

ABSTRACT
The 10E8 antibody targets a helical epitope in the membrane-proximal external region (MPER) and transmembrane domain (TMD) of the envelope glycoprotein (Env) subunit gp41 and is among the broadest known neutralizing antibodies against HIV-1. Accordingly, this antibody and its mechanism of action valuably inform the design of effective vaccines and immunotherapies. 10E8 exhibits unusual adaptations to attain specific, high-affinity binding to the MPER at the viral membrane interface. Reversing the charge of the basic paratope surface (from net positive to net negative) reportedly lowered its neutralization potency. Here, we hypothesized that by increasing the net positive charge in similar polar surface patches, the neutralization potency of the antibody may be enhanced. We found that an increased positive charge at this paratope surface strengthened an electrostatic interaction between the antibody and lipid bilayers, enabling 10E8 to interact spontaneously with membranes. Notably, the modified 10E8 antibody did not gain any apparent polyreactivity and neutralized virus with a significantly greater potency. Binding analyses indicated that the optimized 10E8 antibody bound with a higher affinity to the epitope peptide anchored in lipid bilayers and to Env spikes on virions. Overall, our data provide a proof of principle for the rational optimization of 10E8 via manipulation of its interaction with the membrane element of its epitope. However, the observation that a similar mutation strategy did not affect the potency of the first-generation anti-MPER antibody 4E10 shows possible limitations of this principle. Altogether, our results emphasize the crucial role played by the viral membrane in the antigenicity of the MPER-TMD of HIV-1.
IMPORTANCE The broadly neutralizing antibody 10E8 blocks infection by nearly all HIV-1 isolates, a capacity which vaccine design seeks to reproduce. Engineered versions of this antibody also represent a promising treatment for HIV infection by passive immunization. Understanding its mechanism of action is therefore important to help in developing effective vaccines and biologics to combat HIV/AIDS. 10E8 engages its helical MPER epitope where the base of the envelope spike submerges into the viral membrane. To enable this interaction, this antibody evolved an unusual property: the ability to interact with the membrane surface. Here, we provide evidence that 10E8 can be made more effective by enhancing its interactions with membranes. Our findings strengthen the idea that to elicit antibodies similar to 10E8, vaccines must reproduce the membrane environment where these antibodies perform their function.
KEYWORDS: 10E8, 4E10, anti-MPER antibody, broadly neutralizing antibody, human immunodeficiency virus
INTRODUCTION
Broadly neutralizing antibodies (bnAbs) against human immunodeficiency virus type 1 (HIV-1) have been essential tools in the design of candidate vaccines and therapeutics, from the first-generation bnAbs of the 1990s to the more potent, second-generation bnAbs defined since 2009 (1). The second-generation bnAb 10E8 recognizes the conserved membrane-proximal external region (MPER) of the gp41 subunit of the envelope glycoprotein (Env) (2–5), resulting in one of the highest levels of HIV-1 neutralization reported to date (1, 6–8). Antibodies against this vulnerable site also mediate the neutralization breadth and potency of sera from a subset of HIV-1-infected individuals (8, 9). Despite reported similarities in the epitope binding profile with the first-generation anti-MPER bnAb 4E10, 10E8 displays high neutralization potency and very limited, if any, polyreactivity in comparison (8, 10). These advantageous features have put the focus on 10E8 as a suitable template on which to base vaccine design (11–14) and the rational development of immunotherapeutic agents (15–20).
The antigen responsible for eliciting 10E8-like antibodies and the molecular mechanism underlying effective MPER recognition are not totally understood. Recently reported structural data suggested that the MPER and its connection to the gp41 transmembrane domain (TMD) are organized as a continuous, straight helix that emerges obliquely from the HIV membrane plane (3, 4, 21, 22). The ability to access the helical MPER epitope at the viral membrane interface thus appears to support the neutralizing activity of the most effective anti-MPER antibodies (3, 22). Structural elements of the antibody sustain effective interactions with the lipid bilayer surrounding the viral particle: (i) a long heavy chain complementarity-determining region 3 (CDRH3) loop decorated at the apex with hydrophobic-at-interface aromatic residues critical for function (3, 8, 23, 24) and (ii) a flat surface at the paratope that establishes favorable interactions at the viral Env-membrane interface (4, 22, 25).
Recent studies suggested that the association of anti-MPER antibodies with membranes might be driven by electrostatic interactions between basic residues on the surface of the paratope and anionic phospholipids (4, 22, 25). Despite the inability of 10E8 to partition spontaneously into lipid bilayers (25), cryo-electron microscopy (EM) and X-ray crystallography data suggest that upon engagement with the antigen, a surface patch of its paratope may establish favorable contacts with the membrane-Env interface (2–4). Specifically, in a recent study, anionic phospholipids were found to be attached to this surface (4). Those authors found that the introduction of aspartate or glutamate at basic/polar positions surrounding the phospholipid binding site decreased the neutralization potency to 1/500 of that of wild-type 10E8 (10E8-WT) (4). Thus, the deleterious effects caused by those mutations highlighted the functional relevance of potential electrostatic interactions of 10E8 with the membrane.
Here, we followed the opposite approach, optimizing the function of 10E8 by enhancing the net positive charge of its paratope. We determined the strength of antibody-membrane interactions using liposome flotation assays (a physical separation method). This standard method was complemented with fluorescence-based assays (water-membrane partitioning in intact systems, i.e., without isolation of the bound fractions), namely, confocal microscopy of giant unilamellar vesicles (GUVs) and spectroscopic titration assays. In addition, we determined whether the strength of antibody-membrane interactions was associated with polyreactivity and neutralization potency. We found that the neutralization efficacy of the 10E8 antibody was substantially improved by the manipulation of antibody-membrane interactions. A similar approach taken with a first-generation bnAb, 4E10, did not improve its potency, suggesting that factors other than membrane recognition contribute to neutralization activity. Notably, our observations do not support proportionality between the level of polyreactivity and neutralization potency. In addition, they emphasize that favorable interactions with the membrane are likely crucial for a functional antibody-antigen binding interface and therefore are highly relevant for inducing effective anti-MPER B-cell responses. Finally, in a more general sense, they suggest a possible pathway for improving the potency of antibodies targeting membrane-displayed epitopes.
RESULTS
Design of the 10E8-3R antibody.
To engineer antibody 10E8 to interact more effectively with lipid bilayers, we introduced three basic residues (3R mutation) at strategic, solvent-exposed positions on the corresponding Fab fragment (Fig. 1A). The S30R/N52R/S67R triple substitution, involving light chain residues, increases the positive charge at the surface patch predicted by X-ray crystallography and cryo-EM studies to contact the viral Env-membrane interface (2–4) (Fig. 1A and B). The positioning in the structure of the anionic phospholipid phosphatidylglycerol cocrystallized in complex with Fab 10E8 (4) confirmed the close proximity of its head group moiety to the mutated residues (Fig. 1B, right). Moreover, this arrangement of the antibody with respect to the membrane plane resulted in an insertion angle of the bound MPER helix resembling that deduced from a low-resolution cryo-EM model of 10E8 bound to the Env trimer (2). These changes did not alter binding to the epitope peptide, as judged by the comparable patterns of specific binding observed for mutant and parental Fabs in enzyme-linked immunosorbent assays (ELISAs) (Fig. 1C). Thus, we next studied the membrane binding characteristics and biological functions of the resulting Fab mutant 10E8-3R, in comparison with those of the parental specimen 10E8-WT.
FIG 1.

Design of the 10E8 mutant with 3 Arg residues exposed on the paratope. (A) Surface density charge representation of wild-type Fab 10E8 (PDB accession number 5GHW) and its triple mutant 10E8-3R (bottom views). Negative and positive surface electrostatic potentials are shown in red and blue, respectively. The S30R/N52R/S67R triple substitution was introduced into the light chain (LC). Encircled patches are predicted to establish contact with the membrane interface upon engagement with the MPER epitope (3, 4). (B) Position of the 3R mutation relative to the Env trimer and membrane. (Left) Model for the binding of Fab 10E8 (https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-3308; ribbon representation) to an Env trimer (EMDB accession number EMD-3308) (orange silhouette). Light and heavy chains of the Fab fragment are depicted in brown and tan, respectively. (Right) Closeup view of the mutated positions (Arg side chains in blue) on 10E8. The dotted line indicates the approximate level of the membrane interface. The side chain of W100bHC marks the position of the CDRH3 tip inserted into the membrane (3). Modeled into the structures are a dihexanoyl phosphatidylglycerol molecule, 06:0 PG (PDB accession number 5T85) (colored by atoms), and the epitope peptide MPER(664–690) (PDB accession number 5GHW) (orange helix). (C) Fab binding to the epitope peptide MPER(671–693) in an ELISA (black line). The red dotted line corresponds to the signal obtained for antibody binding to a control epitope peptide that contains the crucial residues 672WF673 replaced by Ala. Abs, absorbance.
Membrane binding properties of 10E8-3R.
Following the approach described in our previous works (24, 25) (see schematics displayed in Fig. 2A), virus-like (VL) vesicle flotation experiments were first used to establish the effect of the 3R mutations on Fab 10E8 partitioning from water into membranes (Fig. 2B). Anti-gp120 antibody PG9, used as a control for the absence of an interaction with membranes, and 10E8-WT showed similar flotation profiles after sucrose centrifugation, with most input antibody being recovered in pellets (i.e., from nonfloating fractions). In sharp contrast, under the same experimental conditions, 67% of 10E8-3R was found cofloating with vesicles (rhodamine [Rho] emission spectra in Fig. 2B, bottom). The antibody signal was detected mainly in the nonfloating fractions when the same experiment was run in the absence of vesicles. To ensure that the observed membrane binding phenomenon was dependent on the specific location of the 3R mutation, three Arg residues were instead placed into solvent-exposed positions S153R, S193R, and S199R of the 10E8 constant-domain light chain. The resultant 10E8-3R(C) mutant was found in the pellet fractions after centrifugation. Thus, the minimal amount of Fab 10E8-3R(C) detected in association with vesicles followed the pattern of parental 10E8-WT. These results indicate that light chain 3R mutation S30R/N52R/S67R specifically endows the 10E8 antibody with the capacity to partition into VL vesicles.
FIG 2.

Partitioning of Fabs into membranes measured by a vesicle flotation assay. (A) Diagram illustrating the method. Fabs (1.5 μM) were incubated with Rho-PE-labeled VL LUVs (1.5 mM) and subsequently deposited onto a stepwise sucrose gradient (time zero [t0]). After ultracentrifugation (t1), four different fractions were collected based on their densities. (B) Partitioning of Fab 10E8 and its derivatives into VL LUVs. The presence of vesicles and Fab in the different fractions was probed by rhodamine emission and Western blotting, respectively. VL LUVs (emission spectra at the bottom right) were found in the third and fourth fractions (i.e., floated fractions). An additional fraction, employing SDS, was collected to recover the material attached to the surface of the tube. Analyzed samples include (from top to bottom) PG9, an anti-gp120 antibody used as a negative control; 10E8-WT; 10E8-3R; 10E8-3R(−LUVs), a 10E8-R3 sample centrifuged in the absence of VL vesicles; and 10E8-3R(C), a control Fab containing a 3R mutation within the constant domain. The values displayed on the right correspond to the percentages of antibody found cofloating with vesicles, calculated by densitometry.
Assays using fluorescently labeled Fabs were additionally performed to prove the membrane binding capacity of 10E8-3R in an intact system (i.e., without physical separation of the vesicle-bound specimens). Figure 3A displays confocal fluorescence microscopy data for GUVs incubated with Fab. To carry out these experiments, GUVs were stained with 6-dodecanoyl-2-dimethylaminonaphthalene (Laurdan) (emission rendered in green), and Fabs were labeled with the Abberior Star Red probe (KK114) (rendered in red), the latter of which was conjugated with heavy-chain residue C216HC of the constant domain. In accordance with the spontaneous partitioning of the 3R mutant into bare membranes, VL GUVs incubated with the KK114-10E8-3R mutant were simultaneously stained with Laurdan and KK114. In contrast, no KK114 staining was detected in GUVs incubated with KK114-10E8-WT.
FIG 3.

Partitioning into membranes measured by fluorescence techniques. (A) Partitioning of fluorescently labeled Fabs KK114-10E8-WT and KK114-10E8-3R into VL GUVs. Micrographs display confocal images of VL GUVs at the equatorial plane. The lipid bilayer was labeled with Laurdan, and bound antibody was imaged following fluorescence emission of KK114 (green and red, respectively). The micrographs of both samples were rendered with equal contrast and brightness to best appreciate the difference in emission intensity. Bars, 2.5 μm. (B) Diagram illustrating the spectroscopic changes, namely, the increase in the emission intensity and maximum shift, that occur after transferring the fluorophore 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD) covalently attached to the CDRH3 tip of Fabs from the solvent to the membrane (3, 24, 25). The thiol-reactive fluorescence probe used was N,N’-dimethyl-N-(iodoacetyl)-N’-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) ethylenediamine (IANBD). (C) Binding of Fab 10E8 to VL LUVs monitored by changes in NBD fluorescence. Emission spectra of NBD-10E8-WT and NBD-10E8-3R were measured in solution (gray solid line) or in the presence of increasing concentrations of VL vesicles (black solid and dotted lines). Prior to spectrum collection, NBD-labeled Fabs (0.5 μM) were incubated with different concentrations of VL LUVs, as indicated. a.u., arbitrary units.
Membrane binding by 10E8-3R was also assessed by using Fabs labeled with the molecular sensor 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD) (Fig. 3B). This approach to probe protein-membrane interactions was described in our previous studies (3, 24, 25) and is based on monitoring changes in fluorescence emission that occur upon transferring the NBD probe from an aqueous solvent to the less polar environment of the lipid bilayer. The changes in NBD emission, i.e., the increase in intensity and blue shift of the emission maximum wavelength, are exemplified by the model spectra displayed in Fig. 3B (left). A shift of the emission maximum wavelength and an increase of the fluorescence intensity were observed for samples incubated with NBD-10E8-3R, which is consistent with a tendency of the 3R mutant to associate spontaneously with VL membranes (Fig. 3C, bottom right). In contrast, incubation of Fab NBD-10E8-WT Fab with VL large unilamellar vesicles (LUVs) did not induce appreciable changes in the emitted fluorescence (Fig. 3C, bottom left).
To confirm the contribution of electrostatic interactions to the observed partitioning of Fab 10E8-3R Fab, we next quantified the antibody-membrane interaction by spectroscopic titration of NBD-labeled Fab against vesicles that combined 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS) lipids at different molar ratios (26, 27) (Fig. 4). The experimental values, adjusted to equation 1, allowed the estimation of a mole fraction partition coefficient (Kx) of 0.2 × 107 for DOPC-DOPS (50:50) vesicles, a value that falls in the range of those observed for peripheral membrane proteins such as phospholipase C-ζ or the juxtamembrane domain of ErbB1 (28, 29). Moreover, the Kx values decreased upon the reduction of the content of the negatively charged lipid DOPS (Fig. 4A). Consistently, the linear plot of partitioning free energies (ΔGobs) as a function of the surface potential (Ψ0) displayed a positive slope (Fig. 4B), following the pattern expected for antibody-membrane interactions being driven by electrostatic forces (30, 31). In conclusion, 10E8-3R followed a membrane binding mechanism that resembled a peripheral membrane interaction, as was described previously for Fab 4E10 (25) (see below).
FIG 4.

Binding of 10E8-3R to DOPC:DOPS LUVs monitored by changes in NBD fluorescence. (A) Titration curves obtained by plotting the fraction of NBD-10E8-3R bound as a function of the concentration of lipid accessible on DOPC:DOPS vesicles (half the total lipid concentration) that contained 50, 25, or 10 mol% of DOPS (data depicted in blue, red, and green, respectively). Each symbol on the plot represents an average of data from three independent experiments (± standard deviations if larger than the symbol). The molar fraction partition coefficients, Kx, displayed were calculated from the best fit of equation 1 to the data (curves). (B) Plot of the free energy of partitioning versus the membrane surface potential in the above-described lipid vesicles, estimated according to equations 2 and 3, respectively. The positive slope of the linear regression is consistent with the favorable contribution of electrostatic interactions to membrane partitioning.
Effects on the biological function of 10E8 imparted by the 3R mutation.
Having determined that the 3R mutations enabled the binding of 10E8 to lipid vesicles, we tested their effect on polyreactivity and neutralizing activity (Fig. 5 and 6). We first assessed the propensity of 10E8 and the cognate 3R mutant to bind to HEp-2 cells, which have been used previously for determining antibody polyreactivity (8, 32). We found that, in contrast to the positive control, 4E10 IgG, neither 10E8-WT nor 10E8-3R bound to HEp-2 epithelial cells (Fig. 5A). We also compared the polyreactivities of 10E8-WT and 10E8-3R in a standard ELISA (8, 33) against various lipids, DNAs, as well as the unrelated proteins ovalbumin (OVA) and bovine serum albumin (BSA). By these assays, neither 10E8-WT nor 10E8-3R showed significant polyreactivity, even at high concentrations, with the exception of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) binding, in which case 10E8 and the 3R mutant showed weak binding signals similar to that of the 4E10 IgG control (Fig. 5B).
FIG 5.

Analysis of 10E8-3R polyreactivity. (A) Immunofluorescence staining experiment 1 using 10E8-3R against HEp-2 cells. Fabs 10E8-WT and 10E8-3R were used at a concentration of 25 μg/ml. 4E10 IgG was used as a positive control. Images shown are at a ×200 magnification. (B) ELISA binding of 10E8-3R against various antigens. Fab PV04 and 4E10 IgG were used as a negative control and a positive control, respectively. dsDNA, double-stranded DNA; ssDNA, single-stranded DNA; mAbs, monoclonal antibodies.
FIG 6.

Neutralization of HIV-1 by 10E8-3R. Fabs 10E8-WT and 10E8-3R were tested in a neutralization assay using TZM-bl target cells and a cross-clade panel of the following HIV-1 isolates: 94UG103 (A), 92RW020 (B), 92BR020 (C), IAVI C22 (D), 16055 (E), and 92TH021 (F).
We next tested the effect of the 3R mutations on the neutralization capacity of 10E8 against a panel of six HIV-1 isolates used previously as an indicator of cross-clade neutralization breadth (34). As illustrated by the data displayed in Fig. 6, 10E8-3R was notably more potent against all six isolates than was 10E8-WT (5- to 35-fold decrease in the 50% inhibitory concentration [IC50]) (Table 1). Ten additional isolates were tested, and 10E8-3R was also more potent against the majority of these isolates, including simian immunodeficiency virus (SIV) strain TAN2 (Table 1). In total, the 3R mutations increased 10E8 neutralization potency against 14 of 16 isolates, with an average 8.9-fold decrease in the IC50; however, the neutralization potency against two isolates (Q23.17 and CH181.12) did not change, so the effect appears to be somewhat isolate dependent.
TABLE 1.
Neutralization of HIV-1 by 10E8-WT and the 3R variant
| Virus | Clade | Tier | 10E8 IC50 (μg/ml) |
Fold changea | |
|---|---|---|---|---|---|
| WT | 3R | ||||
| 92RW020 | A | 2 | 0.53 | 0.038 | 13.9 |
| 94UG103 | A | 2 | 0.46 | 0.094 | 4.9 |
| BG505 | A | 2 | 0.36 | 0.18 | 2.0 |
| Q23.17 | A | 1b | 0.024 | 0.027 | 0.9 |
| 92BR020 | B | 2 | 0.18 | 0.014 | 13.5 |
| Comb-mut | B | 2 | 0.033 | 0.0039 | 8.4 |
| JRFL | B | 2 | 0.058 | 0.013 | 4.5 |
| LAI | B | 1b | 0.090 | 0.012 | 7.5 |
| SF162 | B | 1a | 0.051 | 0.021 | 2.4 |
| 16055 | C | 2 | 0.30 | 0.016 | 18.4 |
| COT6 | C | 2 | 0.0063 | 0.0037 | 1.7 |
| Du422.01 | C | 2 | 0.040 | 0.010 | 4.0 |
| IAVI C22 | C | 2 | 0.035 | 0.0027 | 13.1 |
| CH181.12 | BC | 2 | 0.48 | 0.603 | 0.8 |
| 92TH021 | AE | 2 | 0.025 | 0.0007 | 34.9 |
| TAN2 | SIVcpz | 2 | 0.092 | 0.0078 | 11.9 |
| Avg | 8.9 | ||||
Fold decrease in the IC50 (WT IC50/3R IC50).
10E8-3R antibody engagement of the MPER peptide and HIV-1 Env.
The finding that the 3R mutation did not make 10E8 polyreactive indicates that the differences in the binding and neutralization of 10E8 caused by the mutations are specific. It was shown previously that 10E8 neutralizing activity correlates with its capacity for epitope recognition in a membrane environment (3). Therefore, we sought to establish whether the overall enhanced neutralization observed for 10E8-3R correlated with an improved epitope binding function (Fig. 7 and 8).
FIG 7.

Effect of enhanced electrostatic interactions on epitope recognition at the membrane surface. (A) Partitioning of KK114-labeled Fabs 10E8-WT and 10E8-3R into VL GUVs decorated with the MPER(671–693) epitope peptide. Conditions are otherwise as described in the legend of Fig. 3. (B) Changes of fluorescence emission spectra upon incubation of NBD-labeled 10E8-WT or 10E8-3R with vesicles that contained 1.7 μM MPER(671–693) (black and blue lines, respectively). The gray line represents the emission spectra of the NBD-labeled Fab fragments in solution. (C) Increase in the fractional emission and change in the position of the maximum fluorescence emission wavelength in the presence of increasing concentrations of vesicles that contained 1.7 μM peptide (top and bottom, respectively). The initial value of fluorescence (F0) was determined from the maximum intensity of the labeled Fab fragment in solution, i.e., before the addition of peptide-containing vesicles. Final fluorescence values (F) correspond to the NBD intensity measured after incubation with vesicles. Black and blue symbols correspond to WT and 3R Fabs, respectively. Each data point corresponds to the average of results from three titrations (± standard deviation), as displayed in panel B.
FIG 8.
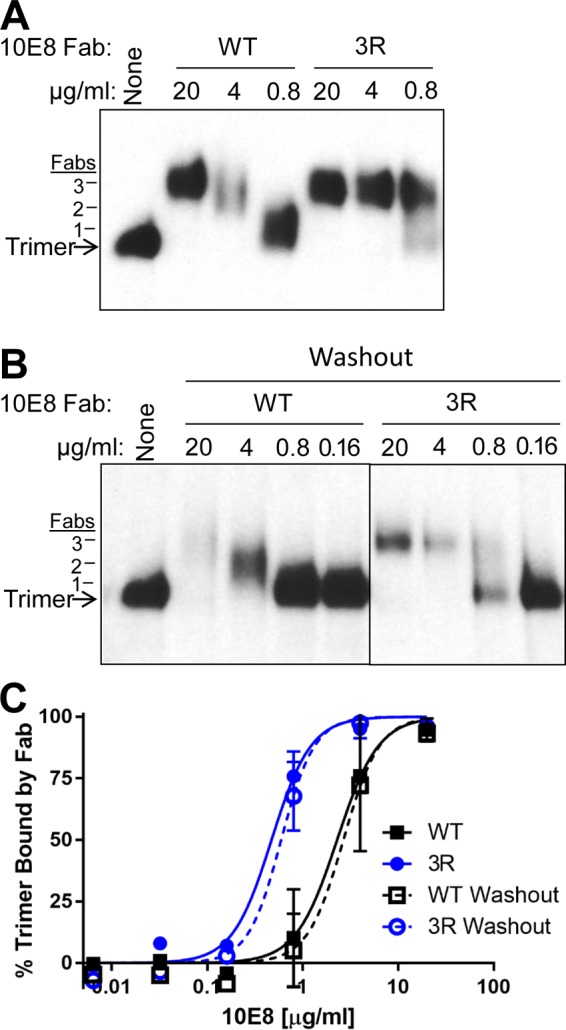
Binding of 10E8-3R to native Env trimers on virions. (A) HIV-1 virions displaying Comb-mut Env were incubated with Fabs 10E8-WT or 10E8-3R, followed by treatment with mild detergent and the resolution of Env-Fab complexes using BN-PAGE Western blotting. Binding of an antibody to the Env trimer causes it to run more slowly on the gel and the cognate band to shift upwards on the blot. Estimation of the migration of a trimer with one, two, or three Fabs bound (left) is based on gel mobility shifts by control Fabs PGT126 (3 Fabs/trimer), PGT151 (2 Fabs), and PG9 (1 Fab). (B) BN-PAGE gel mobility shift analysis was performed as described above for panel A, except that after Fabs and virus were incubated together for 30 min, the virus was pelleted, and unbound Fab was removed. Thus, the gel mobility shift reflects only binding to the trimer while it was incorporated into the virion membrane. (C) Intensities of bands on BN-PAGE Western blots were quantified by using ImageJ software, and the percentage of the trimer bound by antibody was calculated by comparing the intensities of the unliganded trimer band in the presence and absence of Fab. The results shown are the averages of data from four (standard assay) and two (washout) independent BN-PAGE Western blot assays.
Figure 7A (top) displays confocal images of peptide-containing GUVs that were probed by Laurdan labeling and with Fab KK114-10E8-WT. These GUVs exhibited simultaneous staining of the lipid bilayer by both Laurdan and KK114 probes (green and red, respectively), consistent with the previously reported binding of 10E8 to the MPER peptide inserted into membranes (3). The relatively intense KK114 staining of GUVs observed upon incubation with Fab KK114-10E8-3R suggests that the optimized 3R mutant bound with a higher apparent affinity than the parental antibody to GUV membranes decorated with the epitope peptide (Fig. 7A, bottom). These observations were further supported by experiments performed by using NBD-labeled Fabs (Fig. 7B and C). Binding to vesicles containing the MPER peptide resulted in NBD fluorescence changes (Fig. 7B), i.e., increases in emission and maximum blue shift (Fig. 7C), that were overall more prominent in the 10E8-3R variant than in parental Fab 10E8-WT. This observation was again consistent with the more efficient epitope recognition in the membrane milieu by the 3R mutant.
To determine whether the increase in neutralization by 10E8-3R corresponded to an increase in its binding affinity for virion-associated Env trimers, we next turned to a blue native PAGE (BN-PAGE) gel mobility shift assay. Virions were produced from a molecular clone of Comb-mut whose Env protein has been shown to be >95% cleaved and trimeric (35). When a Fab fragment binds to the Env spike, the trimer will run more slowly on BN-PAGE gels, shifting the cognate band upwards on the gel. The 10E8-3R Fab fragment shifted the trimer band at a 5-fold-lower concentration than 10E8-WT, suggesting that the 3R mutations increased the affinity for the trimer (Fig. 8A and C) (shifting 50% effective concentrations [EC50s] of 2.3 μg/ml for the WT and 0.47 μg/ml for 3R). To determine whether the membrane contributed to the enhanced binding capacity of the 10E8-3R Fab fragment, we also performed a “washout” version of the gel mobility shift assay, in which virus was pelleted and unbound antibody was removed prior to the addition of detergent. This experiment yielded results similar to those of the standard assay (Fig. 8B and C). We conclude that the 3R mutations enhance the binding of 10E8 to the unliganded Env trimer in the virus membrane. We note that neutralization by 10E8 and other MPER antibodies typically involves binding to both unliganded Env and receptor-activated forms of Env, and it is possible that the 3R mutations also affect binding to Env in the latter state, but this was not tested here due to the experimental challenges involved.
Effects on 4E10 imparted by a 3R mutation.
Given the potential implications for ongoing efforts in vaccine and biologics design, our optimization approach focused primarily on the highly potent antibody 10E8. To establish whether the same strategy to improve neutralization efficacy by strengthening electrostatic interactions with membranes could be applied to other anti-MPER antibodies, we introduced three Arg mutations into the paratope of the 4E10 antibody (Fig. 9 and Table 2). The first-generation anti-MPER bnAb 4E10 was previously shown to spontaneously partition into membranes (24, 25); exhibits some polyreactivity, particularly with lipids; and is less potent than 10E8 (see reference 8 for a side-by-side comparison of both antibodies).
FIG 9.

Design, membrane binding, and polyreactivity of the 4E10-3R antibody. (A) Surface density charge representation (bottom views) of Fab 4E10-WT (PDB accession number 2FX7) and mutant Fab 4E10-3R (rendered by the introduction of the G27R/S30R/S74R triple substitution into the heavy chain). (B) Model for interaction with an Env trimer and detailed view of the mutated positions (in blue) relative to the membrane (dotted line). Bound dihexanoyl phosphatidic acid molecules (06:0 PA) (colored by atoms) and the MPER helix (in orange) were obtained from structures under PDB accession numbers 4XBG and 5GHW, respectively. The light and heavy chains of the Fabs are shown in brown and tan, respectively. (C) Binding of 4E10-3R (blue traces and symbols) to DOPC-DOPS (50:50) LUVs monitored by changes in NBD fluorescence. The black dotted line follows the binding of 4E10-WT. The fraction of Fab bound as a function of the concentration of lipid accessible (half the total lipid concentration) was plotted after titration of NBD-labeled Fab with increasing concentrations of liposomes. Conditions are otherwise as described in the legend of Fig. 4. (D) Plots of the free energy of partitioning versus the membrane surface potential in the above-described lipid vesicles. The black dotted line adjusts to 4E10-WT data and is included for comparison. (E) Immunofluorescence staining experiment 2 using 4E10-3R against HEp-2 cells. Fab 4E10-WT and mutant Fab 4E10-3R were used at a concentration of 25 μg/ml. Images shown are at a ×200 magnification. (F) Binding of Fab 4E10-3R to native Env trimers on virions. (Top) HIV-1 virions displaying Comb-mut Env were incubated with Fabs 4E10-WT or 4E10-3R, and Env-Fab complexes were resolved by using BN-PAGE Western blotting. (Bottom) The percentage of the trimer bound by antibody was calculated by comparing the intensities of the unliganded trimer band in the presence and absence of Fab. Conditions are otherwise as described in the legend of Fig. 8.
TABLE 2.
Neutralization of HIV-1 by 4E10-WT and the 3R variant
| Virus | Clade | Tier | 4E10 IC50 (μg/ml) |
Fold changea | |
|---|---|---|---|---|---|
| WT | 3R | ||||
| BG505 | A | 2 | 1.3 | 1.4 | 0.91 |
| Comb-mut | B | 2 | 1.6 | 2.9 | 0.57 |
| SF162 | B | 1a | 1.9 | 4.6 | 0.40 |
| COT6 | C | 2 | 0.8 | 2.1 | 0.37 |
| Du422.01 | C | 2 | 0.94 | 1.00 | 0.94 |
| CH181.12 | BC | 2 | 5.4 | 10.1 | 0.53 |
| Avg | 0.6 | ||||
Fold decrease in the IC50 (WT IC50/3R IC50).
Thus, to generate the 4E10-3R mutant, we introduced the G27R/S30R/S74R triple substitution into the heavy chain of Fab 4E10, which resulted in an increased positive charge at the flat paratope surface predicted by crystallography to contact the interface of the lipid bilayer (22) (Fig. 9A and B). These changes did not affect binding to the epitope peptide in ELISAs (not shown). However, Fab 4E10-3R partitioned more effectively into vesicles than did parental Fab 4E10-WT (Fig. 9C), and this effect was mediated by the establishment of stronger electrostatic interactions with membranes (Fig. 9D). The 3R mutation also resulted in significantly enhanced polyreactivity (Fig. 9E) but did not improve 4E10 neutralization relative to that of 4E10-WT against any of the isolates tested and in several cases resulted in up to a 2-fold increase in the IC50 (Table 2). Moreover, in line with the lack of neutralization improvement, the WT and 3R mutant Fab fragments shifted the mobility of the Env trimer in BN-PAGE experiments to similar extents (Fig. 9F).
In conclusion, whereas electrostatic interactions with membranes and polyreactivity were enhanced with 4E10-3R relative to 4E10-WT, the neutralization efficacy and Env trimer engagement were less affected or even slightly diminished. We note that while these results with 4E10-3R are in contrast to those for 10E8-3R, they do not negate the hypothesized mechanism for the improvement of 10E8 by 3R, as these mutations are specific to 10E8, and it is possible that the 4E10 3R mutations are not optimally positioned to augment the neutralization potency of 4E10 by this mechanism.
DISCUSSION
The identification of numerous highly potent bnAbs in the past decade has greatly advanced our understanding of effective humoral responses elicited during HIV-1 infection (1, 11). The knowledge gained has stimulated approaches for rational vaccine development (36–38). The isolated bnAbs have also inspired the rational design of biologics expected to prevent and treat HIV infection (39, 40). Indeed, the passive administration of engineered versions of bnAbs has been shown to prevent HIV infection in animal models (41, 42).
In this context, the potency and breadth of neutralization by 10E8 as well as its effectiveness at conferring cross-protection in vivo in primate models (8, 15–17) make it potentially useful for therapeutic developments. Two complementary strategies have been used to overcome potential limitations of 10E8 for pharmacological use, namely, (i) optimization of function and stability through mutagenesis (19) and (ii) promotion of polyvalence by antibody engineering (43). Following the latter strategy, recently reported works describe antibodies that simultaneously interact with two (bivalent) or three (trivalent) independent Env determinants, which contained the paratope of 10E8 as a basic component (18, 20, 44).
In this work, following the former strategy, we have explored the possibility that the function of 10E8 can be upgraded by promoting its interaction with membranes (Fig. 1). Recent structural studies define a membrane-interacting surface on the light chain of 10E8, which is juxtaposed by its CDRH3 that contacts the MPER helix on Env (Fig. 1) (2–5, 22). In principle at least, the interaction of MPER antibodies through similar patches could be driven by coulombic attraction between exposed basic residues and anionic phospholipids, a possibility that has been formally demonstrated in the case of the 4E10 antibody (25). Likewise, in the case of the 10E8 antibody, the substitution of aspartate or glutamate at positions normally occupied by basic and polar residues on the membrane-contacting patch greatly reduced the neutralization potency of this antibody (4).
Here, we have asked whether the manipulation of the membrane-contacting surface of 10E8 to increase its net charge would enable spontaneous interactions with membranes and what effects this property might have on its biological function. We showed that the S30R/N52R/S67R triple substitution in the light chain of Fab 10E8 (Fig. 1) enabled it to interact spontaneously with lipid bilayers (Fig. 2 to 4). This change importantly also results in more-potent neutralizing activity (Fig. 6 and Table 1). Notably, the improvement in 10E8 function correlated with an enhanced binding affinity not only for the MPER epitope peptide in a membrane environment (Fig. 7) but also for virion-associated Env (Fig. 8) yet had no observable effect on polyreactivity, which remained almost undetectable for the 3R mutant (Fig. 5). This observation implies a lack of a correlation between neutralization potency and polyreactivity. It further highlights a difference between partitioning from water in membranes and lipid polyreactivity, as inferred from the comparison of the results in Fig. 2 to 4 and 5, respectively.
The data obtained with the 4E10 antibody (Fig. 9) further support this concept. Upon the introduction of a cognate 3R mutation (Fig. 9A and B), electrostatic interactions intensified, resulting in a stronger association of 4E10 with the membrane (Fig. 9C and D) and also, notably, apparent polyreactivity (Fig. 9E). However, these enhancing effects translated into neither better nor worse neutralization activity (Table 2). Likewise, the introduction of the 3R mutation into Fab 4E10 did not significantly affect the affinity for detergent-solubilized viral Env (Fig. 9F). Given that 4E10 is less potent than the second-generation bnAbs (1) and therefore has considerable scope for improving its potency, our observations argue against the postulate that 4E10 polyreactivity and neutralization are contingent on each other (32, 45, 46).
We showed that the 3R mutations enhanced neutralization by 10E8 with most isolates, but there was variation in the magnitude of the effect, and the neutralization of two isolates was not improved. Analysis of the Env sequences of the tested isolates revealed no clear signature in or near the MPER that would explain the strain dependence of enhanced neutralization (our unpublished observations). However, it is possible that residues surrounding the MPER alter the effect of the 3R mutations on neutralization. Alternatively, the differential effect of 3R on neutralization between isolates could be related to differences in the kinetics of fusion, proximal glycosylation, or MPER conformation relative to the antibody in different Env backgrounds. Further studies of the Env structure and mechanisms of 10E8 recognition are necessary to address these issues.
Thus, although our data support the idea that interactions with the viral membrane surface occur by the neutralization mechanism of anti-MPER antibodies, they caution that the proposed two-step model in which anti-MPER antibodies attach first to the viral membrane and then to the Env antigen (47–49) might be of limited applicability. The finding that enhanced membrane interactions do not affect 4E10 neutralization potency implies that the accumulation of this antibody at the virus membrane does not necessarily play a role in the mechanism of neutralization. The finding that Fab 10E8, which is more potent than 4E10, does not spontaneously interact with VL membranes (Fig. 2 and 3) also goes against the assumption that the preattachment of this antibody to the viral membrane is strictly required during neutralization (49).
Our data rather indicate that antibody-membrane interactions might function at a step after, or concomitant with, the engagement of the Env antigen. Membrane binding would thus stabilize the antibody-Env complex, resulting in a high-affinity interaction and higher neutralization potency, at least in the case of 10E8 (Fig. 6 to 8). The finding that there is some Env dependence of the magnitude of this effect suggests that elements in Env may influence accessibility to the membrane-interacting region, as in a specific bipartite Env-membrane epitope rather than independent membrane and Env binding sites of a simple two-step model. Since reducing electrostatic interactions with membranes hinders 4E10-induced neutralization (25), but their enhancement has no effect (Fig. 9 and Table 2), it seems possible that the contribution of electrostatic antibody-membrane interactions to the mechanism of 4E10 neutralization has already reached an optimal level. Alternatively, the membrane-contacting surface relevant to 4E10 neutralization might be different from that inferred from previously reported crystal structures and mutagenesis studies (22, 25). Thus, it cannot be excluded that the 3R residues have been incorporated at irrelevant positions on 4E10. On the other hand, the available structures for 10E8 may reflect more precisely its functional binding state.
In conclusion, our observations suggest that electrostatic interactions between anti-MPER antibodies and the lipid bilayer result from structural adaptations to enable functional binding to Env in the membrane milieu (2–4, 22, 24, 25). If this idea is correct, we infer that vaccines targeting the MPER epitope should elicit antibodies that approximate if not reproduce cognate membrane interactions. Our data also demonstrate that with 10E8, there is scope for functional improvement by engineering the membrane-interacting surface of the paratope, a strategy that should be further explored.
MATERIALS AND METHODS
Materials.
The peptides used in this study were synthesized in the C-terminal carboxamide form by solid-phase methods using 9-fluorenylmethoxy carbonyl (Fmoc) chemistry, purified by reverse-phase high-pressure liquid chromatography, and characterized by matrix-assisted time of flight mass spectrometry (purity of >95%). Peptides were routinely dissolved in dimethyl sulfoxide (DMSO), and their concentrations were determined by the bicinchoninic acid microassay (Pierce, Rockford, IL, USA) or by their absorbance at 280 nm. Goat anti-human IgG-Fab antibody was purchased from Sigma (St. Louis, MO). Secondary antibody conjugated to horseradish peroxidase (HRP), mouse anti-goat IgG-HRP and rabbit anti-human IgG-HRP, were purchased from Santa Cruz (Heidelberg, Germany). The fluorescent probes Laurdan and NBD were obtained from Molecular Probes (Eugene, OR, USA). Abberior Star 580 (KK114) was obtained from Abberior (Göttingen, Germany). The lipids DOPC, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), DOPS, sphingomyelin (SM), cholesterol (Chol), and 1,2-dioleoyl-sn-glycerol-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rho-PE) were purchased from Avanti Polar Lipids (Alabaster, AL).
Production, characterization, and labeling of Fabs.
Antibody sequences were cloned into plasmid pColaDuet and expressed in the Escherichia coli T7-shuffle strain. Recombinant expression was induced at 18°C overnight with 0.4 mM isopropyl-d-thiogalactopyranoside when the culture reached an optical density (OD) of 0.8. Cells were harvested and centrifuged at 8,000 × g, after which they were resuspended in a buffer containing 50 mM HEPES (pH 7.5), 500 mM NaCl, 35 mM imidazole, DNase (Sigma-Aldrich, St. Louis, MO), and an EDTA-free protease inhibitor mixture (Roche, Madrid, Spain). Cell lysis was performed by using an Avestin Emulsiflex C5 homogenizer. Cell debris was removed by centrifugation, and the supernatant was loaded onto a nickel-nitrilotriacetic acid (Ni-NTA) affinity column (GE Healthcare). Elution was performed with 500 mM imidazole, and the fractions containing the His-tagged proteins were pooled, concentrated, and dialyzed against a solution containing 50 mM sodium phosphate (pH 8.0), 300 mM NaCl, 1 mM dithiothreitol (DTT), and 0.3 mM EDTA in the presence of purified tobacco etch virus protease (50). Fabs were separated from the cleaved peptides containing the His6 tag by an additional step in a Ni-nitrilotriacetic column. The flowthrough fraction containing the antibody was dialyzed overnight at 4°C against sodium acetate (pH 5.6) supplemented with 10% glycerol and subsequently loaded onto a MonoS ion-exchange chromatography (IEC) column (GE Healthcare). Elution was carried out with a gradient of potassium chloride, and the fractions containing purified Fab were concentrated and dialyzed against a buffer containing 10 mM sodium phosphate (pH 7.5), 150 mM NaCl, and 10% glycerol. For the preparation of mutant Fabs, the KOD-Plus mutagenesis kit (Toyobo, Osaka, Japan) was employed according to the instructions of the manufacturer. Labeling with the polarity-sensitive NBD probe was performed as described previously (51, 52). In brief, a cysteine-substituted Fab derivative (W100bHCC) was first generated by site-directed mutagenesis and modified with a sulfhydryl-specific iodoacetamide derivative of NBD. Thus, this procedure results in the conservative replacement of the Trp indole ring by the similarly bicyclic nitrobenzoxadiazole ring, which makes comparable changes in polarity scored by the NBD label. For confocal microscopy experiments, the fluorescence probe KK114 was introduced in vitro at position C216HC according to the same procedure as the one used for the NBD probe.
Lipid vesicle production.
LUVs made from the VL lipid mixture (DOPC, Chol, SM, DOPE, and DOPS at a molar ratio of 14:46:17:16:7) were produced according to the extrusion method (53). To this end, lipid suspensions were subjected to 10 freeze-thaw cycles prior to extrusion 10 times through 2 stacked polycarbonate membranes with a nominal pore size of 0.1 μm (Nuclepore, Inc., Pleasanton, CA, USA). GUVs with the same composition were produced by spontaneous swelling according to procedures described previously (54, 55). Briefly, lipids were mixed in CHCl3-CH3OH (9:1), and the organic solvent was removed by desiccation for 1 h. Dried silica beads covered with lipid were collected and transferred to a 7.5-g/liter sucrose buffer to induce the spontaneous swelling of GUVs. For the preparation of peptide-GUV complexes, 0.66 μM peptide was added to the observation dish in an isosmotic buffer containing 10 mM HEPES and 150 mM KCl (pH 7.4), followed by the addition of the formed vesicles.
Vesicle flotation assays.
The partition of the antibody into membranes was examined by vesicle flotation experiments in sucrose gradients according to methods described previously by Yethon et al. (56). In brief, 100 μl of VL LUVs labeled with the lipid rhodamine-PE (Avanti Polar Lipids, Alabaster, AL) was incubated with Fabs at 25°C for 15 min under continuous stirring (800 rpm). The mixture was adjusted to a sucrose concentration of 1.4 M in a final volume of 300 μl and subsequently deposited on a stepwise gradient composed of successive solutions containing 0.8 M (400 μl) and 0.5 M (300 μl) sucrose. The gradient was centrifuged at 4°C at 436,000 × g for 3 h in a TLA120.2 rotor (Beckman Coulter, Brea, CA). After centrifugation, four fractions of 250 μl each were collected. The material that adhered to the centrifugation tubes was extracted by washing the tubes with 250 μl of a solution of 1% (wt/vol) SDS at 100°C. Antibody Fab fragments were detected in SDS-PAGE gels by using Western blotting, and the presence of liposomes was monitored by measuring rhodamine fluorescence. Densitometry of the blotted Fab bands was performed by using ImageJ software, and the percentage of Fab bound to vesicles was calculated from the band intensities measured in the vesicle-floating fractions, relative to the sum of the intensities measured in all fractions.
Confocal microscopy.
Images were acquired on a Leica TCS SP5 II microscope (Leica Microsystems GmbH, Wetzlar, Germany) as described previously (55, 57). Laurdan-stained GUVs were excited at 780 nm by using a 63× water immersion objective (numerical aperture [NA] = 1.2), 512- by 512-pixel images were acquired at 400 Hz per scanning line, and emission was imaged at 435 ± 20 nm. The KK114-labeled Fab fragments were excited at 633 nm by using an HeNe laser, and emission was imaged at 775 ± 125 nm.
Spectroscopic titration.
NBD fluorescence emission spectra were recorded with the excitation wavelength fixed at 470 nm. The emission spectrum of a sample lacking the fluorophore was subtracted from the spectrum of the equivalent sample containing the fluorophore. Partitioning curves were computed from the fractional changes in emitted NBD fluorescence when 150 nM NBD-labeled Fab was titrated with increasing lipid concentrations. The mole fraction partition coefficients, Kx, were determined by fitting the experimental values to a hyperbolic function (58):
| (1) |
where [L] is the concentration of accessible lipid and K is the lipid concentration at which the bound peptide fraction is 0.5. Therefore, Kx = [W]/K, where [W] is the molar concentration of water. The observed free energy of water-membrane partitioning was subsequently calculated according to the following expression:
| (2) |
where R is the ideal gas constant and T is the temperature of the binding reaction. For the estimation of the electrical potential at the membrane surface as a function of the PS content, the following equation was used:
| (3) |
where c is the number of ions per volume and σ is the surface charge density (30). To calculate the latter parameter, a surface area per phospholipid of 69.5 Å2 was considered, and net charges of 0 and −1 were assigned to DOPC and DOPS, respectively (59).
HEp-2 cell immunofluorescence assay.
Antibodies (Fabs) were assessed for the ability to stain HIV-1-negative human epithelial HEp-2 (VIRGO ANA/HEp-2) cells on glass slides by indirect-immunofluorescence microscopy. Fabs were diluted to 25 μg/ml, and 10 μl was used for each test, according to the manufacturer's specifications. Fluorescein isothiocyanate (FITC)-conjugated goat anti-human Fab (Jackson) was used as the secondary probe. Slides were photographed by using an Evos fluorescence microscope (Invitrogen). Staining experiment 1 with 10E8-3R and staining experiment 2 with 4E10-3R were performed separately; the intensities of antibody staining can be compared within but not between experiments.
ELISA for antibody polyreactivity.
Ninety-six-well plates were coated with 10 nM cholesterol, DOPS, POPC, or DOPE (Sigma) in 50 μl 100% methanol and left to evaporate overnight at 4°C. Wells were coated with other antigens (500 ng/well in phosphate-buffered saline [PBS]) overnight at 4°C without evaporation. The wells were washed three times with PBS containing 0.05% Tween 20 (PBST) and blocked for 1 h at 37°C with 4% nonfat dry milk (NFDM) in PBS. The wells were washed three times with PBST, Fabs diluted in 0.4% NFDM–PBST were added to the wells, and the wells were incubated for 2 h at 37°C. Following three washes with PBST, a goat anti-human Fab-HRP conjugate (1:500 in 0.4% NFDM–PBST) was added to the wells, and the wells were incubated for 45 min at room temperature (RT). The wells were washed three times with PBST, and the plate was developed by using the 3,3’,5,5’-tetramethylbenzidine (TMB) substrate (Pierce) at 37°C. The OD at 450 nm was determined by using a Synergy H1 plate reader (BioTek).
Virus production.
Pseudotyped HIV-1 was generated by the cotransfection of 293T cells with Env plasmid DNA and HIV-1 backbone plasmid pSG3ΔEnv (60) by using 25,000-molecular-weight (25K) polyethyleneimine (PEI), as previously described (61). Envs of HIV isolates JRFL, LAI, and SF162 were obtained from the NIH AIDS Reagent Program; Envs of 92RW020, 94UG103, 92BR020, 92TH021, IAVI C22, and BG505 were obtained from D. Burton (The Scripps Research Institute); Env of 16055 was obtained from R. Wyatt (Scripps); Env of COT6 was obtained from L. Morris (University of Witwatersrand); Env of Q23.17 was obtained from J. Overbaugh (Fred Hutchinson Cancer Research Center); Envs of Du422.01 and CH181.12 were obtained from M. Seaman (Beth Israel Deaconess Medical Center); and Env of TAN2 was obtained from B. Hahn (University of Pennsylvania). Envs were expressed from the pSVIII plasmid; Comb-mut, by exception, was contained in the pcDNA plasmid. Virions for BN-PAGE analysis were produced by transient transfection using the molecular clone pLAI-Comb-mut (35).
Neutralization assays.
Neutralization was determined by using a single-cycle neutralization assay with CD4+ CXCR4+ CCR5+ TZM-bl cells as target cells. Antibodies were added to virus in cell culture medium (Dulbecco's modified Eagle's medium [DMEM] supplemented with 10% fetal bovine serum [FBS], 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin), and the mixture was incubated for 1 h at 37°C prior to addition to target cells. Following 72 h of incubation at 37°C, cells were lysed, Bright-Glo luciferase reagent (Promega) was added, and luminescence in relative light units (RLUs) was measured by using a Synergy H1 plate reader (BioTek).
BN-PAGE Western blotting.
For BN-PAGE gel mobility shift assays, Comb-mut virions (35) were incubated with Fab fragments of antibodies at RT for 30 min. Env was solubilized from the membrane by using 1% n-dodecyl-β-d-maltoside (DDM) in Tris-glycine native sample buffer (ThermoFisher) supplemented with 0.25% Coomassie G-250 and loaded onto a 3-to-8% gradient Tris-acetate NuPAGE gel (ThermoFisher). For washout assays, following incubation with antibodies for 30 min, the sample volume was increased to 1 ml by using sample buffer, and the virus was centrifuged at 21,000 relative centrifugal force (RCF) for 45 min at 4°C. The supernatant containing unbound Fabs was removed, and the virus pellet was resuspended in sample buffer and processed for BN-PAGE as described above. Electrophoresis was performed for 3 h at 150 V in Tris-glycine native running buffer (ThermoFisher) that was supplemented with 0.002% Coomassie G-250. Proteins were transferred onto a polyvinylidene difluoride (PDVF) membrane by using an XCell II blot module (ThermoFisher) and then probed by using a cocktail of antibodies to gp120 (F105, 2G12, and F425-B4e8 at 2 μg/ml each) and gp41 (2F5, Z13e1, and 7B2 at 1 μg/ml each). Primary antibodies were further probed by using a goat anti-human-Fcγ-HRP secondary antibody (Jackson), and the blot was developed by using the ECL Plus substrate (Pierce).
ACKNOWLEDGMENTS
This study was supported by the Spanish MINECO (BIO2015-64421-R [MINECO/FEDER UE] to J.L.N.) and the Basque Government (IT838-13 to J.L.N.). Support was also provided by the NIH NIAID (R01 AI114401 and R01 AI098602 to M.B.Z.) and the James B. Pendleton Charitable Trust (M.B.Z.). E.R. and S.I. received predoctoral fellowships from the Basque Government.
We acknowledge valuable technical assistance from Rubén Sanchez-Eugenia and Miguel García-Porras.
REFERENCES
- 1.Burton DR, Hangartner L. 2016. Broadly neutralizing antibodies to HIV and their role in vaccine design. Annu Rev Immunol 34:635–659. doi: 10.1146/annurev-immunol-041015-055515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JH, Ozorowski G, Ward AB. 2016. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 351:1043–1048. doi: 10.1126/science.aad2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rujas E, Caaveiro JM, Partida-Hanon A, Gulzar N, Morante K, Apellaniz B, Garcia-Porras M, Bruix M, Tsumoto K, Scott JK, Jimenez MA, Nieva JL. 2016. Structural basis for broad neutralization of HIV-1 through the molecular recognition of 10E8 helical epitope at the membrane interface. Sci Rep 6:38177. doi: 10.1038/srep38177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irimia A, Serra AM, Sarkar A, Jacak R, Kalyuzhniy O, Sok D, Saye-Francisco KL, Schiffner T, Tingle R, Kubitz M, Adachi Y, Stanfield RL, Deller MC, Burton DR, Schief WR, Wilson IA. 2017. Lipid interactions and angle of approach to the HIV-1 viral membrane of broadly neutralizing antibody 10E8: insights for vaccine and therapeutic design. PLoS Pathog 13:e1006212. doi: 10.1371/journal.ppat.1006212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerutti N, Loredo-Varela JL, Caillat C, Weissenhorn W. 2017. Antigp41 membrane proximal external region antibodies and the art of using the membrane for neutralization. Curr Opin HIV AIDS 12:250–256. doi: 10.1097/COH.0000000000000364. [DOI] [PubMed] [Google Scholar]
- 6.Zwick MB, Labrijn AF, Wang M, Spenlehauer C, Saphire EO, Binley JM, Moore JP, Stiegler G, Katinger H, Burton DR, Parren PW. 2001. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J Virol 75:10892–10905. doi: 10.1128/JVI.75.22.10892-10905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Binley JM, Wrin T, Korber B, Zwick MB, Wang M, Chappey C, Stiegler G, Kunert R, Zolla-Pazner S, Katinger H, Petropoulos CJ, Burton DR. 2004. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J Virol 78:13232–13252. doi: 10.1128/JVI.78.23.13232-13252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. 2012. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacob RA, Moyo T, Schomaker M, Abrahams F, Grau Pujol B, Dorfman JR. 2015. Anti-V3/glycan and anti-MPER neutralizing antibodies, but not anti-V2/glycan site antibodies, are strongly associated with greater anti-HIV-1 neutralization breadth and potency. J Virol 89:5264–5275. doi: 10.1128/JVI.00129-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim AS, Leaman DP, Zwick MB. 2014. Antibody to gp41 MPER alters functional properties of HIV-1 Env without complete neutralization. PLoS Pathog 10:e1004271. doi: 10.1371/journal.ppat.1004271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwong PD, Mascola JR. 2012. Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity 37:412–425. doi: 10.1016/j.immuni.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zwick MB. 2005. The membrane-proximal external region of HIV-1 gp41: a vaccine target worth exploring. AIDS 19:1725–1737. doi: 10.1097/01.aids.0000189850.83322.41. [DOI] [PubMed] [Google Scholar]
- 13.Montero M, van Houten NE, Wang X, Scott JK. 2008. The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiol Mol Biol Rev 72:54–84. doi: 10.1128/MMBR.00020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burton DR, Mascola JR. 2015. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat Immunol 16:571–576. doi: 10.1038/ni.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pegu A, Yang ZY, Boyington JC, Wu L, Ko SY, Schmidt SD, McKee K, Kong WP, Shi W, Chen X, Todd JP, Letvin NL, Huang J, Nason MC, Hoxie JA, Kwong PD, Connors M, Rao SS, Mascola JR, Nabel GJ. 2014. Neutralizing antibodies to HIV-1 envelope protect more effectively in vivo than those to the CD4 receptor. Sci Transl Med 6:243ra88. doi: 10.1126/scitranslmed.3008992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Gils MJ, Sanders RW. 2014. In vivo protection by broadly neutralizing HIV antibodies. Trends Microbiol 22:550–551. doi: 10.1016/j.tim.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Barbian HJ, Decker JM, Bibollet-Ruche F, Galimidi RP, West AP Jr, Learn GH, Parrish NF, Iyer SS, Li Y, Pace CS, Song R, Huang Y, Denny TN, Mouquet H, Martin L, Acharya P, Zhang B, Kwong PD, Mascola JR, Verrips CT, Strokappe NM, Rutten L, McCoy LE, Weiss RA, Brown CS, Jackson R, Silvestri G, Connors M, Burton DR, Shaw GM, Nussenzweig MC, Bjorkman PJ, Ho DD, Farzan M, Hahn BH. 2015. Neutralization properties of simian immunodeficiency viruses infecting chimpanzees and gorillas. mBio 6:e00296-. doi: 10.1128/mBio.00296-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asokan M, Rudicell RS, Louder M, McKee K, O'Dell S, Stewart-Jones G, Wang K, Xu L, Chen X, Choe M, Chuang G, Georgiev IS, Joyce MG, Kirys T, Ko S, Pegu A, Shi W, Todd JP, Yang Z, Bailer RT, Rao S, Kwong PD, Nabel GJ, Mascola JR. 2015. Bispecific antibodies targeting different epitopes on the HIV-1 envelope exhibit broad and potent neutralization. J Virol 89:12501–12512. doi: 10.1128/JVI.02097-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon YD, Georgiev IS, Ofek G, Zhang B, Asokan M, Bailer RT, Bao A, Caruso W, Chen X, Choe M, Druz A, Ko SY, Louder MK, McKee K, O'Dell S, Pegu A, Rudicell RS, Shi W, Wang K, Yang Y, Alger M, Bender MF, Carlton K, Cooper JW, Blinn J, Eudailey J, Lloyd K, Parks R, Alam SM, Haynes BF, Padte NN, Yu J, Ho DD, Huang J, Connors M, Schwartz RM, Mascola JR, Kwong PD. 2016. Optimization of the solubility of HIV-1-neutralizing antibody 10E8 through somatic variation and structure-based design. J Virol 90:5899–5914. doi: 10.1128/JVI.03246-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu L, Pegu A, Rao E, Doria-Rose N, Beninga J, McKee K, Lord DM, Wei RR, Deng G, Louder M, Schmidt SD, Mankoff Z, Wu L, Asokan M, Beil C, Lange C, Leuschner WD, Kruip J, Sendak R, Kwon YD, Zhou T, Chen X, Bailer RT, Wang K, Choe M, Tartaglia LJ, Barouch DH, O'Dell S, Todd JP, Burton DR, Roederer M, Connors M, Koup RA, Kwong PD, Yang ZY, Mascola JR, Nabel GJ. 20 September 2017. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science doi: 10.1126/science.aan8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Apellaniz B, Rujas E, Serrano S, Morante K, Tsumoto K, Caaveiro JM, Jimenez MA, Nieva JL. 2015. The atomic structure of the HIV-1 gp41 transmembrane domain and its connection to the immunogenic membrane-proximal external region. J Biol Chem 290:12999–13015. doi: 10.1074/jbc.M115.644351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irimia A, Sarkar A, Stanfield RL, Wilson IA. 2016. Crystallographic identification of lipid as an integral component of the epitope of HIV broadly neutralizing antibody 4E10. Immunity 44:21–31. doi: 10.1016/j.immuni.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scherer EM, Leaman DP, Zwick MB, McMichael AJ, Burton DR. 2010. Aromatic residues at the edge of the antibody combining site facilitate viral glycoprotein recognition through membrane interactions. Proc Natl Acad Sci U S A 107:1529–1534. doi: 10.1073/pnas.0909680107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rujas E, Insausti S, Garcia-Porras M, Sanchez-Eugenia R, Tsumoto K, Nieva JL, Caaveiro JM. 2017. Functional contacts between MPER and the anti-HIV-1 broadly neutralizing antibody 4E10 extend into the core of the membrane. J Mol Biol 429:1213–1226. doi: 10.1016/j.jmb.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rujas E, Caaveiro JM, Insausti S, Garcia-Porras M, Tsumoto K, Nieva JL. 2017. Peripheral membrane interactions boost the engagement by an anti-HIV-1 broadly neutralizing antibody. J Biol Chem 292:5571–5583. doi: 10.1074/jbc.M117.775429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arbuzova A, Wang J, Murray D, Jacob J, Cafiso DS, McLaughlin S. 1997. Kinetics of interaction of the myristoylated alanine-rich C kinase substrate, membranes, and calmodulin. J Biol Chem 272:27167–27177. doi: 10.1074/jbc.272.43.27167. [DOI] [PubMed] [Google Scholar]
- 27.Arbuzova A, Wang L, Wang J, Hangyas-Mihalyne G, Murray D, Honig B, McLaughlin S. 2000. Membrane binding of peptides containing both basic and aromatic residues. Experimental studies with peptides corresponding to the scaffolding region of caveolin and the effector region of MARCKS. Biochemistry 39:10330–10339. doi: 10.1021/bi001039j. [DOI] [PubMed] [Google Scholar]
- 28.Nomikos M, Mulgrew-Nesbitt A, Pallavi P, Mihalyne G, Zaitseva I, Swann K, Lai FA, Murray D, McLaughlin S. 2007. Binding of phosphoinositide-specific phospholipase C-zeta (PLC-zeta) to phospholipid membranes: potential role of an unstructured cluster of basic residues. J Biol Chem 282:16644–16653. doi: 10.1074/jbc.M701072200. [DOI] [PubMed] [Google Scholar]
- 29.McLaughlin S, Smith SO, Hayman MJ, Murray D. 2005. An electrostatic engine model for autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB) family. J Gen Physiol 126:41–53. doi: 10.1085/jgp.200509274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLaughlin S. 1989. The electrostatic properties of membranes. Annu Rev Biophys Biophys Chem 18:113–136. doi: 10.1146/annurev.bb.18.060189.000553. [DOI] [PubMed] [Google Scholar]
- 31.White SH, Wimley WC. 1999. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct 28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 32.Haynes BF, Fleming J, St Clair EW, Katinger H, Stiegler G, Kunert R, Robinson J, Scearce RM, Plonk K, Staats HF, Ortel TL, Liao HX, Alam SM. 2005. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 308:1906–1908. doi: 10.1126/science.1111781. [DOI] [PubMed] [Google Scholar]
- 33.Nelson JD, Brunel FM, Jensen R, Crooks ET, Cardoso RM, Wang M, Hessell A, Wilson IA, Binley JM, Dawson PE, Burton DR, Zwick MB. 2007. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J Virol 81:4033–4043. doi: 10.1128/JVI.02588-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simek MD, Rida W, Priddy FH, Pung P, Carrow E, Laufer DS, Lehrman JK, Boaz M, Tarragona-Fiol T, Miiro G, Birungi J, Pozniak A, McPhee DA, Manigart O, Karita E, Inwoley A, Jaoko W, Dehovitz J, Bekker LG, Pitisuttithum P, Paris R, Walker LM, Poignard P, Wrin T, Fast PE, Burton DR, Koff WC. 2009. Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol 83:7337–7348. doi: 10.1128/JVI.00110-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leaman DP, Zwick MB. 2013. Increased functional stability and homogeneity of viral envelope spikes through directed evolution. PLoS Pathog 9:e1003184. doi: 10.1371/journal.ppat.1003184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haynes BF, Mascola JR. 2017. The quest for an antibody-based HIV vaccine. Immunol Rev 275:5–10. doi: 10.1111/imr.12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwong PD. 2017. What are the most powerful immunogen design vaccine strategies? A structural biologist's perspective. Cold Spring Harb Perspect Biol 9:a029470. doi: 10.1101/cshperspect.a029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burton DR. 3 February 2017. What are the most powerful immunogen design vaccine strategies? Reverse vaccinology 2.0 shows great promise. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein F, Mouquet H, Dosenovic P, Scheid JF, Scharf L, Nussenzweig MC. 2013. Antibodies in HIV-1 vaccine development and therapy. Science 341:1199–1204. doi: 10.1126/science.1241144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.West AP Jr, Scharf L, Scheid JF, Klein F, Bjorkman PJ, Nussenzweig MC. 2014. Structural insights on the role of antibodies in HIV-1 vaccine and therapy. Cell 156:633–648. doi: 10.1016/j.cell.2014.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barouch DH, Whitney JB, Moldt B, Klein F, Oliveira TY, Liu J, Stephenson KE, Chang HW, Shekhar K, Gupta S, Nkolola JP, Seaman MS, Smith KM, Borducchi EN, Cabral C, Smith JY, Blackmore S, Sanisetty S, Perry JR, Beck M, Lewis MG, Rinaldi W, Chakraborty AK, Poignard P, Nussenzweig MC, Burton DR. 2013. Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature 503:224–228. doi: 10.1038/nature12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halper-Stromberg A, Lu CL, Klein F, Horwitz JA, Bournazos S, Nogueira L, Eisenreich TR, Liu C, Gazumyan A, Schaefer U, Furze RC, Seaman MS, Prinjha R, Tarakhovsky A, Ravetch JV, Nussenzweig MC. 2014. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell 158:989–999. doi: 10.1016/j.cell.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montefiori DC. 2016. Bispecific antibodies against HIV. Cell 165:1563–1564. doi: 10.1016/j.cell.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Huang Y, Yu J, Lanzi A, Yao X, Andrews CD, Tsai L, Gajjar MR, Sun M, Seaman MS, Padte NN, Ho DD. 2016. Engineered bispecific antibodies with exquisite HIV-1-neutralizing activity. Cell 165:1621–1631. doi: 10.1016/j.cell.2016.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verkoczy L, Kelsoe G, Haynes BF. 2014. HIV-1 envelope gp41 broadly neutralizing antibodies: hurdles for vaccine development. PLoS Pathog 10:e1004073. doi: 10.1371/journal.ppat.1004073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finton KA, Larimore K, Larman HB, Friend D, Correnti C, Rupert PB, Elledge SJ, Greenberg PD, Strong RK. 2013. Autoreactivity and exceptional CDR plasticity (but not unusual polyspecificity) hinder elicitation of the anti-HIV antibody 4E10. PLoS Pathog 9:e1003639. doi: 10.1371/journal.ppat.1003639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alam SM, Morelli M, Dennison SM, Liao HX, Zhang R, Xia SM, Rits-Volloch S, Sun L, Harrison SC, Haynes BF, Chen B. 2009. Role of HIV membrane in neutralization by two broadly neutralizing antibodies. Proc Natl Acad Sci U S A 106:20234–20239. doi: 10.1073/pnas.0908713106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haynes BF, Nicely NI, Alam SM. 2010. HIV-1 autoreactive antibodies: are they good or bad for HIV-1 prevention? Nat Struct Mol Biol 17:543–545. doi: 10.1038/nsmb0510-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen J, Frey G, Peng H, Rits-Volloch S, Garrity J, Seaman MS, Chen B. 2014. Mechanism of HIV-1 neutralization by antibodies targeting a membrane-proximal region of gp41. J Virol 88:1249–1258. doi: 10.1128/JVI.02664-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawai T, Caaveiro JMM, Abe R, Katagiri T, Tsumoto K. 2011. Catalytic activity of MsbA reconstituted in nanodisc particles is modulated by remote interactions with the bilayer. FEBS Lett 585:3533–3537. doi: 10.1016/j.febslet.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 51.Shepard LA, Heuck AP, Hamman BD, Rossjohn J, Parker MW, Ryan KR, Johnson AE, Tweten RK. 1998. Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an alpha-helical to beta-sheet transition identified by fluorescence spectroscopy. Biochemistry 37:14563–14574. doi: 10.1021/bi981452f. [DOI] [PubMed] [Google Scholar]
- 52.Heuck AP, Hotze EM, Tweten RK, Johnson AE. 2000. Mechanism of membrane insertion of a multimeric beta-barrel protein: perfringolysin O creates a pore using ordered and coupled conformational changes. Mol Cell 6:1233–1242. doi: 10.1016/S1097-2765(00)00119-2. [DOI] [PubMed] [Google Scholar]
- 53.Hope MJ, Bally MB, Webb G, Cullis PR. 1985. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim Biophys Acta 812:55–65. doi: 10.1016/0005-2736(85)90521-8. [DOI] [PubMed] [Google Scholar]
- 54.Mattila J-P, Shnyrova AV, Sundborger AC, Hortelano ER, Fuhrmans M, Neumann S, Muller M, Hinshaw JE, Schmid SL, Frolov VA. 2015. A hemi-fission intermediate links two mechanistically distinct stages of membrane fission. Nature 524:109–113. doi: 10.1038/nature14509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carravilla P, Nieva JL, Goñi FM, Requejo-Isidro J, Huarte N. 2015. Two-photon Laurdan studies of the ternary lipid mixture DOPC:SM:cholesterol reveal a single liquid phase at sphingomyelin:cholesterol ratios lower than 1. Langmuir 31:2808–2817. doi: 10.1021/la504251u. [DOI] [PubMed] [Google Scholar]
- 56.Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW. 2003. Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem 278:48935–48941. doi: 10.1074/jbc.M306289200. [DOI] [PubMed] [Google Scholar]
- 57.Huarte N, Carravilla P, Cruz A, Lorizate M, Nieto-Garai JA, Krausslich HG, Perez-Gil J, Requejo-Isidro J, Nieva JL. 2016. Functional organization of the HIV lipid envelope. Sci Rep 6:34190. doi: 10.1038/srep34190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.White SH, Wimley WC, Ladokhin AS, Hristova K. 1998. Protein folding in membranes: determining energetics of peptide-bilayer interactions. Methods Enzymol 295:62–87. doi: 10.1016/S0076-6879(98)95035-2. [DOI] [PubMed] [Google Scholar]
- 59.Rand RP, Parsegian VA. 1989. Hydration forces between phospholipid bilayers. Biochim Biophys Acta 988:351–376. doi: 10.1016/0304-4157(89)90010-5. [DOI] [Google Scholar]
- 60.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 61.Leaman DP, Kinkead H, Zwick MB. 2010. In-solution virus capture assay helps deconstruct heterogeneous antibody recognition of human immunodeficiency virus type 1. J Virol 84:3382–3395. doi: 10.1128/JVI.02363-09. [DOI] [PMC free article] [PubMed] [Google Scholar]