

ABSTRACT
The inflammasome represents a molecular platform for innate immune regulation and controls proinflammatory cytokine production. The NLRP3 inflammasome is comprised of NLRP3, ASC, and procaspase-1. When the NLRP3 inflammasome is activated, it causes ASC speck formation and caspase-1 activation, resulting in the maturation of interleukin-1β (IL-1β). The NLRP3 inflammasome is regulated at multiple levels, with one level being posttranslational modification. Interestingly, ubiquitination of ASC has been reported to be indispensable for the activation of the NLRP3 inflammasome. Influenza A virus (IAV) infection induces NLRP3 inflammasome-dependent IL-1β secretion, which contributes to the host antiviral defense. However, IAVs have evolved multiple antagonizing mechanisms, one of which is executed by viral NS1 protein to suppress the NLRP3 inflammasome. In this study, we compared IL-1β production in porcine alveolar macrophages in response to IAV infection and found that the 2009 pandemic H1N1 induced less IL-1β than swine influenza viruses (SIVs). Further study revealed that the NS1 C terminus of pandemic H1N1 but not that of SIV was able to significantly inhibit NLRP3 inflammasome-mediated IL-1β production. This inhibitory function was attributed to impaired ASC speck formation and suppression of ASC ubiquitination. Moreover, we identified two target lysine residues, K110 and K140, which are essential for both porcine ASC ubiquitination and NLRP3 inflammasome-mediated IL-1β production. These results revealed a novel mechanism by which the NS1 protein of the 2009 pandemic H1N1 suppresses NLRP3 inflammasome activation.
IMPORTANCE Influenza A virus (IAV) infection activates the NLRP3 inflammasome, resulting in the production of IL-1β, which contributes to the host innate immune response. ASC, an adaptor protein of NLRP3, forms specks that are critical for inflammasome activation. Here, we report that the NS1 C terminus of the 2009 pandemic H1N1 has functions to suppress porcine IL-1β production by inhibiting ASC speck formation and ASC ubiquitination. Furthermore, the ubiquitination sites on porcine ASC were identified. The information gained here may contribute to an in-depth understanding of porcine inflammasome activation and regulation in response to different IAVs, helping to further enhance our knowledge of innate immune responses to influenza virus infection in pigs.
KEYWORDS: ASC, ubiquitination, influenza A virus NS1, NLRP3 inflammasome
INTRODUCTION
Pattern recognition receptors (PRRs) play important roles in host innate immune responses to pathogens (1). Nucleotide-binding domain and leucine-rich repeat-containing proteins (NLRs) are one of the families of PRRs, among which the NLR family pyrin domain-containing protein 3 (NLRP3) has been extensively investigated. With the apoptosis-associated speck-like protein containing caspase recruitment domain (ASC) and procaspase-1, NLRP3 forms a cytosolic protein complex called the NLRP3 inflammasome (1, 2). When the NLRP3 inflammasome is activated in response to a variety of pathogen-associated molecular patterns (PAMPs), it can then regulate the secretion of inflammatory cytokines including interleukin-1β (IL-1β), which is a major mediator for inflammation (1, 2). IL-1β is synthesized as an immature form, pro-IL-1β. Maturation of pro-IL-1β relies on NLRP3 inflammasome activation. This activation occurs by the formation of ASC specks and activation of caspase-1, which is converted from procaspase-1 through an auto-processing mechanism (1, 2).
NLRP3 inflammasome-mediated IL-1β production plays a critical role in modulating host immunity to influenza A virus (IAV) infection and IAV-induced lung pathology (3–5). Sensing viral RNAs by Toll-like receptors or retinoic acid-inducible gene I (RIG-I)-like receptors leads to pro-IL-1β and NLRP3 expression (6, 7). Recently, Z-DNA binding protein 1 was identified as another sensor of IAV for NLRP3 inflammasome activation and inflammatory responses (8). Activation of the NLRP3 inflammasome requires a second signal that includes the recognition of viral components or virus-induced intracellular processes through mechanisms that are less clearly understood (6, 7). While viral RNA itself is shown to activate the NLRP3 inflammasome (4, 5), different viral proteins of IAVs are reported to regulate the NLRP3 inflammasome. For example, PB1-F2 protein could activate or inhibit the NLRP3 inflammasome in a virus strain-dependent manner (9–11), whereas the M2 protein contributes to NLRP3 inflammasome-mediated IL-1β production through its ion channel activity (12). NS1 protein, which is well known for its interferon (IFN)-antagonistic function (13), is reported to inhibit both caspase-1 activation (14) and NLRP3-mediated IL-1β production (15–17).
Recent studies have revealed that the NLRP3 inflammasome is regulated by posttranslational modifications. Among different types of modifications, ubiquitination and deubiquitination are known to play diverse roles in the regulation of inflammasomes (18). Deubiquitinases are required for inflammasome-dependent IL-1β production (19), and, more specifically, deubiquitination of NLRP3 is reported to be important for the activation of the NLRP3 inflammasome (20–25). In contrast, autophagy-mediated or proteasomal degradation of ubiquitinated NLRP3 leads to suppression of the NLRP3 inflammasome (26, 27). Upon activation of several different inflammasomes, caspase-1 can be ubiquitinated, which may downregulate the inflammasome activity (28). Also, pro-IL-1β undergoes ubiquitination, which leads to its maturation into IL-1β or its degradation (29–31).
ASC ubiquitination was first reported in a study of absent in melanoma 2 (AIM2) inflammasome-related autophagy in response to double-stranded DNA (32). More recent studies then revealed an effect of ASC ubiquitination on NLRP3 inflammasome activation. While ASC ubiquitination by an E3 ligase, tumor necrosis factor receptor-associated factor 6 (TRAF6), can limit NLRP3 inflammasome activity (33), deubiquitination of ASC is required for NLRP3 inflammasome activation (34). ASC can also be modified by the linear ubiquitin assembly complex, which is required for the activation of the NLRP3 inflammasome (35). In the context of RNA virus infection, ubiquitination of ASC by the E3 ligase TRAF3 upon infection with vesicular stomatitis virus (VSV) is critical for inflammasome-mediated IL-1β production (36). In spite of these studies, it remains unknown how ASC ubiquitination regulates inflammasome activity during IAV infection.
In this study, we focused on porcine NLRP3 inflammasome regulation upon IAV infection in pigs. We first compared IL-1β production in primary porcine alveolar macrophages (PAMs) infected with either swine influenza viruses (SIVs) or the 2009 pandemic H1N1 virus (pH1N1/09). We found that pH1N1/09 is better capable of inhibiting IL-1β production than the SIVs. Further analyses revealed that the NS1 C terminus of the pH1N1/09 strain inhibits NLRP3 inflammasome-mediated IL-1β production by suppressing ASC speck formation and the ubiquitination of porcine ASC. Mutational analyses identified the target lysine residues, K110 and K140, found in porcine ASC to be critical for NLRP3 inflammasome-mediated IL-1β production. Thus, these results revealed a new mechanism of innate immune evasion achieved by the NS1 protein in pH1N1/09.
RESULTS
IAV infection induces NLRP3 inflammasome-mediated IL-1β production in PAMs.
To assess the levels of IL-1β produced from PAMs in response to IAV infection, PAMs were infected with two different subtypes of IAVs isolated from pigs, A/swine/Saskatchewan/18789/2002/H1N1 (Sk02) (H1N1) and A/swine/Texas/4199-2/1998/H3N2 (Tx98) (H3N2), at the multiplicities of infection (MOIs) indicated in the legend to Fig. 1A. At 24 h postinfection (hpi), cell-free supernatants were harvested for porcine IL-1β enzyme-linked immunosorbent assay (ELISA), and the cells were lysed for Western blotting to measure the expression of pro-IL-1β and viral proteins. PAMs from different piglets showed variable levels of IL-1β production in response to SIV infection. However, the differences in IL-1β levels in response to SIVs at different MOIs were reproducible when cells from different piglets were used, and we show representative data using PAMs isolated from a piglet with the lowest background level of IL-1β. Pro-IL-1β levels from PAMs infected at an MOI of 1 or 10 were comparable and were significantly higher than those in mock-infected cells or in cells infected at an MOI of 0.1 (Fig. 1A). In contrast, IL-1β levels in virus-infected cells were significantly higher than those of mock-infected cells and were virus dose dependent. Accordingly, the expression levels of the viral NP and NS1 proteins were also virus dose dependent, confirming that PAMs were infected with SIVs. Based on these results, we chose an MOI of 1 for 24 h in the following infection experiments with PAMs.
FIG 1.

IAV infection induces NLRP3 inflammasome-mediated IL-1β production in PAMs. (A) PAMs were infected with two SIV strains, Sk02 and Tx98, at MOIs of 0.1, 1, and 10. At 24 hpi, cell-free supernatants were harvested for porcine IL-1β ELISA, and cells were lysed for Western blotting to measure the expression of pro-IL-1β and viral proteins. ***, P < 0.001. (B) PAMs were infected with WT or UV-inactivated virus of two SIV strains at an MOI of 1 for 24 h. IL-1β production and pro-IL-1β expression were measured as described for panel A. ***, P < 0.001. (C) PAMs were treated with DMSO or an NLRP3 inhibitor, MNS, and infected with Sk02 at an MOI of 1 for 24 h. IL-1β production was measured as described for panel A. ***, P < 0.001. (D) HEK293T cells were transfected with different combinations of plasmids expressing porcine NLRP3, ASC, procaspase-1, and pro-IL-1β as indicated. At 12 hpt, the cells were mock infected or infected with Sk02 at an MOI of 5. Porcine IL-1β from the cell-free supernatants at 12 hpi was measured by ELISA. The expression of pro-IL-1β, the active caspase-1, p20, and viral proteins was measured by Western blotting in the cell lysates (***, P < 0.001). Results are representative of three independent experiments using PAMs from different piglets.
Since the above results suggested that viral replication is required for IL-1β production in PAMs, we wanted to further confirm this finding by inactivating the two virus strains by UV irradiation. The effect of UV inactivation on virus replication was confirmed by the NS1 protein expression level. Compared to wild-type (WT) viruses, UV-inactivated viruses lost the ability to induce IL-1β production in PAMs (Fig. 1B), indicating that viral replication is necessary for the conversion of pro-IL-1β into mature IL-1β.
Next, we tested whether the NLRP3 inflammasome is involved in IL-1β production in PAMs in response to SIV infection. PAMs were first treated with an NLRP3 inhibitor, 3,4-methylenedioxy-β-nitrostyrene (MNS), which inhibits inflammasome activation by binding to NLRP3 (37), and were then infected with Sk02. Compared to the IL-1β levels of vehicle-treated and virus-infected PAMs, a dose-dependent decrease in IL-1β induction upon inhibitor treatment was observed (Fig. 1C), indicating that porcine IL-1β production in SIV-infected PAMs is mediated by the NLRP3 inflammasome. At the tested concentration of MNS, cells did not show noticeable cytotoxicity (data not shown).
Since primary macrophages are difficult to be transfected and, thus, knockdown or overexpression of genes in PAMs is less practical and inefficient, we sought to reconstitute the porcine NLRP3 inflammasome in HEK293T cells that are deficient in the endogenous human NLRP3 inflammasome (38). We cloned porcine NLRP3, ASC, procaspase-1, and pro-IL-1β genes from PAMs and made corresponding mammalian expression constructs. Cotransfection of all four plasmids led to a moderate level of IL-1β in the supernatant owing to the engagement of NLRP3, ASC, and caspase-1. IL-1β was significantly reduced when pro-IL-1β was expressed alone (Fig. 1D). These results validated the successful setup of our reconstitution system, showing that IL-1β maturation and secretion predominantly rely on the porcine NLRP3 inflammasome. To confirm the dependency of IL-1β production on the NLRP3 inflammasome in the context of IAV infection, the porcine NLRP3 inflammasome was reconstituted in HEK293T cells for 12 h, and then cells were infected with Sk02 at an MOI of 5 for another 12 h. In the presence of all the NLRP3 inflammasome components and pro-IL-1β, infection with Sk02 induced over 5,000 pg/ml of IL-1β and a noticeable level of cleaved caspase-1, p20, which were much higher than those of its counterpart in the absence of virus infection (Fig. 1D). In all samples, viral protein expression and cellular β-actin levels were monitored.
Pandemic influenza virus induces significantly less IL-1β than SIVs.
To examine whether other IAV strains in addition to SIVs also induce IL-1β in PAMs, the 2009 pandemic strain, A/Halifax/210/2009/H1N1 (Hf09), was used. Surprisingly, IL-1β production at 24 hpi from Hf09-infected PAMs was significantly less than that from Sk02-infected PAMs (Fig. 2A). The time course experiments showed that IL-1β production in response to Sk02 increased sharply between 8 and 16 hpi, after the virus established its productive replication at around 8 hpi, as indicated by the viral growth curve (Fig. 2B). In contrast, this increase in IL-1β was not observed in Hf09-infected PAMs, while the viral titers of Hf09 were comparable to those of Sk02 during the time course. These results show that less IL-1β production by Hf09 is not due to the inefficient replication of Hf09 in PAMs, which was also confirmed by viral NS1 expression levels (Fig. 2A). In spite of the fact that the pro-IL-1β level in Sk02-infected PAMs was slightly higher than that in Hf09-infected PAMs (Fig. 2A), the remarkable difference in IL-1β levels was possibly attributed to the suppression of NLRP3 inflammasome activity in cells infected by Hf09.
FIG 2.
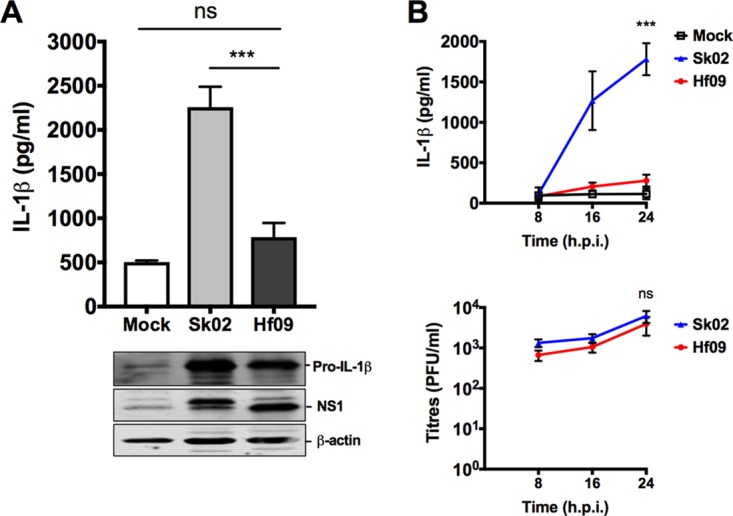
Pandemic H1N1 induces significantly less IL-1β than SIVs. (A) PAMs were infected with an SIV strain, Sk02, or a human pandemic strain, Hf09, at an MOI of 1. At 24 hpi, cell-free supernatants were harvested for IL-1β ELISA, and the cells were lysed for Western blotting to measure the expression of pro-IL-1β and viral NS1 protein. ns, not significant, ***, P < 0.001. (B) PAMs were infected with Sk02 or Hf09 at an MOI of 1, and the supernatants were harvested at 8, 16, and 24 hpi. IL-1β was measured by ELISA, and viral titers were determined by plaque assay on MDCK cells. Two-way ANOVA with Tukey's multiple-comparison test was used to compare IL-1β levels (***, P < 0.001), and a Mann-Whitney test was used to compare virus titers (ns, not significant). Results are representative of three independent experiments.
The NS1 C terminus of the pandemic virus, but not SIV, inhibits the production of IL-1β.
The NS1 protein of IAV is well known for its function of antagonizing host immune responses (13, 15–17). However, the NS1 protein of the 2009 pandemic H1N1 was reported to be inefficient in controlling host gene expression (39). Our unexpected results that Hf09 has a strong inhibitory effect on porcine IL-1β production prompted us to further study whether Hf09 NS1 regulates NLRP3 inflammasome activity. To investigate this, we constructed several mutant viruses derived from WT Sk02 or Hf09 containing mutations in the NS1 protein (Fig. 3A).
FIG 3.

NS1 C terminus of pandemic H1N1, but not SIV, inhibits the IL-1β production. (A) A schematic representation of genome composition of WT and mutant viruses. The NS1 protein encoded by different viruses is depicted. (B) PAMs were infected with Sk02 WT or Sk02-NS1/1–99 virus at an MOI of 1. At 24 hpi, cell-free supernatants were harvested for IL-1β ELISA, and the cells were lysed for Western blotting to measure the expression of pro-IL-1β and viral proteins. ns, not significant. (C) PAMs were infected with Hf09 WT or two NS1-mutant viruses, Hf09-816 and Hf09-817, at an MOI of 1 for 24 h. IL-1β ELISA and Western blotting were done as described for panel B. *, P = 0.0201; **, P = 0.0015. (D) HEK293T cells were transfected with plasmids expressing porcine NLRP3, ASC, procaspase-1, and pro-IL-1β. At 12 hpt, the cells were infected with Hf09 WT or NS1-mutant virus at an MOI of 5. IL-1β production at 12 hpi was measured by ELISA, and the expression of p20, pro-IL-1β, and viral proteins was measured by Western blotting. ***, P < 0.001. (E) PAMs were infected with Sk02, Sk02-616, or Sk02-930 at an MOI of 1 for 24 h. IL-1β ELISA and Western blotting were done as described for panel B. *, P = 0.0104; **, P = 0.0012. (F) HEK293T cells were transfected as described for panel D and infected with Sk02 or Sk02-930 at an MOI of 5 for 12 h. IL-1β ELISA and Western blotting were done as described for panel D. **, P = 0.0056. (G) HEK293T cells were transfected with plasmids expressing porcine NLRP3 inflammasome components and pro-IL-1β along with Flag-vector or Flag-tagged WT/N-terminal amino acids of Hf09 NS1. IL-1β levels and protein expression at 18 hpt were measured. **, P = 0.002; ***, P < 0.001. (H) HEK293T cells were transfected with plasmids expressing porcine NLRP3 inflammasome components and pro-IL-1β along with Myc-vector or the Myc-tagged NS1 C terminus from Hf09 or Sk02. IL-1β levels and protein expression at 18 hpt were measured as described for panel G. ***, P < 0.001.
First, PAMs were infected with WT Sk02 or an Sk02 virus expressing NS1 consisting of amino acids (aa) 1 to 99 (NS11–99), Sk02-NS1/1–99 virus. Deletion of the NS1 C terminus in Sk02 resulted in moderate upregulation of the pro-IL-1β level but did not alter the IL-1β level. Specifically, Sk02-NS1/1–99 virus induced only 1.07-fold more IL-1β than that induced by WT Sk02 (Fig. 3B).
Next, we constructed two mutant Hf09 viruses, Hf09-816 and Hf09-817, encoding a truncated NS1 (aa 1 to 73 or aa 1 to 99, respectively). The mutant viruses grew to titers 0.7 to 1 log lower on MDCK-NS1 cells than the WT Hf09 on MDCK cells (titers were 7 × 106 PFU/ml for Hf09-816 and 1.25 × 107 PFU/ml for Hf09-817 versus 7.5 × 107 PFU/ml for WT Hf09). Upon infection of PAMs, the Hf09-816 and Hf09-817 viruses induced 2.53- and 3.05-fold more IL-1β production than WT Hf09, respectively, yet they did not significantly affect pro-IL-1β expression levels (Fig. 3C). In the reconstitution system, Hf09-816 and Hf09-817 infection led to a striking increase of IL-1β production above the WT Hf09 level although they did not markedly alter the pro-IL-1β level. The level of active caspase-1, p20, another indicator for NLRP3 inflammasome activation, was also consistently increased in the cells infected with NS1 mutant viruses (Fig. 3D).
To demonstrate conclusively that the C terminus of Hf09 NS1 is responsible for inhibition of IL-1β production, we constructed another two mutant viruses in the background of Sk02 virus: Sk02-616 that has the entire segment 8 from Hf09 and Sk02-930 that has a chimeric NS1 with the N terminus from Sk02 and C terminus from Hf09. Their titers were comparable to the titer of WT Sk02 on MDCK cells: 6.25 × 107 PFU/ml for Sk02-616 and 8.75 × 107 PFU/ml for Sk02-930 versus 4 × 107 PFU/ml for WT Sk02. Infection of PAMs with Sk02-616 or Sk02-930 led to a significant reduction of IL-1β, while pro-IL-1β expression was not significantly impaired (Fig. 3E). To further demonstrate that the C terminus of Hf09 NS1 has a greater effect on inflammasome activation than on the pro-IL-1β level, the expression of pro-IL-1β and p20 upon infection with Sk02 WT and Sk02-930 was monitored in the reconstitution system. Indeed, Sk02-930 could inhibit both caspase-1 activation and IL-1β production while not affecting pro-IL-1β expression (Fig. 3F).
Finally, we examined caspase-1 activity and IL-1β production in the reconstitution system by including WT or mutant NS1 constructs. This experiment showed that coexpression of the full-length Hf09 NS1 significantly inhibited IL-1β production, whereas coexpression of the truncated NS1 with a C-terminal deletion had little or no inhibitory effect (Fig. 3G). Further, coexpression of the C terminus of Hf09 NS1 aa 100 to 219 led to a reduction of IL-1β due to less caspase-1 activation, whereas this effect was not observed with the NS1 C terminus derived from Sk02 virus (Fig. 3H). All of these results evidently demonstrate that the C terminus of the Hf09 NS1 has an inhibitory effect on NLRP3 inflammasome activation.
NS1 C terminus of pandemic virus inhibits ASC speck formation.
To further investigate the inhibition mechanism, we examined the roles of WT and NS1 mutant Hf09 virus on the formation of ASC specks, a hallmark of NLRP3 inflammasome activation. In noninfected cells, transfection of either ASC or NLRP3 alone did not induce a noticeable number of specks (Fig. 4A, top three rows). In contrast, cotransfection of porcine ASC and NLRP3 led to the aggregation of two proteins, forming the specks in the perinuclear area. Furthermore, in virus-infected cells as indicated by NP staining, Hf09-817 led to 2-fold more ASC speck formation than WT Hf09 (Fig. 4A, bottom three rows). Specifically, while 6.8% of NP-positive cells displayed ASC specks in Hf09-infected cells, 14.3% of NP-positive cells exhibited ASC specks in Hf09-817-infected cells (Fig. 4B). These results further demonstrate that the Hf09 NS1 C terminus inhibits NLRP3 inflammasome activation.
FIG 4.

NS1 C terminus of pandemic H1N1 inhibits ASC speck formation. (A) HEK293T cells were transfected with Myc-tagged NLRP3 or Myc-vector and Flag-tagged ASC or Flag-vector for 24 h and left uninfected (top three rows). The cells were transfected with Myc-tagged NLRP3 and Flag-tagged ASC for 12 h and left uninfected or infected with Hf09 or Hf09-817 at an MOI of 5 for 12 h (bottom three rows). The cells were fixed, permeabilized, blocked, and probed with appropriate antibodies, followed by DAPI staining. NLRP3-Myc (gray), Flag-ASC (green), viral NP (red), and nuclei (blue) were visualized by confocal microscopy. Scale bar, 10 μm. (B) ASC speck-forming cells and NP-stained cells infected by Hf09 or Hf09-817 were quantified from five randomly selected fields, and the percentages of ASC speck- and NP-positive cells in total NP-positive cells were shown. Results are representative of three independent experiments (**, P = 0.0025, Mann-Whitney test).
NS1 C terminus of pandemic virus inhibits the ubiquitination of porcine ASC.
Next, we were interested in investigating the mechanism by which Hf09 NS1 protein regulates the porcine NLRP3 inflammasome at the posttranslational level. We focused on the regulation of NLRP3 and ASC ubiquitination. HEK293T cells were cotransfected with plasmids expressing hemagglutinin (HA)-tagged ubiquitin and Flag-tagged NLRP3 or Flag-tagged ASC and were then infected with Hf09 WT or NS1 mutant viruses. NLRP3 was ubiquitinated in this setting; however, the ubiquitination levels were not affected by infection with the Hf09 WT or the NS1 mutant viruses (Fig. 5A). In contrast, while ASC was slightly ubiquitinated in mock- or WT Hf09-infected cells, the ubiquitination levels of ASC were increased in cells infected with NS1 mutant viruses (Fig. 5B). The total cellular ubiquitination levels were not considerably altered by viral infection. These results suggested that the NS1 C terminus of Hf09 plays a role in inhibiting the ubiquitination of ASC but has no effect on NLRP3 ubiquitination. To further confirm the inhibitory function of NS1 on ASC ubiquitination, we conducted a compensation assay. In this assay, Myc-tagged WT NS1 or truncated NS1 (NS1 aa 1 to 99) was preexpressed together with ASC and ubiquitin in HEK293T cells. After 24 h, cells were infected with an NS1 mutant virus, Hf09-817, to induce the ubiquitination of ASC. While preexpression of green fluorescent protein (GFP) or NS11–99 protein did not have any impact on ASC ubiquitination, WT NS1 protein could diminish the ubiquitination induced by Hf09-817 virus, reaffirming that the NS1 C terminus of Hf09 suppresses ASC ubiquitination (Fig. 5C). In addition, we questioned whether the ability of Hf09 NS1 to suppress ASC ubiquitination is limited to porcine immunity by expanding our ubiquitination assay by using human ASC. Interestingly, the ubiquitination level of human ASC was increased in cells infected with Hf09-817 compared to that in mock-infected or WT Hf09-infected cells (Fig. 5D), showing that the Hf09 NS1 C terminus can inhibit the ubiquitination of both porcine and human ASC.
FIG 5.

NS1 C terminus of pandemic H1N1 inhibits ASC ubiquitination. (A and B) HEK293T cells were transfected with HA-vector or HA-ubiquitin (Ub) and Flag-vector or Flag-NLRP3/ASC constructs. At 24 hpt, the cells were infected with WT or NS1-mutant Hf09 virus at an MOI of 10 for 9 h. Cell lysates under denaturing conditions were subjected to co-IP with beads conjugated with anti-Flag antibody. IP and input samples were used for Western blotting with antibodies indicated. (C) HEK293T cells were transfected with HA-vector or HA-ubiquitin and Flag-ASC constructs along with plasmids expressing GFP, Myc-tagged WT, or mutant NS1 of Hf09. At 24 hpt, the cells were infected with Hf09-817 at an MOI of 10 for 9 h. Co-IP was done as described for panel A, and Western blotting was done with the antibodies indicated. (D) HEK293T cells were transfected with HA-ubiquitin and Flag-tagged human ASC (Flag-huASC) constructs for 24 h and infected with WT Hf09 or Hf09-817 at an MOI of 10 for 9 h. Co-IP was done as described for panel A, and Western blotting was done with the antibodies indicated. The asterisks indicate IgG heavy chains. Results are representative of three independent experiments.
K110 and K140 in porcine ASC are the target sites for ubiquitination.
Ubiquitination generally occurs by conjugation of ubiquitin molecules on the lysine(s) in target proteins. To investigate which lysine in porcine ASC is targeted for ubiquitination, we compared the positions of lysine residues in porcine ASC to those in human ASC. As illustrated in Fig. 6A, porcine ASC has 16 lysines, among which 7 lysines (K79, K87, K91, K96, K101, K106, and K183) (identified above the bar in Fig. 6A) are unique for porcine ASC. The K175 residue in porcine ASC corresponds to K174 in human ASC, which was reported to be the target site for ubiquitination upon an RNA virus infection (36). Thus, we first tested whether these sites are targeted for ubiquitination. We made single lysine-to-arginine mutation constructs; however, none of the mutations led to a reduced level of ASC ubiquitination upon Hf09-817 infection (Fig. 6B, right panel). We then expanded the mutations to the remaining lysines. While individual mutations were made on K55, K110, K140, and K159, we simultaneously mutated a cluster of lysines (K21, K22, K24, and K26) due to their close proximity. However, the replacement of these remaining lysines with arginines did not reduce the ubiquitination levels (Fig. 6B, left panel). ASC has an N-terminal pyrin domain (PYD) and a C-terminal caspase activation and recruitment domain (CARD). Based on the reports that CARD is targeted for different modifications (36, 40, 41), we tested whether multiple lysines in the CARD of porcine ASC may be targeted for ubiquitination. Thus, we constructed two truncated ASCs, ASC consisting of aa 1 to 109 (ASC1–109) and ASC consisting of aa 1 to 158 (ASC1–158) (Fig. 6A). Upon Hf09-817 infection, the ubiquitination level of ASC1–158 was not altered, while the level on ASC1–109 was significantly reduced compared to that of the WT ASC (Fig. 6C). This finding suggested that the two lysines, K110 and K140, within aa 110 to 158, may be the target for ubiquitination. Indeed, ASC with a K110R/K140R mutation had an appreciably reduced level of ubiquitination compared to that of WT ASC in response to Hf09-817 infection (Fig. 6D). In contrast, other combinatorial mutations did not show any impairment on ASC ubiquitination (data not shown).
FIG 6.

K110 and K140 in the C terminus of porcine ASC are targeted for ubiquitination. (A) A schematic representation of WT and two mutant ASC constructs (aa 1 to 109 and aa 1 to 158) is illustrated with lysine (K) residues. (B to D) HEK293T cells were transfected with HA-ubiquitin and Flag-tagged WT or mutant ASC constructs. At 24 hpt, the cells were infected with Hf09 WT or Hf09-817 at an MOI of 10 for 9 h. Cell lysates under denaturing conditions were subjected to co-IP with beads conjugated with anti-Flag antibody. IP and input samples were used for Western blotting with antibodies indicated. (E) HEK293T cells were transfected with HA-tagged WT or mutant ubiquitin and Flag-ASC constructs for 24 h and infected as described for panel B. Co-IP and Western blotting were done as described for panel B. The asterisks indicate IgG heavy chains. Results are representative of three independent experiments.
Which lysine residue(s) among seven in the ubiquitin molecules is involved in the ubiquitination of target proteins may determine the fate of target proteins. We further conducted ubiquitination assays using mutant ubiquitin constructs with lysines replaced by arginines. Ubiquitination levels were not diminished when either lysine 48 or 63 was replaced by arginine in ubiquitin (Fig. 6E), suggesting that lysine 48 or 63 alone is not linked to ASC ubiquitination, but, rather, mixed lysines are involved.
ASC ubiquitination at K110/K140 is essential for procaspase-1 cleavage and IL-1β production.
In an attempt to observe the impact of ubiquitination at K110/K140 on the ASC protein itself and on inflammasome activity, we first examined whether ubiquitination on K110/K140 would affect ASC speck formation by immunofluorescence staining. Elimination of ASC ubiquitination by the K110R/K140R mutation did not alter the overall number or the perinuclear localization pattern of ASC specks (Fig. 7A). In NLRP3 inflammasome reconstitution followed by Hf09-817 infection, the NLRP3 inflammasome consisting of WT ASC generated an appreciable level of active caspase-1, p20, and a large amount of IL-1β. In contrast, the inflammasome consisting of ASC K110R/K140R produced lower caspase-1 activity, as evidenced by the reduced level of both p20 and IL-1β (Fig. 7B). None of the single mutations on ASC had any effect on caspase-1 activation and IL-1β maturation.
FIG 7.

ASC ubiquitination at K110 and K140 is critical for porcine NLRP3 inflammasome-mediated IL-1β production. (A) HEK293T cells were transfected with plasmids expressing Myc-tagged NLRP3 and Flag-tagged WT/mutant ASC. At 24 hpt, the cells were fixed, permeabilized, immunostained with appropriate antibodies, and counterstained with DAPI. ASC specks formed by colocalization of NLRP3-Myc (gray) and Flag-ASC (green) were seen in the perinuclear region. Nuclei were visualized in blue. Scale bar, 5 μm. (B) HEK293T cells were transfected with plasmids expressing porcine NLRP3, Flag-tagged WT/mutant ASC, procaspase-1, and pro-IL-1β. At 12 hpt, the cells were infected with Hf09-817 at an MOI of 5 for 12 h. Porcine IL-1β was measured by ELISA with cell-free supernatants, and the expression of pro-IL-1β, p20, and viral proteins was measured by Western blotting with cell lysates. Expression of pro-IL-1β and p20 was normalized to that of β-actin using ImageJ. Results are representative of three independent experiments. **, P = 0.002.
DISCUSSION
The NLRP3 inflammasome exerts important roles in the regulation of innate immunity. In this study, we focused on porcine NLRP3 inflammasome regulation in response to IAV infection. While SIVs generally induce inflammatory responses in pigs (42, 43), Ma et al. (44) showed that pH1N1/09-infected pigs had upregulated expression of genes involved in the inflammatory and immune responses. In terms of the cytokine response in human cells infected by pH1N1/09, there have been some controversial reports. Both upregulation of inflammatory cytokines (45–47) and poor inflammatory responses were reported (48, 49). Nevertheless, there is little information concerning the regulation of IL-1β production, which requires the activation of the NLRP3 inflammasome upon IAV infection in pigs. Since alveolar macrophages are essential for the regulation of lung inflammation and are required for protection against IAV infection (50–52), we first utilized primary PAMs to characterize how porcine IL-1β is produced in response to IAV infection. We showed that IAVs isolated from swine induced IL-1β production, which is mediated by the NLRP3 inflammasome, in a viral dose- and replication-dependent manner. In contrast, pH1N1/09 (Hf09) induced a much lower level of IL-1β production. The fact that Hf09 and Sk02 replicate to similar titers in PAMs and that they induce similar levels of pro-IL-1β suggested that the NLRP3 inflammasome is less active or is suppressed in Hf09-infected cells. Infection with mutant Hf09 encoding an NS1 protein with a C-terminal deletion, but not infection with a mutant of SIV (Sk02), resulted in an increased level of IL-1β compared to that of each of their WT viruses. Moreover, a mutant Sk02 virus (Sk02-930) whose NS1 C terminus was replaced by that from Hf09 induced less IL-1β production than WT Sk02 due to inhibited caspase-1 activity. These results strongly demonstrate an inhibitory function of the Hf09 NS1 C terminus on NLRP3 inflammasome activity.
IAV NS1 protein has been reported to inhibit NLRP3 inflammasome-mediated IL-1β production in a murine macrophage cell line and human THP-1 cells (16, 17). NS1 physically interacts with NLRP3 and suppresses inflammasome activation in THP-1 cells (16). NS1 also suppresses IL-1β secretion by inhibiting NLRP3/ASC-induced single speck formation. Using ectopically expressed NS1 constructs derived from the A/Puerto Rico/8/34 (PR8) strain, the inhibitory function was mapped to the RNA-binding domain and tripartite motif-containing protein 25 (TRIM25)-binding domain within the N terminus of NS1 (17). Consistent with these studies, we observed that NS1 protein derived from Hf09 disrupted porcine NLRP3 and ASC aggregation and speck formation. In contrast to the aforementioned reports, we found that mutant pH1N1/09 virus carrying an NS1 C-terminal deletion from aa 100 to 219 induced a strikingly higher level of IL-1β and more ASC specks than the WT virus. Expression of the C-terminal aa 100 to 219 alone led to a decreased level of IL-1β secretion. Thus, our results demonstrated that the C terminus of NS1 from pH1N1/09 has an inhibitory function on inflammasome activation.
It is noted that NS1 from pH1N1/09 is 219 aa in length and is highly conserved among the 2009 pandemic isolates, whereas the NS1 from Sk02 and PR8 consists of 230 aa. Whether the discrepancy in the responsible inhibitory domain of NS1 is due to the host species difference between human and porcine NLRP3 inflammasomes or to the strain difference warrants further investigation. Interestingly, all of the other pandemic influenza viruses encode longer NS1 proteins than pH1N1/09. The 1918 pandemic NS1 consists of 230 aa, and both the 1957 and 1968 pandemic viruses encode an NS1 protein of 237 aa in length. Whether the described mechanism to regulate the immune response is unique to pH1N1/09 also needs further examination. Nonetheless, the capacity of pH1N1/09 to inhibit inflammatory cytokines may be beneficial to the virus, allowing it to evolve in pigs and to keep the ability to reassort with other viruses.
Posttranslational modifications of the inflammasome components play key roles in regulation of inflammasome activity. To seek the mechanism by which Hf09 NS1 suppresses porcine NLRP3 inflammasome activity, we explored whether NS1 modifies ASC or NLRP3 ubiquitination. Our data showed that infection with mutant Hf09 viruses with an NS1 C-terminal deletion caused more ubiquitination of ASC than the WT virus but did not affect the ubiquitination of NLRP3, implying an inhibitory role of the Hf09 NS1 C terminus on porcine ASC ubiquitination. We eliminated the speculation of an enhancing role of the NS1 C terminus on ASC deubiquitination since WT virus infection induced a low level of ubiquitination, which was similar to that in mock-infected cells. One possible speculation could be that NS1 influences E3 ligase activity. This type of regulation is reported in RIG-I ubiquitination by an E3 ligase, TRIM25 that is targeted by NS1 to suppress IFN production (53). The NS1 C-terminal aa 126 to 225 of avian H5N1 was recently reported to downregulate RIG-I-mediated IFN signaling through interacting with TRAF3 and inhibiting the K63-linked ubiquitination of TRAF3 (54). This shows that the inhibitory functions of the NS1 C terminus on the host immunity is not limited to the suppression of the NLRP3 inflammasome. Guan et al. discovered that ASC undergoes ubiquitination via the E3 ligase TRAF3 by forming the complex ASC-TRAF3-MAVS (where MAVS is mitochondrial antiviral signaling protein) upon VSV infection (36). In that study, MAVS contributes to the stability of ASC and increases the interaction of ASC with TRAF3. In this context, it will be of interest to examine if NS1 interacts with another adaptor protein that associates with an E3 ligase responsible for the ubiquitination of porcine ASC. In addition to the K63-linked or linear ubiquitination previously reported (35, 36), we found that ASC was modified by mixed lysine-linked ubiquitination. This does not seem to be a rare event since NLRP3 is modified by both K48- and K63-linked ubiquitination and is deubiquitinated upon activation (21, 24, 25, 27). Different types of ubiquitination are thought to modulate the activities of inflammasomes for tight regulation of innate immune signaling. We found that K110 and K140 in porcine ASC were the ubiquitin acceptor sites, suggesting that modifications of the C-terminal CARD of ASC, which interacts with another CARD of procaspase-1 (55, 56), are crucial for inflammasome activation. We also showed that removal of ASC ubiquitination by a K-to-R mutation on the acceptor sites impaired caspase-1 activity during virus infection but did not affect the stability of ASC or localization and number of ASC specks, suggesting that the ubiquitination of porcine ASC contributes to the fully functional activation of the NLRP3 inflammasome. It is noteworthy that deubiquitination of ASC can lead to inflammasome activation (34), which contradicts our observation. The ambivalence may be due to the diverse interplay between virus and the host signaling pathways that is unlike how conventional agonists stimulate the immune system. IAV infection can manipulate not only host gene expression but also the host ubiquitin system (57).
In conclusion, we demonstrated a novel role of NS1 protein in innate immune regulation in pigs. Porcine IL-1β production in response to pH1N1/09 was significantly repressed, and this was due to the inhibitory function of pH1N1/09 NS1 protein on NLRP3 inflammasome activation. The NS1 C terminus of pH1N1/09 performed this function by suppressing ASC speck formation and ASC ubiquitination. Furthermore, two lysines in the ASC CARD, K110 and K140, were identified as target sites for ubiquitination which is critical for NLRP3 inflammasome activation. These findings enable a better understanding of the mechanism by which IAV NS1 protein evades porcine immunity.
MATERIALS AND METHODS
Cells and viruses.
PAMs were isolated from bronchoalveolar lavage fluid of 4- to 7-week-old piglets that were confirmed to be seronegative against H1N1 and H3N2 subtypes of SIVs. All experimental procedures were in accordance with the guidelines of the Canadian Council on Animal Care and were approved by the University of Saskatchewan Animal Research Ethics Committee. PAMs were characterized by flow cytometry as previously described (58) and were cultured with HyClone RPMI 1640 medium (SH30027.01; GE Healthcare) supplemented with 20% fetal bovine serum (FBS) (16000-044; Thermo Fisher), 50 μg/ml gentamicin (BS724; Bio Basic Canada), and 1× antibiotic-antimycotic (15240-062; Thermo Fisher). Madin-Darby canine kidney (MDCK) cells were cultured with minimum essential medium (MEM) (M4655; Sigma) supplemented with 10% FBS and 50 μg/ml gentamicin. Human embryonic kidney 293T (HEK293T) cells were cultured with Dulbecco's modified Eagle's medium (DMEM) (D5796; Sigma) supplemented with 10% FBS and 50 μg/ml gentamicin.
The wild-type (WT) IAVs used in this study, which were all grown in MDCK cells, were influenza A/swine/Saskatchewan/18789/2002/H1N1 (Sk02), influenza A/swine/Texas/4199-2/1998/H3N2 (Tx98), and influenza A/Halifax/210/2009/H1N1 (Hf09). A mutant Sk02 virus (Sk02-NS1/1–99) containing only amino acids (aa) 1 to 99 of the N-terminal NS1 was described previously (58). The following mutant viruses were generated by reverse genetics: Sk02-616 and Sk02-930, two isogenic Sk02 viruses, with Sk02-616 containing segment 8 of Hf09 and Sk02-930 containing a chimeric segment 8 that encodes NS1 with aa 1 to 99 from Sk02 and aa 100 to 219 from Hf09. Hf09-816 and Hf09-817 are two isogenic Hf09 viruses that encode a truncated NS1, i.e., aa 1 to 73 or 1 to 99, respectively. All mutant viruses with truncated NS1 proteins were propagated in MDCK cells stably expressing the NS1 protein of influenza A/Puerto Rico/8/1934/H1N1 (PR8) (MDCK-NS1), while Sk02-616 and Sk02-930 were propagated in MDCK cells. Viral titers were determined by plaque assay in either MDCK or MDCK-NS1 cells.
Antibodies and reagents.
Antibodies used for Western blotting and immunoprecipitation (IP) are as follows. Rabbit polyclonal antibodies against NP and NS1 were generated as previously described (59). Goat polyclonal anti-porcine IL-1β antibody (BAF681; R&D Systems), rabbit polyclonal anti-porcine caspase-1 (p20) antibody (PAB592Po01; Cloud-Clone Corp), mouse monoclonal anti-β-actin antibody (3700; Cell Signaling Technology [CST]), mouse monoclonal anti-GFP antibody (2955; CST), mouse monoclonal anti-Myc tag antibody (2276; CST), mouse monoclonal anti-FLAG M2 antibody (F3165; Sigma), rabbit polyclonal anti-HA tag antibody (ab9110; Abcam), and mouse monoclonal anti-ubiquitin antibody (sc-8017; Santa Cruz) were purchased from different sources. For the secondary antibodies, IRDye 680RD donkey anti-rabbit (926-68073), IRDye 800CW donkey anti-mouse (926-32212), and IRDye 800CW donkey anti-goat (926-32214) antibodies were purchased from Li-Cor Biosciences.
Transfection was conducted using TransIT-LT1 transfection reagent (MIR2300; Mirus Bio). EDTA-free protease inhibitor cocktail tablets (4693132001; Roche) were used in cell lysis and IP. An NLRP3 inhibitor, 3,4-methylenedioxy-β-nitrostyrene (MNS) (574713; EMD Millipore), was used as previously reported (37), and dimethyl sulfoxide ([DMSO] D2660; Sigma) was used as the vehicle.
Plasmid construction.
Total RNA was extracted from PAMs stimulated with lipopolysaccharides (L3024; Sigma) and ATP (A1852; Sigma), and then cDNA was synthesized using oligo(dT). The full-length genes of porcine NLRP3, ASC, procaspase-1, and pro-IL-1β were amplified using primers that were designed based on the GenBank sequences (accession numbers NM_001256770 for NLRP3, AB873106 for ASC, NM_214162 for procaspase-1, and NM_214055 for pro-IL-1β). Primer sequences are available upon request. The genes were cloned into pcDNA3.1, pCMV-3×Flag (N-terminal tag), or pcDNA3.1-3×Myc (C-terminal tag) vector. The resulting plasmids were designated pcDNA-NLRP3, pcDNA-NLRP3-Myc, pCMV-Flag-NLRP3, pCMV-Flag-ASC, pCMV-Flag-procaspase-1, and pcDNA-pro-IL-1β. Mutant ASC constructs with lysines replaced by arginines were generated by site-directed mutagenesis using pCMV-Flag-ASC as the template. Truncated ASC with aa 1 to 109 or 1 to 158 was cloned into pCMV-3×Flag vector generating pCMV-Flag-ASC1–109 and pCMV-Flag-ASC1–158. The full-length gene of human ASC (huASC) was cloned into pCMV-3×Flag vector to generate pCMV-Flag-huASC using pCA7-ASC (60) as the template and primers that were designed based on the GenBank sequence (accession number NM_013258).
WT and truncated NS1 of Hf09 were cloned into pCMV-3×Flag vector, and the resulting plasmids were designated pCMV-Flag-NS1, pCMV-Flag-NS11–73, and pCMV-Flag-NS11–99, encoding the full-length, N-terminal aa 1 to 73, or N-terminal aa 1 to 99 of NS1, respectively. The WT and the N-terminal aa 1 to 99 of Hf09 NS1 were also cloned into pcDNA3.1-3×Myc to generate pcDNA-NS1-Myc and pcDNA-NS11–99-Myc. The NS1 C termini of Hf09 and Sk02 were cloned into pcDNA3.1-3×Myc and designated pcDNA-Hf09 NS1100–219-Myc and pcDNA-Sk02 NS1100–230-Myc, respectively.
The HA-tagged ubiquitin construct, pCMV-HA-ubiquitin, with eight tandem sequences of HA-ubiquitin (61), was used in ubiquitination assays. pcDNA-HA-ubiquitin (62) was used as a template to generate ubiquitin with the mutation K48R [pcDNA-HA-ubiquitin (K48R)] or K63R [pcDNA-HA-ubiquitin (K63R)]. The mutations and truncations were confirmed by DNA sequencing.
Infection of PAMs.
The culture supernatant of isolated PAMs from each individual piglet was subjected to enzyme-linked immunosorbent assay (ELISA) to test for background levels of IL-1β. The cells from six individual piglets with low background levels of IL-1β were selected for further use. PAMs were seeded at 1 × 106 cells per well on 24-well plates and infected with viruses for 24 h at the multiplicities of infection (MOIs) indicated in the figure legends. Cell-free supernatants were harvested for ELISA while the cells were lysed with 1× sodium dodecyl sulfate (SDS) sample buffer for Western blotting. For time course experiments, the supernatants collected at 8, 16, and 24 h postinfection (hpi) were used for both ELISA and plaque assay in MDCK cells.
NLRP3 inflammasome reconstitution assay.
HEK293T cells were seeded at 1.8 × 105 cells per well on 24-well plates and transfected with pcDNA-NLRP3 (30 ng), pcDNA-ASC or pCMV-Flag-ASC (10 or 20 ng), pCMV-Flag-procaspase-1 (20 ng), and pcDNA-pro-IL-1β (100 ng). At 12 h posttransfection (hpt), cells were infected with viruses. At 12 hpi, cell-free supernatants were harvested for porcine IL-1β ELISA, and cells were lysed with 1× SDS sample buffer for Western blotting. For reconstitution with coexpressed WT/mutant NS1, the cells were transfected as above along with 300 ng of an empty vector or tagged WT/mutant NS1 constructs. At 18 hpt, the supernatants were harvested, and cell lysates were prepared.
Porcine IL-1β ELISA.
Immulon 2 HB U plates (3655; Thermo Fisher) were coated with 2 μg/ml of mouse monoclonal anti-porcine IL-1β antibody (MAB6811; R&D Systems) in phosphate-buffered saline (PBS) at room temperature overnight. All of the following steps were done at room temperature, and all reagents or samples were applied at 100 μl per well. The plates were washed four times with Tris-buffered saline (TBS) with 0.05% Tween 20 (P1379; Sigma) (TBST) between each step. Blocking was done with 1% bovine serum albumin (BSA) (A7030; Sigma) in PBS for 1 h. Twofold serial dilutions of recombinant porcine IL-1β protein (681-PI-010; R&D Systems) in diluent (0.1% BSA in TBST) was used as the standard. The plates were incubated with samples or the standard for 2 h. The plates were then incubated with 50 ng/ml of goat polyclonal anti-porcine IL-1β biotinylated antibody (BAF681; R&D Systems) in diluent for 1 h, followed by the incubation with alkaline phosphatase streptavidin (016-050-084; Jackson ImmunoResearch) diluted to 1/5,000 in diluent for another 1 h. For color development, the plates were incubated with 1 mg/ml of p-nitrophenyl phosphate in diethanolamine buffer (1 M diethanolamine, 0.5 M MgCl2, pH 9.8) until the optical density of the first dilution of the standard reached around 2.0. The plates were read at 405 nm with a reference at 490 nm using an xMark microplate absorbance spectrophotometer (Bio-Rad).
Western blotting.
Cell lysates or IP samples were fractionated by SDS-polyacrylamide gel electrophoresis (PAGE) and blotted onto nitrocellulose membranes. The membranes were blocked with 5% skim milk in TBS with 0.1% Tween 20 for 1 h and incubated with primary antibodies at 4°C overnight. After incubation with the secondary antibodies at room temperature for 1 h, the membranes were scanned with an Odyssey infrared imager (Li-Cor Biosciences). ImageJ software (National Institutes of Health, USA) was used for the normalization of expression levels.
Ubiquitination assay.
HEK293T cells were seeded at 1 × 106 cells per well on six-well plates and transfected with 1 μg of pCMV-HA-ubiquitin or pcDNA-HA-WT/mutant ubiquitin together with one of the following constructs encoding target proteins: 3×Flag-tagged NLRP3 (pCMV-Flag-NLRP3), WT/mutant ASC (pCMV-Flag-WT/mutant ASC), or human ASC (pCMV-Flag-huASC). At 24 hpt, the cells were infected with viruses at an MOI of 10 for 9 h. For the compensation assay, the cells were transfected with 1 μg of pCMV-HA-ubiquitin and pCMV-Flag-ASC along with a plasmid expressing either GFP, pcDNA-NS1-Myc, or pcDNA-NS11–99-Myc. At 24 hpt, the cells were infected with Hf09-817 as described above. Cell lysis and IP were performed as previously described with some modifications (63). Briefly, the cells were lysed with 200 μl of SDS lysis buffer (20 mM Tris, pH 8.0, 250 mM NaCl, 3 mM EDTA, 10% glycerol, 1% SDS, 0.5% Nonidet P-40 substitute) freshly supplemented with 20 mM N-ethylmaleimide (E3876; Sigma), 5 mM 1,10-phenanthroline monohydrate (P9375; Sigma), and 1× protease inhibitor cocktail, while 5% of the cells were lysed with 1× SDS sample buffer for input. The clarified cell lysates were mixed with 600 μl of SDS IP buffer (20 mM Tris, pH 8.0, 250 mM NaCl, 3 mM EDTA, 10% glycerol, 0.1% SDS, 0.5% Nonidet P-40 substitute) freshly supplemented with 20 mM N-ethylmaleimide, 5 mM 1,10-phenanthroline monohydrate, and 1× protease inhibitor cocktail and incubated with 40 μl of a slurry of mouse anti-FLAG M2 affinity agarose (A2220; Sigma) at 4°C for 4 h with agitation. After being washed four times with 500 μl of SDS IP buffer, the agarose was mixed with 60 μl of 2× SDS sample buffer and boiled at 95°C for 5 min for elution. The eluted proteins were subjected to SDS-PAGE and Western blotting.
Immunofluorescence microscopy.
HEK293T cells were seeded at 6 × 104 cells per well on a LabTek II CC2 chamber slide (154941; Thermo Fisher). The cells were transfected with 50 ng of pcDNA-NLRP3-Myc or Myc-vector and 30 ng of pCMV-Flag-ASC or Flag-vector for 24 h to validate the specificity of antibodies. To observe the effects of WT/mutant Hf09 virus on ASC specks, the cells were transfected with pcDNA-NLRP3-Myc (50 ng) and pCMV-Flag-ASC (30 ng) for 12 h and infected with Hf09 or Hf09-817 at an MOI of 5 for 12 h. To compare the specks formed by WT and mutant ASC, the cells were transfected with pcDNA-NLRP3-Myc (50 ng) and pCMV-Flag-ASC (WT or K110R/K140R; 30 ng) for 24 h. Cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.2% Triton X-100 for 5 min at room temperature. The cells were incubated with 5% BSA in Dulbecco's phosphate-buffered saline (DPBS) (14040; Thermo Fisher) for 1 h at room temperature and probed with the primary antibodies (rabbit monoclonal anti-Myc-tag antibody [2278; CST], goat polyclonal anti-DDDDK tag antibody [ab1257; Abcam], and mouse monoclonal anti-IAV NP antibody [MCA400; Bio-Rad]) overnight at 4°C followed by an incubation with the secondary antibodies (Alexa Fluor 546 donkey polyclonal anti-rabbit IgGH+L [A10040, Invitrogen], Alexa Fluor 633 donkey polyclonal anti-goat IgGH+L [A21082; Invitrogen], and Alexa Fluor 488 donkey polyclonal anti-mouse IgGH+L [A21202; Invitrogen]) for 1 h at room temperature. The cells were washed with DPBS between each step. The cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (D1306; Invitrogen) for 5 min, and the slide was mounted with ProLong Diamond Antifade Mountant (P36961; Invitrogen) overnight. Images were visualized by using a confocal laser scanning microscope (TCS SP8; Leica). Where applicable, the numbers of ASC speck-positive cells and IAV NP-positive cells were counted from five randomly selected fields per each condition, and the percentages of speck- and NP-positive cells in NP-positive cells are shown.
Statistical analysis.
The data were analyzed using GraphPad Prism, version 7.0a, by one-way analysis of variance (ANOVA) with Tukey's multiple-comparison test. Two-way ANOVA with Tukey's multiple-comparison test or Mann-Whitney test was used where specified in the figure legends. The bars indicate the means ± standard deviations (SD). A P value of less than 0.05 was considered to be statistically significant.
ACKNOWLEDGMENTS
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) to Y.Z. H.-S.P. was partially supported by a scholarship and fellowship from the Western College of Veterinary Medicine, University of Saskatchewan.
We thank Donna Dent of the Vaccine and Infectious Disease Organization-International Vaccine Centre (VIDO-InterVac) for help with ELISAs. We are also grateful to Takeshi Ichinohe (University of Tokyo) for providing the human ASC construct, and Dirk Bohmann (University of Rochester Medical Center) and Edward Harhaj (Johns Hopkins School of Medicine) for sharing the HA-tagged ubiquitin constructs.
REFERENCES
- 1.Chen IY, Ichinohe T. 2015. Response of host inflammasomes to viral infection. Trends Microbiol 23:55–63. doi: 10.1016/j.tim.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 2.Guo H, Callaway JB, Ting JP. 2015. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Núñez G. 2006. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem 281:36560–36568. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 4.Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP. 2009. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas PG, Dash P, Aldridge JR, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD. 2009. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwasaki A, Pillai PS. 2014. Innate immunity to influenza virus infection. Nat Rev Immunol 14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ong JD, Mansell A, Tate MD. 2017. Hero turned villain: NLRP3 inflammasome-induced inflammation during influenza A virus infection. J Leukoc Biol 101:863–874. doi: 10.1189/jlb.4MR0616-288R. [DOI] [PubMed] [Google Scholar]
- 8.Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, Kanneganti TD. 2016. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 1:aaq2045. doi: 10.1126/sciimmunol.aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McAuley JL, Tate MD, MacKenzie-Kludas CJ, Pinar A, Zeng W, Stutz A, Latz E, Brown LE, Mansell A. 2013. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog 9:e1003392. doi: 10.1371/journal.ppat.1003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshizumi T, Ichinohe T, Sasaki O, Otera H, Kawabata S, Mihara K, Koshiba T. 2014. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat Commun 5:4713. doi: 10.1038/ncomms5713. [DOI] [PubMed] [Google Scholar]
- 11.Pinar A, Dowling JK, Bitto NJ, Robertson AA, Latz E, Stewart CR, Drummond GR, Cooper MA, McAuley JL, Tate MD, Mansell A. 2017. PB1-F2 peptide derived from avian influenza A virus H7N9 induces inflammation via activation of the NLRP3 inflammasome. J Biol Chem 292:826–836. doi: 10.1074/jbc.M116.756379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ichinohe T, Pang IK, Iwasaki A. 2010. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11:404–410. doi: 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krug RM. 2015. Functions of the influenza A virus NS1 protein in antiviral defense. Curr Opin Virol 12:1–6. doi: 10.1016/j.coviro.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stasakova J, Ferko B, Kittel C, Sereinig S, Romanova J, Katinger H, Egorov A. 2005. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1β and 18. J Gen Virol 86:185–195. doi: 10.1099/vir.0.80422-0. [DOI] [PubMed] [Google Scholar]
- 15.Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, Vidal SM. 2013. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog 9:e1003256. doi: 10.1371/journal.ppat.1003256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheong WC, Kang HR, Yoon H, Kang SJ, Ting JP, Song MJ. 2015. Influenza A virus NS1 protein inhibits the NLRP3 inflammasome. PLoS One 10:e0126456. doi: 10.1371/journal.pone.0126456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moriyama M, Chen IY, Kawaguchi A, Koshiba T, Nagata K, Takeyama H, Hasegawa H, Ichinohe T. 2016. The RNA- and TRIM25-binding domains of influenza virus NS1 protein are essential for suppression of NLRP3 inflammasome-mediated interleukin-1β secretion. J Virol 90:4105–4114. doi: 10.1128/JVI.00120-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bednash JS, Mallampalli RK. 2016. Regulation of inflammasomes by ubiquitination. Cell Mol Immunol 13:722–728. doi: 10.1038/cmi.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez-Castejon G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch S, Kirov A, Prudovsky I, Swanton E, Brough D. 2013. Deubiquitinases regulate the activity of caspase-1 and interleukin-1β secretion via assembly of the inflammasome. J Biol Chem 288:2721–2733. doi: 10.1074/jbc.M112.422238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. 2012. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. 2013. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49:331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Han S, Lear TB, Jerome JA, Rajbhandari S, Snavely CA, Gulick DL, Gibson KF, Zou C, Chen BB, Mallampalli RK. 2015. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J Biol Chem 290:18124–18133. doi: 10.1074/jbc.M115.645549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yen H, Sugimoto N, Tobe T. 2015. Enteropathogenic Escherichia coli uses NleA to inhibit NLRP3 inflammasome activation. PLoS Pathog 11:e1005121. doi: 10.1371/journal.ppat.1005121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, Zheng M, Zhang X, Xia D, Ke Y, Lu L, Wang D. 2016. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 45:944. doi: 10.1016/j.immuni.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Kawashima A, Karasawa T, Tago K, Kimura H, Kamata R, Usui-Kawanishi F, Watanabe S, Ohta S, Funakoshi-Tago M, Yanagisawa K, Kasahara T, Suzuki K, Takahashi M. 2017. ARIH2 ubiquitinates NLRP3 and negatively regulates NLRP3 inflammasome activation in macrophages. J Immunol 199:3614–3622. doi: 10.4049/jimmunol.1700184. [DOI] [PubMed] [Google Scholar]
- 26.Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, Zhou R. 2015. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160:62–73. doi: 10.1016/j.cell.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 27.Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, Han L, Jiang G, Zhang L, Gao C, Zhao W. 2016. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun 7:13727. doi: 10.1038/ncomms13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti TD, Lamkanfi M. 2014. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun 5:3209. doi: 10.1038/ncomms4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC. 2011. Autophagy controls IL-1β secretion by targeting pro-IL-1β for degradation. J Biol Chem 286:9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ainscough JS, Frank Gerberick G, Zahedi-Nejad M, Lopez-Castejon G, Brough D, Kimber I, Dearman RJ. 2014. Dendritic cell IL-1α and IL-1β are polyubiquitinated and degraded by the proteasome. J Biol Chem 289:35582–35592. doi: 10.1074/jbc.M114.595686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duong BH, Onizawa M, Oses-Prieto JA, Advincula R, Burlingame A, Malynn BA, Ma A. 2015. A20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses NLRP3 inflammasome activity. Immunity 42:55–67. doi: 10.1016/j.immuni.2014.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. 2012. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiu HW, Chen CH, Chang JN, Hsu YH. 2016. Far-infrared promotes burn wound healing by suppressing NLRP3 inflammasome caused by enhanced autophagy. J Mol Med (Berl) 94:809–819. doi: 10.1007/s00109-016-1389-0. [DOI] [PubMed] [Google Scholar]
- 34.Lee JY, Seo D, You J, Chung S, Park JS, Lee JH, Jung SM, Lee YS, Park SH. 2017. The deubiquitinating enzyme, ubiquitin-specific peptidase 50, regulates inflammasome activation by targeting the ASC adaptor protein. FEBS Lett 591:479–490. doi: 10.1002/1873-3468.12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, Amatya R, Kelly TJ, Iwai K, Ting J, Jung JU. 2014. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med 211:1333–1347. doi: 10.1084/jem.20132486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guan K, Wei C, Zheng Z, Song T, Wu F, Zhang Y, Cao Y, Ma S, Chen W, Xu Q, Xia W, Gu J, He X, Zhong H. 2015. MAVS promotes inflammasome activation by targeting ASC for K63-linked ubiquitination via the E3 ligase TRAF3. J Immunol 194:4880–4890. doi: 10.4049/jimmunol.1402851. [DOI] [PubMed] [Google Scholar]
- 37.He Y, Varadarajan S, Muñoz-Planillo R, Burberry A, Nakamura Y, Núñez G. 2014. 3,4-Methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem 289:1142–1150. doi: 10.1074/jbc.M113.515080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. 2009. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 182:3173–3182. doi: 10.4049/jimmunol.0802367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hale BG, Steel J, Medina RA, Manicassamy B, Ye J, Hickman D, Hai R, Schmolke M, Lowen AC, Perez DR, García-Sastre A. 2010. Inefficient control of host gene expression by the 2009 pandemic H1N1 influenza A virus NS1 protein. J Virol 84:6909–6922. doi: 10.1128/JVI.00081-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, Mizuguchi J, Schweighoffer E, Tybulewicz V, Mitsuyama M. 2013. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol 14:1247–1255. doi: 10.1038/ni.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin YC, Huang DY, Wang JS, Lin YL, Hsieh SL, Huang KC, Lin WW. 2015. Syk is involved in NLRP3 inflammasome-mediated caspase-1 activation through adaptor ASC phosphorylation and enhanced oligomerization. J Leukoc Biol 97:825–835. doi: 10.1189/jlb.3HI0814-371RR. [DOI] [PubMed] [Google Scholar]
- 42.Khatri M, Dwivedi V, Krakowka S, Manickam C, Ali A, Wang L, Qin Z, Renukaradhya GJ, Lee CW. 2010. Swine influenza H1N1 virus induces acute inflammatory immune responses in pig lungs: a potential animal model for human H1N1 influenza virus. J Virol 84:11210–11218. doi: 10.1128/JVI.01211-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pomorska-Mól M, Markowska-Daniel I, Kwit K, Czyżewska E, Dors A, Rachubik J, Pejsak Z. 2014. Immune and inflammatory response in pigs during acute influenza caused by H1N1 swine influenza virus. Arch Virol 159:2605–2614. doi: 10.1007/s00705-014-2116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma W, Belisle SE, Mosier D, Li X, Stigger-Rosser E, Liu Q, Qiao C, Elder J, Webby R, Katze MG, Richt JA. 2011. 2009 pandemic H1N1 influenza virus causes disease and upregulation of genes related to inflammatory and immune responses, cell death, and lipid metabolism in pigs. J Virol 85:11626–11637. doi: 10.1128/JVI.05705-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakabe S, Iwatsuki-Horimoto K, Takano R, Nidom CA, Le M, Nagamura-Inoue T, Horimoto T, Yamashita N, Kawaoka Y. 2011. Cytokine production by primary human macrophages infected with highly pathogenic H5N1 or pandemic H1N1 2009 influenza viruses. J Gen Virol 92:1428–1434. doi: 10.1099/vir.0.030346-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee N, Wong CK, Chan PK, Chan MC, Wong RY, Lun SW, Ngai KL, Lui GC, Wong BC, Lee SK, Choi KW, Hui DS. 2011. Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PLoS One 6:e26050. doi: 10.1371/journal.pone.0026050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu X, Zhang X, Zhao B, Wang J, Zhu Z, Teng Z, Shao J, Shen J, Gao Y, Yuan Z, Wu F. 2011. Intensive cytokine induction in pandemic H1N1 influenza virus infection accompanied by robust production of IL-10 and IL-6. PLoS One 6:e28680. doi: 10.1371/journal.pone.0028680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osterlund P, Pirhonen J, Ikonen N, Rönkkö E, Strengell M, Mäkelä SM, Broman M, Hamming OJ, Hartmann R, Ziegler T, Julkunen I. 2010. Pandemic H1N1 2009 influenza A virus induces weak cytokine responses in human macrophages and dendritic cells and is highly sensitive to the antiviral actions of interferons. J Virol 84:1414–1422. doi: 10.1128/JVI.01619-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng H, Pappas C, Katz JM, Tumpey TM. 2011. The 2009 pandemic H1N1 and triple-reassortant swine H1N1 influenza viruses replicate efficiently but elicit an attenuated inflammatory response in polarized human bronchial epithelial cells. J Virol 85:686–696. doi: 10.1128/JVI.01568-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim HM, Lee YW, Lee KJ, Kim HS, Cho SW, van Rooijen N, Guan Y, Seo SH. 2008. Alveolar macrophages are indispensable for controlling influenza viruses in lungs of pigs. J Virol 82:4265–4274. doi: 10.1128/JVI.02602-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, Vogel J, Kopf M. 2014. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog 10:e1004053. doi: 10.1371/journal.ppat.1004053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cardani A, Boulton A, Kim TS, Braciale TJ. 2017. Alveolar macrophages prevent lethal influenza pneumonia by inhibiting infection of type-1 alveolar epithelial cells. PLoS Pathog 13:e1006140. doi: 10.1371/journal.ppat.1006140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, García-Sastre A. 2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qian W, Wei X, Guo K, Li Y, Lin X, Zou Z, Zhou H, Jin M. 2017. The C-terminal effector domain of non-structural protein 1 of influenza A virus blocks IFN-β production by targeting TNF receptor-associated factor 3. Front Immunol 8:779. doi: 10.3389/fimmu.2017.00779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. 2002. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem 277:21119–21122. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 56.Wang L, Manji GA, Grenier JM, Al-Garawi A, Merriam S, Lora JM, Geddes BJ, Briskin M, DiStefano PS, Bertin J. 2002. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-κB and caspase-1-dependent cytokine processing. J Biol Chem 277:29874–29880. doi: 10.1074/jbc.M203915200. [DOI] [PubMed] [Google Scholar]
- 57.Tisoncik JR, Billharz R, Burmakina S, Belisle SE, Proll SC, Korth MJ, García-Sastre A, Katze MG. 2011. The NS1 protein of influenza A virus suppresses interferon-regulated activation of antigen-presentation and immune-proteasome pathways. J Gen Virol 92:2093–2104. doi: 10.1099/vir.0.032060-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu G, Park HS, Pyo HM, Liu Q, Zhou Y. 2015. Influenza A virus panhandle structure is directly involved in RIG-I activation and interferon induction. J Virol 89:6067–6079. doi: 10.1128/JVI.00232-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shin YK, Liu Q, Tikoo SK, Babiuk LA, Zhou Y. 2007. Effect of the phosphatidylinositol 3-kinase/Akt pathway on influenza A virus propagation. J Gen Virol 88:942–950. doi: 10.1099/vir.0.82483-0. [DOI] [PubMed] [Google Scholar]
- 60.Komune N, Ichinohe T, Ito M, Yanagi Y. 2011. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1β secretion. J Virol 85:13019–13026. doi: 10.1128/JVI.05942-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Treier M, Staszewski LM, Bohmann D. 1994. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 78:787–798. doi: 10.1016/S0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- 62.Xiao G, Cvijic ME, Fong A, Harhaj EW, Uhlik MT, Waterfield M, Sun SC. 2001. Retroviral oncoprotein Tax induces processing of NF-κB2/p100 in T cells: evidence for the involvement of IKKα. EMBO J 20:6805–6815. doi: 10.1093/emboj/20.23.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Palazón-Riquelme P, López-Castejón G. 2016. Method to measure ubiquitination of NLRs. Methods Mol Biol 1417:223–229. doi: 10.1007/978-1-4939-3566-6_16. [DOI] [PubMed] [Google Scholar]