

ABSTRACT
Kaposi's sarcoma (KS) tumors are derived from endothelial cells and express Kaposi's sarcoma-associated herpesvirus (KSHV) microRNAs (miRNAs). Although miRNA targets have been identified in B cell lymphoma-derived cells and epithelial cells, little has been done to characterize the KSHV miRNA targetome in endothelial cells. A recent innovation in the identification of miRNA targetomes, cross-linking, ligation, and sequencing of hybrids (CLASH), unambiguously identifies miRNAs and their targets by ligating the two species while both species are still bound within the RNA-induced silencing complex (RISC). We developed a streamlined quick CLASH (qCLASH) protocol that requires a lower cell input than the original method and therefore has the potential to be used on patient biopsy samples. Additionally, we developed a fast-growing, KSHV-negative endothelial cell line derived from telomerase-immortalized vein endothelial long-term culture (TIVE-LTC) cells. qCLASH was performed on uninfected cells and cells infected with either wild-type KSHV or a mutant virus lacking miR-K12-11/11*. More than 1,400 cellular targets of KSHV miRNAs were identified. Many of the targets identified by qCLASH lacked a canonical seed sequence match. Additionally, most target regions in mRNAs originated from the coding DNA sequence (CDS) rather than the 3′ untranslated region (UTR). This set of genes includes some that were previously identified in B cells and some new genes that warrant further study. Pathway analysis of endothelial cell targets showed enrichment in cell cycle control, apoptosis, and glycolysis pathways, among others. Characterization of these new targets and the functional consequences of their repression will be important in furthering our understanding of the role of KSHV miRNAs in oncogenesis.
IMPORTANCE KS lesions consist of endothelial cells latently infected with KSHV. Cells that make up these lesions express KSHV miRNAs. Identification of the targets of KSHV miRNAs will help us understand their role in viral oncogenesis. The cross-linking and sequencing of hybrids (CLASH) protocol is a method for unambiguously identifying miRNA targetomes. We developed a streamlined version of CLASH, called quick CLASH (qCLASH). qCLASH requires a lower initial input of cells than for its parent protocol. Additionally, a new fast-growing KSHV-negative endothelial cell line, named TIVE-EX-LTC cells, was established. qCLASH was performed on TIVE-EX-LTC cells latently infected with wild-type (WT) KSHV or a mutant virus lacking miR-K12-11/11*. A number of novel targets of KSHV miRNAs were identified, including targets of miR-K12-11, the ortholog of the cellular oncogenic miRNA (oncomiR) miR-155. Many of the miRNA targets were involved in processes related to oncogenesis, such as glycolysis, apoptosis, and cell cycle control.
KEYWORDS: CLASH, CLIP, HHV-8, Kaposi's sarcoma-associated herpesvirus, miRNA, microRNA, qCLASH, ribonomics, targetome
INTRODUCTION
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV) is the etiological agent of KS, primary effusion lymphoma, and a subset of multicentric Castleman's diseases (1–3). KS is a disease of global significance (4, 5), yet much work remains to be done to better understand the mechanisms by which KSHV infection leads to cancer. KS tumors are composed of spindle cells that are latently infected with the virus (6, 7). During latency, KSHV expresses only a small subset of genes, which are mainly confined to the KSHV latency-associated region (KLAR) of the genome (8, 9). Located in this region are 12 microRNA (miRNA) genes that give rise to 25 mature miRNAs (10–12). miRNAs are 19- to 22-nucleotide (nt) RNAs that perform important regulatory functions by exerting posttranscriptional control over gene expression. They bind to imperfectly complementary sequences of target mRNAs, often found within the 3′ untranslated region (UTR), thereby preventing translation and mediating the eventual degradation of certain transcripts (reviewed in reference 13). There is already evidence that KSHV miRNAs contribute to oncogenesis. One of the earliest targets of KSHV miRNAs identified was THBS1, a tumor suppressor and antiangiogenic factor (14). miR-K12-5 is known to target BCLAF1, which has been shown to promote apoptosis (15). In addition, one of the KSHV miRNAs, miR-K12-11, is an ortholog of the cellular oncomiR miR-155 (16, 17). The exogenous expression of miR-K12-11 or miR-155 in hematopoietic progenitors leads to the expansion of splenic B cells in NOD/LtSz-scid IL2Rγ(null) mice (18). Most of the work aimed at identifying KSHV miRNA targets has been performed in B cells, yet KS tumors are composed of cells principally of endothelial origin (19). KSHV miRNA expression has been shown to vary widely between different cell types (20); thus, there is a need to determine which mRNAs are targeted by the virus miRNAs in endothelial cells. Doing so will help us to better understand the process by which KSHV infection leads to KS.
Studies to determine which mRNAs are targeted by miRNAs on a genome-wide scale were pioneered by Chi et al. with the development of high-throughput sequencing, cross-linking, and immunoprecipitation (HITS-CLIP) (21). Put simply, protein and nucleic acid are cross-linked in living cells, the RNA-binding protein Argonaute (Ago) is immunoprecipitated, and the bound RNA is isolated, reverse transcribed, and sequenced. This yields two data sets: Ago-bound miRNAs and Ago-bound mRNAs. mRNAs and miRNAs must then be assigned to each other bioinformatically based on whether the mRNA contains a perfect or near-perfect complement to the seed sequence (nucleotides 2 to 8) of the miRNA (21). Although miRNAs and mRNAs may interact in this way much of the time, there is also evidence that noncanonical base pairing is significant as well (reviewed in reference 22). These nonstandard interactions are not easily detected by HITS-CLIP or the related photoactivatable ribonucleoside cross-linking and immunoprecipitation (PAR-CLIP) method (23). Recently, Helwak et al. developed cross-linking and sequencing of hybrids (CLASH), a method for the identification of miRNA targets which builds on the HITS-CLIP approach. CLASH adds an RNA ligation step prior to the removal of the Ago protein. Separate RNAs (miRNA and the mRNA target), both of which are bound to the same Ago protein, are joined to become a single hybrid RNA. This removes the ambiguity from miRNA target discovery (24, 25).
While CLASH is an improvement upon HITS-CLIP, both methods contain several postimmunoprecipitation cleanup steps that result in large losses of RNA and as a result require large numbers of cells as the input. However, in CLASH, the hybrids are formed prior to the cleanup steps (24). We reasoned that, given current increased sequencing capabilities, it was not strictly necessary to separate extraneous RNAs from the hybrids; they could all be sequenced, and the nonhybrids could simply be ignored. In keeping with this line of thinking, we have developed a shortened CLASH protocol that omits the steps resulting in the largest losses of RNA. We have named the new variant protocol quick CLASH, or qCLASH, as it takes significantly less time than the original method. It can also be completed by using fewer cells as the input, making it preferable for cell types that are difficult to grow in large quantities and for clinical samples. We successfully used qCLASH to identify targets of KSHV miRNAs in endothelial cells. Almost 1,500 genes were identified as high-confidence targets of viral miRNAs, some of which were previously shown to be targeted by KSHV miRNAs in B cells. Additionally, we identified 54 high-confidence targets of the viral miRNA miR-K12-11. A number of the target genes were found to be components of several cancer-related pathways, including cell cycle control, angiogenesis, and glycolysis. These results provide a unique resource, broaden our understanding of the roles played by KSHV miRNAs in endothelial cells, and point to a number of novel targets and pathways that can be studied more extensively in the future.
RESULTS
qCLASH.
The interactions between KSHV miRNAs and cellular mRNAs were investigated by performing qCLASH on cells latently infected with KSHV, with 5 × 107 endothelial cells per sample. The endothelial cells used were derived from telomerase-immortalized human umbilical vein endothelial long-term culture (TIVE-LTC) cells. TIVE-LTC cells are KSHV positive and relatively fast growing. This makes them easier to work with than primary endothelial cells or their parent TIVE cells, which are slow growing and dependent on exogenous growth factors for survival. At very high passage numbers, TIVE-LTC cells begin to lose the KSHV genome. We performed single-cell dilutions on high-passage-number TIVE-LTC cells in order to isolate KSHV-negative clones. A single clone was selected for all future experiments. We named the new cells, which grow quickly and can be reinfected with KSHV, TIVE-EX-LTC cells.
The TIVE-EX-LTC cells used for qCLASH analysis were either uninfected, infected with wild-type (WT) KSHV, or infected with a KSHV miR-K12-11 knockout virus, with three biological replicates for each treatment. The basic procedure for qCLASH (Fig. 1A) was derived from the one reported previously by Haecker et al., with the important addition of intermolecular ligation as described previously by Helwak and Tollervy (20, 25). Cell lysates were thawed, treated with DNase, treated with a low concentration of RNase T1, and then incubated with beads coated in an Ago-specific antibody. This made it possible to pull down the endogenous protein rather than overexpressed tagged Ago. After immunoprecipitation, the samples were again treated with RNase T1. Two RNase digestions were performed in order to remove as much non-Ago-bound RNA from the samples as possible. The samples were 5′ phosphorylated with T4 polynucleotide kinase (PNK) and then incubated with T4 RNA ligase. Intermolecular ligation was performed in a large volume as described previously by Moore et al. (26). This was done to ensure that Ago-RNA complexes were well separated from each other and from any remaining extraneous RNAs in solution, making it more likely that hybrids would be formed only by RNAs bound to the same Ago. After intermolecular ligation, the samples were treated with alkaline phosphatase before ligation to the 3′ adapter. At this point, the qCLASH protocol diverges sharply from the original CLASH protocol. Normal CLASH calls for radiolabeling, SDS-PAGE, and the transfer of Ago-RNA complexes to nitrocellulose, after which labeled RNAs running at a molecular mass of >110 kDa are cut and eluted from the membrane. Rather than performing these steps, which result in the loss of large amounts of RNA, we proceeded directly to proteinase K digestion of Ago, phosphorylation, and 5′ adapter ligation. From there, reverse transcription (RT) and PCR were performed, followed by Illumina sequencing (Fig. 1A).
FIG 1.
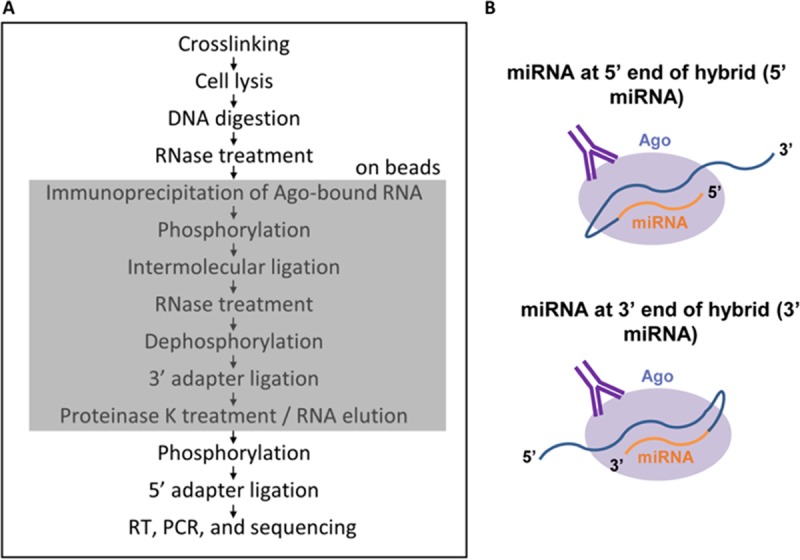
(A) Schematic of the qCLASH method. The portion of the method that takes place on beads is indicated by the shaded box. (B) Diagram of the two ways in which miRNAs and mRNAs can join during intermolecular ligation.
Hybrid library characteristics.
The cDNA libraries were sequenced on a HiSeq 2500 instrument with a read length of 100 nt. There were three libraries per flow cell lane, and each library yielded between 10 million and 20 million reads. Sequencing reads were preprocessed by using Trimmomatic (27) and then analyzed with Hyb (28). Approximately 0.3% to 1% of unique, filtered reads were classified as hybrids (Table 1). On average, approximately 23% of hybrids consisted of miRNA and mRNA. The remaining 77% largely consisted of mRNA-mRNA, miRNA-miRNA, or rRNA hybrids. In approximately 95% of the miRNA-mRNA hybrids, the miRNA was located at the 5′ end of the hybrid (Fig. 1B). This is consistent with what was observed previously by others (24, 26). Approximately 6% of miRNA-mRNA hybrids consisted of a KSHV miRNA and a cellular mRNA in WT-infected cells (Table 1). Even though only a minute percentage of total reads were KSHV miRNA-cellular mRNA hybrids, this still resulted in a range of approximately 6,000 to 12,000 such hybrids per WT biological replicate and 2,200 to 4,000 hybrids per ΔmiR-K12-11 biological replicate. There were on average 10 KSHV hybrids identified in each biological replicate performed on uninfected cells. This indicates that the false-positive discovery rate of Hyb is very low. None of the hybrids found in uninfected cells occurred in more than one biological replicate. Moreover, only one hybrid containing miR-K12-11 was identified across all biological replicates of ΔmiR-K12-11-infected endothelial cells.
TABLE 1.
Characteristics of qCLASH hybrids
| Expta | % of reads that are hybrids | % of hybrids that are miRNA-mRNA | % of miRNA-mRNA hybrids that have 5′ miRNA | % of miRNA-mRNA hybrids that have 3′ miRNA | % of miRNA-mRNA hybrids that have cellular miRNA | % of miRNA-mRNA hybrids that have KSHV miRNA |
|---|---|---|---|---|---|---|
| WT BR1 | 1.02 | 31.13 | 95.65 | 4.35 | 94.84 | 5.16 |
| WT BR2 | 0.84 | 35.88 | 95.59 | 4.41 | 93.92 | 6.08 |
| WT BR3 | 0.57 | 26.77 | 95.64 | 4.36 | 94.26 | 5.74 |
| ΔmiR-K12-11 BR1 | 0.58 | 23.69 | 95.15 | 4.85 | 97.68 | 2.32 |
| ΔmiR-K12-11 BR2 | 0.57 | 19.58 | 94.78 | 5.22 | 97.88 | 2.12 |
| ΔmiR-K12-11 BR3 | 0.53 | 19.78 | 95.42 | 4.58 | 96.78 | 3.22 |
| UI BR1 | 0.44 | 18.34 | 94.35 | 5.65 | 99.99 | 0.01 |
| UI BR2 | 0.45 | 17.40 | 94.82 | 5.18 | 100.00 | 0.00 |
| UI BR3 | 0.32 | 13.91 | 95.11 | 4.89 | 99.99 | 0.01 |
BR1, biological replicate 1; UI, uninfected.
The frequencies of miRNA-mRNA hybrids involving specific KSHV miRNAs are shown in Fig. 2 for cells infected with WT KSHV and with KSHV ΔmiR-K12-11 and are expressed as the number of hybrids obtained from each biological replicate. The five KSHV miRNAs most commonly found in WT-infected cell hybrids were, in descending order, miR-K12-3, -K12-4-3p, -K12-12*, -K12-10a, and -K12-6-3p (Fig. 2A). The same miRNAs made up the top five miRNAs in ΔmiR-K12-11-infected cells (Fig. 2B). Three of these miRNAs, miR-K12-3, -K12-10a, and -K12-4-3p, were previously found to be expressed at high levels in two B cell lymphoma lines, BCBL-1 and BC-3, using HITS-CLIP (20). The KSHV miRNAs were present in hybrids at similar frequencies relative to each other in ΔmiR-K12-11-infected cells as in WT-infected cells, but the number of viral hybrids was lower overall in ΔmiR-K12-11-infected cells. This is consistent with our previous observation that the expression levels of all KSHV miRNAs are lower in cells infected with this knockout virus (29).
FIG 2.

KSHV miRNAs are found in hybrids at different frequencies. The number of hybrids containing each of the 25 mature KSHV miRNAs was quantified for wild-type-infected (A) and ΔmiR-K12-11-infected (B) TIVE-EX-LTC cells. BR1, biological replicate 1.
Region of hybrid mRNA origin.
Next, we investigated which region of the mRNAs was targeted by the viral and cellular miRNAs. First, we considered all the miRNAs present in WT KSHV-infected cells, in ΔmiR-K12-11-infected cells, and in uninfected cells (Fig. 3A). When the mRNA portion of each hybrid was mapped to an ENSEMBL transcript database, more than 50% of hybrid mRNAs mapped to the coding DNA sequence (CDS) of their respective transcripts in each case. This is similar to what was noted previously by others based on CLASH data (24, 26). Targeting was assessed for all KSHV miRNAs pooled from WT- and ΔmiR-K12-11-infected cells and for all cellular miRNAs pooled from all three samples (Fig. 3B). mRNAs from KSHV miRNA-cellular mRNA hybrids had a slightly greater likelihood of mapping to the CDS than did those from cellular miRNA-cellular mRNA hybrids. Of the KSHV hybrids, 27.7% contained an mRNA that mapped to a 3′ UTR, and 65% contained an mRNA that mapped to a CDS. These figures were 33.6% and 61.2%, respectively, for cellular hybrids (Fig. 3B). It is unclear why KSHV miRNAs would have a greater propensity than cellular miRNAs to target the CDS of mRNAs as opposed to the 3′ UTR. Nevertheless, this difference is statistically significant.
FIG 3.

A majority of miRNA target sites are located within the CDS. The transcript region from which each hybrid mRNA fragment originated was determined. The regions were categorized as 5′ UTR, 5′ UTR-CDS, CDS, CDS-3′ UTR, or 3′ UTR. Hybrid mRNAs for which the transcript was not annotated in the database were omitted. (A) Comparison of transcript regions from wild-type-infected (WT), ΔmiR-K12-11-infected (Δ11), and uninfected (UI) TIVE-EX-LTC cells. (B) Comparison of hybrids containing KSHV miRNAs from either WT- or ΔmiR-K12-11-infected cells against hybrids containing cellular miRNAs from WT-infected cells, ΔmiR-K12-11-infected cells, and uninfected cells. The total number of hybrids is indicated above each bar.
Seed pairing characteristics.
We plotted the frequency with which each nucleotide of each miRNA was base paired with the mRNA portion of the hybrid (Fig. 4). This yielded a broad picture of intermolecular base pairing within the data sets. Plots were generated for cellular and viral miRNAs, with 5′ and 3′ hybrids (miRNAs at the 5′ and 3′ ends of the hybrids, respectively) combined and for 5′ and 3′ hybrids alone (Fig. 4A to F). In general, nucleotides 2 to 8, the canonical seed region, were bound at relatively high frequencies. This was true for both viral and cellular miRNAs, although canonical seed pairing was more common in cellular hybrids relative to 3′ binding (Fig. 4A) than it was for KSHV hybrids (Fig. 4D). Nevertheless, both cellular and viral miRNAs showed a high frequency of base pairing toward the 3′ end, particularly between nucleotides 11 to 13 and 16 to 18. An appreciable number of 3′ interactions have been observed in other, similar experiments (24, 26, 30). Interestingly, when 5′ and 3′ hybrids were compared, a greater proportion of hybrids with the miRNA 3′ to the mRNA had noncanonical binding (Fig. 4B, C, E, and F). This agrees with what is thought to occur at the molecular level. If the extreme 5′ end of the miRNA is bound to the mRNA, steric hindrance may prevent it from joining with the 3′ tail of the mRNA. If the 5′ end is unbound, however, this could provide more flexibility of movement for ligation to proceed.
FIG 4.

miRNAs exhibit binding both in the seed region and at the 3′ end. Based on the Vienna diagrams generated through Hyb, the status of each nucleotide (bound or unbound) along the length of the miRNA portion of each hybrid was determined. (A) All cellular miRNAs. (B) All cellular miRNAs located at the 5′ end of the hybrid. (C) All cellular miRNAs located at the 3′ end of the hybrid. (D) All KSHV miRNAs. Only KSHV hybrids from WT-infected TIVE-EX-LTC cells were included in this analysis. (E) All KSHV miRNAs located at the 5′ end of the hybrid. (F) All KSHV miRNAs located at the 3′ end of the hybrid. (G) KSHV miR-K12-1. (H) KSHV miR-K12-3. (I) KSHV miR-K12-6-5p.
When the KSHV miRNAs were considered separately, distinct patterns of seed or nonseed base pairing became evident. Plots are shown for miR-K12-1 (Fig. 4G), miR-K12-3 (Fig. 4H), and miR-K12-6-5p (Fig. 4I). For miR-K12-1, a clear M-shaped plot was generated. However, miR-K12-3 showed a distinct propensity for 3′ interactions to the exclusion of most 5′ binding. This is similar to what was reported previously by Grosswendt et al. when they reanalyzed existing CLIP data sets with KSHV-infected cells in order to look for spontaneously formed hybrids that occur at a very low frequency (30). miR-K12-6-5p, in contrast, had mainly canonical seed interactions. Binding characteristics for all individual viral miRNAs for which hybrids were detected are depicted in Fig. 5.
FIG 5.

miRNAs exhibit binding both in the seed region and at the 3′ end. Based on the Vienna diagrams generated through Hyb, the status of each nucleotide (bound or unbound) along the length of the miRNA portion of each hybrid was determined. Only KSHV hybrids from WT-infected TIVE-EX-LTC cells were included in this analysis. KSHV miRNAs for which there were fewer than 20 hybrids total are omitted. (A) miR-K12-2; (B) miR-K12-4-3p; (C) miR-K12-4-5p; (D) miR-K12-5; (E) miR-K12-6-3p; (F) miR-K12-7; (G) miR-K12-8; (H) miR-K12-8*; (I) miR-K12-9*; (J) miR-K12-10a; (K) miR-K12-10b; (L) miR-K12-11; (M) miR-K12-12; (N) miR-K12-12*.
In order to look at miRNA-mRNA interactions in a different way, hybrids were placed into groups based on how their miRNA seed region is predicted to bind to the mRNA portion of the hybrid. The categories were as follows: pairing at nt 2 to 8 with no mismatches, pairing at nt 2 to 7 with no mismatches, pairing at nt 2 to 8 with one mismatch, pairing at nt 2 to 8 with two mismatches, and “other,” for interactions that did not fall into any of the former categories (Fig. 6). The seed pairing characteristics of the hybrid miRNAs were considered in different groupings. First, hybrids from WT-infected cells, hybrids from ΔmiR-K12-11-infected cells, and hybrids from uninfected cells were compared side by side (Fig. 6A). In order to understand how KSHV and cellular hybrids differ, all KSHV hybrids from WT- and ΔmiR-K12-11-infected cells were compared with all cellular hybrids from WT- and ΔmiR-K12-11-infected cells as well as uninfected cells (Fig. 6B). Finally, all hybrids with the miRNA located at the 5′ end were compared with all hybrids with the miRNA located at the 3′ end (Fig. 6C). Interestingly, a large proportion of miRNAs fell into the “other” category; i.e., they showed noncanonical binding. This was true for both uninfected and infected cells. Moreover, when hybrids containing viral and cellular miRNAs were considered separately, viral miRNAs were significantly more likely than cellular miRNAs to lack a canonical seed match (Fig. 6B). The proportion of cellular miRNAs in the “other” category was 46%, compared to 56% for viral miRNAs. Additionally, hybrids with the miRNA at the 5′ end fell into the “other” category 44% of the time, while 3′ miRNA hybrids did so 68% of the time. The prevalence of noncanonical seed pairing in our hybrids agrees with recent findings of others indicating that canonical seed matches are not necessarily the only determinants of which genes a given miRNA will target (24, 26).
FIG 6.

Nearly half of the miRNAs do not have any recognizable seed pairing. Hybrids were sorted into categories based on the type of seed pairing determined for the miRNA portion of each hybrid. These categories were no mismatches (mm) for nucleotides 2 to 8, no mismatches for nucleotides 2 to 7, one mismatch for nucleotides 2 to 8, 2 mismatches for nucleotides 2 to 8, and other. (A) Comparison of hybrids (viral and cellular) from WT-infected, ΔmiR-K12-11-infected, and uninfected TIVE-EX-LTC cells. (B) Comparison of all hybrids containing KSHV miRNAs from WT- and ΔmiR-K12-11-infected TIVE-EX-LTC cells and all hybrids containing cellular miRNAs from WT- and ΔmiR-K12-11-infected as well as uninfected TIVE-EX-LTC cells. (C) Comparison of all hybrids (viral and cellular) with the miRNA at the 5′ end with all hybrids with the miRNA at the 3′ end.
In light of this information, we chose to examine more closely how the 3′ ends of the miRNAs, which we classified as anything downstream of nucleotide 8, interacted with the mRNA ends of the hybrids. We wanted to ask if compensatory base pairing toward the 3′ end of the miRNA was more prevalent in cases where a traditional seed sequence was absent. Interactions involving the 3′ end of the miRNA were considered “absent” if they had no paired bases from nt 8 to the 3′ end of the miRNA, “weak” if they had between 1 and 4 bound nucleotides, “moderate” if they had between 5 and 8 bound nucleotides, and “strong” if they had more than 8 bound nucleotides. The proportions of the four classes of pairing at the 3′ end of the miRNA are shown in Fig. 7 for each of the seed pairing classes. Viral (Fig. 7A) and cellular (Fig. 7B) hybrid miRNAs showed the same basic binding pattern. miRNAs with perfect seed pairing at nt 2 to 8 or nt 2 to 7 had proportionally more weak and absent 3′ interactions than the other categories of seed pairing. Conversely, miRNAs falling into the “other” category had a greater propensity for strong 3′ interactions and fewer interactions classified as being weak or absent. It would appear that the lack of canonical binding to the seed sequence is compensated for with a larger amount of base pairing at the 3′ end of the miRNA.
FIG 7.

miRNAs with no recognizable seed pairing have stronger interactions at the 3′ end. Hybrids in each of the seed pairing categories were further sorted based on the number of nucleotides bound in the miRNA portion of the hybrid downstream from the seed sequence. These categories were strong (>8 bound nucleotides), moderate (5 to 8 bound nucleotides), weak (1 to 4 bound nucleotides), and absent (0 bound nucleotides) interactions. (A) KSHV hybrids from WT- and ΔmiR-K12-11-infected cells; (B) cellular hybrids from all samples.
KSHV miRNA targets.
We have chosen to define “target” as any gene for which a portion of the mRNA transcript was found in a hybrid with a KSHV miRNA. In total, there were 27,022 KSHV miRNA-cellular mRNA hybrids across all three biological replicates in WT-infected endothelial cells, representing 6,497 unique genes. Of these genes, 3,324 were found in at least two of three biological replicates, while 1,433 genes were found in three of three biological replicates (see Table S2 in the supplemental material). Since it was impractical to follow up on all of these targets, we chose to focus specifically on targets of miR-K12-11. We selected genes that were present in hybrids in at least two of three biological replicates for WT-infected cells and absent in at least two of three biological replicates for ΔmiR-K12-11-infected cells. This yielded 54 genes that we considered to be high-confidence miR-K12-11 targets. One of the genes on our list, STIP1, has already been validated as a target of miR-K12-11 in B cells (20). Two more genes, CTNND1 and PKN2, were also previously confirmed to be targeted by a KSHV miRNA, although that miRNA was miR-K12-10 (31). We chose to verify 30 of the 54 genes by RT-quantitative PCR (qPCR), along with 10 others present in at least one biological replicate for the WT and absent from at least one biological replicate for ΔmiR-K12-11. The genes selected represented hybrids with a variety of characteristics. In order to validate our targets, ΔmiR-K12-11-infected TIVE-EX-LTC cells were transfected with either an miR-K12-11 mimic or a control mimic. After RNA extraction, cDNAs were prepared, and the transcript levels of the selected targets were determined by RT-qPCR. Of the 40 genes, 26 had significantly lower expression levels in the presence of the miR-K12-11 mimic, while 14 showed no significant change in expression (Fig. 8A). Genes that were not expected to be targets of miR-K12-11 did not show reduced expression levels (data not shown). The genes that were positive for repression had hybrids with characteristics distinct from those of hybrids from genes that were not repressed. Unsurprisingly, positive genes were more likely to be represented by hybrids in a larger number of biological replicates. They were also more likely than negative genes to have hybrids with mRNAs originating from the 3′ UTR (Fig. 8B). Nevertheless, all genes analyzed still showed a high proportion of hybrids in which the mRNA target originated from the CDS. Hybrids from genes that were positive for repression had a greater likelihood of having a perfect (nt 2 to 8) seed match, while those that were negative showed more noncanonical seed pairing (Fig. 8C). The strengths of pairing at the 3′ end of hybrid miRNAs were similar for both subsets (Fig. 8D). While the presence of seed sequences and targeting of the 3′ UTR had predictive value toward suppression, we note that we also identified CDS-targeted genes that were efficiently suppressed by miR-K12-11 mimic transfection.
FIG 8.

Repression of selected mRNA targets validated by RT-qPCR. TIVE-EX-LTC cells infected with ΔmiR-K12-11 were transfected with either a KSHV miR-K12-11-3p mimic or a nontargeting control mimic. RNA was prepared from the cells, and expression levels of specific mRNA targets of miR-K12-11 discovered by qCLASH were measured by RT-qPCR. (A) Fold changes in expression levels of the indicated mRNAs in miR-K12-11-3p mimic-transfected cells compared to control mimic-transfected cells. Data shown are the averages of results from two biological replicates. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. (B and C) Genes that were positive for repression in the presence of the miR-K12-11-3p mimic were compared to those that were negative for repression. (B) Percentages of hybrids that contain an mRNA fragment originating from the CDS or the 3′ UTR. (C) Percentages of hybrids exhibiting the different types of indicated seed matches (2-8 0 mm, nucleotides 2 to 8 with no mismatches; 2-7 0 mm, nucleotides 2 to 7 with no mismatches; 2-8 1 mm, nucleotides 2 to 8 with 1 mismatch; 2-8 2 mm nucleotides 2 to 8, with 2 mismatches). (D) Comparison of genes that were positive for repression versus those that were negative based on binding strength at the 3′ end of the hybrid miRNA. Strong, >8 bound nucleotides; moderate, 5 to 8 bound nucleotides; weak, 1 to 4 bound nucleotides; absent, 0 bound nucleotides.
Hybrids in B cells.
As mentioned above, a small number of hybrids also forms when regular HITS-CLIP is performed. We ran Hyb on previously reported HITS-CLIP data (20) using two KSHV-infected B cell lymphoma lines in order to search for hybrids formed by endogenous ligases, a phenomenon first observed by Grosswendt et al. (30). On average, 0.01% of reads were identified as hybrids, indicating that the natural formation of hybrids is a vanishingly rare event. Even so, KSHV miRNA hybrids made up a much higher percentage of hybrids overall in B cells than in endothelial cells (see Table S3 in the supplemental material). There were a total of 833 KSHV miRNA-cellular mRNA hybrids in BCBL-1 cells and a total of 3,065 such hybrids in BC-3 cells. These hybrids were analyzed in the same way as for hybrids from endothelial cells. In contrast to hybrids from endothelial cells, it was found that more than 50% of mRNAs from B cell hybrids originated from the 3′ UTR (Fig. 9A and B). This also differs from the percentage of mRNAs from 3′ UTRs in the original HITS-CLIP analysis, which was closer to 30% (20). Another surprising finding was that approximately 90% of B cell hybrids lacked canonical seed pairing (Fig. 9C to E). It is unclear whether this actually represents the reality of miRNA-mRNA interactions in B cells, is a characteristic of hybrids formed by endogenous ligases, or is simply an artifact of having a small sample size. When the hybrids were analyzed for binding toward the 3′ ends of the miRNAs, a pattern similar to that in endothelial cells was observed, with noncanonical seed pairing correlating with strong 3′ interactions (data not shown). The raw numbers used to create data in Fig. 3, 5, 6, and 9 are available in Table S4 in the supplemental material.
FIG 9.

B cell hybrids have unique characteristics. Spontaneously occurring hybrids in B cells from previously reported CLIP data were analyzed in the same manner as for hybrids identified by qCLASH in TIVE-EX-LTC cells. (A and B) Comparisons of mRNA regions of origin in viral and cellular hybrids from BCBL-1 cells with those of BC-3 cell hybrids (A) and hybrids containing KSHV miRNAs from either BCBL-1 or BC-3 cells with hybrids containing cellular miRNAs from either cell type (B). Hybrid mRNAs for which the transcript was not annotated in the database are omitted. (C to E) Comparisons of seed pairing types in viral and cellular hybrids from BCBL-1 cells with those from BC-3 cell hybrids (C), hybrids containing KSHV miRNAs from either BCBL-1 or BC-3 cells with hybrids containing cellular miRNAs from either cell type (D), and all hybrids in both BCBL-1 and BC-3 cells (viral and cellular) with the miRNA at the 5′ end with all hybrids with the miRNA at the 3′ end (E).
Comparison with targets in B cells.
KSHV miRNA targets previously identified through HITS-CLIP on two types of KSHV-infected B cells, BCBL-1 and BC3 (20), were compared with targets found with CLASH on KSHV-infected endothelial cells. We expected to find at least some overlap between the results of the different experiments. Indeed, when lists of genes identified as targets of KSHV miRNAs in three out of three biological replicates of HITS-CLIP on BCBL-1 or BC3 cells were compared with genes found in three of three biological replicates of qCLASH in WT-infected cells, 223 and 169 targets, respectively, were shared. There were 105 shared targets when all three cell lines were compared simultaneously (see Table S5 in the supplemental material). It would seem that it is important for KSHV to reduce the levels of these particular mRNAs regardless of the cell type infected. Additionally, since these targets were identified in three different cell types in all biological replicates using two different protocols, they are particularly promising for further study.
In addition to the targets discovered previously by Haecker et al., KSHV miRNA targets identified by qCLASH in endothelial cells were also compared with previously reported, validated targets of KSHV miRNAs from a variety of sources. A number of the previously reported targets were also found in our qCLASH data set (Table 2) (15–17, 20, 31–39). It is interesting to note that some of these genes, while targeted in both data sets, were associated with a larger number of KSHV miRNAs in the qCLASH data. These additional miRNAs were most likely missed by other methods because either they do not undergo canonical seed pairing or they target a region of the mRNA other than the 3′ UTR. Nevertheless, we deem it highly likely that the genes listed in Table 2, which were initially identified by a variety of methods in different cell types, are highly significant for KSHV regardless of the cell type infected. When we focused specifically on endothelial cells for comparison to our data, we found encouraging results. For example, in one recent study carried out with human umbilical vein endothelial cells (HUVECs), genes were screened for repressed mRNA and protein levels in the presence of KSHV miRNAs (40). Out of the 13 genes from those experiments found to be most repressed, and therefore most responsive to KSHV miRNAs, 12 were also present in our data.
TABLE 2.
Comparison of KSHV miRNA targets identified by HITS-CLIP or PAR-CLIP in B cells or by proteomics analysis in primary endothelial cells against qCLASH dataa
| Validated target (reference[s]) | Targeting miRNA(s) (reported previously) | Targeting miRNA(s) (qCLASH) |
|---|---|---|
| BACH1 (16, 17) | miR-K12-11 | miR-K12-4-3p, -10a, -12* |
| BCLAF1 (15) | miR-K12-5, -9, -10a, -10b | miR-K12-8*, -12* |
| CASP3 (30) | miR-K12-1, -3, -4-3p | miR-K12-4-3p, -10a, -10b |
| CDKN1A (31) | miR-K12-1 | miR-K12-1, -3, -10b, -12, -12* |
| NFIB (32) | miR-K12-3 | miR-K12-1, -10b |
| NFKBIA (33) | miR-K12-1 | miR-K12-1, -4-3p |
| RBL2 (34) | miR-K12-4-5p | miR-K12-3, -4-3p |
| TNFRSF12A (35) | miR-K12-10a | miR-K12-3, -4-3p, -4-5p, -6-5p, -7, -8*, -9*, -10a, -10b, -12, -12* |
| MYD88 (36) | miR-K12-5 | miR-K12-8 |
| ZFYVE9 (29) | miR-K12-9, -10 | miR-K12-2, -4-3p, -12* |
| SOS1 (29) | miR-K12-10 | miR-K12-7, -11, -12* |
| CTNND1 (29) | miR-K12-10 | miR-K12-3, -4-3p, -8*, -10a, -11, -12* |
| CDKN1B (29) | miR-K12-10 | miR-K12-3, -4-3p, -8*, -11, -12* |
| PKN2 (29) | miR-K12-10 | miR-K12-10a, -11 |
| TPD52 (20, 29) | miR-K12-4, -10 | miR-K12-4-3p, -11 |
| ANXA2 (20) | miR-K12-1, -11 | miR-K12-1, -3, -4-3p, -6-3p, -8, -9*, -10a, -11, -12* |
| CEBPB (20) | miR-K12-11 | miR-K12-9*, -10b, -12 |
| HMGA1 (20) | miR-K12-10, -12 | miR-K12-2, -6-5p, -8, -9*, -10a, -12 |
| IRF2BP2 (20) | miR-K12-11 | miR-K12-4-3p, -12* |
| PTPN11 (20) | miR-K12-11 | miR-K12-3, -4-3p, -10a, -10b, -11, -12* |
| STIP1 (20) | miR-K12-11 | miR-K12-1, -3, -6-3p, -7, -11 |
| TP53INP1 (20) | miR-K12-11 | miR-K12-1, -8 |
| YWHAE (20) | miR-K12-11 | miR-K12-3, 4-3p, -6-3p, -8, -8*, -10b, -11, -12* |
| ADRBK1 (37) | miR-K12-3 | miR-K12-3, -4-3p, -6-5p, -10a |
| HMGCS1 (40) | miR-K12-9*, -11 | miR-4-3p, -11, -12* |
| ROCK2 (40) | miR-K12-4-3p, -10a, -10b | miR-K12-4-3p, -6-3p, -6-5p, -9*, -12* |
| GRB2 (40) | miR-K12-1, -2, 4-3p, -9, -9*, -10a, -11 | miR-K12-2, -3, -4-3p, -8, -9*, -10a, -10b |
The selected targets have all been validated by luciferase reporter assays or comparable experiments.
Pathway and gene enrichment analysis.
The 1,433 genes that appeared in three of three biological replicates for WT-infected cells were further examined in order to determine if there are any major cellular processes that are targeted by KSHV miRNAs. By using Enrichr, KEGG pathways that contain a significant number of our genes were identified (see Table S6 in the supplemental material). Among these pathways were several with particular relevance to cancer and/or the biology of the virus. These pathways were, among others, the vascular endothelial growth factor (VEGF) pathway, apoptosis, cell cycle control (Fig. 10), and glycolysis (Fig. 11). Apoptosis and glycolysis were previously identified in a similar analysis of HITS-CLIP data in B cells.
FIG 10.

Ingenuity Pathway Analysis (IPA) diagram of the cell cycle control pathway. IPA software was used to identify pathways that are enriched in the set of 1,433 genes that were present in three of three biological replicates for WT-infected TIVE-EX-LTC cells. Genes that are targeted by KSHV miRNAs are highlighted in purple. Ovals, transcription regulators; downward-pointing triangles, kinases; upward-pointing triangles, phosphatases; dotted squares, growth factors; circles, other; double-walled shapes, complexes/groups; diamonds, enzymes. TGF-β, transforming growth factor β; HDAC, histone deacetylase.
FIG 11.

Ingenuity Pathway Analysis diagram of the glycolysis pathway. IPA software was used to identify pathways that are enriched in the set of 1,433 genes that were present in three of three biological replicates on WT-infected TIVE-EX-LTC cells. Genes that are targeted by KSHV miRNAs are highlighted in purple. Ovals, transcription regulators; downward-pointing triangles, kinases; upward-pointing triangles, phosphatases; dotted squares, growth factors; circles, other; double-walled shapes, complexes/groups; diamonds, enzymes.
Controls for the cell cycle become suppressed or inactivated in cancer, allowing the unimpeded proliferation of cells. As an oncogenic virus, it is therefore unsurprising that KSHV has the ability to dysregulate the cell cycle. It has been demonstrated that this can occur in multiple ways. For example, latency-associated nuclear antigen (LANA) has the ability to inhibit both tumor protein 53 (p53) and RB transcriptional corepressor 1 (Rb), two important checks on cell cycle progression (41–43). vCyclin, the KSHV homolog of cellular cyclin D2, can also inhibit Rb (44). Viral FLICE-inhibitory protein (vFLIP) prevents the formation of active caspase 8, a proapoptotic protein (45, 46). KSHV miRNAs have also been implicated in cell cycle dysregulation. miR-K12-1 was previously found to target cyclin-dependent kinase inhibitor 1A (CDKN1A) (also called p21), thereby preventing cell cycle arrest (33).
KSHV miRNA targets identified in the cell cycle control pathway are depicted in Fig. 8. We found an interaction between miR-K12-1 and p21. Indeed, we observed that suppressors of cyclins and cyclin-dependent kinases were frequent targets of KSHV miRNAs. These suppressors included glycogen synthase kinase 3 beta (GSK3B), a suppressor of cyclin D1 (47); cyclin-dependent kinase inhibitor 1B (CDKN1B) (also called p27), which blocks the activation of cyclin E-CDK2 and cyclin D-CDK4 complexes (48); and WEE1 G2 checkpoint kinase (WEE1), an inhibitor of mitosis (49). In addition, we found cMYC and SMAD to be targeted by multiple KSHV miRNAs. We also found that miR-K12-3 and miR-K12-8, like LANA and vCyclin, target Rb. Overall, the KSHV miRNA targets identified by qCLASH appear to interact broadly with many components of the cell cycle control machinery (Fig. 10).
The Warburg effect is the preference of cancer cells to produce ATP through glycolysis rather than oxidative phosphorylation. It was shown previously that KSHV induces the Warburg effect in infected cells (50). Indeed, the exogenous expression of the KSHV miRNA cluster by itself in lymphatic endothelial cells was sufficient to induce this effect (51). Consistent with this, our findings show an extensive involvement of KSHV miRNAs at various stages in the glycolysis pathway (Fig. 11). The targets include phosphofructokinase, the inhibition of which confers a growth advantage on cancer cells (52), and phosphopyruvate hydratase (ENO1), which activates monocytes in its soluble form (53).
In summary, pathway analysis of high-confidence KSHV miRNA targets identified by qCLASH confirmed the importance of various cellular pathways for the biology of KSHV. Further experimentation on the impacts of the KSHV miRNAs in the context of specific cellular pathways may be a fruitful line of inquiry in the future.
DISCUSSION
We have successfully established a shortened version of CLASH, named qCLASH, and demonstrated that the resulting libraries have a composition very similar to that of libraries generated by the standard CLASH protocol. There are two major advantages to our approach. First, it can be completed more quickly than the original method, in as few as 4 days. Second, qCLASH requires far fewer cells as the input. To date, it has been successfully performed with as few as 1 × 107 cells (data not shown). This opens up the important possibility of applying this protocol to patient biopsy specimens, which would be an unprecedented opportunity to examine KSHV miRNA targets in vivo.
The establishment of the TIVE-EX-LTC cell line also creates more opportunities in the field of KSHV research. Since the discovery that SLK cells are not actually endothelial cells (54), the relevance of KSHV studies with this cell type has become more limited. TIVE-EX-LTC cells grow quickly, require no exogenous growth factors, and can be readily infected with KSHV. As such, they have the potential to fill the void left by SLK cells.
Using qCLASH on TIVE-EX-LTC cells infected with WT KSHV, we identified a number of high-confidence cellular targets of KSHV miRNAs. There were 1,433 genes that appeared in three of three biological replicates for WT-infected cells. While following up on all of these targets is well beyond the scope of the present study, this information has the potential to inform the work of other researchers. We also performed qCLASH on ΔmiR-K12-11-infected TIVE-EX-LTC cells. As the viral ortholog of the known human oncomiR miR-155, miR-K12-11 is of particular interest to studies of KSHV and cancer (17). Our experiment allowed us to look for miR-K12-11 targets found in WT-infected cells that were absent in ΔmiR-K12-11-infected cells. There were 54 such high-confidence targets of miR-K12-11. Of the 30 of these genes tested by RT-qPCR, 65% were found to have reduced expression levels in the presence of an miR-K12-11 mimic. Hybrids of genes that scored positive for reduced expression had characteristics distinct from those that did not. Notably, the hybrids of positive genes were more likely to contain a perfect seed match and more often contained an mRNA fragment originating from the 3′ UTR. This parallels the well-accepted knowledge that a 3′-UTR-binding site and complementarity at nucleotides 2 to 8 are usually required for an miRNA to be functional (55). However, we note that we also identified multiple miR-K12-11-binding sites within coding regions of target genes that were efficiently suppressed (Fig. 8). It cannot be ignored that we and others continue to find miRNAs precipitated from Ago that do not have canonical matches to their partner mRNAs and do not bind to 3′ UTRs (24, 26, 30, 56–58). These miRNAs do not seem to be incorporated into the RNA-induced silencing complex (RISC) at random, as the same miRNA-mRNA pairs can be found repeatedly across different experiments. One possible explanation is that some of these miRNAs serve a purpose other than repression. One alternative model is competing endogenous RNAs by which mRNAs or other RNAs that interact with miRNAs are not regulated by miRNAs but rather regulate other mRNAs indirectly by sponging miRNAs, thereby derepressing miRNA targets (recently reviewed in reference 59). For example, hepatitis C virus RNA requires a specific interaction with hsa-miR-122 to replicate (60) and at the same time sponges hsa-miR122, which derepresses liver-specific genes that are usually repressed by miR-122 (61). We identified a number of miR-K12-11-containing hybrids for which the levels of the targeted mRNA were not repressed by an miR-K12-11 mimic. This may suggest a converse regulatory relationship where host RNAs have evolved to act as sponges for viral miRNAs, hence preventing the targeting of antiviral host genes by viral miRNAs. In this context, it is also interesting to note that KSHV miRNAs interact with many long noncoding RNAs, some of which have been proposed to function in innate immunity (62, 63). Understanding whether both classical miRNA-mRNA interactions and sponging interactions are important for KSHV pathogenesis will be an important line of research in the future.
MATERIALS AND METHODS
Cell lines, tissue culture, and generation of TIVE-EX-LTC cells.
TIVE-LTC cells (64), which had been passaged many times (the exact passage number is not known), were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cells were trypsinized, diluted to a concentration of 10 cells per ml, and plated into a 96-well plate at 100 μl per well. Cells from wells that did not express green fluorescent protein (GFP) were expanded. The absence of virus was confirmed by qPCR for open reading fame 73 (ORF73), using plasmid LANA-pcDNA3.1 as a standard. One KSHV-negative culture was selected for all future experiments, and the cell line is referred to here as TIVE-EX-LTC. Cells were maintained in DMEM containing 10% FBS and 1% penicillin-streptomycin. For infections, 90 to 100% confluent TIVE-EX-LTC cells were incubated with either WT bacterial artificial chromosome 16 (BAC16) (65) or ΔmiR-K12-11 BAC16 (66) for 24 h with the equivalent of 200 genomes per cell. Infected cells were maintained with 100 ng/μl hygromycin B in the culture medium.
qCLASH.
TIVE-EX-LTC cells were expanded in 15-cm plates until they reached approximately 80 to 90% confluence. Working with 6 plates at a time, cells were trypsinized, resuspended in medium, and rinsed twice with phosphate-buffered saline (PBS). After the second rinse, the cells were resuspended in 10 ml PBS and transferred to a clean 10-cm cell culture plate on ice. Cells were UV irradiated in a cross-linker at a wavelength of 250 nm, receiving a total energy of 600 J/cm2. After cross-linking, the cells were counted by using a hemocytometer and distributed into aliquots of 50 million cells. The cells were pelleted at 4°C, the supernatants were removed, and the cells were frozen at −80°C until further use. The cells for the three separate biological replicates were thawed, expanded, and prepared separately and sequentially, using a different stock of cells each time.
The preparation of cell lysates and antibody-coated beads and immunoprecipitation were performed as described previously by Haecker et al. (20), with only slight modifications. All quantities given are for one experiment with 5 × 107 cells. Six milligrams of Dynabeads protein G (catalog number 10004D; Invitrogen) was rinsed three times with PBS-T (pH 7.2) (1× PBS [pH 7.4] without Ca2+/Mg2+, 0.02% Tween 20). Beads were briefly pelleted between rinses at ≤3,000 rpm. The beads were resuspended in PBS-T, and 72 μl AffiniPure rabbit anti-mouse IgG antibody (catalog number 315-005-008; Jackson ImmunoResearch) was added for a final concentration of 200 μg/ml. The mixture was incubated while spinning for 50 min at room temperature. The beads were rinsed three times with PBS-T and resuspended in PBS-T, and 10 μl of 2A8 anti-Ago antibody (a generous gift of Zissimos Mourelatos) was added. The mixture was incubated while spinning at 4°C overnight. The next day, the beads were rinsed four times with 1× PXL (1× PBS [pH 7.4] without Ca2+/Mg2+, 0.1% SDS, 0.5% Na-deoxycholate, 0.5% NP-40), resuspended in 1× PXL, and stored on ice until the cell lysates were ready.
To prepare cell lysates, 5 × 107 UV-cross-linked TIVE-EX-LTC cells were thawed on ice and resuspended in 500 μl lysis buffer (50 mM HEPES-KOH [pH 7.5], 150 mM KCl, 2 mM EDTA, 1 mM NaF, 0.5% NP-40, 0.5 mM dithiothreitol [DTT] [added immediately before use], 1× complete protease inhibitor [catalog number 11836170001; Roche] [added immediately before use]). Cells were allowed to lyse for 15 min on ice. Ten microliters of RQ1 DNase (catalog number M610A; Promega) was added, and the lysate was incubated for 5 min at 37°C with shaking at 1,000 rpm. Afterwards, the lysate was centrifuged for 15 min at 21,000 × g at 4°C. The supernatant was transferred to a new tube, while the pellet was discarded. A total of 0.5 μl of RNase T1 (catalog number EN0541; ThermoFisher Scientific) was added to the lysate, and it was incubated for 15 min at 22°C. A 5-μl aliquot was transferred to a new tube and stored at −20°C to be used later for Western blotting.
For immunoprecipitation, 1× PXL buffer was removed from the antibody-coated beads, and the lysate was added. The mixture was incubated while spinning at 4°C overnight. After incubation, the supernatant was removed and stored at −20°C for Western blot analysis. The beads were rinsed three times with lysis buffer and resuspended in 1 ml lysis buffer. After the addition of 0.5 μl RNase T1, the beads were incubated at 22°C for 12 min with continuous shaking at 700 rpm. The beads were washed four times with each of the following buffers: 1× PXL, 5× PXL (5× PBS [pH 7.4] without Ca2+/Mg2+, 0.1% SDS, 0.5% Na-deoxycholate, 0.5% NP-40), high-stringency buffer (15 mM Tris-HCl [pH 7.5], 5 mM EDTA, 2.5 mM EGTA, 1% Triton X-100, 1% Na-deoxycholate, 0.1% SDS, 120 mM NaCl, 25 mM KCl), high-salt buffer (15 mM Tris-HCl [pH 7.5], 5 mM EDTA, 2.5 mM EGTA, 1% Triton X-100, 1% Na-deoxycholate, 0.1% SDS, 1 M NaCl), and PNK (50 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 0.5% NP-40). The beads were left in the final wash buffer while the T4 PNK mixture was prepared.
Phosphorylation and intermolecular ligation were performed as follows. To prepare the T4 PNK mixture, the following components were combined: 8 μl 10× PNK buffer, 2 μl RNasin Plus (40 U/μl) (catalog number N2615; Promega), 0.8 μl 100 mM ATP (catalog number R0041; ThermoFisher Scientific), 65.2 μl autoclaved double-distilled water (ddH2O), and 4 μl T4 PNK (10 U/μl) (catalog number M0201; NEB). Eighty microliters of this mixture was added to the beads, and they were incubated for 40 min at 10°C. After incubation, the beads were washed three times with PNK. To prepare the ligation mixture, the following components were combined: 50 μl 10× T4 RNA ligase buffer, 60 μl 50% polyethylene glycol 8000 (PEG-8000), 1.25 μl 4 M KCl, 12.5 μl RNasin Plus, 5 μl 100 mM ATP, 321.25 μl autoclaved ddH2O, and 50 μl T4 RNA ligase 1 (catalog number M0204; NEB). Five hundred microliters of the ligation mixture was added to the beads. The beads were incubated with rotation at 4°C overnight.
Dephosphorylation and 3′ linker addition were performed as follows. The beads were washed three times with PNK. The dephosphorylation mixture was prepared with the following components: 8 μl 10× dephosphorylation buffer, 2 μl RNasin Plus, 67 μl autoclaved ddH2O, and 3 μl alkaline phosphatase (1 U/μl) (catalog number 10713023001; Roche). Eighty microliters of the dephosphorylation mixture was added to the beads, and they were incubated at 10°C for 40 min with shaking every 2 min for 15 s at 1,000 rpm. The beads were washed twice with PNK-EGTA (50 mM Tris-HCl [pH 7.5], 20 mM EGTA, 0.5% NP-40) and three times with PNK. The ligation mix was prepared by combining the following components: 42 μl autoclaved ddH2O, 8 μl 10× T4 RNA ligase buffer, 16 μl 50% PEG-8000, 2 μl RNasin Plus, 8 μl 10 μM miRCat-33 3′ linker (5′-TGGAATTCTCGGGTGCCAAGG-3′), and 4 μl T4 RNA ligase 2, truncated K227Q. Eighty microliters of the mixture was added to the beads, and they were incubated overnight at 16°C with shaking every 2 min for 15 s at 1,000 rpm.
To elute complexes, the beads were washed three times with PNK. The elution buffer was prepared by combining the following components: 40 μl 500 mM NaHCO3, 20 μl 10% SDS, and 140 μl autoclaved ddH2O. One hundred microliters was added to the beads, and they were incubated for 15 min at room temperature with continuous shaking at 1,400 rpm. This step was repeated a second time. On both occasions, the supernatant was transferred to a new tube. Two microliters of the supernatant was transferred to a new tube and stored at −20°C for later Western blot analysis.
For proteinase K treatment and RNA extraction, the following components were combined: 10 μl 5× PK buffer (500 mM Tris-HCl [pH 7.5], 250 mM NaCl, 50 mM EDTA), 10 μl proteinase K (final concentration, 4 mg/ml) (catalog number 03115887001; Roche), and 30 μl autoclaved ddH2O. The proteinase K solution was incubated at 37°C for 20 min. Fifty microliters of the solution was added to the 200 μl of the supernatant from the previous step, and the mixture was incubated for 20 min at 37°C. Two hundred fifty microliters of phenol-chloroform-isoamyl alcohol (25:24:1) was added, and the sample was incubated for 8 min at room temperature with continuous shaking at 1,400 rpm. The sample was then centrifuged for 10 min at 18,000 × g at 4°C. The aqueous supernatant was transferred to a new tube and combined with 20 μl 3 M sodium acetate (NaOAc) (pH 5.2), 2 μl GlycoBlue (catalog number AM9516; Invitrogen), and 500 μl of a 1:1 mixture of ethanol and isopropanol. The sample was incubated overnight at −20°C.
The sample was centrifuged for 30 min at 21,000 × g at 4°C. The supernatant was removed, and the RNA pellet was washed once with 950 μl and once with 200 μl of 80% ice-cold ethanol. The sample was centrifuged at 18,000 × g for 10 min at 4°C after each wash. The pellet was allowed to air dry for approximately 10 min before being resuspended in 10.5 μl autoclaved ddH2O. The T4 PNK mix was prepared with the following components: 1.5 μl 10× T4 PNK buffer, 0.5 μl RNasin Plus, 1.5 μl 10 mM ATP, and 1 μl T4 PNK. A total of 4.5 μl of the mixture was added to the RNA, and it was incubated for 40 min at 10°C. A ligation solution was prepared by combining the following components: 0.5 μl 10× T4 RNA ligase buffer, 2 μl bovine serum albumin (BSA), 0.5 μl 10 mM ATP, 1 μl 5′ RNA linker (100 pM/μl), and 1 μl T4 RNA ligase. Five microliters of the ligation mixture was added to the RNA, and the sample was incubated overnight at 16°C. After incubation, the RNA was extracted with phenol-chloroform-isoamyl alcohol as described above.
The sample was centrifuged for 30 min at 21,000 × g at 4°C. The supernatant was removed, and the RNA pellet was washed twice with 200 μl of 80% ice-cold ethanol. After each wash, the sample was centrifuged at 18,000 × g for 10 min at 4°C. The pellet was air dried for approximately 10 min before it was resuspended in 11 μl autoclaved ddH2O. One microliter of 10 μM reverse transcription primer (Illumina TruSeq Small RNA Sample Prep kit RTP) and 1 μl of 10 mM deoxynucleoside triphosphates (dNTPs) were added to the RNA. The mixture was incubated at 65°C for 5 min and then chilled on ice and centrifuged briefly. The RT mix was prepared with the following components: 4 μl 5× SuperScript RT buffer, 1 μl 0.1 M DTT, 1 μl RNasin Plus, and 1 μl SuperScript III (200 U/μl) (catalog number 18080093; Invitrogen). Seven microliters of the RT mix was added to the RNA. The mixture was incubated at 50°C for 45 min, 55°C for 15 min, and 95°C for 5 min and then chilled on ice while the PCR mix was prepared. The following components were combined to make the PCR mix: 10 μl 2× Phusion high-fidelity master mix; 1 μl 10 μM primer 1 (Illumina TruSeq Small RNA Sample Prep kit RP1); 1 μl index primer 1, 2, or 3 (Illumina TruSeq Small RNA Sample Prep kit RPI1, RPI2, or RPI3); and 6 μl autoclaved ddH2O. Eighteen microliters of the PCR mix was combined with 2 μl of the RT product. The PCR block was preheated to 98°C before the following cycle was run: 98°C for 30 s; 98°C for 10 s, 52°C for 30 s, and 72°C for 30 s for 19 to 24 cycles; and 72°C for 5 min.
A mixture containing 30% PEG-8000 and 29.25 mM MgCl2 was prepared by combining 19.2 μl 50% PEG-8000, 0.94 μl 1 M MgCl2, and 11.86 μl autoclaved ddH2O. Sixty microliters of Tris-EDTA (TE) buffer was added to the PCR product, followed by 40 μl of the PEG-8000 mixture. The sample was vortexed, incubated for 10 min at room temperature, and then centrifuged for 15 min at 10,000 × g at room temperature. The supernatant was transferred to a new tube, to which 12 μl 3 M NaOAc, 300 μl 100% ethanol, and 1 μl GlycoBlue were added. The sample was incubated overnight at −20°C and precipitated the following day as described above. The pellet was resuspended in 15 μl autoclaved ddH2O. The sample was submitted for sequencing on an Illumina HiSeq 2500 instrument with a read length of 100 bp.
The efficacy of the Ago immunoprecipitation was confirmed by Western blotting. The equivalents of material from 5 × 105 cells before immunoprecipitation (“input”), after immunoprecipitation (“flowthrough”), and after elution (“eluate”) were run side by side on a polyacrylamide gel and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was probed with anti-Ago2 antibody 11A9 (catalog number MABE253; Millipore) diluted 1:1,000, followed by horseradish peroxidase (HRP)-tagged anti-rat secondary antibody (catalog number 312-036-045; Jackson ImmunoResearch) diluted 1:5,000.
Mimic transfections and qPCR.
TIVE-EX-LTC cells infected with KSHV ΔmiR-K12-11 were grown to confluence in 10-cm plates, trypsinized, and resuspended in medium without serum at a density of 2 × 105 cells/ml. A total of 2 × 106 cells were used per transfection treatment. Cells were transfected with either an miR-K12-11-3p mimic (Syn-kshv-miR-K12-11-3p miScript miRNA mimic, catalog number MSY0002181; Qiagen) or a control mimic (miRIDIAN microRNA mimic negative control 1, catalog number CN-0010000-01-05; Dharmacon) by using Lipofectamine RNAiMax transfection reagent (catalog number 13778150; Invitrogen) according to the manufacturer's instructions and plated in a clean 10-cm plate. Each transfection was performed in duplicate. Forty-eight hours after transfection, the cells were harvested, and RNA was extracted by using RNA-Bee (catalog number CS-501B; Amsbio). cDNA was prepared by using SuperScript III and treated with DNase prior to qPCR. qPCR for selected transcripts was performed by using Fast SYBR green master mix (catalog number 4385610; Applied Biosystems). Primers were designed by using IDT PrimerQuest. Primer sequences are listed in Table S1 in the supplemental material.
Sequencing and bioinformatics analysis.
The libraries were sequenced on a HiSeq 2500 instrument with a read length of 100 bp. The raw sequences were preprocessed with Trimmomatic (27) to remove adapter sequences and then analyzed with Hyb, a bioinformatics pipeline developed by Travis et al. specifically for the analysis of CLASH data (28). Determination of base pairing along the length of the miRNA, categorization of miRNA seed pairing and 3′ end pairing, and determination of mRNA transcript region origin were all carried out with custom scripts (available at GitHub [https://github.com/RenneLab/qCLASH-Analysis]).
Accession number(s).
The data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus (GEO) (67) and are accessible through GEO series accession number GSE101978.
Supplementary Material
ACKNOWLEDGMENTS
We thank Zissimos Mourelatos (University of Pennsylvania) for the 2A8 anti-Argonaute antibody and Alberto Riva for assistance with the gene pathway analysis. Scott Tibbetts (University of Florida) and Erik Flemington (Tulane University) contributed valuable assistance with pathway analysis and manuscript organization.
This work was supported by grant R01 CA119917 from the NIH/NCI to R.R. and grant F31 CA180522 to L.A.G.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.02138-17.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280. [PubMed] [Google Scholar]
- 3.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 4.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. 2015. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 5.Bray F, Ren JS, Masuyer E, Ferlay J. 2013. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer 132:1133–1145. doi: 10.1002/ijc.27711. [DOI] [PubMed] [Google Scholar]
- 6.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol 71:715–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sturzl M, Blasig C, Schreier A, Neipel F, Hohenadl C, Cornali E, Ascherl G, Esser S, Brockmeyer NH, Ekman M, Kaaya EE, Tschachler E, Biberfeld P. 1997. Expression of HHV-8 latency-associated T0.7 RNA in spindle cells and endothelial cells of AIDS-associated, classical and African Kaposi's sarcoma. Int J Cancer 72:68–71. doi: 10.1002/(SICI)1097-0215(19970703)72:1<68::AID-IJC10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 8.Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J Virol 72:8309–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kellam P, Boshoff C, Whitby D, Matthews S, Weiss RA, Talbot SJ. 1997. Identification of a major latent nuclear antigen, LNA-1, in the human herpesvirus 8 genome. J Hum Virol 1:19–29. [PubMed] [Google Scholar]
- 10.Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. 2005. Identification of microRNAs of the herpesvirus family. Nat Methods 2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 11.Samols MA, Hu J, Skalsky RL, Renne R. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J Virol 79:9301–9305. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai X, Cullen BR. 2006. Transcriptional origin of Kaposi's sarcoma-associated herpesvirus microRNAs. J Virol 80:2234–2242. doi: 10.1128/JVI.80.5.2234-2242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ambros V. 2004. The functions of animal microRNAs. Nature 431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 14.Samols MA, Hu J, Skalsky RL, Maldonado AM, Riva A, Lopez MC, Baker HV, Renne R. 2007. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog 3:e65. doi: 10.1371/journal.ppat.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziegelbauer JM, Sullivan CS, Ganem D. 2009. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat Genet 41:130–134. doi: 10.1038/ng.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, Braich R, Manoharan M, Soutschek J, Ohler U, Cullen BR. 2007. A viral microRNA functions as an orthologue of cellular miR-155. Nature 450:1096–1099. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, Lopez MC, Baker HV, Renne R. 2007. Kaposi's sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol 81:12836–12845. doi: 10.1128/JVI.01804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boss IW, Nadeau PE, Abbott JR, Yang Y, Mergia A, Renne R. 2011. A Kaposi's sarcoma-associated herpesvirus-encoded ortholog of microRNA miR-155 induces human splenic B-cell expansion in NOD/LtSz-scid IL2Rgammanull mice. J Virol 85:9877–9886. doi: 10.1128/JVI.05558-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schenk P, Konrad K. 1985. Ultrastructure of Kaposi's sarcoma in acquired immune deficiency syndrome (AIDS). Arch Otorhinolaryngol 242:305–313. doi: 10.1007/BF00453555. [DOI] [PubMed] [Google Scholar]
- 20.Haecker I, Gay LA, Yang Y, Hu J, Morse AM, McIntyre L, Renne R. 2012. Ago-HITS-CLIP expands understanding of Kaposi's sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog 8:e1002884. doi: 10.1371/journal.ppat.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chi SW, Zang JB, Mele A, Darnell RB. 2009. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp A-C, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helwak A, Kudla G, Dudnakova T, Tollervey D. 2013. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Helwak A, Tollervey D. 2014. Mapping the miRNA interactome by cross-linking ligation and sequencing of hybrids (CLASH). Nat Protoc 9:711–728. doi: 10.1038/nprot.2014.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore MJ, Scheel TK, Luna JM, Park CY, Fak JJ, Nishiuchi E, Rice CM, Darnell RB. 2015. miRNA-target chimeras reveal miRNA 3′-end pairing as a major determinant of Argonaute target specificity. Nat Commun 6:8864. doi: 10.1038/ncomms9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Travis AJ, Moody J, Helwak A, Tollervey D, Kudla G. 2014. Hyb: a bioinformatics pipeline for the analysis of CLASH (crosslinking, ligation and sequencing of hybrids) data. Methods 65:263–273. doi: 10.1016/j.ymeth.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plaisance-Bonstaff K, Choi HS, Beals T, Krueger BJ, Boss IW, Gay LA, Haecker I, Hu J, Renne R. 2014. KSHV miRNAs decrease expression of lytic genes in latently infected PEL and endothelial cells by targeting host transcription factors. Viruses 6:4005–4023. doi: 10.3390/v6104005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grosswendt S, Filipchyk A, Manzano M, Klironomos F, Schilling M, Herzog M, Gottwein E, Rajewsky N. 2014. Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol Cell 54:1042–1054. doi: 10.1016/j.molcel.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gottwein E, Corcoran DL, Mukherjee N, Skalsky RL, Hafner M, Nusbaum JD, Shamulailatpam P, Love CL, Dave SS, Tuschl T, Ohler U, Cullen BR. 2011. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 10:515–526. doi: 10.1016/j.chom.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suffert G, Malterer G, Hausser J, Viiliainen J, Fender A, Contrant M, Ivacevic T, Benes V, Gros F, Voinnet O, Zavolan M, Ojala PM, Haas JG, Pfeffer S. 2011. Kaposi's sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS Pathog 7:e1002405. doi: 10.1371/journal.ppat.1002405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gottwein E, Cullen BR. 2010. A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J Virol 84:5229–5237. doi: 10.1128/JVI.00202-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu CC, Li Z, Chu CY, Feng J, Feng J, Sun R, Rana TM. 2010. MicroRNAs encoded by Kaposi's sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep 11:784–790. doi: 10.1038/embor.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. 2010. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol 12:193–199. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu F, Stedman W, Yousef M, Renne R, Lieberman PM. 2010. Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J Virol 84:2697–2706. doi: 10.1128/JVI.01997-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abend JR, Uldrick T, Ziegelbauer JM. 2010. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi's sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J Virol 84:12139–12151. doi: 10.1128/JVI.00884-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, Ziegelbauer JM. 2012. Kaposi's sarcoma-associated herpesvirus microRNAs target IRAK1 and MYD88, two components of the Toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J Virol 86:11663–11674. doi: 10.1128/JVI.01147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu M, Wang C, Li W, Lu W, Bai Z, Qin D, Yan Q, Zhu J, Krueger BJ, Renne R, Gao SJ, Lu C. 2015. A KSHV microRNA directly targets G protein-coupled receptor kinase 2 to promote the migration and invasion of endothelial cells by inducing CXCR2 and activating AKT signaling. PLoS Pathog 11:e1005171. doi: 10.1371/journal.ppat.1005171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallaher AM, Das S, Xiao Z, Andresson T, Kieffer-Kwon P, Happel C, Ziegelbauer J. 2013. Proteomic screening of human targets of viral microRNAs reveals functions associated with immune evasion and angiogenesis. PLoS Pathog 9:e1003584. doi: 10.1371/journal.ppat.1003584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friborg J Jr, Kong W, Hottiger MO, Nabel GJ. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889–894. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- 42.Si H, Verma SC, Robertson ES. 2006. Proteomic analysis of the Kaposi's sarcoma-associated herpesvirus terminal repeat element binding proteins. J Virol 80:9017–9030. doi: 10.1128/JVI.00297-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radkov SA, Kellam P, Boshoff C. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat Med 6:1121–1127. doi: 10.1038/80459. [DOI] [PubMed] [Google Scholar]
- 44.Godden-Kent D, Talbot SJ, Boshoff C, Chang Y, Moore P, Weiss RA, Mittnacht S. 1997. The cyclin encoded by Kaposi's sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol 71:4193–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belanger C, Gravel A, Tomoiu A, Janelle ME, Gosselin J, Tremblay MJ, Flamand L. 2001. Human herpesvirus 8 viral FLICE-inhibitory protein inhibits Fas-mediated apoptosis through binding and prevention of procaspase-8 maturation. J Hum Virol 4:62–73. [PubMed] [Google Scholar]
- 46.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B. 2006. Immortalization of primary endothelial cells by the K1 protein of Kaposi's sarcoma-associated herpesvirus. Cancer Res 66:3658–3666. doi: 10.1158/0008-5472.CAN-05-3680. [DOI] [PubMed] [Google Scholar]
- 48.Koff A, Polyak K. 1995. p27KIP1, an inhibitor of cyclin-dependent kinases. Prog Cell Cycle Res 1:141–147. doi: 10.1007/978-1-4615-1809-9_11. [DOI] [PubMed] [Google Scholar]
- 49.Potapova TA, Sivakumar S, Flynn JN, Li R, Gorbsky GJ. 2011. Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited. Mol Biol Cell 22:1191–1206. doi: 10.1091/mbc.E10-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delgado T, Carroll PA, Punjabi AS, Margineantu D, Hockenbery DM, Lagunoff M. 2010. Induction of the Warburg effect by Kaposi's sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc Natl Acad Sci U S A 107:10696–10701. doi: 10.1073/pnas.1004882107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yogev O, Lagos D, Enver T, Boshoff C. 2014. Kaposi's sarcoma herpesvirus microRNAs induce metabolic transformation of infected cells. PLoS Pathog 10:e1004400. doi: 10.1371/journal.ppat.1004400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA III, Peters EC, Driggers EM, Hsieh-Wilson LC. 2012. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guillou C, Freret M, Fondard E, Derambure C, Avenel G, Golinski ML, Verdet M, Boyer O, Caillot F, Musette P, Lequerre T, Vittecoq O. 2016. Soluble alpha-enolase activates monocytes by CD14-dependent TLR4 signalling pathway and exhibits a dual function. Sci Rep 6:23796. doi: 10.1038/srep23796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sturzl M, Gaus D, Dirks WG, Ganem D, Jochmann R. 2013. Kaposi's sarcoma-derived cell line SLK is not of endothelial origin, but is a contaminant from a known renal carcinoma cell line. Int J Cancer 132:1954–1958. doi: 10.1002/ijc.27849. [DOI] [PubMed] [Google Scholar]
- 55.Agarwal V, Bell GW, Nam JW, Bartel DP. 2015. Predicting effective microRNA target sites in mammalian mRNAs. eLife 4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chi SW, Hannon GJ, Darnell RB. 2012. An alternative mode of microRNA target recognition. Nat Struct Mol Biol 19:321–327. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, Leslie CS, Rudensky AY. 2012. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol Cell 48:760–770. doi: 10.1016/j.molcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khorshid M, Hausser J, Zavolan M, van Nimwegen E. 2013. A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat Methods 10:253–255. doi: 10.1038/nmeth.2341. [DOI] [PubMed] [Google Scholar]
- 59.Tay Y, Rinn J, Pandolfi PP. 2014. The multilayered complexity of ceRNA crosstalk and competition. Nature 505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 61.Luna JM, Scheel TK, Danino T, Shaw KS, Mele A, Fak JJ, Nishiuchi E, Takacs CN, Catanese MT, de Jong YP, Jacobson IM, Rice CM, Darnell RB. 2015. Hepatitis C virus RNA functionally sequesters miR-122. Cell 160:1099–1110. doi: 10.1016/j.cell.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sethuraman S, Gay LA, Jain V, Haecker I, Renne R. 2017. MicroRNA dependent and independent deregulation of long non-coding RNAs by an oncogenic herpesvirus. PLoS Pathog 13:e1006508. doi: 10.1371/journal.ppat.1006508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Atianand MK, Caffrey DR, Fitzgerald KA. 2017. Immunobiology of long noncoding RNAs. Annu Rev Immunol 35:177–198. doi: 10.1146/annurev-immunol-041015-055459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.An FQ, Folarin HM, Compitello N, Roth J, Gerson SL, McCrae KR, Fakhari FD, Dittmer DP, Renne R. 2006. Long-term-infected telomerase-immortalized endothelial cells: a model for Kaposi's sarcoma-associated herpesvirus latency in vitro and in vivo. J Virol 80:4833–4846. doi: 10.1128/JVI.80.10.4833-4846.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jain V, Plaisance-Bonstaff K, Sangani R, Lanier C, Dolce A, Hu J, Brulois K, Haecker I, Turner P, Renne R, Krueger B. 2016. A toolbox for herpesvirus miRNA research: construction of a complete set of KSHV miRNA deletion mutants. Viruses 8:E54. doi: 10.3390/v8020054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.