

ABSTRACT
Multiple steps of the life cycle of hepatitis B virus (HBV) are known to be coupled to hepatic metabolism. However, the details of involvement of the hepatic metabolic milieu in HBV infection remain incompletely understood. Hepatic lipid metabolism is controlled by a complicated transcription factor network centered on retinoid X receptor alpha (RXRα). Here, we report that RXRα negatively regulates HBV infection at an early stage in cell cultures. The RXR-specific agonist bexarotene inhibits HBV in HepG2 cells expressing the sodium taurocholate cotransporting polypeptide (NTCP) (HepG2-NTCP), HepaRG cells, and primary Tupaia hepatocytes (PTHs); reducing RXRα expression significantly enhanced HBV infection in the cells. Transcriptome sequencing (RNA-seq) analysis of HepG2-NTCP cells with a disrupted RXRα gene revealed that reduced gene expression in arachidonic acid (AA)/eicosanoid biosynthesis pathways, including the AA synthases phospholipase A2 group IIA (PLA2G2A), is associated with increased HBV infection. Moreover, exogenous treatment of AA inhibits HBV infection in HepG2-NTCP cells. These data demonstrate that RXRα is an important cellular factor in modulating HBV infection and implicate the participation of AA/eicosanoid biosynthesis pathways in the regulation of HBV infection.
IMPORTANCE Understanding how HBV infection is connected with hepatic lipid metabolism may provide new insights into virus infection and its pathogenesis. By a series of genetic studies in combination with transcriptome analysis and pharmacological assays, we here investigated the role of cellular retinoid X receptor alpha (RXRα), a crucial transcription factor for controlling hepatic lipid metabolism, in de novo HBV infection in cell cultures. We found that silencing of RXRα resulted in elevated HBV covalently closed circular DNA (cccDNA) formation and viral antigen production, while activation of RXRα reduced HBV infection efficiency. Our results also showed that silencing phospholipase A2 group IIA (PLA2G2A), a key enzyme of arachidonic acid (AA) synthases, enhanced HBV infection efficiency in HepG2-NTCP cells and that exogenous AA treatment reduced de novo HBV infection in the cells. These findings unveil RXRα as an important cellular factor in modulating HBV infection and may point to a new strategy for host-targeted therapies against HBV.
KEYWORDS: HBV, RXRα, NTCP, arachidonic acid, inhibition
INTRODUCTION
Hepatitis B virus (HBV) infection remains a major public health problem, with more than 240 million people chronically infected worldwide. These individuals are at high risk of developing cirrhosis, liver failure, or hepatocellular carcinoma (HCC) (1–3). Currently, the available therapies against HBV are limited to the immune modulator alpha interferon (IFN-α) (4), which has a low antiviral efficacy of less than 40% (5, 6), and viral reverse transcription inhibitors such as lamivudine, adefovir, dipivoxil, entecavir, and tenofovir (7–10). Given this limited set of therapies and considering the vast scale of global HBV infection, novel treatment strategies against HBV are needed.
HBV is a small, enveloped DNA virus with a 3.2-kb partially double-stranded DNA genome. The only confirmed organ targeted by HBV is the liver. During its life cycle, HBV closely couples its gene expression to hepatic metabolism, for example, by recruiting transcription factors and coactivators from host cells to the viral genome during virus replication. Most of these transcription factors are liver-enriched nuclear receptors such as farnesoid X receptor (FXR), hepatocyte nuclear factor 4α (HNF-4α), peroxisome proliferator-activated receptor alpha (PPARα), retinoid X receptor alpha (RXRα), and proliferator-activated receptor γ coactivator 1α (PGC-1α), which are known to be key hepatic metabolic genes (11–18). The identification of sodium taurocholate cotransporting polypeptide (NTCP) as the essential receptor for human HBV (19) has significantly advanced our understanding of HBV/hepatitis delta virus (HDV) infection and has suggested that bile acid-related metabolism may be intimately linked to the HBV infection process. In addition, accumulating evidence has indicated that hepatic cholesterol is important for HBV infection. Cholesterol within the viral envelope is indispensable for the entry process of HBV (20), and decreasing cellular cholesterol levels resulted in reduced levels of secreted HBV virions (21) and HBV surface antigen (HBsAg) in Huh7 cells transfected with a plasmid that contained HBV genome (22). Although these observations have implied important relationships, it remains unclear how the metabolism of bile acids and/or hepatic lipids may influence HBV infection.
Retinoid X receptor alpha (RXRα) acts as a master regulator of hepatic metabolism homeostasis (23). It is expressed abundantly in the liver and acts as an obligate heterodimer partner for other liver-enriched nuclear receptors (24). Here, we utilized HepG2-NTCP and HepaRG cells, as well as primary Tupaia hepatocytes (PTHs), all of which are susceptible to human HBV infection, to study the potential role of RXRα in de novo HBV infection. We found that bexarotene, a specific agonist of RXR, inhibited HBV infection while knockdown of RXRα expression enhanced viral infection, indicating that RXRα levels are inversely correlated to the efficiency of early-stage HBV infection. We further performed transcriptome analysis (RNA-seq) of HepG2-NTCP cell clones with a disrupted endogenous RXRα gene and found that the expression of genes in arachidonic acid (AA)/eicosanoid biosynthesis pathways was downregulated in RXRα knockdown cells. By combining targeted silencing of the genes with inhibitor treatment of key enzymes involved in the biosynthesis of AA/eicosanoids, we show that AA can suppress HBV infection in cell cultures. Blocking the biosynthesis of prostanoids but not of leukotriene could enhance HBV infection.
RESULTS
Activation of RXRs inhibited HBV infection in HepG2-NTCP cells, HepaRG cells, and primary Tupaia hepatocytes.
RXRs are nuclear receptors. Bexarotene is a retinoid that selectively activates RXRs; it is used clinically for the treatment of cutaneous T cell lymphoma (25). To assess the role of RXRs on HBV infection, HepG2-NTCP cells were coincubated with bexarotene and HBV for 24 h, and control cells were coincubated with HBV and dimethyl sulfoxide (DMSO). A myristoylated pre-S1 peptide containing the first N-terminal 59 amino acids (Myr-59), which is an efficient entry inhibitor for both HBV and HDV infection, was used as a positive control for viral entry inhibition. Bexarotene inhibited HBV infection in a dose-dependent manner. Compared to levels in the control, bexarotene (5 μM) treatment led to a 70% reduction in the levels of two HBV antigens, HBV e antigen (HBeAg) and HBsAg (Fig. 1A). Consistently, the levels of viral RNA, including the HBV total RNA and the 3.5-kb RNA (for HBV pre-C and pregenomic RNA [pgRNA]), were significantly reduced in the bexarotene-treated cells (Fig. 1B). Moreover, immunofluorescence staining revealed a marked reduction in the intracellular levels of HBcAg (Fig. 1C, upper panel) and HBsAg (Fig. 1C, lower panel). These three lines of evidence all demonstrate that bexarotene treatment during the early stages inhibits HBV infection. The hepatitis delta virus (HDV) is a satellite of HBV, and it utilizes HBV envelope proteins to assemble virions and enter hepatocytes through the HBV-specific receptor NTCP (19). HDV infection was also inhibited by bexarotene in a dose-dependent manner. As shown in Fig. S1A in the supplemental material, the expression of HDV delta antigen (Fig. S1A, left) and copies of HDV total RNA (Fig. S1A, right) were reduced in bexarotene-treated cells. In contrast, infection with control lenti-VSV-G-EGFP virus (an HIV-1 pseudovirus enveloped by glycoprotein of vesicular stomatitis virus [VSV-G] and expressing enhanced green fluorescent protein [EGFP]) was not altered by bexarotene treatment (Fig. 1D). Importantly, bexarotene showed no deleterious effects on cell viability, even at the highest-tested concentration (Fig. 1E, left panel). Additionally, we observed that bexarotene treatment resulted in activation of RXRs in HepG2-NTCP cells: the expression levels of the liver-type fatty acid binding protein (L-FABP) gene, a known downstream target of RXRs, was induced by more than 5-fold in bexarotene-treated cells (Fig. 1E, right panel), confirming the activation of RXRs.
FIG 1.

Activation of RXRs inhibited HBV infection in HepG2-NTCP cells. (A to C) Cells were inoculated with HBV in the presence of various concentrations of bexarotene (Bexa), Myr-59 (250 nM), or DMSO for 24 h. Culture medium samples were collected at the indicated times, and HBV viral antigens were measured by ELISA (A). The copy numbers of HBV total RNA and the HBV 3.5-kb RNA (for HBV pre-C and pregenomic RNA [pgRNA]) were measured at 7 dpi (B). At 7 dpi, intracellular HBcAg (green) and HBsAg (red) were stained with 1C10 and 17B9 antibodies, respectively, and nuclei were stained with DAPI (blue) (C). (D) Bexarotene treatment during VSV-G control virus incubation did not affect viral infection in HepG2-NTCP cells. Cells were inoculated with lenti-VSV-G-EGFP virus in the presence of bexarotene, 250 nM Myr-59, or DMSO for 24 h. The expression of EGFP was analyzed by fluorescence microscopy at 48 h postinfection. (E) In HepG2-NTCP cells, bexarotene treatment can activate RXRs. HepG2-NTCP cells were treated with bexarotene at the indicated concentration in PMM for 24 h. Cells were then inoculated with 10% alamarBlue in PMM for 1 h. Cellular health was evaluated based fluorescence absorbance at 590 nm. The absorbance of DMSO-treated cells was set to 100% (left). L-FABP gene expression was evaluated by qRT-PCR (right). (F) The inhibitory effect of bexarotene on HBV infection occurred mainly in the first 24 h after viral incubation. A schematic diagram of the time course of bexarotene treatment is shown (top). HepG2-NTCP cells were incubated with HBV for 24 h. In different groups, compounds (bexarotene, DMSO, and Myr-59) were added to cell culture at different stages of viral infection and incubated with cells for different lengths of time. Culture medium samples were collected at 7 dpi, and HBeAg (middle) and HBsAg (bottom) were measured by ELISA. (G) Bexarotene treatment at 1 day post-HBV incubation had little effect on viral infection in HepG2-NTCP cells. Cells were inoculated with HBV virus for 24 h. At 2 dpi, cells were treated with bexarotene, 250 nM Myr-59, or DMSO for another 24 h. The copy numbers of HBV total RNA (left) and the 3.5-kb RNA (right) were measured at the end of experiment. (*, P < 0.05; **, P < 0.01, ns, not significant; Student's t test). OD450, optical density at 450 nm.
These initial coincubation assays probed only the first 24 h of the HBV infection process. To more precisely determine the stage at which bexarotene exhibits its inhibitory effects on HBV infection, we performed HBV incubation assays in expanded time course experiments. HepG2-NTCP cells were divided into five groups, and all groups were incubated with HBV for the initial 24 h. Some of the groups were treated with bexarotene for the first 24 h; other groups were treated with bexarotene at later phases throughout a 72-h time course (Fig. 1F, top). The extent of viral infection in cells was assessed by enzyme-linked immunosorbent assay (ELISA). Comparison of group 1 (24-h bexarotene) and group 2 (72-h bexarotene) revealed that prolonged bexarotene treatment did not result in a greater extent of inhibition than treatment for 24 h. Consistently, the groups that received bexarotene treatment after the cells had been exposed to HBV for 24 h (groups 3 to 5) exhibited either slight or no inhibitory effects on HBV infection (Fig. 1F). Reverse transcription-PCR (RT-PCR) analysis indicated that there were no significant differences in viral RNA levels between control cells and the cells treated with bexarotene only on the final day of the experiment (i.e., treated at 48 h, which is 24 h after the completion of the HBV incubation) (Fig. 1G). Likewise, bexarotene treatment after the cells had been exposed to HDV for 24 h revealed no inhibitory effect on HDV infection (Fig. S1B).
We next used other susceptible cell types, including HepaRG cells and primary Tupaia hepatocytes (PTHs), to confirm the inhibitory effects of bexarotene on HBV infection and to confirm that these effects occur at the early stages of HBV exposure. These experiments tested coincubation assays and assays that treated cells following an initial exposure to HBV (postinoculation). Bexarotene and HBV coincubation in HepaRG and PTHs reduced the levels of viral antigens (Fig. 2A, B, and C, left panel). However, bexarotene had little effect on HBV viral antigen levels when the drug was added to the culture after the initial 24 h of HBV exposure on PTHs (Fig. 2C, right panel). We also used 9-cis retinoic acid (9cRA), another known agonist of RXRs, to treat PTHs during HBV infection (Fig. 2D) and found that 9cRA treatment during HBV incubation for the first 24 h significantly inhibited viral infection and did so in a dose-dependent manner (Fig. 2D, left panel). Consistent with our results for bexarotene, adding 9cRA at 24 h post-HBV infection (i.e., at hour 48 of the experiment) had only minimal effects on the extent of viral infection (Fig. 2D, right panel). These results demonstrate that an RXR agonist like bexarotene inhibits early-stage HBV infection in three different HBV-susceptible cells, implying that RXRs are critically important in this phase of HBV infection.
FIG 2.

Activation of RXRs inhibited HBV infection in both HepaRG cells and primary tupaia hepatocytes (PTHs). (A) Bexarotene treatment during HBV inoculation inhibited viral infection in HepaRG cells. Cells were treated with bexarotene or DMSO for 24 h. Culture medium samples were collected at the indicated times, and HBV viral antigens were measured by ELISA. (B to D) Activation of RXRs by bexarotene and 9cRA inhibited HBV infection at an early stage in PTHs. A schematic diagram for the time course of pharmaceutical treatment is shown (B). PTHs were treated with bexarotene (C) or 9cRA (D) at different concentrations during HBV inoculation (left) or at 1 dpi (right) for 16 h. Culture medium samples were collected at 7 dpi, and HBV viral antigens were measured by ELISA. (*, P < 0.05; **, P < 0.01, ns, not significant; Student's t test).
Decreased RXRα expression enhanced HBV infection in HepG2-NTCP cells and PTHs.
There are three known subtypes of the RXR family in humans: RXRα, RXRβ, and RXRγ; RXRα is the most abundant of these in the adult liver (24). We conducted HBV infection assays in cells with small interfering RNA (siRNA)-induced knockdown of RXRα gene expression. We evaluated the efficiency of RXRα-specific siRNAs (RXRα-si1 and RXRα-si2) in knocking down RXRα mRNA and protein expression in HepG2-NTCP cells. The RXRα-si1 transfected cells had an 80% reduction in the mRNA levels of RXRα, and RXRα protein expression was almost undetectable (Fig. 3A). As shown in Fig. 3B, compared to levels in control cells, the levels of both HBsAg and HBeAg were significantly increased (>2-fold) in the RXRα-silenced cells on day 5 and day 7 after HBV infection. We also compared viral RNA levels (Fig. 3C) and the intracellular expression of HBcAg/HBsAg (Fig. 3D) and found that all of these were increased in RXRα knockdown cells. Moreover, the magnitude of these increases was correlated with the efficiency of RXRα siRNA knockdown.
FIG 3.

Decreased RXRα expression enhanced HBV infection in HepG2-NTCP and PTHs. (A to D) Knocking down RXRα expression prior to viral inoculation enhanced HBV infection in HepG2-NTCP cells. HepG2-NTCP cells were transfected with siRNAs against hRXRα or a control siRNA. Three days after transfection, RXRα gene expression was measured by RT-PCR (A, left) and Western blotting (A, right). Cells were then inoculated with HBV; culture medium samples were collected at the indicated times, and levels of viral antigens were measured by ELISA (B). The copy numbers of HBV total RNA and the 3.5-kb RNA were measured at 7 days after HBV infection (C). Intracellular HBcAg (green) and HBsAg (red) were stained with 1C10 and 17B9 antibodies, respectively (D). (E) Knocking down RXRα prior to viral inoculation enhanced HBV infection in PTHs. Freshly isolated PTHs were transfected with siRNAs against tsRXRα or a control siRNA. Forty-eight hours after transfection, cells were inoculated with HBV. Expression of the tsRXRα gene was measured by RT-PCR (left). Culture medium samples were collected at 7 dpi, and HBV viral antigens were measured by ELISA (right). (F) Knocking down RXRα expression post-viral inoculation did not affect HBV infection in HepG2-NTCP cells. Cells were transfected with siRNAs against hRXRα or a control siRNA 1 day post-HBV inoculation. The copy numbers of HBV total RNA and the 3.5-kb RNA were measured at 7 dpi. (G and H) RXRα stable knockdown monoclone cell lines were more susceptible to HBV infection than wild-type (WT) HepG2-NTCP cells. A TALEN-based strategy was used to target the endogenous RXRα locus (G, left). TALEN targeting arms were designed to bind to target sequence within the fourth exon of the RXRα genome. The donor plasmid contained the phosphoglycerate kinase-puromycin resistance gene expression cassette, which was flanked with two arms that are homologous with the 5′-to-3′ regions of the TALEN target sequence. Western blot analysis of the expression of endogenous RXRα was performed in both the RXRα-KD monoclone cell lines and in wild-type HepG2-NTCP cells (G, right). After HBV infection, at 7 dpi, intracellular HBcAg (green) was stained with the 1C10 antibody (H). (*, P < 0.05; **, P < 0.01, ns, not significant; Student's t test).
Experiments using PTHs also revealed that HBV infection was enhanced in cells transfected with siRNAs targeting Tupaia (tree shrew [ts]) RXRα (Fig. 3E). In tsRXRα-si1 transfected cells where the tsRXRα mRNA level was reduced by ∼75%, the HBeAg level was more than 2-fold greater than that of the control group. Consistent with results from the bexarotene and 9cRA inhibition assays, viral infection was not affected if the silencing of RXRα was initiated after the viral incubation phase (Fig. 3F). Meanwhile, HDV infection was also increased in both HepG2-NTCP cells (Fig. S1C) and PTHs (Fig. S1D) after RXRα depletion, as evidenced by elevated expression levels of delta antigen and copies of HDV total RNA.
To further confirm the role of RXRα in HBV infection, we generated a TALEN (transcription activator-like effector nuclease) pair targeting a sequence in the fourth exon of the RXRα gene (Fig. 3G, left panel). We screened 150 clones with puromycin selection assays and chose two of the RXRα monoclones with confirmed disruption of RXRα expression as assessed by Western blot analysis (Fig. 3G, right panel). We examined HBV infection in these RXRα stable knockdown cells. As shown in Fig. 3H, HBV infection was significantly enhanced in both of the RXRα stable knockdown clones compared to the infection level in the wild-type HepG2-NTCP (HepG2-NTCP/WT) cells. These results further confirmed that reduced RXRα expression increased the extent of HBV infection.
To determine whether RXRα modulates HBV infection at an early stage by regulating NTCP expression, we evaluated both mRNA and protein levels of NTCP in HepG2-NTCP cells after bexarotene treatment or RXRα silencing. As shown in Fig. S2, activation of RXRα by bexarotene did not alter the mRNA level of NTCP in HepG2-NTCP cells (Fig. S2A). Compared to expression in control cells, knocking down RXRα affected neither mRNA expression (Fig. S2B) nor cell surface protein expression of NTCP (Fig. S2C and D). These results suggest that another factor(s) in addition to the surface expression of NTCP is involved in modulating the early-stage infection efficiency of the viruses in the cells.
Arachidonic acid biosynthesis was downregulated in RXRα knockdown cells.
RXRα plays a pivotal role in regulating liver metabolism by forming heterodimers with nuclear receptors in adult liver. We hypothesized that metabolism pathways regulated by RXRs may affect the efficiency of HBV infection. We therefore performed transcriptome analysis to compare the gene expression levels between the wild-type HepG2-NTCP cells and cells with a TALEN-disrupted RXRα allele. As expected, the RNA-seq data showed that a large number of genes responsible for lipid homeostasis were altered in the RXRα stable knockdown cells, including genes involved in the metabolism of cholesterol, fatty acid, bile acid, and xenobiotic and eicosanoid biogenesis (Fig. 4). Correspondingly, global changes in gene expression regulated by RXRα and its heterodimers (including PPAR, FXR, liver X receptor [LXR], vitamin D receptor [VDR], retinoic acid receptor [RAR], constitutive androstane receptor [CAR], and pregnane X receptor PXR) were observed in HepG2-NTCP cells after RXRα depletion (Fig. S3A). Increased mRNA levels of some key lipogenic enzymes, including acetyl-coenzyme A (CoA) carboxylase alpha (ACACA) and stearoyl-CoA desaturase (SCD), were found in RXRα stable knockdown cells, while the expression levels of genes (including AGPAT1, -2, -3, -4, and -6 and GPAM) involved in the first two steps of de novo phospholipid biosynthesis (which uses fatty acids as substrates) were reduced (Fig. 4B). These findings are in line with the previous in vivo studies that highlight a critical role of RXRα in hepatic fatty acid homeostasis (26, 27). In RXRα stable knockdown cells, the expression levels of several genes involved in cholesterol synthesis was upregulated, such as HMGCS1, LSS, IDI1, and CYP51A1, etc. Conversely, the expression levels of LRP1, which prevents intracellular cholesterol accumulation (28), and ABCG5/ABCG8, which promote cholesterol removal from the body, were downregulated (Fig. 4C). These data indicate that RXRα ablation resulted in disruption of cholesterol homeostasis and are consistent with the finding that hepatocyte-specific RXRα mutant mice exhibited an inability to process and eliminate cholesterol (27). Since RXRα functions as a master regulator of xenobiotic metabolism, we also compared the expression levels of CAR/RXRα and PXR/RXRα target genes encoding phase I enzymes and phase II enzymes. As shown in Fig. 4D, RXRα deficiency resulted in reduced basal levels of CYP2B and CYP3A, which are downregulated in RXRα-null hepatocytes as well (29, 30). Intriguingly, among these genes involved in xenobiotic metabolism, RXRα deficiency alters homeostasis of glutathione (GSH), a common conjugate employed in the phase II detoxification process, as manifested by slightly decreased expression levels of glutathione-metabolizing enzymes, such as GSTP (where GST is glutathione S-transferase), GSTM, and glutamate-cysteine ligase catalytic subunit (GCLC) (Fig. 4E). These results are consistent with previously reported works demonstrating that the regulation of hepatic GSH levels by RXRα is essential to protect hepatocytes from oxidative stress (31). In addition, the expression levels of many retinoid target genes (such as ALDH1A1, CYP26B1, and CRABP2) regulated by RXR/RAR were decreased after RXRα depletion (Fig. 4F). Nevertheless, an interesting result in the analysis of hepatic lipid metabolism was the downregulation of the expression of the genes of arachidonic acid (AA) synthesis and related metabolism pathways in the RXRα stable knockdown cells (Fig. 4A). Mass spectrometry analysis confirmed a compromised cellular level of AA in the RXRα stable knockdown cells (Fig. S3B).
FIG 4.

Arachidonic acid biosynthesis was downregulated in RXRα knockdown cells. Gene expression in an RXRα stable knockdown (RXRα-KD) monoclone cell line and wild-type HepG2-NTCP (WT) cells was analyzed by RNA-seq. Expression profiles of selected genes involved in the eicosanoid (A), fatty acid and phospholipid (B), cholesterol and bile acid (C), xenobiotic metabolism (D and E), and vitamin A metabolism (F) pathways are presented in heat map. The expression levels of genes (reads per kilobase per million of >0.5; total of 21,172 genes) in RXRα stable knockdown and wild-type HepG2-NTCP cells are compared to the averages among them. The color coding in the heat maps represents log2-fold changes in gene expression levels.
AA inhibited HBV infection in HepG2-NTCP and HepaRG cells.
Given the decreased expression levels of arachidonic acid (AA) synthesis genes in the RXRα stable knockdown cells, we reasoned that the AA synthesis pathway may influence HBV infection. AA is released from plasma membrane phospholipids by members of the phospholipase A2 (PLA2) family; the Ca2+-dependent cytosolic protein PLA2 (cPLA2) is known to be the dominant functional member of this family (32). PLA2G2A, which functions in arachidonic acid synthesis, exhibited decreased expression in the RXRα stable knockdown cells. To directly assess the potential function of PLA2G2A in HBV infection, HepG2-NTCP cells were transfected with a PLA2G2A-specific siRNA or control siRNA prior to viral infection. As shown in Fig. 5A, gene silencing of PLA2G2A resulted in elevated HBsAg and HBeAg levels during viral infection. Northern blot analysis also revealed increased levels of the HBV 3.5-kb RNA and 2.1-kb RNA after viral infection (Fig. 5B). To examine whether the inhibitory effect of RXRα on HBV infection could be abrogated by disruption of the AA biosynthesis pathway, HepG2-NTCP cells were transfected with a PLA2G2A-specific or a control siRNA, and then the HBV infection was carried out in the presence or absence of bexarotene. As shown in Fig. 5H, bexarotene suppressed HBV infection, but the effect could be partially abrogated by silencing PLA2G2A in the cells. However, the restorative effect was not observed in HDV infection (Fig. S1E). These results indicate that PLA2G2A may have contributed to the inhibitory effect of RXRα on HBV infection.
FIG 5.

Arachidonic acid (AA) inhibited HBV infection in HepG2-NTCP and HepaRG cells. (A) Knocking down PLA2G2A expression prior to viral inoculation enhanced HBV infection in HepG2-NTCP cells. Cells were transfected with siRNAs against human PLA2G2A (hPLA2G2A) or control siRNA. At three days posttransfection, cells were incubated with HBV for 24 h. Culture medium samples were collected at 5 dpi, and HBV viral antigens were measured by ELISA (right).Gene expression was measured by real-time PCR (left). (B) Total RNA was extracted from infected cells at 5 dpi and analyzed by Northern blotting. (C to E) Arachidonic acid treatment during HBV incubation reduced viral infection in HepG2-NTCP cells. Cells treated with various doses of AA (or a methanol control) were incubated with HBV for 24 h. Culture medium samples were collected at the indicated times, and HBV viral antigens were measured by ELISA (C). At 7 dpi, intracellular HBcAg (green) and HBsAg (red) were stained with 1C10 and 17B9 antibodies, respectively (D). The copy numbers of HBV total RNA and the 3.5-kb RNA (E) were measured at 7 dpi. (F) IFN-α treatment during HBV incubation slightly reduced viral infection in HepG2-NTCP cells. Cells were inoculated with HBV virus in the presence of IFN-α (or a PBS control) for 24 h. (G) AA treatment during HBV inoculation reduced viral infection in HepaRG cells. Cells treated with various doses of AA (or with a methanol control) were incubated with HBV for 16 h. Culture medium samples were collected at 7 dpi, and HBV viral antigens were measured by ELISA. (H) Silencing PLA2G2A partially relieved the inhibitory effect of bexarotene on HBV infection in HepG2-NTCP cells. Cells were transfected with siRNAs against hPLA2G2A or a control siRNA. At 3 days posttransfection, cells were incubated with HBV in the presence of 5 μM bexarotene for 24 h. Intracellular HBcAg (green) was stained with the 1C10 at 7 dpi. (*, P < 0.05; **, P < 0.01; ns, not significant; Student's t test).
We also tested whether the application of exogenous AA could inhibit HBV infection. As shown in Fig. 5C, the addition of 2 μM AA to cell cultures during viral incubation resulted in significantly reduced infection, while the addition of 10 μM AA led to yet further reductions in infection. This 10 μM AA treatment also resulted in an ∼70% reduction of the levels of both HBeAg and HBsAg in infected cells. Intracellular immunofluorescence staining showed that in the vehicle-treated control group, more than 80% of cells were infected, while less than 30% of AA-treated cells (10 μM) were infected (Fig. 5D). Consistently, the levels of viral RNA, including the HBV total RNA and the 3.5-kb RNA (Fig. 5E), were reduced by AA treatment in a dose-dependent manner. For comparison, we also tested the effect of IFN-α on HBV infection in HepG2-NTCP cells with the same experimental procedure. There was less than a 50% reduction of the infection at a concentration of IFN-α up to 20 ng/ml (Fig. 5F). We also examined the effect of AA on HepaRG cells; as expected, the AA reduced the levels of both HBeAg and HBsAg in a dose-dependent manner (Fig. 5G). A high concentration of 10 μM AA led to more than a 50% decrease of HBsAg at 7 days postinfection (dpi). AA did not affect cell viability or NTCP expression at the tested concentrations (data not shown). We also assessed the effect of AA on HDV infection; the agent produced only a slight or no reduction in HDV infection on HepG2-NTCP cells (Fig. S1F).
The prostanoid biosynthesis pathway moderated HBV infection in HepG2-NTCP and HepaRG cells.
To examine whether metabolic pathways downstream of AA biosynthesis (e.g., cyclooxygenases [COXs] and lipoxygenases [LOXs]) affect HBV infection, we performed HBV infection assays with cells treated with indomethacin (COX inhibitor), zileuton (LOX5 inhibitor), baicalein (LOX12 inhibitor), and PD146176 (LOX15 inhibitor) (Fig. 6A). None of these drugs caused toxicity in HepG2-NTCP cells at the tested concentrations. Treatment with the LOX inhibitors (zileuton/baicalien/PD146176) did not alter HBV infection. However, treatment with the COX inhibitor indomethacin increased HBV infection in a dose-dependent manner. Indomethacin treatment (25 μM) increased HBsAg and HBeAg levels by about 1-fold in HBV-infected cells (Fig. S4A). The levels of HBV RNA (Fig. 6B and E, left) and intracellular HBcAg and HBsAg levels (Fig. 6C) were also increased following indomethacin treatment, and the level of cccDNA was increased to a much less extent in HepG2-NTCP cells (Fig. 6E, right). The effect was also observed in HepaRG cells in a dose-dependent manner (Fig. 6D). Taken together, these results showed that blocking prostanoid biosynthesis, but not leukotriene synthesis, increases HBV infection. Furthermore, indomethacin treatment partially rescued HBV infection suppressed by bexarotene (Fig. 6F). Similar to the effect of AA on HDV infection, none of these drugs had a significant effect on HDV infection in HepG2-NTCP cells (Fig. S1G and H). To further address whether the products of AA metabolism generated by COX could directly affect HBV infection, HepG2-NTCP cells were coincubated with PGD2/PGE2 and HBV. As shown in Fig. S4B and C, 5 μM PGE2 treatment reduced HBV RNAs to about 30% of levels in vehicle-treated cells, while PGD2 was less effective in inhibiting HBV infection.
FIG 6.

Prostanoid biosynthesis, but not leukotriene biosynthesis, modulated HBV infection in HepG2-NTCP and HepaRG cells. (A to C) Indomethacin treatment during HBV incubation enhanced viral infection in HepG2-NTCP cells. A simplified scheme of AA-related metabolism pathways is shown. Selected lipid mediators are boxed in color, and enzyme inhibitors appear in red (A). Cells were inoculated with HBV in the presence of drugs or DMSO for 24 h. At 7 dpi, the copy numbers of HBV total RNA and the 3.5-kb RNA (right) were measured (B). Intracellular HBcAg and HBsAg (C) were stained with 1C10 (in green) and 17B9 (in red) antibodies, respectively. (D) Indomethacin treatment during HBV incubation enhanced viral infection in HepaRG cells. Cells were inoculated with HBV and treated with various doses of indomethacin (or DMSO) for 16 h. HBeAg and HBsAg levels in the culture medium were collected at the indicated times and measured by ELISA. (E) Total RNA (left) and cccDNA (right) were extracted from infected cells at 7 dpi and detected by Northern or Southern blot analysis, respectively. (F) Indomethacin treatment partially relieved the inhibitory effect of bexarotene on HBV infection in HepG2-NTCP cells. Cells were incubated with HBV in the presence of 5 μM bexarotene or DMSO plus 25 μM indomethacin (Indo) or vehicle for 24 h. Intracellular HBcAg (green) was stained with the 1C10 at 7 dpi (upper), and HBsAg in culture medium was measured by ELISA (lower). (*, P < 0.05; **, P < 0.01; Student's t test).
Silencing of RXRα and PLA2G2A modestly increases the level of cccDNA.
As cccDNA plays a central role in establishing HBV infection and as the formation of cccDNA is a hallmark of the hepadnavirus replication cycle, we attempted to assess the effects of modulating RXRα and the arachidonic acid(AA)/eicosanoid biosynthesis pathways on HBV cccDNA formation. We infected HepG2-NTCP (AC12) cells, a highly susceptible HepG2-NTCP cell clone, and examined cccDNA by Southern blotting (33, 34). HBV cccDNA from infected cells was isolated using the Hirt method and examined by Southern blotting. After the sample was heated at 95°C, in contrast to the band corresponding to relaxed circular DNA (rcDNA), the cccDNA band remained and shifted to a position corresponding to a 3.2-kb DNA after further digestion with EcoRI (a unique restriction site in the HBV genome) (Fig. 7A). Silencing RXRα with an siRNA markedly enhanced the amounts of HBV RNA (Fig. 7B, left) and, to a lesser extent, cccDNA in HepG2-NTCP (AC12) cells (Fig. 7B, right). HBV cccDNA was reduced in the cells treated with bexarotene (Fig. 7C).
FIG 7.

Silencing of RXRα and PLA2G2A modestly increases the level of cccDNA. (A) HBV cccDNA was isolated by the Hirt method and confirmed by Southern blot analysis. Lane M, molecular mass marker. (B) Knocking down RXRα and PLA2G2A expression in HepG2-NTCP (AC12) cells prior to viral inoculation enhanced HBV RNA and cccDNA formation. Cells were transfected with siRNAs against RXRα, PLA2G2A, or a control siRNA. At 3 days posttransfection, cells were incubated with HBV for 24 h; total RNA and cccDNA were extracted from infected cells at 7 dpi and detected by Northern and Southern blot analysis, respectively. (C) HBV cccDNA was detected in infected HepG2-NTCP (AC12) cells treated with bexarotene (0.5 or 0.25 μM) or DMSO during viral inoculation. (D) Total DNAs were isolated from infected cells at the indicated times, and HBV cccDNA were quantified by real-time qPCR assays. (E) AA treatment during HBV incubation reduced HBV cccDNA formation in HepG2-NTCP cells. Cells were treated with different doses of AA (or methanol control [Me]) during HBV incubation for 24 h (coincubation) or treated with AA after HBV inoculation (postinoculation). Total RNA or HBV cccDNA was extracted and detected by Northern and Southern blot analysis.
We also examined the cccDNA level in cells with diminished expression of the AA synthase PLA2G2A. As shown in Fig. 7B, silencing PLA2G2A in HepG2-NTCP (AC12) cells increased the amounts of HBV RNA and cccDNA. Consistently, cccDNA was increased in RXRα or PLA2G2A knockdown cells based on the quantitative analysis of cccDNA using a more sensitive real-time PCR assay during infection (Fig. 7D). Moreover, HBV RNAs were reduced when AA treatment occurred during coincubation or at 1 day postinoculation (Fig. 7E, left). Meanwhile coincubation of AA led to impaired HBV cccDNA formation in a dose-dependent manner in HepG2-NTCP cells (Fig. 7E, right). These studies demonstrated an intriguing role of the AA/eicosanoid biosynthesis pathways in modulating cccDNA establishment and viral infection.
DISCUSSION
HBV utilizes the hepatocyte sodium-dependent bile acid transporter NTCP for viral entry and couples its life cycle to hepatic metabolic events. The virus relies heavily on liver-enriched nuclear receptors for its propagation. In this study, we investigated the role of RXRα, a master regulator of hepatic metabolism homeostasis, in de novo HBV infection of cell cultures.
Our study differs from previous reports that used HBV genome or reporter-viral DNA transfection systems. Earlier transfection studies showed that RXRα, coupled as a heterodimer with either PPARα (16, 17, 35–37) or FXR (13, 14), two nuclear receptors, activates HBV transcription and replication in human hepatoma cell lines (e.g., Huh7 and HepG2). Our study, in contrast, used genuine HBV infection systems, and we found that RXRα suppresses early-stage HBV infection in HepG2-NTCP cells, PTHs, and HepaRG cells. Previous reports are all based on exogenous overexpression of RXRα (plus agonist stimulation), whereas the role of endogenous RXRα in modulating de novo HBV infection remains unexplored. Indeed, the expression of HBV genes is regulated by numerous ubiquitous and liver-enriched transcriptional factors, such as COUP-TF, Sp1, HNF-1α, HNF-3β, HNF-4α, and C/EBP (38). RXRα is one of the multiple host factors which forms heterodimers with FXR (13) and PPARs (36) to increase the activity of enhancer II (Enh II)/core promoter and Enh I/X promoter, respectively. In addition, binding motifs for host factors are enormously overlapped (e.g., binding sites for HNF-4α and RXRα/PPARα within the Enh II/core promoter) (16, 38) and even competitive (e.g., COUP-TF competes with RXRα and HNF-4 for binding to the same site on Enh I) (16, 39, 40). Moreover, addition of agonists (RAs plus PPARα agonist) or nuclear receptors (cotransfection of RXRα and FXR or PPAR expression plasmids) alone only slightly increased or had little effect on transcription from HBV promoters (13, 36). Together, although reasons for the discrepant results from previous studies and this work are not completely clear, they may due to a different cellular system (viral genome/reporter-viral DNA transfection versus genuine HBV infection), a different strategy (exogenous overexpression versus endogenous depletion), and a different evaluation standard (promoter activity versus HBV antigen expression and viral DNA/RNA synthesis). Nevertheless, our results from three HBV-susceptible cells lines demonstrated that the overall outcome of RXRα silencing is to enhance HBV infection in bona fide infection experimental systems.
Mice harboring an RXRα-null mutation are embryonic lethal due to cardiac failure at embryonic days 13.5 (E13.5) to E16.5 (41). Mouse RXRα is ubiquitously expressed from at least day 10.5 postconception and then becomes more highly expressed in the liver during late fetal development (42, 43). These studies are consistent with the critical role RXRα plays during development and in the maintenance of liver function (27, 44) and also raise an intriguing question whether the developmental changes in hepatic RXRα gene expression have any role in the transmission efficiency of neonatal HBV. If fetal and/or postnatal livers have relatively low levels of expression of RXRα, they may support more efficient viral infection. The association between RXRα expression and HBV infection in neonates merits further investigation.
We conducted RNA-seq analysis of RXRα-silenced cells. The transcriptome data showed that the expression of a large number of genes involved in lipid homeostasis was altered in the HepG2-NTCP cells with a TALEN-disrupted RXRα allele. Of note, a subset of genes in the AA/eicosanoid biosynthesis pathways was downregulated in these cells. Among them, the secretory phospholipase A2 group IIA (sPLA2-IIA) gene PLA2G2A was decreased after RXRα depletion. PLA2G2A encodes secreted phospholipase A2 (PLA2) that catalyzes the hydrolysis of the sn-2 position of membrane glycerophospholipid to liberate arachidonic acid. It has been reported that HBV could induce upregulation of PLA2G2A in primary human hepatocytes and that PLA2G2A participates in NKT cell-dependent protective immunity against HBV (45). To test the functional importance of PLA2G2A in de novo HBV infection, we silenced this gene with siRNAs in the HepG2-NTCP cells, and we observed an enhanced HBV infection with augmented viral antigen production. Our data suggest a significant role of secretory phospholipase in controlling HBV infection. However, rigorous investigation of a potential causal link between sPLA2 expression and inhibited HBV infection should be the subject of future studies.
The transcriptome analysis in our study has illustrated that RXRα regulates numerous fundamental metabolic pathways that are dependent on class II nuclear receptors (including PPARα, FXR, LXR, CAR, PXR, and RAR). This function fits well with RXRα's previously defined roles as central coordinators of cholesterol, fatty acid, bile acid, steroid, and xenobiotic metabolism and homeostasis. We observed that AA/eicosanoid biosynthesis pathways are downregulated in RXRα-silenced HepG2-NTCP cells. PLA2G2A is highly expressed in hepatocytes, and knocking down PLA2G2A expression could partially rescue HBV infection suppressed by RXR agonist. These data suggest that RXRα could inhibit HBV infection by downregulating the expression of sPLA2-IIA. In line with this, there is a binding motif for LXR/RXRα heterodimers within the sPLA2 promoter, and sPLA2 promoter is synergistically activated by the combination of agonists for LXR and RXR (9cRA) (46). Nonetheless, given the global changes in diverse physiological pathways by specifically disrupting RXRα expression and the divergent effects of RXRα and AA biosynthesis/metabolism pathways, it is likely that RXRα modulates early HBV/HDV infection through multiple mechanisms. Other metabolic pathways contributing to the effect of RXRα on HBV infection need further investigation.
As we discussed above, AA is produced by PLA2 and is subsequently metabolized by two types of enzymes: cyclooxygenases (COXs) and lipoxygenases (LOXs). AA exerts its effects by different mechanisms: either via the direct action of the AA molecule itself or via an effect of the AA metabolites. It has been reported that AA killed Schistosoma mansoni and Schistosoma haematobium juvenile and adult worms (47, 48). Mechanistically, the authors of these reports speculated that this lethality was likely due to excessive activation of parasite sphingomyelinase (nSMase), leading to the hydrolysis of sphingomyelin into ceramide and phosphorylcholine (49). Here, we showed that AA treatment during HBV incubation dose-dependently impaired HBV cccDNA formation and antigen production. We also demonstrated that blocking the cyclooxygenase pathway, but not the alternative pathway (lipoxygenases pathway), could enhance HBV infection, indicating that AA may inhibit HBV infection via a COX-dependent mechanism. Interestingly, previous studies suggested that AA could bind to the RXRα ligand-binding domain and efficiently stimulate RXRα (50, 51). Whether AA inhibited HBV infection via activating RXRα (even if only partially) and whether there is a positive feedback loop between RXRα activity and AA production contributing to reduced viral infection deserve further investigation.
In this study, we elucidated a prominent regulatory role of endogenous RXRα in HBV infection. Silencing of RXRα remarkably enhances HBV infection, which is at least partially due to downregulated AA/eicosanoid metabolism pathways after RXRα depletion. The liver is the most important organ for the storage and metabolism of retinol (vitamin A), with hepatic stellate cells (HSC) as the central cellular site for retinoid storage (52). Hepatic nonparenchymal cells (especially Kupffer cells and endothelial cells) are the main producers of eicosanoids, and prostaglandins are key mediators of cell signaling between Kupffer cells and hepatocytes (53). Hepatocytes contribute significantly to retinoid activation to retinoic acid, and they are also target cells for the action of eicosanoids (54). Our findings shed new light on the importance of hepatic lipid homeostasis for modulating viral infection (Fig. 8) and raise the intriguing possibility that metabolic components of liver microenvironment are actively involved in the initiation and progression of HBV infection.
FIG 8.
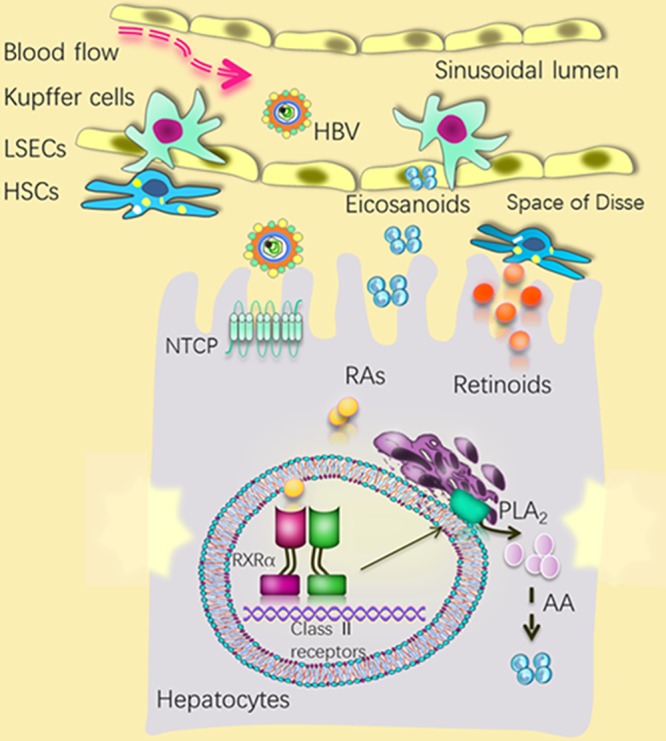
A speculative scheme for the role of hepatic RXRα and related metabolic homeostasis in modulating HBV infection within the liver microenvironment. Silencing RXRα in hepatocytes enhanced HBV infection at early stages, whereas activation of RXRα by natural ligands (e.g., retinoic acid) inhibited HBV infection at early stages. Altered retinoid homeostasis during HSC activation and increased production of eicosanoids (from Kupffer cells) in a response to various inflammatory stimuli, such as HBV infection, may affect HBV infection. LSECs, liver sinusoidal endothelial cells.
MATERIALS AND METHODS
Cell cultures.
The human embryonic kidney cell line 293T and the human hepatocellular carcinoma cell line HepG2 were from the American Type Culture Collection (ATCC). The human hepatocellular carcinoma cell line Huh-7 was from the cell bank of the Type Culture Collection of the Chinese Academy of Sciences. HepG2-NTCP cell clones were generated by expressing human NTCP (hNTCP) with a C-terminal tag (C9; TETSQVAPA) and were selected with G418 treatment (500 μg/ml). These cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Gibco). Primary tupaia hepatocytes were obtained from anesthetized tupaia with a two-step perfusion method and were cultured as described previously (19). The studies were performed in accordance with institutionally approved protocols (09001T) and adhered to guidelines of the National Institute of Biological Sciences (58). HepaRG cells were purchased from Biopredic International (Rennes, France) and were cultured according to the manufacturer's protocol. All cells were maintained at 37°C in a 5% CO2 humidified incubator. For viral infection, cells were pretreated with PTH maintenance medium (PMM) for 24 h as described previously (19).
Reagents and antibodies.
Bexarotene (4-[1-(5,6,7,8-tetrahydro-3,5,5,8,8-pentamethyl-2-naphthalenyl)ethenyl]benzoic acid) was purchased from Biovision. GW4064 (G5172), 9cRA (R4643), GW3965 (G6295), arachidonic acid (A8798), indomethacin (I7378), zileuton (Z4277), baicalein (465119), and PD 146176 (P4620) were purchased from Sigma. Mouse monoclonal antibodies (MAbs) 1C10 and 4G5 were generated in our laboratory at the National Institute of Biological Sciences (NIBS); MAb 17B9 was a kind gift from Lin Jiang of the China National Biotech Group. Rabbit polyclonal antibody D-20, which targets human RXRα (hRXRα), was purchased from Santa Cruz.
Virus and infection.
HBV and HDV were produced as described previously (19). Briefly, for HDV production, Huh7 cells were first transfected with the HDV RNP generation plasmid pCMV-HDV3.0 (which contains a head-to-tail trimer of 1.0× HDV cDNA of a genotype I virus under the control of a cytomegalovirus [CMV] promoter), cultured, and propagated for 2 weeks; cells were then transfected with HBV envelope expressing plasmid pUC-HBV-LMS (which contains nucleotides 2431 to 1990 of HBV genotype D). For HBV production, a plasmid containing 1.05 copies of the HBV genome (genotype D; Gene bank accession number U95551.1) was transfected into Huh-7 cells. The virus-containing cell culture supernatant was harvested every 3 days posttransfection, clarified from cell debris via centrifugation, and stored at −80°C.
Lenti-VSV-G-EGFP was produced by simultaneous transfection of 293T cells with three plasmids: one for VSV-G expression (pMD2.G), one for HIV genome packaging (psPAX2), and one for EGFP expression (pLKO.3G), as previously described (19). The virus-containing cell culture supernatant was harvested and passed through a 0.45-μm-pore-size filter to remove cell debris and was stored at −80°C.
For viral infection of HepG2-NTCP and HepaRG cells, ∼5% polyethylene glycol (PEG) 8000 was included during the inoculation period (for 24 h). Viral infection of PTHs was conducted in the absence of PEG 8000 for 16 h. The culture medium was changed every 2 to 3 days, and fresh PMM was added.
ELISA of viral antigens.
The cell culture supernatants from infected cells were harvested at the time points indicated in the figures. The levels of HBeAg and HBsAg in the culture medium were evaluated using commercial ELISA kits (Wantai Pharm, Inc.), according to the manufacturer's instructions.
Quantitation of RNA and cccDNA.
Total RNA was isolated with TRIzol reagent (Invitrogen) and transcribed into cDNA with random primers (PrimeScript RT kit; TaKaRa). cDNA (2 μl) was used for real-time PCR assays (SYBR Premix Ex Taq kit; TaKaRa). HDV total RNA, the HBV 3.5-kb RNA, and HBV total RNA were evaluated as described previously (19). HBV cccDNA was quantified by real-time quantitative PCR (qPCR) assays as described previously (33). Primers used in this study are listed in Table 1.
TABLE 1.
Primers for real-time PCR analysis
| Target | Forward sequence | Reverse sequence |
|---|---|---|
| hRXRα | 5′-CTCCTCAGGCAAGCACTATG-3′ | 5′-TTGTCAATCAGGCAGTCCTT-3′ |
| tsRXRα | 5′-CCCAAGACCGACACCTATG-3′ | 5′-CCACTCCACAAGGGTGAAC-3′ |
| hLFABP | 5′-GTGGTTCAGTTGGAAGGTGA-3′ | 5′-TGTCACCCAATGTCATGGT-3′ |
| hNTCP, tsNTCP | 5′-TTCAGCAAGATCAAGGC-3′ | 5′-TGGAGCAGGTGGTCATC-3′ |
| hPLA2G2A | 5′-CCTCCTACTGTTGGCAGTGA-3′ | 5′-ATAACTGAGTGCGGCTTCCT-3′ |
| hGAPDH | 5′-GAAGGTGAAGGTCGGAGTCA-3′ | 5′-TGGAATCATATTGGAACATGT-3′ |
Immunofluorescence staining analysis.
HBV-infected cells were fixed with 3.7% paraformaldehyde (PFA) for 10 min and permeabilized with 0.5% Triton-X100 (in phosphate-buffered saline [PBS]) for 10 min at room temperature. Cells were then blocked with 3% bovine serum albumin (BSA) at 37°C for 1 h. Cells were stained with 5 μg/ml mouse MAbs 1C10 and 17B9, which recognize HBcAg and HBsAg, respectively, followed by staining with fluorescein isothiocyanate (FITC)-conjugated secondary antibody. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI dilactate). The cells were analyzed under a Zeiss LSM 510 Meta confocal microscope or a Nikon Eclipse Ti fluorescence microscope.
Gene-silencing experiments on HepG2-NTCP cells and primary hepatocytes.
HepG2-NTCP cells were transfected with SMART pool siRNAs (Dharmacon) or with specific siRNAs, as indicated in the figures, along with a nonspecific control (5′-UUCUCCGAACGUGUCACGUdTdT-3′; Ctrl-si) using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer's instructions. Transfected cells were reseeded into the wells of new 48-well plates after 48 h and were inoculated with HBV for 24 h. The mRNA levels of multiple target genes were measured with qRT-PCR at 72 h post-siRNA transfection. siRNAs that specifically targeted Tupaia genes were designed using siDESIGN Center software. PTHs were transfected with the Ctrl-si or gene-specific siRNAs using Lipofectamine 2000 (Invitrogen) at 48 h posttransfection, and cells were inoculated with HBV for 16 h.
RXR gene knockout with TALEN.
A custom TALEN (transcription activator-like effector nuclease) plasmid targeting the human RXRα gene (NR2B1) was constructed as described previously (55). It targeted the following sequences: 5′-TGCGCAAGGACCTGACCTAC-3′ and 5′-TTCCTGACGGACTAACTGTT-3′. Donor DNA was generated by inserting two arms that are homologous with the RXRα gene sequence into the PLKO.1 plasmid, which contains a puromycin resistance gene expression cassette.
Cell viability assay using alamarBlue reagent.
alamarBlue (Invitrogen) was added to cells pretreated with different types of drugs in PMM for 24 h. The cells were then cultured in an incubator at 37°C for 2 h. Cellular viability was evaluated via measurement of fluorescence with an excitation wavelength of 530 nm and an emission wavelength of 590 nm. The absorbance of vehicle-treated cells was set to 100%.
Western blot analysis.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, pH 7.4) containing 1× protease inhibitor cocktail (Roche) by boiling for 10 min. The cell lysates were clarified via centrifugation at 12,000 × g for 20 min. The proteins in the supernatant were quantified using a bicinchoninic acid assay (Bio-Rad); equal amounts were separated by SDS-PAGE (12%) and were then transferred to polyvinylidene difluoride (PVDF) membranes for Western blot analysis. The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control.
RNA-seq analysis.
Wild-type HepG2 cells expressing NTCP (HepG2-NTCP/WT) or HepG2 cells with a disrupted RXRα gene (HepG2-NTCP/RXR-KD, where KD is knockdown) were seeded on 10-cm plates 3 h after cell attachment, and the original DMEM (10% FBS) medium was replaced with PMM. Total RNAs were isolated from the cells after incubation in PMM for 24 h. The samples were sequenced with an Illumina Genome Analyzer IIx at the genome center of the National Institute of Biological Sciences, Beijing (NIBS). Reads were mapped to the human reference genome (release GRCh37) with TopHat2 (version 2.0.12) (56). Cufflinks, version 2.1.1 (57), was used for transcript assembly, abundance estimation, and differential expression analysis using a known set of reference transcripts from Ensembl, version 73. All heat maps were generated using R (R Development Core Team Team, Vienna, Austria).
Lipid extraction.
Cells cultured in 10-cm dishes were collected by scraping in 1 ml of cold PBS (total cell numbers were counted) and transferred into glass tubes containing 4 ml of a chloroform-methanol (2:1) mixture. Cell pellets were extracted with the extraction mixture three times and centrifuged at 4,000 rpm for 5 min. The upper aqueous phase was discarded, and the lower layer (chloroform phase) was collected and evaporated under a nitrogen stream.
Liquid chromatography (LC)-MS/MS.
An ultraperformance liquid chromatography (UPLC) system was coupled to a Q-Exactive Orbitrap mass spectrometer (MS) (Thermo Fisher, CA) equipped with a heated electrospray ionization (HESI) probe. Lipid extracts were separated by a Hypersil Gold C18 column (100 by 2.1 mm; 1.9-μm particle size; Thermo Fisher, CA). A binary solvent system was used, in which mobile phase A consisted of ACN-H2O (60:40) and 10 mM ammonium acetate, and mobile phase B of isopropanol (IPA)-ACN (90:10) and 10 mM ammonium acetate. A 30-min gradient with a flow rate of 250 μl/min was used. Column chamber and sample tray were held at 45°C and 10°C, respectively. Data with mass ranges of m/z 200 to 2,000 were acquired at negative ion mode with data-dependent tandem mass spectrometry (MS/MS) acquisition. The full scan and fragment spectra were collected with resolutions of 70,000 and 17,500, respectively. The source parameters are as follows: spray voltage, 3,000 V; capillary temperature, 320°C; heater temperature, 300°C; sheath gas flow rate, 35 arbitrary units (Arb); auxiliary gas flow rate, 10 Arb. Data analysis and lipid identification were performed by the software LipidSearch, version 4.0 (Thermo Fisher, CA). All molecular identifications were based on MS2, with an MS1 mass error of <5 ppm and MS2 mass error of <8 ppm. Subsequently, the lipids with a Mascot (m) score of <15 and peak area of <1e7 were excluded.
Northern blot and Southern blot analysis.
Total RNA or HBV cccDNA was extracted from one well of six-well plates of HBV-infected HepG2-NTCP cells (AC12) at 7 dpi by TRIzol or the Hirt method (33, 34); AC12 cells were transfected with the siRNAs indicated in the figures or treated with different compounds during HBV infection. For detection of HBV RNA by Northern blotting, 8 μg of total RNA from each sample was loaded onto a 1.2% formaldehyde agarose gel; for cccDNA detection by Southern blotting, about 35 μg of extracted HBV DNA from each sample was loaded onto a 1.2% agarose gel. The RNA or DNA on agarose gels was transferred onto nylon membrane with 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) by the capillary transfer method. Following overnight transfer, the RNA or DNA was fixed to dry membranes by UV exposure with energy dosage at 1,200 mJ in a UV Stratalinker and then detected with [α-32P]dCTP-labeled HBV genotype D linear full-length genomic DNA (39) or digoxigenin-dUTP-labeled probe, respectively. Hybridization was performed in 5 ml of Perfect Hyb Plus hybridization buffer as per the manufacturer's instructions. The hybridized probes were immunodetected with anti-digoxigenin-alkaline phosphatase (AP) and visualized with the chemiluminescence substrate CSPD. Ten picograms each of the 3.2-kb, 2.1-kb, and 1.7-kb HBV DNA fragments was prepared by PCR amplification and used as a DNA marker.
Accession number(s).
RNA-seq data have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE108366.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (NSFC81525018), Ministry of Science and Technology, China (2014CB849601), and the Science and Technology Bureau of the Beijing Municipal Government.
We thank Xiaohui Liu and Xueying Wang from the Metabolomics Facility at Technology Center for Protein Sciences, Tsinghua University, for the LC-MS/MS analysis.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01771-17.
REFERENCES
- 1.MacLachlan JH, Cowie BC. 2015. Hepatitis B virus epidemiology. Cold Spring Harb Perspect Med 5(5):a021410. doi: 10.1101/cshperspect.a021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lavanchy D. 2004. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 11:97–107. doi: 10.1046/j.1365-2893.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- 3.Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 4.Hoofnagle JH, di Bisceglie AM. 1997. The treatment of chronic viral hepatitis. N Engl J Med 336:347–356. doi: 10.1056/NEJM199701303360507. [DOI] [PubMed] [Google Scholar]
- 5.Marcellin P, Bonino F, Yurdaydin C, Hadziyannis S, Moucari R, Kapprell HP, Rothe V, Popescu M, Brunetto MR. 2013. Hepatitis B surface antigen levels: association with 5-year response to peginterferon alfa-2a in hepatitis B e-antigen-negative patients. Hepatol Int 7:88–97. doi: 10.1007/s12072-012-9343-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dusheiko G. 2013. Treatment of HBeAg positive chronic hepatitis B: interferon or nucleoside analogues. Liver Int 33(Suppl 1):S137–S150. doi: 10.1111/liv.12078. [DOI] [PubMed] [Google Scholar]
- 7.Dienstag JL, Schiff ER, Wright TL, Perrillo RP, Hann HW, Goodman Z, Crowther L, Condreay LD, Woessner M, Rubin M, Brown NA. 1999. Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med 341:1256–1263. doi: 10.1056/NEJM199910213411702. [DOI] [PubMed] [Google Scholar]
- 8.Marcellin P, Chang TT, Lim SG, Tong MJ, Sievert W, Shiffman ML, Jeffers L, Goodman Z, Wulfsohn MS, Xiong S, Fry J, Brosgart CL. 2003. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N Engl J Med 348:808–816. doi: 10.1056/NEJMoa020681. [DOI] [PubMed] [Google Scholar]
- 9.Lei J, Wang Y, Wang LL, Zhang SJ, Chen W, Bai ZG, Xu LY. 2013. Profile of hepatitis B virus resistance mutations against nucleoside/nucleotide analogue treatment in Chinese patients with chronic hepatitis B. Virol J 10:313. doi: 10.1186/1743-422X-10-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lok AS, McMahon BJ, Brown RS Jr, Wong JB, Ahmed AT, Farah W, Almasri J, Alahdab F, Benkhadra K, Mouchli MA, Singh S, Mohamed EA, Abu Dabrh AM, Prokop LJ, Wang Z, Murad MH, Mohammed K. 2016. Antiviral therapy for chronic hepatitis B viral infection in adults: a systematic review and meta-analysis. Hepatology 63:284–306. doi: 10.1002/hep.28280. [DOI] [PubMed] [Google Scholar]
- 11.Shlomai A, Shaul Y. 2008. The “metabolovirus” model of hepatitis B virus suggests nutritional therapy as an effective anti-viral weapon. Med Hypotheses 71:53–57. doi: 10.1016/j.mehy.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 12.Locarnini S. 2004. Molecular virology of hepatitis B virus. Semin Liver Dis 24(Suppl 1):S3–S10. doi: 10.1055/s-2004-828672. [DOI] [PubMed] [Google Scholar]
- 13.Ramiere C, Scholtes C, Diaz O, Icard V, Perrin-Cocon L, Trabaud MA, Lotteau V, Andre P. 2008. Transactivation of the hepatitis B virus core promoter by the nuclear receptor FXRα. J Virol 82:10832–10840. doi: 10.1128/JVI.00883-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reese VC, Oropeza CE, McLachlan A. 2013. Independent activation of hepatitis B virus biosynthesis by retinoids, peroxisome proliferators, and bile acids. J Virol 87:991–997. doi: 10.1128/JVI.01562-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doitsh G, Shaul Y. 2004. Enhancer I predominance in hepatitis B virus gene expression. Mol Cell Biol 24:1799–1808. doi: 10.1128/MCB.24.4.1799-1808.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huan B, Kosovsky MJ, Siddiqui A. 1995. Retinoid X receptor alpha transactivates the hepatitis B virus enhancer 1 element by forming a heterodimeric complex with the peroxisome proliferator-activated receptor. J Virol 69:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ondracek CR, Reese VC, Rushing CN, Oropeza CE, McLachlan A. 2009. Distinct regulation of hepatitis B virus biosynthesis by peroxisome proliferator-activated receptor γ coactivator 1α and small heterodimer partner in human hepatoma cell lines. J Virol 83:12545–12551. doi: 10.1128/JVI.01624-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shlomai A, Paran N, Shaul Y. 2006. PGC-1α controls hepatitis B virus through nutritional signals. Proc Natl Acad Sci U S A 103:16003–16008. doi: 10.1073/pnas.0607837103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bremer CM, Bung C, Kott N, Hardt M, Glebe D. 2009. Hepatitis B virus infection is dependent on cholesterol in the viral envelope. Cell Microbiol 11:249–260. doi: 10.1111/j.1462-5822.2008.01250.x. [DOI] [PubMed] [Google Scholar]
- 21.Dorobantu C, Macovei A, Lazar C, Dwek RA, Zitzmann N, Branza-Nichita N. 2011. Cholesterol depletion of hepatoma cells impairs hepatitis B virus envelopment by altering the topology of the large envelope protein. J Virol 85:13373–13383. doi: 10.1128/JVI.05423-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin YL, Shiao MS, Mettling C, Chou CK. 2003. Cholesterol requirement of hepatitis B surface antigen (HBsAg) secretion. Virology 314:253–260. doi: 10.1016/S0042-6822(03)00403-3. [DOI] [PubMed] [Google Scholar]
- 23.Evans RM, Mangelsdorf DJ. 2014. Nuclear Receptors, RXR, and the Big Bang. Cell 157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, De Lera AR, Lotan R, Mangelsdorf DJ, Gronemeyer H. 2006. International Union of Pharmacology. LXIII. Retinoid X receptors. Pharmacol Rev 58:760–772. [DOI] [PubMed] [Google Scholar]
- 25.Duvic M, Hymes K, Heald P, Breneman D, Martin AG, Myskowski P, Crowley C, Yocum RC. 2001. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J Clin Oncol 19:2456–2471. doi: 10.1200/JCO.2001.19.9.2456. [DOI] [PubMed] [Google Scholar]
- 26.Gyamfi MA, Tanaka Y, He L, Klaassen CD, Wan YJ. 2009. Hepatic effects of a methionine-choline-deficient diet in hepatocyte RXRα-null mice. Toxicol Appl Pharmacol 234:166–178. doi: 10.1016/j.taap.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan YJ, An D, Cai Y, Repa JJ, Hung-Po Chen T, Flores M, Postic C, Magnuson MA, Chen J, Chien KR, French S, Mangelsdorf DJ, Sucov HM. 2000. Hepatocyte-specific mutation establishes retinoid X receptor alpha as a heterodimeric integrator of multiple physiological processes in the liver. Mol Cell Biol 20:4436–4444. doi: 10.1128/MCB.20.12.4436-4444.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Terrand J, Bruban V, Zhou L, Gong W, El Asmar Z, May P, Zurhove K, Haffner P, Philippe C, Woldt E, Matz RL, Gracia C, Metzger D, Auwerx J, Herz J, Boucher P. 2009. LRP1 controls intracellular cholesterol storage and fatty acid synthesis through modulation of Wnt signaling. J Biol Chem 284:381–388. doi: 10.1074/jbc.M806538200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai T, Wu Y, Leng AS, Ao Y, Robel RC, Lu SC, French SW, Wan YJ. 2003. RXRα-regulated liver SAMe and GSH levels influence susceptibility to alcohol-induced hepatotoxicity. Exp Mol Pathol 75:194–200. doi: 10.1016/S0014-4800(03)00091-1. [DOI] [PubMed] [Google Scholar]
- 30.Cai Y, Konishi T, Han G, Campwala KH, French SW, Wan YJ. 2002. The role of hepatocyte RXR alpha in xenobiotic-sensing nuclear receptor-mediated pathways. Eur J Pharm Sci 15:89–96. doi: 10.1016/S0928-0987(01)00211-1. [DOI] [PubMed] [Google Scholar]
- 31.Wu Y, Zhang X, Bardag-Gorce F, Robel RC, Aguilo J, Chen L, Zeng Y, Hwang K, French SW, Lu SC, Wan YJ. 2004. Retinoid X receptor alpha regulates glutathione homeostasis and xenobiotic detoxification processes in mouse liver. Mol Pharmacol 65:550–557. doi: 10.1124/mol.65.3.550. [DOI] [PubMed] [Google Scholar]
- 32.Smith WL, DeWitt DL, Garavito RM. 2000. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem 69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 33.Sun Y, Qi Y, Peng B, Li W. 2017. NTCP-reconstituted in vitro HBV infection system. Methods Mol Biol 1540:1–14. doi: 10.1007/978-1-4939-6700-1_1. [DOI] [PubMed] [Google Scholar]
- 34.Qi Y, Gao Z, Xu G, Peng B, Liu C, Yan H, Yao Q, Sun G, Liu Y, Tang D, Song Z, He W, Sun Y, Guo JT, Li W. 2016. DNA polymerase κ is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog 12:e1005893. doi: 10.1371/journal.ppat.1005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dubuquoy L, Louvet A, Hollebecque A, Mathurin P, Dharancy S. 2009. Peroxisome proliferator-activated receptors in HBV-related infection. PPAR Res 2009:145124. doi: 10.1155/2009/145124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raney AK, Johnson JL, Palmer CN, McLachlan A. 1997. Members of the nuclear receptor superfamily regulate transcription from the hepatitis B virus nucleocapsid promoter. J Virol 71:1058–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang H, McLachlan A. 2001. Transcriptional regulation of hepatitis B virus by nuclear hormone receptors is a critical determinant of viral tropism. Proc Natl Acad Sci U S A 98:1841–1846. doi: 10.1073/pnas.98.4.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim DH, Kang HS, Kim KH. 2016. Roles of hepatocyte nuclear factors in hepatitis B virus infection. World J Gastroenterol 22:7017–7029. doi: 10.3748/wjg.v22.i31.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia AD, Ostapchuk P, Hearing P. 1993. Functional interaction of nuclear factors EF-C, HNF-4, and RXRα with hepatitis B virus enhancer I. J Virol 67:3940–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li M, Xie Y, Wu X, Kong Y, Wang Y. 1995. HNF3 binds and activates the second enhancer, ENII, of hepatitis B virus. Virology 214:371–378. doi: 10.1006/viro.1995.0046. [DOI] [PubMed] [Google Scholar]
- 41.Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR, Evans RM. 1994. RXRα mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev 8:1007–1018. doi: 10.1101/gad.8.9.1007. [DOI] [PubMed] [Google Scholar]
- 42.Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Oro AE, Kakizuka A, Evans RM. 1992. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev 6:329–344. doi: 10.1101/gad.6.3.329. [DOI] [PubMed] [Google Scholar]
- 43.Dolle P, Fraulob V, Kastner P, Chambon P. 1994. Developmental expression of murine retinoid X receptor (RXR) genes. Mech Dev 45:91–104. doi: 10.1016/0925-4773(94)90023-X. [DOI] [PubMed] [Google Scholar]
- 44.Kastner P, Grondona JM, Mark M, Gansmuller A, LeMeur M, Decimo D, Vonesch JL, Dolle P, Chambon P. 1994. Genetic analysis of RXRα developmental function: convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 78:987–1003. doi: 10.1016/0092-8674(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 45.Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, Bosse E, Iqbal J, Hussain MM, Balschun K, Rocken C, Arlt A, Gunther R, Hampe J, Schreiber S, Baron JL, Moody DB, Liang TJ, Blumberg RS. 2012. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat Med 18:1060–1068. doi: 10.1038/nm.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Antonio V, Janvier B, Brouillet A, Andreani M, Raymondjean M. 2003. Oxysterol and 9-cis-retinoic acid stimulate the group IIA secretory phospholipase A2 gene in rat smooth-muscle cells. Biochem J 376:351–360. doi: 10.1042/bj20030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El Ridi R, Aboueldahab M, Tallima H, Salah M, Mahana N, Fawzi S, Mohamed SH, Fahmy OM. 2010. In vitro and in vivo activities of arachidonic acid against Schistosoma mansoni and Schistosoma haematobium. Antimicrob Agents Chemother 54:3383–3389. doi: 10.1128/AAC.00173-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El Ridi R, Tallima H, Salah M, Aboueldahab M, Fahmy OM, Al-Halbosiy MF, Mahmoud SS. 2012. Efficacy and mechanism of action of arachidonic acid in the treatment of hamsters infected with Schistosoma mansoni or Schistosoma haematobium. Int J Antimicrob Agents 39:232–239. doi: 10.1016/j.ijantimicag.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 49.El Ridi R, Tallima H. 2006. Equilibrium in lung schistosomula sphingomyelin breakdown and biosynthesis allows very small molecules, but not antibody, to access proteins at the host-parasite interface. J Parasitol 92:730–737. doi: 10.1645/GE-745R1.1. [DOI] [PubMed] [Google Scholar]
- 50.Lengqvist J, Mata De Urquiza A, Bergman AC, Willson TM, Sjovall J, Perlmann T, Griffiths WJ. 2004. Polyunsaturated fatty acids including docosahexaenoic and arachidonic acid bind to the retinoid X receptor alpha ligand-binding domain. Mol Cell Proteomics 3:692–703. doi: 10.1074/mcp.M400003-MCP200. [DOI] [PubMed] [Google Scholar]
- 51.Calderon F, Kim HY. 2007. Role of RXR in neurite outgrowth induced by docosahexaenoic acid. Prostaglandins, leukotrienes, and essential fatty Acids 77:227–232. doi: 10.1016/j.plefa.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blomhoff R, Blomhoff HK. 2006. Overview of retinoid metabolism and function. J Neurobiol 66:606–630. doi: 10.1002/neu.20242. [DOI] [PubMed] [Google Scholar]
- 53.Puschel GP, Kirchner C, Schroder A, Jungermann K. 1993. Glycogenolytic and antiglycogenolytic prostaglandin E2 actions in rat hepatocytes are mediated via different signalling pathways. Eur J Biochem 218:1083–1089. doi: 10.1111/j.1432-1033.1993.tb18468.x. [DOI] [PubMed] [Google Scholar]
- 54.Shirakami Y, Lee SA, Clugston RD, Blaner WS. 2012. Hepatic metabolism of retinoids and disease associations. Biochim Biophys Acta 1821:124–136. doi: 10.1016/j.bbalip.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. 2012. A transcription activator-like effector toolbox for genome engineering. Nat Protoc 7:171–192. doi: 10.1038/nprot.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.National Institute of Biological Sciences, Beijing: 2009. Guide for the care and use of laboratory animals. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.