate that small differences, even at residues that differ between strains, can impact viral infectivity.
Mutations at conserved sites of the capsid generally would be expected to compromise a critical step in the virus infectious life cycle. Thus, it was not surprising that only 1 of the 10 conserved surface residues that were mutated was viable (A454S). The remaining nine mutants could be divided into three groups according to their capacity to assemble VLPs and bind fJAM-A. The first group includes those who could not assemble VLPs (Fig. 7B, left). For these mutants (P431A and G433A), the failure to recover recombinant viruses is likely explained by either a failure to assemble VLPs or instability of the VLPs. These two residues were 100% conserved among the aligned VP1 sequences. The second group assembled VLPs but did not bind to fJAM-A on CHO cells (N459A and N462A) (Fig. 7B, middle). Both of these residues lie within the predicted fJAM-A binding region and can be concluded to be required for fJAM binding by rFCV-5–Urbana. However, it is clear from our data that not all of the conserved residues within the region predicted to contact fJAM-A by Bhella and Goodfellow are required for binding (25). Members of the third group assemble VLPs and bind to fJAM-A on CHO cells (I437A, K479A, T484A, D513A, and H515A) (Fig. 7B, right). The reason these mutants were nonviable could be due to the capsid mutation preventing a postbinding step required for infection, such as a capsid conformation change required for delivery of the viral genome into the cytoplasm. Alternatively, because we were unable to quantify and compare the binding of these mutant VLPs to fJAM-A to that of WT VLPs, it is possible that small differences in receptor binding reduced the efficiency of infection below the threshold required to recover viable recombinant viruses.
FIG 7.

Proposed model of receptor-induced conformational change for FCV infection. The red polygon represents the FCV capsid, and viral genome is represented by the black circular line, with VpG shown as a black dot. fJAM-A molecule is shown as a blue dimer at the tight junction between cells. (A) After FCV binds to fJAM-A, it undergoes a conformational change(s) that allows delivery of the genome into the cytoplasm. (B) Possible blocks to infection of the nonviable VP1 capsid mutants: 1, mutant did not assemble VLPs or the VLPs were unstable (P431A and G433A); 2, mutant formed VLP but did not bind to fJAM-A (N459A and N462A); 3, mutants assembled VLPs and bound to fJAM-A but were unable to undergo postbinding steps (perhaps conformational changes) required for subsequent genome delivery (I437A, K479A, V481I, T484A, K493E, D513A, and H515A).
In this study, we used the full-length precursor of VP1 and VP2 to generate VLPs based on the first publication of FCV VLP production (32). VP1 is normally cleaved during FCV infection and the N-terminal 124 amino acids are removed (33). Although the WT FCV-5 VLP resembled the virus under the electron microscope, we noticed that some mutant VLPs were larger and were slightly deformed (such as I437A and H515A). This could be because of the accommodation of the extra 124 amino acids, most likely within the VLP, as the N-terminal arm lies internally. We did not observe any T=1 VLP structures that have been described by others when mature VP1 sequences (amino acids 125 to 668) were expressed in insect cells (34). These differences in size and shape of VLPs may also have affected the interaction of these particles with fJAM-A. It is possible that different results would be obtained if VLPs were expressed using expression constructs that omit the leader region of the capsid.
Because nonenveloped viruses lack a lipid envelope, they are unable to fuse with cellular membranes. After attachment to cell membranes, it has been proposed that nonenveloped viral capsids undergo conformational changes that expose hydrophobic regions or release amphipathic peptides that can disrupt or form pores in cellular membranes (35–37). Our data suggest that a receptor-induced conformational change is triggered in the FCV capsid following initial binding to fJAM-A. For virulent FCV isolates exposed to soluble receptor, this conformational change leads to loss of infectivity and increased capsid hydrophobicity (16). However, for other isolates, the conformational change may be less extreme or represent an intermediate conformation. The model we propose for FCV receptor interaction and subsequent infection (Fig. 7) involves at least two steps: an initial binding reaction to the cell surface receptor followed by a receptor-induced conformational change. It is possible that the substantial loss of infectivity seen following incubation of FCV-5 with fJAM-A represents an irreversible conformational change, whereas intermediate reversible conformational changes may be induced by interactions of lower-virulence isolates with soluble fJAM-A. The enhanced kinetics of growth of virulent FCV isolates may be a consequence of this irreversible change. In summary, our data suggest that conserved surface residues on the P2 subdomain of the capsid are important in regulating fJAM-A interactions and downstream conformational changes that likely influence the kinetics of infection in vitro and the virulence of different FCV isolates in vivo.
MATERIALS AND METHODS
Cells, plasmids, and viruses.
Crandell-Reese feline kidney (CRFK; ATCC CCL-94) cells were grown in Eagle's minimal essential medium (EMEM; CellGro) supplemented with 5% fetal bovine serum (FBS; HyClone), 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 0.25 μg ml−1 amphotericin B, 1 mM sodium pyruvate, and nonessential amino acids (Cellgro). Suspension Chinese hamster ovary (CHO-S) cells (Life Technologies) were grown in CHO-S-SFM II medium (Gibco) supplemented with 0.5× hypoxanthine thymidine (HT) and 8 mM l-glutamine (Life Technologies). Insect Spodoptera frugiperda (Sf9) suspension cells were maintained in Sf-900 SFM medium (Life Technologies) at 28°C.
The virulent FCV-5 isolate was previously characterized (23). Recombinant FCV-Urbana, rFCV-5–Urbana, and mutant chimeric viruses were generated from a full-length infectious cDNA clone, pQ14, as previously described (38). Briefly, CRFK cells were infected with a replication-deficient recombinant vaccinia virus that expresses the T7 RNA polymerase (MVA-T7) at an MOI of 2 for 1 h. One hour after MVA-T7 infection, CRFK cells were transfected with pQ14 constructs using FuGene 6 (Promega). Cells were lysed at 48 h posttransfection by freeze-thawing three times, and viral plaques were isolated on monolayers of CRFK cells overlaid with 2% Bacto agar in CRFK growth medium. Third-passage viral stocks were prepared from viruses that were plaque purified twice and amplified in CRFK cells. Recombinant baculoviruses based on Autographa californica nuclear polyhedrosis virus (AcNPV) were propagated in Sf9 cells as described previously (17, 18).
Plasmids.
A stretch of 46 adenosine nucleotides was synthesized by DNA 2.0 (Life Technologies) and added to the 3′ end of ORFs 2 and 3, derived from FCV-5, and inserted into the shuttle vector pF151 using standard cloning methods to produce pF151 poly(A). The addition of the poly(A) tail to the shuttle vector simplified subsequent subcloning. Site-directed mutagenesis of the FCV-5 VP1 sequences was performed using pF151 poly(A). Sequence from pF151 poly(A) containing FCV-5 ORFs 2 and 3 plus the poly(A) region was used to replace the FCV-Urbana sequences in pQ14. The restriction sites BstBI and NotI were used to remove and replace ORFs 2 and 3 to generate the chimeric infectious plasmid cDNA clone (39).
Site-directed mutagenesis.
We obtained 273 amino acid sequences of the FCV major capsid protein VP1 FCV from NCBI and aligned them using CLUSTAL. We used this alignment together with the structure of the FCV-5 capsid to predict the degree of conservation of surface residues using ConSurf 3.0 (40, 41). We selected 14 surface-exposed residues of FCV-5 VP1 for mutagenesis on the basis of their conservation or predicted contact with fJAM-A. Residues were mutated to alanine or the corresponding residue in the VP1 sequence of FCV-Urbana. The residue at position 454 was highly conserved (269 out of 273 sequences were alanine and the remaining residues were serine) and was changed to serine. The FCV-5 VP1 point mutants were generated using the QuikChange site-directed mutagenesis kit (Agilent) in pF151 poly(A); the PCR mutagenesis primers are available on request. All point mutants were validated by sequencing prior to subcloning into the infectious pQ14 chimeric clone to generate mutant chimeric viruses.
Generation of single- and multiple-cycle growth curves.
Single- and multiple-cycle growth kinetics of recombinant chimeric virus or mutant viruses were performed as described previously (23). Briefly, CRFK cells were infected with viruses at multiplicities of 5 or 0.01 for single- and multiple-cycle growth curves, respectively. Virus was adsorbed to cells for 1 h (time zero) and supplemented with CRFK growth medium. At various times postinfection (p.i.), samples were frozen and stored at −80°C for later titration. Virus growth at each time point was expressed as the change in log plaque titer and calculated by subtracting the log10(PFU ml−1) at time zero from the log10(PFU ml−1) measured at the time point. The means and standard deviations (SD) from three independent experiments are shown.
Inactivation of FCV by soluble fJAM-A-GST protein.
The soluble ectodomain of fJAM-A fused at its C terminus to glutathione S-transferase (fJAM-A-GST) or GST control protein was expressed in Escherichia coli and affinity purified as described previously (7). The virus soluble receptor inactivation assays were performed as described before (16). A total of 106 PFU of rFCV-Urbana, FCV-5, rFCV-5–Urbana, or viable chimeric mutant viruses were incubated with 27.5 μM fJAM-A-GST, GST, or Dulbecco's modified Eagle's medium (DMEM) only for 30 min at 4°C or 37°C, and the remaining infectivity was then determined by plaque assay in CRFK cells.
Construction of recombinant baculoviruses and purification of VLPs.
Full-length precursor capsid VP1 and VP2 sequences from nine nonviable mutants and wild-type FCV-5 in pF151 poly(A) were excised with NotI and ligated into pFastBac1 vector (Life Technologies), which was cut with NotI and treated with recombinant shrimp alkaline phosphatase to prevent self-ligation (New England BioLabs). The resulting pFastBac1-FCV-5 WT and mutant plasmids were used to insert the recombinant genes into baculovirus genomes by Tn7 transposition in DH10Bac competent cells (Life Technologies) according to the manufacturer's instructions. Recombinant baculovirus AcNPV–FCV-5, expressing soluble WT or mutant capsid proteins, was generated in Sf9 cells.
Purified WT or mutant FCV-5 VLPs were prepared using a protocol for human norovirus VLPs, with the modification that VLPs were collected from cell pellets (42). Briefly, the baculovirus titer was first determined by infecting Sf-9 Easy Titer cells using a 96-well plate format endpoint dilution assay (43). Subsequently, 2 × 106 cells ml−1 of Sf9 suspension were infected at an MOI of 1. Three days after infection cells were pelleted by centrifugation, and the pellets were frozen and thawed twice and then suspended in hypotonic lysis buffer (20 mM phosphate, 150 mM NaCl, pH 6, with addition of Pierce protease inhibitor; Thermo Scientific). The suspensions were then sonicated until fully emulsified and centrifuged at 13,000 × g for 45 min, and the clarified supernatants containing FCV VLPs were loaded into 30 to 60% sucrose gradients for ultracentrifugation at 71,000 × g overnight at 4°C. The densest band after centrifugation contained VLPs and was collected. The purity of the expressed VLPs was analyzed by 10% SDS-PAGE followed by Coomassie blue staining, and expression of VP1 was confirmed by immunoblotting using monoclonal 8D1A antibody against VP1 (Custom Monoclonal Antibodies International). To assess the capacity of the nonviable mutant VP1 molecules to assemble into VLPs, aliquots of purified VLPs were applied to Formvar-coated copper grids (Electron Microscopy Sciences) and negatively stained using 1% phosphotungstic acid (Sigma-Aldrich). Specimens were examined by FEI Tecnai Spirit transmission EM (FEI Company).
VLP receptor binding.
To assay the capacity of VLPs to bind fJAM-A, we transiently transfected suspension CHO cells (CHO-S; 106 cells per sample) with a control expression vector (pCI-Neo) or with a vector to express full-length fJAM-A (pCI-fJAM-A) using Fugene 6 (Promega). Transfected CHO-S cells were incubated at 37°C for 24 h prior to binding equal concentrations of mutant VLP proteins on ice for 30 min. After washing (twice), the cells were incubated with fJAM-A rabbit antisera for 30 min on ice, washed three times, and then fixed with 2% paraformaldehyde in PBS for 30 min on ice. Bound VLPs were detected by staining with an FCV monoclonal antibody, S1-9 (Custom Monoclonal Antibodies International), for 30 min. After further washing, the cells were incubated with secondary antibodies Alexa 488-conjugated goat-anti-rabbit and Alexa 594-conjugated goat-anti-mouse IgG for 30 min to detect expression of fJAM-A and binding of VLPs to the cell surface. Immunostained cells were mounted on glass slides with coverslips and examined by fluorescence microscopy. Images were obtained using a Nikon TE2000 inverted microscope equipped with fluorescence optics through a PlanApo 60× 1.40-numerical-aperture oil objective with type A immersion liquid (Nikon) with or without a ×1.5 optical zoom. Images were collected digitally with a Coolsnap HQ charge-coupled-device (CCD) camera (Roper) and NIS Elements software (version 4.30; Nikon) and were processed using Photoshop (CS6; Adobe) and Illustrator (CS6; Adobe) software.
Statistical analysis.
Comparison of viral titers between sets of three replicates were analyzed with GraphPad Prism 7 software using two-way analysis of variance (ANOVA).
ACKNOWLEDGMENT
This research was supported by a grant from the Feline Health Center, Cornell University, and the Morris Animal Foundation (D12FE-002).
REFERENCES
- 1.Hoover EA, Kahn DE. 1975. Experimentally induced feline calicivirus infection: clinical signs and lesions. J Am Vet Med Assoc 166:463–468. [PubMed] [Google Scholar]
- 2.Reubel GH, Hoffmann DE, Pedersen NC. 1992. Acute and chronic faucitis of domestic cats. A feline calicivirus-induced disease. Vet Clin North Am Small Anim Pract 22:1347–1360. doi: 10.1016/S0195-5616(92)50131-0. [DOI] [PubMed] [Google Scholar]
- 3.Coyne KP, Jones BR, Kipar A, Chantrey J, Porter CJ, Barber PJ, Dawson S, Gaskell RM, Radford AD. 2006. Lethal outbreak of disease associated with feline calicivirus infection in cats. Vet Rec 158:544–550. doi: 10.1136/vr.158.16.544. [DOI] [PubMed] [Google Scholar]
- 4.Pedersen NC, Elliott JB, Glasgow A, Poland A, Keel K. 2000. An isolated epizootic of hemorrhagic-like fever in cats caused by a novel and highly virulent strain of feline calicivirus. Vet Microbiol 73:281–300. doi: 10.1016/S0378-1135(00)00183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hurley KE, Pesavento PA, Pedersen NC, Poland AM, Wilson E, Foley JE. 2004. An outbreak of virulent systemic feline calicivirus disease. J Am Vet Med Assoc 224:241–249. doi: 10.2460/javma.2004.224.241. [DOI] [PubMed] [Google Scholar]
- 6.Schorr-Evans EM, Poland A, Johnson WE, Pedersen NC. 2003. An epizootic of highly virulent feline calicivirus disease in a hospital setting in New England. J Feline Med Surg 5:217–226. doi: 10.1016/S1098-612X(03)00008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ossiboff RJ, Parker JS. 2007. Identification of regions and residues in feline junctional adhesion molecule required for feline calicivirus binding and infection. J Virol 81:13608–13621. doi: 10.1128/JVI.01509-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makino A, Shimojima M, Miyazawa T, Kato K, Tohya Y, Akashi H. 2006. Junctional adhesion molecule 1 is a functional receptor for feline calicivirus. J Virol 80:4482–4490. doi: 10.1128/JVI.80.9.4482-4490.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ebnet K, Schulz CU, Meyer Zu Brickwedde MK, Pendl GG, Vestweber D. 2000. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem 275:27979–27988. [DOI] [PubMed] [Google Scholar]
- 10.Mandell KJ, Parkos CA. 2005. The JAM family of proteins. Adv Drug Deliv Rev 57:857–867. doi: 10.1016/j.addr.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, Parkos CA. 2000. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci 113(Part 13):2363–2374. [DOI] [PubMed] [Google Scholar]
- 12.Mandell KJ, McCall IC, Parkos CA. 2004. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J Biol Chem 279:16254–16262. doi: 10.1074/jbc.M309483200. [DOI] [PubMed] [Google Scholar]
- 13.Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E. 1998. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozaki H, Ishii K, Horiuchi H, Arai H, Kawamoto T, Okawa K, Iwamatsu A, Kita T. 1999. Cutting edge: combined treatment of TNF-alpha and IFN-gamma causes redistribution of junctional adhesion molecule in human endothelial cells. J Immunol 163:553–557. [PubMed] [Google Scholar]
- 15.Pesavento PA, Chang KO, Parker JS. 2008. Molecular virology of feline calicivirus. Vet Clin North Am Small Anim Pract 38:775–786. doi: 10.1016/j.cvsm.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Ossiboff RJ, Zhou Y, Lightfoot PJ, Prasad BV, Parker JS. 2010. Conformational changes in the capsid of a calicivirus upon interaction with its functional receptor. J Virol 84:5550–5564. doi: 10.1128/JVI.02371-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Martino B, Marsilio F, Roy P. 2007. Assembly of feline calicivirus-like particle and its immunogenicity. Vet Microbiol 120:173–178. doi: 10.1016/j.vetmic.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 18.Geissler K, Schneider K, Fleuchaus A, Parrish CR, Sutter G, Truyen U. 1999. Feline calicivirus capsid protein expression and capsid assembly in cultured feline cells. J Virol 73:834–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arita M, Koike S, Aoki J, Horie H, Nomoto A. 1998. Interaction of poliovirus with its purified receptor and conformational alteration in the virion. J Virol 72:3578–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belnap DM, McDermott BM Jr, Filman DJ, Cheng N, Trus BL, Zuccola HJ, Racaniello VR, Hogle JM, Steven AC. 2000. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc Natl Acad Sci U S A 97:73–78. doi: 10.1073/pnas.97.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colston E, Racaniello VR. 1994. Soluble receptor-resistant poliovirus mutants identify surface and internal capsid residues that control interaction with the cell receptor. EMBO J 13:5855–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strauss M, Filman DJ, Belnap DM, Cheng N, Noel RT, Hogle JM. 2015. Nectin-like interactions between poliovirus and its receptor trigger conformational changes associated with cell entry. J Virol 89:4143–4157. doi: 10.1128/JVI.03101-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ossiboff RJ, Sheh A, Shotton J, Pesavento PA, Parker JSL. 2007. Feline caliciviruses (FCVs) isolated from cats with virulent systemic disease possess in vitro phenotypes distinct from those of other FCV isolates. J Gen Virol 88:506–517. doi: 10.1099/vir.0.82488-0. [DOI] [PubMed] [Google Scholar]
- 24.Pesavento PA, MacLachlan NJ, Dillard-Telm L, Grant CK, Hurley KF. 2004. Pathologic, immunohistochemical, and electron microscopic findings in naturally occurring virulent systemic feline calicivirus infection in cats. Vet Pathol 41:257–263. doi: 10.1354/vp.41-3-257. [DOI] [PubMed] [Google Scholar]
- 25.Bhella D, Goodfellow IG. 2011. The cryo-electron microscopy structure of feline calicivirus bound to junctional adhesion molecule A at 9-angstrom resolution reveals receptor-induced flexibility and two distinct conformational changes in the capsid protein VP1. J Virol 85:11381–11390. doi: 10.1128/JVI.05621-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhella D, Gatherer D, Chaudhry Y, Pink R, Goodfellow IG. 2008. Structural insights into calicivirus attachment and uncoating. J Virol 82:8051–8058. doi: 10.1128/JVI.00550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spriggs DR, Bronson RT, Fields BN. 1983. Hemagglutinin variants of reovirus type-3 have altered central nervous-system tropism. Science 220:505–507. doi: 10.1126/science.6301010. [DOI] [PubMed] [Google Scholar]
- 28.Forrest JC, Dermody TS. 2003. Reovirus receptors and pathogenesis. J Virol 77:9109–9115. doi: 10.1128/JVI.77.17.9109-9115.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smelt SC, Borrow P, Kunz S, Cao W, Tishon A, Lewicki H, Campbell KP, Oldstone MB. 2001. Differences in affinity of binding of lymphocytic choriomeningitis virus strains to the cellular receptor alpha-dystroglycan correlate with viral tropism and disease kinetics. J Virol 75:448–457. doi: 10.1128/JVI.75.1.448-457.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hueffer K, Parker JS, Weichert WS, Geisel RE, Sgro JY, Parrish CR. 2003. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J Virol 77:1718–1726. doi: 10.1128/JVI.77.3.1718-1726.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parrish CR, Kawaoka Y. 2005. The origins of new pandemic viruses: the acquisition of new host ranges by canine parvovirus and influenza A viruses. Annu Rev Microbiol 59:553–586. doi: 10.1146/annurev.micro.59.030804.121059. [DOI] [PubMed] [Google Scholar]
- 32.DeSilver DA, Guimond PM, Gibson JK, Thomsen DR, Wardley RC, Lowery DE. 1997. Expression of the complete capsid and the hypervariable region of feline calicivirus in the baculovirus expression system, p 131–143 In Chasey R, Gaskell M, Clarke IN (ed), Proceedings of the 1st International Symposium on Calicivirus. European Society for Veterinary Virology, Madrid, Spain. [Google Scholar]
- 33.Sosnovtsev SV, Sosnovtseva SA, Green KY. 1998. Cleavage of the feline calicivirus capsid precursor is mediated by a virus-encoded proteinase. J Virol 72:3051–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burmeister WP, Buisson M, Estrozi LF, Schoehn G, Billet O, Hannas Z, Sigoillot C, Poulet H. 2015. Structure determination of feline calicivirus virus-like particles in the context of a pseudo-octahedral arrangement. PLoS One 10:e0119289. doi: 10.1371/journal.pone.0119289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai B. 2007. Penetration of nonenveloped viruses into the cytoplasm. Annu Rev Cell Dev Biol 23:23–43. doi: 10.1146/annurev.cellbio.23.090506.123454. [DOI] [PubMed] [Google Scholar]
- 36.Chandran K, Farsetta DL, Nibert ML. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein micro 1 mediates membrane disruption. J Virol 76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hogle JM. 2002. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu Rev Microbiol 56:677–702. doi: 10.1146/annurev.micro.56.012302.160757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sosnovtsev SV, Sosnovtseva SA, Green KY. 1997. Recovery of feline calicivirus from plasmid DNA containing a full-length copy of the genome, p 125–130. In Chasey R, Gaskell M, Clarke IN (ed), Proceedings of the 1st International Symposium on Calicivirus. European Society for Veterinary Virology, Madrid, Spain. [Google Scholar]
- 39.Sosnovtsev S, Green KY. 1995. RNA transcripts derived from a cloned full-length copy of the feline calicivirus genome do not require VpG for infectivity. Virology 210:383–390. doi: 10.1006/viro.1995.1354. [DOI] [PubMed] [Google Scholar]
- 40.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. 2010. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38:W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Celniker G, Nimrod G, Ashkenazy H, Glaser F, Martz E, Mayrose I, Pupko T, Ben-Tal N. 2013. ConSurf: using evolutionary data to raise testable hypotheses about protein function. Israel J Chem 53:199–206. doi: 10.1002/ijch.201200096. [DOI] [Google Scholar]
- 42.Huhti L, Blazevic V, Nurminen K, Koho T, Hytonen VP, Vesikari T. 2010. A comparison of methods for purification and concentration of norovirus GII-4 capsid virus-like particles. Arch Virol 155:1855–1858. doi: 10.1007/s00705-010-0768-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hopkins R, Esposito D. 2009. A rapid method for titrating baculovirus stocks using the Sf-9 Easy Titer cell line. Biotechniques 47:785–788. doi: 10.2144/000113238. [DOI] [PubMed] [Google Scholar]