

ABSTRACT
JC polyomavirus (JCPyV) establishes a lifelong persistence in roughly half the human population worldwide. The cells and tissues that harbor persistent virus in vivo are not known, but renal tubules and other urogenital epithelial cells are likely candidates as virus is shed in the urine of healthy individuals. In an immunosuppressed host, JCPyV can become reactivated and cause progressive multifocal leukoencephalopathy (PML), a fatal demyelinating disease of the central nervous system. Recent observations indicate that JCPyV may productively interact with cells in the choroid plexus and leptomeninges. To further study JCPyV infection in these cells, primary human choroid plexus epithelial cells and meningeal cells were challenged with virus, and their susceptibility to infection was compared to the human glial cell line, SVG-A. We found that JCPyV productively infects both choroid plexus epithelial cells and meningeal cells in vitro. Competition with the soluble receptor fragment LSTc reduced virus infection in these cells. Treatment of cells with neuraminidase also inhibited both viral infection and binding. Treatment with the serotonin receptor antagonist, ritanserin, reduced infection in SVG-A and meningeal cells. We also compared the ability of wild-type and sialic acid-binding mutant pseudoviruses to transduce these cells. Wild-type pseudovirus readily transduced all three cell types, but pseudoviruses harboring mutations in the sialic acid-binding pocket of the virus failed to transduce the cells. These data establish a novel role for choroid plexus and meninges in harboring virus that likely contributes not only to meningoencephalopathies but also to PML.
IMPORTANCE JCPyV infects greater than half the human population worldwide and causes central nervous system disease in patients with weakened immune systems. Several recent reports have found JCPyV in the choroid plexus and leptomeninges of patients with encephalitis. Due to their role in forming the blood-cerebrospinal fluid barrier, the choroid plexus and leptomeninges are also poised to play roles in virus invasion of brain parenchyma, where infection of macroglial cells leads to the development of progressive multifocal leukoencephalopathy, a severely debilitating and often fatal infection. In this paper we show for the first time that primary choroid plexus epithelial cells and meningeal cells are infected by JCPyV, lending support to the association of JCPyV with meningoencephalopathies. These data also suggest that JCPyV could use these cells as reservoirs for the subsequent invasion of brain parenchyma.
KEYWORDS: choroid plexus, JCPyV, meninges, PML, Polyomaviridae, receptors
INTRODUCTION
Progressive multifocal leukoencephalopathy (PML) is a rapidly progressing and often fatal neurodegenerative disease caused by the human polyomavirus JCPyV (1–4). The mode of transmission of JCPyV is unknown, since the initial infection is asymptomatic. After initial infection, lifelong persistent infection is thought to be established in the kidneys and urinary tracts of immunocompetent hosts, and about 20% of these infected individuals exhibit viruria (5, 6). Additional sites of virus persistence are likely since both JCPyV DNA and viral proteins have been detected in other tissues, including B lymphocytes in the bone marrow, tonsillar stromal cells, lungs, spleen, choroid plexus, leptomeninges, and brain (7–16).
JCPyV is composed of a 5,130-bp circular, double-stranded DNA genome enclosed by a nonenveloped icosahedral capsid, which is made up of three proteins: VP1, VP2, and VP3 (17). The virus engages sialic acid on the cell surface via the capsid protein VP1 (18–20). Previous crystallographic and functional studies have demonstrated that the α2,6-linked glycan lactoseries tetrasaccharide c (LSTc) is the molecule utilized by JCPyV VP1 as its host cell attachment receptor (21). Virus attachment to LSTc is necessary, but not sufficient, for productive infection. Following attachment, JCPyV entry requires the presence of the serotonin receptor 2 (5-HT2R) family of proteins, which are comprised of three members: 5-HT2A, 5-HT2B, and 5-HT2C. Although these proteins do not appear to be involved with initial virus attachment, virus infection is blocked by pretreating cells with inhibitors to 5-HT2 receptor family subtypes, and JCPyV entry is markedly enhanced when any of the three 5-HT2 receptors are expressed in nonpermissive cells (22, 23).
PML develops in immunosuppressed individuals when JCPyV gains access to brain parenchyma and infects and destroys the myelin-producing oligodendrocytes (24, 25). Initially, PML was described as a very rare disease in individuals with B cell lymphoproliferative disorders (26, 27). The incidence of PML in patients rose significantly during the AIDS pandemic, with 3 to 5% of HIV/AIDS patients developing PML (28, 29). More recently, the incidence of PML in HIV/AIDS patients has declined due to the introduction of combined antiretroviral therapy (cART), although it has decreased less than other opportunistic infections (30). In recent years, PML has emerged as a significant and life-threatening complication in patients treated with immunomodulatory drugs designed to treat autoimmune diseases, such as multiple sclerosis, rheumatoid arthritis, psoriasis, and Crohn's disease (4, 31–36). In addition to PML, JCPyV has been shown to cause other diseases of the central nervous system (CNS), including JCPyV encephalopathy (37), JCPyV granule cell neuronopathy (38), and meningoencephalitis (16, 39).
It is not yet known how JCPyV is transmitted from its original site(s) of infection and persistence to its main site of pathogenesis, the CNS. Neural tissue is well protected from the periphery by the blood-brain barrier and the blood-cerebrospinal fluid (CSF) barrier (BCSFB). The BCSFB includes the choroid plexus (CP), a villous structure found in the CSF-filled ventricles of the brain. The core of the CP is the stroma, composed of connective tissue, fibroblasts, immune cells, and fenestrated blood vessels, which create an interface between the peripheral circulation and the CNS. The stroma is surrounded by a layer of specialized epithelium, the choroid plexus epithelium (CPE). CPE cells are bound by tight junctions, forming a barrier to separate the blood from the CSF (40). An additional component of the BCSFB is found in the meninges. Covering the brain are three layers of meninges: the dura mater, the arachnoid mater, and the pia mater. Leptomeningeal cells, which are comprised of the arachnoid and pia mater cells, line the subarachnoid spaces that are filled with CSF. Like CPE cells, the leptomeningeal cells are bound by tight junctions to restrict access to the CNS from the blood that circulates in the dura mater. These cellular barriers impede the migration of both cells and microbes between the peripheral circulation, brain, and CSF. The mechanism by which JCPyV evades these obstacles to gain entry to the CNS is currently unknown. Recently, the CPE and leptomeningeal cells of a patient with meningitis were shown to be infected with JCPyV (16, 39). Moreover, human CPE and leptomeninges express both the attachment and entry receptors for JCPyV in vivo, and JCPyV binding to these tissues is dependent on the attachment receptor LSTc (41–43). These observations suggest that JCPyV infection of these barrier tissues could provide JCPyV access to the brain parenchyma and are a potential site of virus persistence.
The present study focused on determining whether JCPyV could infect primary choroid plexus and meningeal cells in vitro. Primary human choroid plexus epithelial cells (CPEpiC) and human meningeal cells (HMC) were obtained from ScienCell Research Labs, validated, and then challenged with JCPyV. We found that both choroid plexus epithelial cells and meningeal cells in culture were susceptible to virus infection and that the infection was dependent on expression of the major virus attachment receptor LSTc.
RESULTS
JCPyV infection of choroid plexus epithelial cells and meningeal cells is sialic acid dependent.
To assess whether CPEpiC and HMC are susceptible to infection by JCPyV in vitro, these cells were challenged with virus and infection scored 72 h later by indirect immunofluorescent analysis. CPEpiC and HMC expressed both the early viral protein large T antigen and the late viral protein V-antigen (VP1), indicating that JCPyV established a productive infection in both cell types (Fig. 1A and B). Initial JCPyV attachment to these cells was analyzed by flow cytometry using fluorescently labeled virus (JC-488). We found that CPEpiC and HMC bound significantly more labeled JCPyV than SVG-A cells (Fig. 1C). To determine whether the infected cells were capable of producing infectious virus, we harvested supernatants from each cell type and used them to reinfect naive cells. These same supernatants were also subjected to digital droplet PCR (ddPCR) to quantify the amount of virus produced. Supernatants from CPEpiC and HMC cells readily produced infectious virus capable of infecting naive cells (Fig. 1C). These same supernatants also contained significant numbers protected viral genomes, as determined by ddPCR (Fig. 1D).
FIG 1.

JCPyV infects CPEpiC and HMC. (A) Quantification of the numbers of T antigen- and V antigen-positive cells per visual field in SVG-A, CPEpiC, and HMC. (B) Representative images of T antigen- and V antigen-positive cells. The infected cells were stained with antibodies to T antigen (Ab-2; Oncogene) and V antigen (Pab597), and then these were detected with goat anti-mouse Alexa Fluor 488-labeled secondary antibodies. (C) Reinfection assay demonstrating each cell line produces infectious virus. (D) ddPCR quantification of the amount of complete virus released by each cell line following one round of infection. (E) Flow cytometric analysis of the binding of Alexa Fluor 488-labeled JCPyV (JC-488) and the LSTc binding lectin PSL to SVG-A, CPEpiC, and HMC as indicated. Error bars represent standard errors of the mean for three independent experiments performed in triplicate.
To explore the nature of JCPyV initial attachment and determine the involvement of the known attachment receptor LSTc, we used a lectin from the mushroom Polyporus squamosus or PSL. PSL recognizes the terminal three sugars of LSTc and detects LSTc-like oligosaccharides on cells and in tissues (43, 44). JCPyV directly interacts with the terminal α2,6-linked sialic acid on LSTc to facilitate virus attachment to cells, and mutations in the sialic acid-binding pocket of VP1 prevent this interaction and abolish infection by purified JCPyV virions (21, 45). Using biotinylated PSL (PSL-biotin), we found that these cells bound higher levels of PSL than did SVG-A cells, consistent with the higher-level binding of JCPyV (Fig. 1E). To assay the contribution of sialic acid to receptor-dependent infection in CPEpiC and HMC, we treated cells with neuraminidase (NA) and then incubated the cells with highly purified and labeled JCPyV virions. Similar to SVG-A cells, treatment with NA significantly reduced binding to both cell types (Fig. 2A). The reduction in virus binding correlated with a significant decrease in the ability of JCPyV to infect both CPEpiC and HMC (Fig. 2B), demonstrating that the presence of sialic acid on the plasma membrane is necessary for infection in these cells.
FIG 2.

Infection of CPEpiC and HMC is sialic acid dependent. (A) Cells were either untreated or treated with type II neuraminidase to remove cell surface sialic acid receptors. The cells were then incubated with Alexa Fluor 488-labeled JCPyV and analyzed by flow cytometry. The histograms are shown in the insets. (B) This same treatment inhibits JCPyV infection of all three cell types as indicated. Error bars represent standard errors of the mean for three independent experiments performed in triplicate.
Attachment and entry receptor involvement in JCPyV infection.
To determine the specific mechanism by which JCPyV attaches to CPEpiC and HMC, we analyzed binding in the presence of the soluble attachment receptor LSTc. Purified and labeled JCPyV was preincubated with the soluble receptor fragment, LSTc, and cells were assayed for virus and lectin attachment by flow cytometry. Similar to SVG-A control cells, preincubation of JCPyV with LSTc significantly inhibited virus binding to the HMC. In contrast, JCPyV binding to CPEpiC was not affected by the presence of high concentrations of soluble LSTc (Fig. 3A). Competition with LSTc significantly reduced PSL binding to both cell types (Fig. 3B). Significantly, JCPyV infection in all cell types was inhibited by competition with LSTc (Fig. 3C). These data suggest that LSTc is the attachment receptor in CPEpiC and HMC cells that mediates productive infection by JCPyV.
FIG 3.

LSTc competes with PSL and virus for binding to cells and inhibits infection. (A) Purified, labeled JCPyV was incubated with soluble receptor LSTc, and then binding to cells was assayed by flow cytometry. LSTc inhibited JCPyV binding to SVG-A and HMC but did not inhibit binding to CPEpiC. (B) The LSTc binding lectin, PSL, was incubated with LSTc, and then PSL binding to cells was assayed by flow cytometry. LSTc inhibited binding of PSL to all three cell types. (C) Purified JCPyV was incubated with LSTc and then used to directly infect cells. LSTc inhibited infection of all three cell types. (D) The 5-HT2R antagonist inhibits infection of SVG-A and HMC by JCPyV. Cells were treated with 25 μM and 10 μM ritanserin and then challenged with JCPyV. The 25 μM dose of ritanserin abolished infection of SVG-A but was toxic to the primary CPEpiC and HMC. The lower dose, 10 μM, was not sufficient to inhibit infection of SVG-A or CPEpiC but did inhibit infection of HMC. (E) HTR2A mRNA levels measured by quantitative PCR (qPCR) are represented. Error bars represent standard errors of the mean for three independent experiments performed in triplicate.
We then analyzed the role of JCPyV entry receptors in infection of CPEpiC and HMC. To establish a productive infection, JCPyV first attaches to LSTc, and then the presence of 5-HT2Rs facilitates virus entry (23). To determine whether 5-HT2Rs were important for the infection in CPEpiC and HMC, the cells were pretreated with different concentrations of the 5-HT2R antagonist, ritanserin, and then challenged with virus. Ritanserin treatment inhibited infection of SVG-A cells and HMC, but it did not inhibit infection of CPEpiC (Fig. 3D). We could not use doses of ritanserin greater than 10 μM on the primary cells due to toxicity. Consistent with previous reports, CPEpiC and HMC both express 5-HT2A (Fig. 3E).
Choroid plexus epithelial cells and leptomeningeal cells are readily transduced by wild type but not mutant JCPyV pseudoviruses.
Luciferase-expressing pseudoviruses were prepared by transfecting HEK293FT producer cells with plasmids encoding wild-type VP1, VP2, and VP3 of JCPyV (Mad-1 strain) or with the L54F or S268F mutant VP1s (Fig. 4). The L54F and S268F mutations are frequently found in the CSF and parenchyma of patients with PML (46). These mutations render the virus incapable of binding to the sialic acid containing receptor LSTc, indicating that they may infect cells by alternative mechanisms (45, 47). We therefore examined whether the L54F and S268S mutant pseudoviruses were capable of transducing CPEpiC and HMC. The pseudoviruses were extensively purified, and equal amounts of each were used to transduce the cells. Pseudovirus transduction was measured by quantifying secreted luciferase activity at 6 days postransduction. We found that wild-type pseudovirus readily transduced each cell type but that the mutant pseudoviruses failed to transduce any of the cells.
FIG 4.
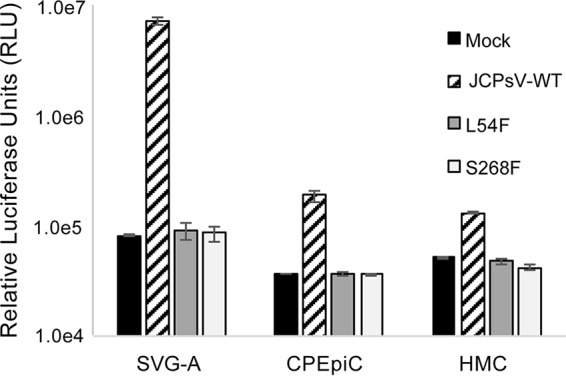
Wild-type JC pseudovirus but not L54F or S268F mutants transduce SVG-A, CPEpiC, and HMC. Luciferase-expressing pseudoviruses were prepared using wild-type VP1 (JCPsV-WT) or with two sialic acid pocket mutants of VP1 (L54F and S268F). The wild-type pseudovirus readily transduced all three cell types, but neither of the mutants transduced any of the cells above the mock control. Error bars represent standard errors of the mean for three independent experiments performed in triplicate.
DISCUSSION
Human polyomaviruses establish long-term, persistent infections in their natural host and rarely cause disease. Due to the lack of an experimental animal model capable of supporting the full virus life cycle, the initial sites of virus infection and the tissues that harbor persistent virus are unknown. The urogenital system has long been suspected as a site of persistence for JCPyV as the virus is excreted in the urine and can lead to renal dysfunction in immunosuppressed patients. When JCPyV migrates from sites of persistence to the CNS, it can cause the often fatal neurological disease, progressive multifocal leukoencephalopathy. Of central interest in understanding the pathogenesis of JCPyV is determining how JCPyV crosses the barriers that protect the CNS from the periphery to cause disease in the brain. In a study of the distribution of JCPyV receptors in human tissues it was found that two different cell types essential for the creation of barriers between the blood and CSF, the choroid plexus epithelium and the leptomeninges, express the known attachment and entry components of the JCPyV receptor complex (4). These data, as well as recent reports of HIV-negative patients with JCPyV-associated meningitis (16, 39), prompted us to examine the susceptibility of these cell types to virus infection in vitro since they are potential entry points for the virus from the periphery to the brain parenchyma.
Here, we demonstrate an in vitro model of JCPyV infection of primary human choroid plexus epithelial and leptomeningeal cells. We found that both CPEpiC and HMC were readily susceptible to infection, with CPEpiC infected at nearly the same level as the control SVG-A cell line. Both cell types expressed early and late viral proteins. The expression of the late protein occurs in conjunction with viral DNA replication indicating that the cells fully support the viral life cycle. To determine whether the mechanisms of viral attachment are the same in these primary cells as a control glial cell line, we tested whether they expressed the major attachment receptor for JCPyV, LSTc, and whether they were capable of binding virus. The crystal structure of LSTc complexed with JCPyV capsid protein VP1 has been solved, and the interactions between these molecules have been extensively characterized both structurally and functionally (45, 48, 49). We found that competition of JCPyV with soluble LSTc effectively inhibited infection of all three cell types, indicating that LSTc is a critical attachment receptor for JCPyV that leads to productive infection. Preincubation with LSTc also inhibited binding of JCPyV to SVG-A and HMC cells, indicating that initial virus engagement with HMC is mediated specifically by LSTc. However, LSTc pretreatment did not inhibit binding to CPEpiC even at the highest concentrations of LSTc that could be reasonably used. This finding suggests that while the JCPyV infection of CPEpiC requires LSTc, the majority of virus binding sites on CPEpiC are nonspecific and that only a fraction of that binding is to the specific attachment receptor LSTc. Sialic acid is necessary for virus binding, since treatment of all three cell types with neuraminidase significantly inhibited both virus binding and infection. Based on these data and previous observations we can reasonably conclude that neuraminidase-mediated removal of sialic acid from LSTc was responsible for the inhibition of infection seen in these experiments.
It has been shown previously that JCPyV infects SVG-A cells via 5-HT2Rs (22). After virus engagement of attachment receptors on the cell surface, the 5-HT2Rs facilitate entry by clathrin-dependent endocytosis into a productive infection pathway (50–52). In addition, the expression of 5-HT2Rs in BCSFB cells is well documented (42, 43, 53). Since it was not yet known whether BCSFB cells were involved in JCPyV infection, primary human CPEpiC and HMC were treated with different concentrations of the specific 5-HT2R antagonist ritanserin. Although both BCSFB cell types demonstrate abundant receptor expression, the meningeal cells were inhibited at lower concentrations of ritanserin compared to either CPEpiC or the control SVG-A cells. The primary CPEpiC and HMC cells appear to be more sensitive to toxicity of ritanserin compared to our immortalized SVG-A control cell line. At a nontoxic dose of ritanserin, HMC infection was inhibited, indicating that JCPyV infection is mediated by 5-HT2Rs in leptomeningeal cells. We did not observe a decrease in infection in CPEpiC treated with ritanserin at this nontoxic concentration. While it is likely that SVGA and CPE cells need more ritanserin to be inhibited, it does not exclude the possibility that JCPyV may enter CPEpiC in the absence of 5-HT2R activity, perhaps via the robust clathrin-mediated endocytosis that takes place in these cells as part of receptor-mediated transcytosis pathways for large molecules to cross the BCSFB (reviewed in reference 54). Unfortunately, blocking antibodies to these receptors are no longer available, and knockdown approaches using these primary cells with limited life spans are not possible. We are currently immortalizing the cells to perform these experiments in the future.
A confirmatory diagnosis of PML is made when JCPyV DNA is found in the CSF of symptomatic patients. Sequencing of JCPyV genomes from CSF and brain parenchyma show the presence of a swarm of viruses, including wild-type and mutant genomes (46). Several of these mutations cluster in and around the sialic acid-binding site of VP1, and it has been suggested that these mutants are better able to spread and cause disease in the CNS (55–59). In vitro these mutants, when introduced into the Mad-1 strain of JCPyV, render the virus noninfectious in SVG-A cells (21). Reconstitution of these mutations into pseudoviruses, however, has shown that they are capable of infecting some cell types (47). For this reason, we wanted to determine whether the mutants, when reconstituted as pseudoviruses, were capable of infecting CPEpiC and HMC. Similar to our previous results, wild-type pseudovirus transduced all of the cell types, but the mutants failed to transduce any of these cells. Transduction of the CPEpiC and HMC was less efficient than in SVG-A cells; therefore, we cannot rule out low-level transduction of these cells below the sensitivity of the assay.
In summary, we have described the first instance of JCPyV infection of primary BCSFB cells in vitro. These data also suggest that choroid plexus and leptomeninges could be a potential pathway involved in the trafficking of infectious JCPyV to brain parenchyma. Given the specificity of the virus to the human host, in vitro systems using human cell lines are critical for understanding viral infection and pathogenesis. Previous to this study, JCPyV infection in vitro has been limited to cell types found in human brain parenchymal cells, including the cell lines derived from human fetal glial cells, SVG and SVG-A (60–63), progenitor-derived astrocytes (64–66), hESC-derived oligodendrocyte progenitors (67), glial progenitor cells and astrocytes (68), and a recently described glioblastoma-derived oligodendrocyte precursor line (69). These cells do not always proliferate well and have varying degrees of virus susceptibility. Primary cultures of human renal tubule epithelial cells are also susceptible, but infection is highly restricted (70, 71). The fact that CPEpiC and HMC are susceptible to JCPyV infection and that these cells express relevant receptors, bind virus, and are infected in vivo opens up a new chapter in our understanding of the pathobiology of JCPyV-induced disease. It will be interesting to determine whether JCPyV persists in these tissues in more individuals than previously observed or whether the virus is only present during disease processes. We are currently investigating the prevalence of viral genomes in the CSF of individuals with and without JCPyV-associated disease. It will be critical to determine whether there is an increase in JCPyV infiltration of CSF in patients who are at risk for developing PML, including multiple sclerosis (MS), and HIV-infected populations.
MATERIALS AND METHODS
Cells, viruses and pseudoviruses.
Human choroid plexus epithelial cells (CPEpiC) and human meningeal cells (HMC) were obtained from ScienCell Research Labs and cultured in cell line-specific complete media, as indicated by the manufacturer (ScienCell) in a humidified incubator at 37°C with 5% CO2. SVG-A cells were grown in minimum essential medium supplemented to contain 10% fetal bovine serum and 1% penicillin-streptomycin (Mediatech, Inc.) in a humidified incubator at 37°C with 5% CO2. 293FT cells (Invitrogen) were grown in Dulbecco modified Eagle medium supplemented to contain 10% fetal bovine serum (Mediatech, Inc.), 0.1 mM nonessential amino acids, 6 mM l-glutamine, 1 mM sodium pyruvate, and 1% penicillin-streptomycin. Generation and purification of the Mad-1 strain of JC virus and of the wild-type and mutant pseudoviruses were performed as described previously (72, 73). Alexa Fluor 488-labeled carboxylic acid-succinimidyl ester (Invitrogen) was used to label purified virus according to the manufacturer's protocol. Briefly, 5.0 mg of CsCl-purified JCPyV was dialyzed against labeling buffer (0.1 M NaHCO3; pH 8.3) at 4°C overnight in 10,000 MWCO cartridges (Pierce). The virus was then incubated for 1 h on a platform rocker at room temperature, protected from light, with 0.5 μg of Alexa Fluor 488-labeled (AF488) succinimidyl ester in 100 μl of dimethyl sulfoxide (DMSO). The AF488-labeled virus was extensively dialyzed in 10,000 MWCO cartridges against two changes of buffer A (10 mM Tris-HCl, 50 mM NaCl, 0.1 mM CaCl2) at 4°C for an additional 48 h to remove excess dye.
Viral infections.
At 24 h prior to infection, SVG-A, CPEpiC, and HMC were seeded to poly-l-lysine-coated 96-well dishes at a density of 10,000 cells/cm2. Purified JCPyV (1.6 × 105 fluorescent forming units) was used to infect cells for 1 h at 37°C in cell type-specific basal media. After infection, the virus was aspirated, and cells were fed with complete media appropriate to each cell type. At 48 or 72 h postinfection, the samples were fixed in ice-cold methanol and stained for large T-antigen (AB-2; Oncogene) or V-antigen (VP1) using the monoclonal antibody Pab597. Goat anti-mouse Alexa Fluor 488-labeled secondary antibody (Invitrogen) was used to detect the binding of the primary antibodies. Infection was scored by counting T antigen- and VP1-positive nuclei. Nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole).
Virus production assays.
To determine whether infection of CPEpiC and HMC resulted in the production of infectious virus, supernatants from these cells were collected and used to reinfect naive cells (a reinfection assay). At 72 h postinfection, the samples were fixed in ice-cold methanol and stained for VP1 as described above. We also quantified the amount of virus produced from the infected cells using ddPCR. The same supernatants used in the reinfection assay were treated with DNase I (Invitrogen) for 1 h at 37°C to remove nonencapsidated DNA and then shifted to 75°C for 10 min to inactivate the DNase. DNase-treated samples were then processed using a blood and tissue nucleic acid extraction kit (Qiagen) according to the manufacturer's instructions. The isolated nucleic acids were linearized with BamHI (New England BioLabs), and ddPCR was performed using Bio-Rad reagents and TaqMan primer/probe sets for JCPyV VP1 (IDT; probe, 5′-/56-FAM/TGTGGCCAG/ZEN/AATTCCACTACCCAA/3IABkfq/-3′; primer 1, 5′-AGGGACATGCTTCCTTGTTAC-3′; primer 2, 5′-CAGCCTCCACATGAGTATATTT-3′).
Pseudovirus transductions.
At 24 h prior to infection, SVG-A, CPEpiC and HMC were seeded to poly-l-lysine-coated 96-well dishes at a density of 10,000 cells/well, in phenol-red free media as appropriate per cell type. Phenol red-free medium was used through the duration of the experiment. Purified pseudovirus containing the pSV40Cluc reporter plasmid (New England BioLabs) with wild-type VP1, mutant VP1, or a mock preparation containing no capsid protein was used to transduce cells for 2 h at 37°C. After transduction, the cells were washed and fed with complete medium as appropriate per cell type to a total volume of 100 μl. Sample supernatant was quantified for secreted luciferase (GloMax plate reader; Promega) using a BioLuxCyprindina luciferase assay kit (New England BioLabs).
Neuraminidase and ritanserin inhibition assays.
For neuraminidase (NA) treatment, cells were treated with 1 U/ml of NA Type II from Vibrio cholerae (Sigma) for 1 h at 37°C at pH 6.0 or with phosphate-buffered saline (PBS) at pH 6.0 as a control. After incubation, the cells were washed twice with cell type-specific basal media to remove the NA. Cesium chloride-purified JCPyV was used to infect cells for 1 h at 37°C in cell type-specific basal media. After infection, virus was aspirated, and the cells were supplemented with complete medium appropriate to each cell type. At 72 h postinfection, the samples were fixed in ice-cold methanol and stained for VP1 using Pab597 primary antibody and Alexa Fluor 488-labeled secondary antibody (Invitrogen). Infection was scored by counting VP1-positive nuclei. For the ritanserin experiment SVG-A, CPEpiC, and HMC were seeded to poly-l-lysine-coated 96-well dishes at a density of 10,000 cells/well. Cells were pretreated for 24 h prior to infection with 10 μM or 25 μM ritanserin, or vehicle control (DMSO) in the appropriate media. Purified JCPyV (1.6 × 105 fluorescent forming units) was used to infect cells for 1 h at 37°C in cell type-specific basal media. After infection, virus was aspirated, and the cells were fed with complete medium plus ritanserin or vehicle control at the indicated doses. Infection was scored at 72 h by counting VP1-positive nuclei.
Quantitative real-time PCR.
Total RNA from CPEpiC, HMC, SVGA, and 293FT cells was isolated by using an RNeasy minikit (Qiagen) according to the manufacturer's instructions. Quantitative real-time PCR was conducted with iScript reverse transcription supermix for quantitative reverse transcription-PCR (qRT-PCR) and SYBR SsoAdvanced Universal Sybr green SuperMix (Bio-Rad) according to the manufacturer's instructions. qRT-PCR was performed by a Bio-RadCFX96 qRT-PCR detection system. β-Actin served as an internal control (HTR2A, forward [5′-GAATCGTCCTGTAGCCCAAA-3′] and reverse [5′-CAGAATCCCATCCACCACAG-3′]; β-actin, forward [5′-GTTGTCGACGACGAGCG-3′] and reverse [5′-GCACAGAGCCTCGCCTT-3′]). Each qRT-PCR was performed in triplicate for qRT-PCR yield validation. Error bars correspond to standard errors of the mean.
LSTc competition assay.
To inhibit infection with LSTc, CsCl-purified virus was pretreated with 20 mM LSTc (V Labs, Inc.) in media without supplementation on ice for 1 h. Cells in 96-well plates were prechilled at 4°C for 30 min prior to infection. Virus-pentasaccharide complexes were added to cells, followed by incubation at 4°C for an additional 1 h. After infection, the cells were washed with PBS, complete medium was added as appropriate per cell type, and the cells were incubated at 37°C for 72 h. Cells were fixed and stained by indirect immunofluorescence for VP1 as described above.
Flow cytometry.
To assess binding to each cell type, the cells were harvested using Cellstripper (Mediatech) and washed in 1× PBS, and CsCl-purified Alexa Fluor 488-labeled JCPyV or PSL-biotin (E-Y Labs) was incubated with 106 SVG-A, CPEpiC, or HMC for 1 h on ice. PSL-biotin was detected by incubation for an additional hour on ice with streptavidin-488 secondary antibody (Invitrogen). Samples were washed in 1× PBS, fixed in 1% paraformaldehyde, and analyzed by flow cytometry (BD FACSCanto II). For competition assays, purified Alexa Fluor 488-labeled JCPyV was incubated for 1 h on ice in the presence or absence of 20 mM LSTc or PSL-biotin. Virus-compound complexes were then bound to cells for an additional hour on ice. Cells were washed in PBS, fixed with 1% paraformaldehyde, and analyzed by flow cytometry. For NA treatment, cells were incubated with 1 U/ml of NA Type II from Vibrio cholerae (Sigma) for 1.5 h at 37°C and pH 6.0 or in PBS at pH 6.0 as a control. After treatment, the cells were washed with PBS and bound with either Alexa Fluor 488-labeled JCPyV or PSL-biotin for an additional hour on ice. The cells were washed again in PBS, fixed in 1% paraformaldehyde, and analyzed.
ACKNOWLEDGMENTS
We thank all members of the laboratory for helpful discussions.
Work in our laboratory is supported in part by National Institutes of Health grants R01NS043097 and P01NS065719 to W.J.A.
REFERENCES
- 1.Kean JM, Rao S, Wang M, Garcea RL. 2009. Seroepidemiology of human polyomaviruses. PLoS Pathog 5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knowles WA, Pipkin P, Andrews N, Vyse A, Minor P, Brown DW, Miller E. 2003. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol 71:115–123. doi: 10.1002/jmv.10450. [DOI] [PubMed] [Google Scholar]
- 3.Assetta B, Atwood WJ. 2017. The biology of JC polyomavirus. Biol Chem 398:839–855. doi: 10.1515/hsz-2016-0345. [DOI] [PubMed] [Google Scholar]
- 4.Haley SA, Atwood WJ. 2017. Progressive multifocal leukoencephalopathy: endemic viruses and lethal brain disease. Annu Rev Virol 4:349–367. doi: 10.1146/annurev-virology-101416-041439. [DOI] [PubMed] [Google Scholar]
- 5.Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, Gosert R, Hirsch HH. 2009. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis 199:837–846. doi: 10.1086/597126. [DOI] [PubMed] [Google Scholar]
- 6.Dorries K. 1998. Molecular biology and pathogenesis of human polyomavirus infections. Dev Biol Stand 94:71–79. [PubMed] [Google Scholar]
- 7.Monaco MC, Jensen PN, Hou J, Durham LC, Major EO. 1998. Detection of JC virus DNA in human tonsil tissue: evidence for site of initial viral infection. J Virol 72:9918–9923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monaco MC, Atwood WJ, Gravell M, Tornatore CS, Major EO. 1996. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: implications for viral latency. J Virol 70:7004–7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grinnell B, Padgett B, Walker D. 1983. Distribution of non-integrated DNA from JC papovavirus in organs of. J Infect Dis 147:669–675. doi: 10.1093/infdis/147.4.669. [DOI] [PubMed] [Google Scholar]
- 10.Newman JT, Frisque RJ. 1997. Detection of archetype and rearranged variants of JC virus in multiple tissues from a pediatric PML patient. J Med Virol 52:243–252. doi: 10.1002/(SICI)1096-9071(199707)52:3<243::AID-JMV2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Atwood WJ, Amemiya K, Traub R, Harms J, Major EO. 1992. Interaction of the human polyomavirus, JCV, with human B lymphocytes. Virology 190:716–723. doi: 10.1016/0042-6822(92)90909-9. [DOI] [PubMed] [Google Scholar]
- 12.Major EO, Amemiya K, Elder G, Houff SA. 1990. Glial cells of the human developing brain and B cells of the immune system share a common DNA binding factor for recognition of the regulatory sequences of the human polyomavirus, JCV. J Neurosci Res 27:461–471. doi: 10.1002/jnr.490270405. [DOI] [PubMed] [Google Scholar]
- 13.Caldarelli-Stefano R, Vago L, Omodeo-Zorini E, Mediati M, Losciale L, Nebuloni M, Costanzi G, Ferrante P. 1999. Detection and typing of JC virus in autopsy brains and extraneural organs of AIDS patients and non-immunocompromised individuals. J Neurovirol 5:125–133. doi: 10.3109/13550289909021994. [DOI] [PubMed] [Google Scholar]
- 14.Bayliss J, Karasoulos T, McLean CA. 2013. Immunosuppression increases JC polyomavirus large T antigen DNA load in the brains of patients without progressive multifocal leukoencephalopathy. J Infect Dis 207:133–136. doi: 10.1093/infdis/jis668. [DOI] [PubMed] [Google Scholar]
- 15.Tan CS, Ellis LC, Wuthrich C, Ngo L, Broge TA Jr, Saint-Aubyn J, Miller JS, Koralnik IJ. 2010. JC virus latency in the brain and extraneural organs of patients with and without progressive multifocal leukoencephalopathy. J Virol 84:9200–9209. doi: 10.1128/JVI.00609-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agnihotri SP, Wuthrich C, Dang X, Nauen D, Karimi R, Viscidi R, Bord E, Batson S, Troncoso J, Koralnik IJ. 2014. A fatal case of JC virus meningitis presenting with hydrocephalus in a human immunodeficiency virus-seronegative patient. Ann Neurol 76:140–147. doi: 10.1002/ana.24192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah KV. 1996. Polyomaviruses, p 2027–2043. In Fields BN, Knipe DM, Howley PM. (eds), Fields virology. Lippincott-Raven, Philadelphia, PA. [Google Scholar]
- 18.Dugan AS, Gasparovic ML, Atwood WJ. 2008. Direct correlation between sialic acid binding and infection of cells by two human polyomaviruses (JC virus and BK virus). J Virol 82:2560–2564. doi: 10.1128/JVI.02123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komagome R, Sawa H, Suzuki T, Suzuki Y, Tanaka S, Atwood WJ, Nagashima K. 2002. Oligosaccharides as receptors for JC virus. J Virol 76:12992–13000. doi: 10.1128/JVI.76.24.12992-13000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu CK, Wei G, Atwood WJ. 1998. Infection of glial cells by the human polyomavirus JC is mediated by an N-linked glycoprotein containing terminal α2,6-linked sialic acids. J Virol 72:4643–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neu U, Maginnis MS, Palma AS, Stroh LJ, Nelson CD, Feizi T, Atwood WJ, Stehle T. 2010. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 8:309–319. doi: 10.1016/j.chom.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, Dugan A, Stanifer M, Bhatnagar A, Kroeze WK, Roth BL, Atwood WJ. 2004. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 306:1380–1383. doi: 10.1126/science.1103492. [DOI] [PubMed] [Google Scholar]
- 23.Assetta B, Maginnis MS, Gracia Ahufinger I, Haley SA, Gee GV, Nelson CD, O'Hara BA, Allen Ramdial SA, Atwood WJ. 2013. 5-HT2 receptors facilitate JC polyomavirus entry. J Virol 87:13490–13498. doi: 10.1128/JVI.02252-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gheuens S, Wuthrich C, Koralnik IJ. 2013. Progressive multifocal leukoencephalopathy: why gray and white matter. Annu Rev Pathol 8:189–215. doi: 10.1146/annurev-pathol-020712-164018. [DOI] [PubMed] [Google Scholar]
- 25.Berger JR, Aksamit AJ, Clifford DB, Davis L, Koralnik IJ, Sejvar JJ, Bartt R, Major EO, Nath A. 2013. PML diagnostic criteria: consensus statement from the AAN Neuroinfectious Disease Section. Neurology 80:1430–1438. doi: 10.1212/WNL.0b013e31828c2fa1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Astrom K, Mancall E, Richardson EP Jr. 1958. Progressive multifocal leukoencephalopathy. Brain 81:93–127. doi: 10.1093/brain/81.1.93. [DOI] [PubMed] [Google Scholar]
- 27.Brooks B, Walker D. 1984. Progressive multifocal leukoencephalopathy. Neurol Clin 2:299–313. [PubMed] [Google Scholar]
- 28.Cinque P, Koralnik IJ, Gerevini S, Miro JM, Price RW. 2009. Progressive multifocal leukoencephalopathy in HIV-1 infection. Lancet Infect Dis 9:625–636. doi: 10.1016/S1473-3099(09)70226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO. 2012. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev 25:471–506. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grabar S, Lanoy E, Allavena C, Mary-Krause M, Bentata M, Fischer P, Mahamat A, Rabaud C, Costagliola D, Clinical Epidemiology Group of the French Hospital Database on HIV. 2008. Causes of the first AIDS-defining illness and subsequent survival before and after the advent of combined antiretroviral therapy. HIV Med 9:246–256. doi: 10.1111/j.1468-1293.2008.00554.x. [DOI] [PubMed] [Google Scholar]
- 31.Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, Bennett CL. 2009. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol 10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 32.Major EO. 2009. Reemergence of PML in natalizumab-treated patients–new cases, same concerns. N Engl J Med 361:1041–1043. doi: 10.1056/NEJMp0906248. [DOI] [PubMed] [Google Scholar]
- 33.Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. 2005. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N Engl J Med 353:362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- 34.Kleinschmidt-DeMasters BK, Tyler KL. 2005. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med 353:369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- 35.Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. 2005. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med 353:375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- 36.Zaheer F, Berger JR. 2012. Treatment-related progressive multifocal leukoencephalopathy: current understanding and future steps. Ther Adv Drug Safety 3:227–239. doi: 10.1177/2042098612453849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wuthrich C, Dang X, Westmoreland S, McKay J, Maheshwari A, Anderson MP, Ropper AH, Viscidi RP, Koralnik IJ. 2009. Fulminant JC virus encephalopathy with productive infection of cortical pyramidal neurons. Ann Neurol 65:742–748. doi: 10.1002/ana.21619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koralnik IJ, Wuthrich C, Dang X, Rottnek M, Gurtman A, Simpson D, Morgello S. 2005. JC virus granule cell neuronopathy: a novel clinical syndrome distinct from progressive multifocal leukoencephalopathy. Ann Neurol 57:576–580. doi: 10.1002/ana.20431. [DOI] [PubMed] [Google Scholar]
- 39.Ballesta B, Gonzalez H, Martin V, Ballesta JJ. 2017. Fatal ruxolitinib-related JC virus meningitis. J Neurovirol 23:783–785. doi: 10.1007/s13365-017-0558-4. [DOI] [PubMed] [Google Scholar]
- 40.Banks WA, Erickson MA. 2010. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis 37:26–32. doi: 10.1016/j.nbd.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 41.Pasqualetti M, Ori M, Castagna M, Marazziti D, Cassano GB, Nardi I. 1999. Distribution and cellular localization of the serotonin type 2C receptor messenger RNA in human brain. Neuroscience 92:601–611. doi: 10.1016/S0306-4522(99)00011-1. [DOI] [PubMed] [Google Scholar]
- 42.Nichols DE, Nichols CD. 2008. Serotonin receptors. Chem Rev 108:1614–1641. doi: 10.1021/cr078224o. [DOI] [PubMed] [Google Scholar]
- 43.Haley SA, O'Hara BA, Nelson CD, Brittingham FL, Henriksen KJ, Stopa EG, Atwood WJ. 2015. Human polyomavirus receptor distribution in brain parenchyma contrasts with receptor distribution in kidney and choroid plexus. Am J Pathol 185:2246–2258. doi: 10.1016/j.ajpath.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang B, Palcic MM, Mo H, Goldstein IJ, Hindsgaul O. 2001. Rapid determination of the binding affinity and specificity of the mushroom Polyporus squamosus lectin using frontal affinity chromatography coupled to electrospray mass spectrometry. Glycobiology 11:141–147. doi: 10.1093/glycob/11.2.141. [DOI] [PubMed] [Google Scholar]
- 45.Maginnis MS, Stroh LJ, Gee GV, O'Hara BA, Derdowski A, Stehle T, Atwood WJ. 2013. Progressive multifocal leukoencephalopathy-associated mutations in the JC polyomavirus capsid disrupt lactoseries tetrasaccharide c binding. mBio 4:e00247-13. doi: 10.1128/mBio.00247-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wharton KA Jr, Quigley C, Themeles M, Dunstan RW, Doyle K, Cahir-McFarland E, Wei J, Buko A, Reid CE, Sun C, Carmillo P, Sur G, Carulli JP, Mansfield KG, Westmoreland SV, Staugaitis SM, Fox RJ, Meier W, Goelz SE. 2016. JC polyomavirus abundance and distribution in progressive multifocal leukoencephalopathy (PML) brain tissue implicates myelin sheath in intracerebral dissemination of infection. PLoS One 11:e0155897. doi: 10.1371/journal.pone.0155897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geoghegan EM, Pastrana DV, Schowalter RM, Ray U, Gao W, Ho M, Pauly GT, Sigano DM, Kaynor C, Cahir-McFarland E, Combaluzier B, Grimm J, Buck CB. 2017. Infectious entry and neutralization of pathogenic JC polyomaviruses. Cell Rep 21:1169–1179. doi: 10.1016/j.celrep.2017.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neu U, Maginnis MS, Palma AS, Ströh L, Feizi T, Atwood WJ, Stehle T. 2010. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 8:309–319. doi: 10.1016/j.chom.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stroh LJ, Maginnis MS, Blaum BS, Nelson CD, Neu U, Gee GV, O'Hara BA, Motamedi N, DiMaio D, Atwood WJ, Stehle T. 2015. The greater affinity of JC polyomavirus capsid for α2,6-linked lactoseries tetrasaccharide c than for other sialylated glycans is a major determinant of infectivity. J Virol 89:6364–6375. doi: 10.1128/JVI.00489-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pho MT, Ashok A, Atwood WJ. 2000. JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J Virol 74:2288–2292. doi: 10.1128/JVI.74.5.2288-2292.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Querbes W, Benmerah A, Tosoni D, Di Fiore PP, Atwood WJ. 2004. A JC virus-induced signal is required for infection of glial cells by a clathrin- and eps15-dependent pathway. J Virol 78:250–256. doi: 10.1128/JVI.78.1.250-256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Assetta B, Maginnis MS, Gracia Ahufinger I, Haley SA, Gee GV, Nelson CD, O'Hara BA, Allen Ramdial SA, Atwood WJ. 2013. 5-Ht2 Receptors facilitate Jc polyomavirus entry. J Virol 87:13490–13498. doi: 10.1128/JVI.02252-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith RL, Canton H, Barrett RJ, Sanders-Bush E. 1998. Agonist properties of N,N-dimethyltryptamine at serotonin 5-HT2A and 5-HT2C receptors. Pharmacol Biochem Behav 61:323–330. doi: 10.1016/S0091-3057(98)00110-5. [DOI] [PubMed] [Google Scholar]
- 54.Strazielle N, Ghersi-Egea JF. 2016. Potential pathways for CNS drug delivery across the blood-cerebrospinal fluid barrier. Curr Pharm Des 22:5463–5476. doi: 10.2174/1381612822666160726112115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng HY, Ikegaya H, Takasaka T, Matsushima-Ohno T, Sakurai M, Kanazawa I, Kishida S, Nagashima K, Kitamura T, Yogo Y. 2005. Characterization of the VP1 loop mutations widespread among JC polyomavirus isolates associated with progressive multifocal leukoencephalopathy. Biochem Biophys Res Commun 333:996–1002. doi: 10.1016/j.bbrc.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 56.Sunyaev SR, Lugovskoy A, Simon K, Gorelik L. 2009. Adaptive mutations in the JC virus protein capsid are associated with progressive multifocal leukoencephalopathy (PML). PLoS Genet 5:e1000368. doi: 10.1371/journal.pgen.1000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delbue S, Branchetti E, Bertolacci S, Tavazzi E, Marchioni E, Maserati R, Minnucci G, Tremolada S, Vago G, Ferrante P. 2009. JC virus VP1 loop-specific polymorphisms are associated with favorable prognosis for progressive multifocal leukoencephalopathy. J Neurovirol 15:51–56. doi: 10.1080/13550280802425467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reid CE, Li H, Sur G, Carmillo P, Bushnell S, Tizard R, McAuliffe M, Tonkin C, Simon K, Goelz S, Cinque P, Gorelik L, Carulli JP. 2011. Sequencing and analysis of JC virus DNA from natalizumab-treated PML patients. J Infect Dis 204:237–244. doi: 10.1093/infdis/jir256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gorelik L, Reid C, Testa M, Brickelmaier M, Bossolasco S, Pazzi A, Bestetti A, Carmillo P, Wilson E, McAuliffe M, Tonkin C, Carulli JP, Lugovskoy A, Lazzarin A, Sunyaev S, Simon K, Cinque P. 2011. Progressive multifocal leukoencephalopathy (PML) development is associated with mutations in JC virus capsid protein VP1 that change its receptor specificity. J Infect Dis 204:103–114. doi: 10.1093/infdis/jir198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Padgett BL, Rogers CM, Walker DL. 1977. JC Virus, a human polyomavirus associated with progressive multifocal leukoencephalopathy: additional biological characteristics and antigenic relationships. Infect Immun 15:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. 1971. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet i:1257–1260. [DOI] [PubMed] [Google Scholar]
- 62.Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. 1985. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc Natl Acad Sci U S A 82:1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mandl C, Walker DL, Frisque RJ. 1987. Derivation and characterization of POJ cells, transformed human fetal glial cells that retain their permissivity for JC virus. J Virol 61:755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferenczy MW, Johnson KR, Marshall LJ, Monaco MC, Major EO. 2013. Differentiation of human fetal multipotential neural progenitor cells to astrocytes reveals susceptibility factors for JC virus. J Virol 87:6221–6231. doi: 10.1128/JVI.00396-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Messam CA, Hou J, Gronostajski RM, Major EO. 2003. Lineage pathway of human brain progenitor cells identified by JC virus susceptibility. Ann Neurol 53:636–646. doi: 10.1002/ana.10523. [DOI] [PubMed] [Google Scholar]
- 66.Major EO, Vacante DA. 1989. Human fetal astrocytes in culture support the growth of the neurotropic human polyomavirus, JCV. J Neuropathol Exp Neurol 48:425–436. doi: 10.1097/00005072-198907000-00004. [DOI] [PubMed] [Google Scholar]
- 67.Schaumburg C, O'Hara BA, Lane TE, Atwood WJ. 2008. Human embryonic stem cell-derived oligodendrocyte progenitor cells express the serotonin receptor and are susceptible to JC virus infection. J Virol 82:8896–8899. doi: 10.1128/JVI.00406-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kondo Y, Windrem MS, Zou L, Chandler-Militello D, Schanz SJ, Auvergne RM, Betstadt SJ, Harrington AR, Johnson M, Kazarov A, Gorelik L, Goldman SA. 2014. Human glial chimeric mice reveal astrocytic dependence of JC virus infection. J Clin Invest 124:5323–5336. doi: 10.1172/JCI76629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peterson JN, Lin B, Shin J, Phelan PJ, Tsichlis P, Schwob JE, Bullock PA. 2017. Replication of JC virus DNA in the G144 oligodendrocyte cell line is dependent upon Akt. J Virol 91:e00735-17. doi: 10.1128/JVI.00735-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Assetta B, De Cecco M, O'Hara B, Atwood WJ. 2016. JC polyomavirus infection of primary human renal epithelial cells is controlled by a type I IFN-induced response. mBio 7:e00903-16. doi: 10.1128/mBio.00903-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miyamura T, Yoshiike K, Takemoto K. 1980. Characterization of JC papovavirus adapted to grow in human embryonic kidney cells. J Virol 35:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gee GV, O'Hara BA, Derdowski A, Atwood WJ. 2013. Pseudovirus mimics cell entry and trafficking of the human polyomavirus JCPyV. Virus Res 178:281–286. doi: 10.1016/j.virusres.2013.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nelson CD, Carney DW, Derdowski A, Lipovsky A, Gee GV, O'Hara B, Williard P, DiMaio D, Sello JK, Atwood WJ. 2013. A retrograde trafficking inhibitor of ricin and Shiga-like toxins inhibits infection of cells by human and monkey polyomaviruses. mBio 4:e00729-13. doi: 10.1128/mBio.00729-13. [DOI] [PMC free article] [PubMed] [Google Scholar]