

Abstract
Objective
To test the association of antemortem CSF biomarkers with postmortem pathology in Lewy body disorders (LBD).
Methods
Patients with autopsy-confirmed LBD (n = 24) and autopsy-confirmed Alzheimer disease (AD) (n = 23) and cognitively normal (n = 36) controls were studied. In LBD, neuropathologic criteria defined Lewy body α-synuclein (SYN) stages with medium/high AD copathology (SYN + AD = 10) and low/no AD copathology (SYN − AD = 14). Ordinal pathology scores for tau, β-amyloid (Aβ), and SYN pathology were averaged across 7 cortical regions to obtain a global cerebral score for each pathology. CSF total tau (t-tau), phosphorylated tau at threonine181, and Aβ1-42 levels were compared between LBD and control groups and correlated with global cerebral pathology scores in LBD with linear regression. Diagnostic accuracy for postmortem categorization of LBD into SYN + AD vs SYN − AD or neocortical vs brainstem/limbic SYN stage was tested with receiver operating curves.
Results
SYN + AD had higher CSF t-tau (mean difference 27.0 ± 8.6 pg/mL) and lower Aβ1-42 (mean difference −84.0 ± 22.9 g/mL) compared to SYN − AD (p < 0.01, both). Increasing global cerebral tau and plaque scores were associated with higher CSF t-tau (R2 = 0.15–0.16, p < 0.05, both) and lower Aβ1-42 (R2 = 0.43–0.49, p < 0.001, both), while increasing cerebral SYN scores were associated with lower CSF Aβ1-42 (R2 = 0.31, p < 0.001) and higher CSF t-tau/Aβ1-42 ratio (R2 = 0.27, p = 0.01). CSF t-tau/Aβ1-42 ratio had 100% specificity and 90% sensitivity for SYN + AD, and CSF Aβ1-42 had 77% specificity and 82% sensitivity for neocortical SYN stage.
Conclusions
Higher antemortem CSF t-tau/Aβ1-42 and lower Aβ1-42 levels are predictive of increasing cerebral AD and SYN pathology. These biomarkers may identify patients with LBD vulnerable to cortical SYN pathology who may benefit from both SYN and AD-targeted disease-modifying therapies.
Roughly 40% of all patients with Lewy body disorders (LBD) (i.e., Parkinson disease [PD], PD with dementia [PDD]; dementia with Lewy bodies [DLB]) have sufficient amyloid plaque and tau tangle pathology for a concomitant diagnosis of Alzheimer disease (AD) at autopsy.1–6 Furthermore, increasing levels of cerebral tau and amyloid pathology in postmortem brains of patients with LBD are associated with shorter survival, rapider time to develop dementia in relation to onset of motor parkinsonism, and higher burden of cortical α-synuclein (SYN) pathology.2 Thus, AD copathology has significant consequences in LBD, and antemortem identification of this patient subgroup has important implications for prognosis and clinical management in LBD.7 CSF levels of the protein constituents of AD neuropathology (i.e., total tau [t-tau], phosphorylated tau [p-tau], and β-amyloid [Aβ1-42]) have been extensively studied in the context of AD,8 but CSF biomarker work in LBD is largely restricted to living cohorts without postmortem validation. Furthermore, neuroimaging modalities to visualize cerebral SYN pathology during life are lacking, and CSF SYN analytes are yet to be validated,9 so in vivo measures that predict cerebral SYN pathology are urgently needed.
Here, we test the hypothesis that AD CSF biomarkers measured during life predict postmortem cortical severity of both AD-associated tau tangles and Aβ plaques, as well as SYN Lewy pathology, in a cohort of 24 patients with autopsy-confirmed LBD. We find evidence of a direct association of CSF Aβ1-42 and CSF t-tau/Aβ1-42 ratio with cerebral SYN Lewy pathology.
Methods
Patients
Patients were followed up in clinical cores of the Penn Udall Center for Parkinson's Disease Research, Frontotemporal Degeneration Center, or Alzheimer's Disease Core Center and selected from the Penn Integrated Neurodegenerative Disease Database10 as of April 1, 2017, to identify patients with an antemortem clinical diagnosis of LBD (PD, PDD, DLB), autopsy confirmation through the Penn Center for Neurodegenerative Disease brain bank of SYN pathology (brainstem, limbic, or neocortical stage), and baseline antemortem CSF samples for study. We included reference cohorts with available CSF data and age-matched normal cognition (n = 36) or autopsy-confirmed AD neuropathology without SYN copathology (n = 23) (table e-1, links.lww.com/WNL/A257).
Standard protocol approvals, registrations, and patient consents
All procedures were performed with informed consent under institutional review board approval.
CSF collection and analysis
CSF was collected under standard operating procedures and analyzed with a Luminex xMAP immunoassay platform (Luminex, Austin, TX) to measure CSF t-tau, p-tau (phosphorylated at threonine-181), and Aβ1-42 as described.8
Neuropathologic examination
Autopsy procedures were performed as previously described11 with sampling of fresh brain tissue according to a standardized atlas and fixed in 10% neutral buffered formalin or 70% ethanol with 150 mmol/L NaCl overnight, processed, and embedded in paraffin for sectioning. For neuropathologic diagnosis, 6-μm sections were cut and stained with immunohistochemistry with established antibodies specific for hyperphosphorylated tau (PHF-1), SYN (SYN303), Aβ (Nab228), and phosphorylated TDP-43 (p409-410) and chemically stained with the amyloid-binding dye Thioflavin-S to detect neuritic plaques. Expert neuropathologists (E.B.L., J.Q.T.) applied currently validated diagnostic criteria12,13 to assign Braak tau, Thal amyloid, Consortium to Establish a Registry for Alzheimer's Disease neuritic plaque, and SYN Lewy body stages, as well as the final diagnosis for each case.
We categorized patients with medium- or high-level AD as having AD copathology (SYN + AD) and patients with no or low-level AD pathology as those without significant AD copathology (SYN − AD) according to neuropathologic criteria12 as described.1,2 Alternative analyses compared patients with low/medium/high AD copathology (n = 18) to SYN with no AD (n = 6).
To obtain a continuous measure of global neuropathologic severity for tau-positive tangles, amyloid-positive plaques, and SYN-positive Lewy bodies/Lewy neurites, we calculated an average of ordinal scores (i.e., 0 = rare/none, 1 = mild, 2 = moderate, 3 = severe) obtained at autopsy using diagnostic criteria in 7 cortical regions. Briefly, the medial temporal lobe severity was calculated by averaging the ordinal scores in the amygdala, hippocampal entorhinal cortex, and cornu ammonis/subiculum regions. The global cerebral scores were derived from the average of ordinal scores in the medial temporal lobe, superior/midtemporal lobe, angular cortex, midfrontal cortex, and anterior cingulate gyrus as described.1,2 We also calculated a global subcortical score for SYN pathology from sections of medulla, substantia nigra, nonnigral midbrain, pons, striatum, globus pallidus, and thalamus.
Statistical analysis
Demographics were compared between groups by use of χ2 analysis for categorical data and cerebral neuropathology scores with the Mann-Whitney U test. CSF analyte measurements did not have a normal distribution and thus were natural log–transformed for analysis. Transformed CSF biomarker values and demographics at CSF collection were compared across groups with a 1-way analysis of variance with planned post hoc t tests for individual group comparisons. We performed analysis of covariance models to adjust for disease duration and time to autopsy at CSF collection (table e-2, links.lww.com/WNL/A257).
Linear regression with transformed CSF biomarker values as the dependent variable was used to test the independent association of the global cerebral score for each pathology. To account for demographic variables (i.e., age at CSF), time interval from onset of disease to CSF collection (years), time from CSF collection to autopsy (years), sex, and clinical diagnosis (PD/PDD vs DLB), we tested univariate models to predict each CSF analyte. Demographic variables with significant associations were added to the univariate base model including global cerebral pathology score with a stepwise approach, and bayesian information criteria14 were used to derive a final demographic-adjusted model for comparison with univariate pathology base models. Because demographic data were not influential in our models (table e-3, links.lww.com/WNL/A257), we report univariate model data (table e-4).
Diagnostic accuracy for postmortem pathology in LBD was tested with receiver operating curve (ROC) analysis to predict SYN + AD pathology (compared to SYN − AD) and neocortical SYN stage (compared to brainstem/limbic stage). To avoid overfitting, we performed a bootstrapping random sampling procedure with 1,000 bootstrap samples to generate a 95% confidence interval (CI) for the area under the curve (AUC) value for each analyte and report both the sensitivity and specificity for optimal cut points from this study and a previously cross-validated diagnostic CSF t-tau/Aβ1-42 ratio of 0.34 established in a different autopsy-confirmed neurodegenerative disease cohort (i.e., frontotemporal degeneration) sensitive and specific for AD pathology.15
All analyses were 2 tailed with α = 0.05 and performed with SPSS version 23.0 (IBM, Chicago, IL) or STATA version 12.1 (StataCorp, College Station, TX).
Results
Table 1 gives LBD patient data and table e-1 (links.lww.com/WNL/A257) shows data for our reference cohorts of autopsy-confirmed AD and normal controls. CSF analysis finds groupwise differences in all CSF analytes across normal controls, SYN − AD, SYN + AD, and AD (table 2 and figure 1). After adjusting for disease duration or interval to autopsy at the time of CSF collection, we found similar results of higher CSF t-tau and t-tau/Aβ1-42 and p-tau/Aβ1-42 ratios and lower CSF Aβ1-42 in SYN + AD compared to SYN − AD (table e-2, links.lww.com/WNL/A257). An alternative analysis comparing patients with LBD with any level of AD copathology (low, medium. or high, n = 18) to the minority with SYN pathology in the absence of any AD copathology (n = 6) yielded similar results (table e-5).
Table 1.
Autopsy-confirmed LBD cohort clinical, demographic, and neuropathologic data
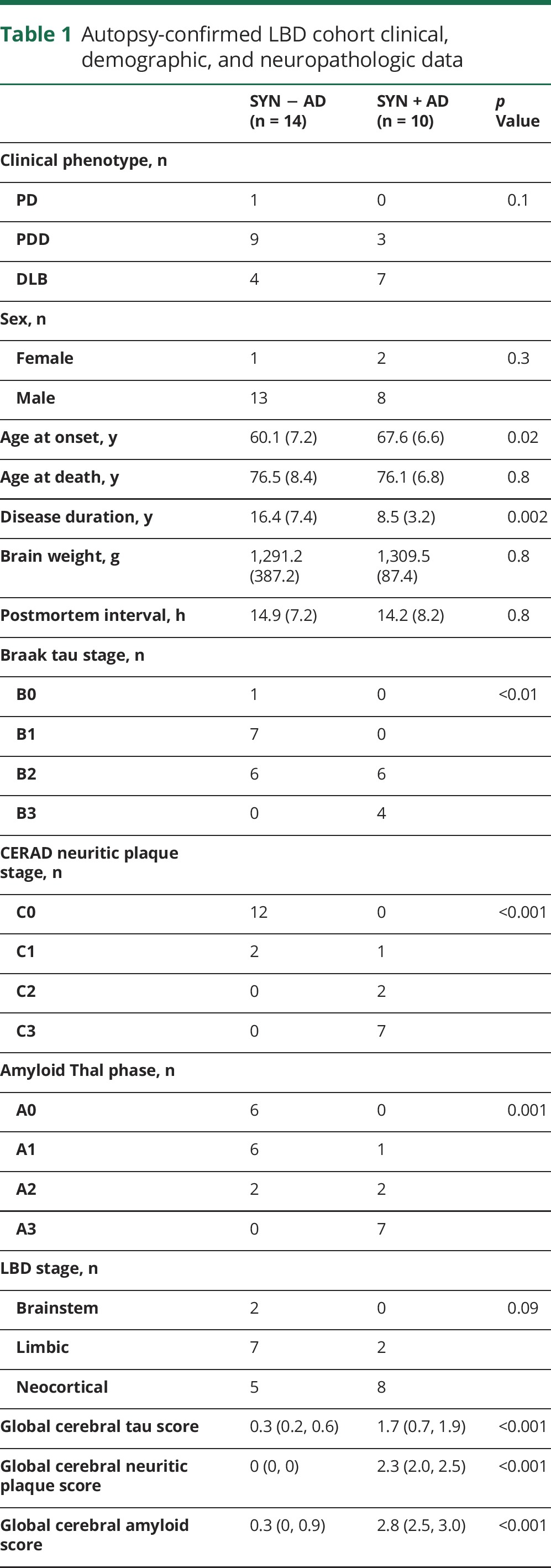
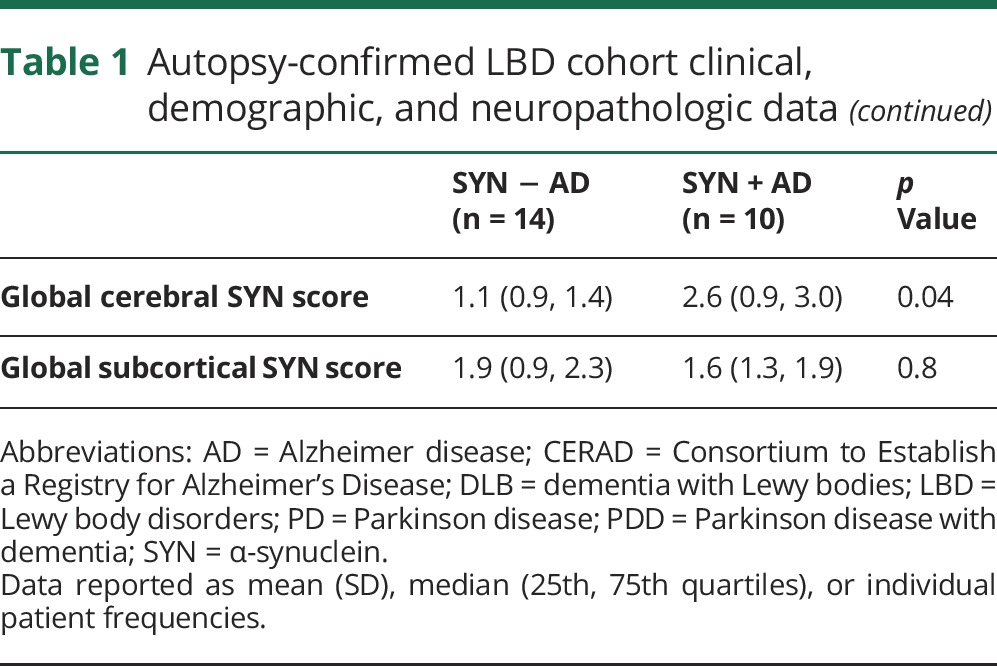
Table 2.
Autopsy-confirmed LBD CSF data
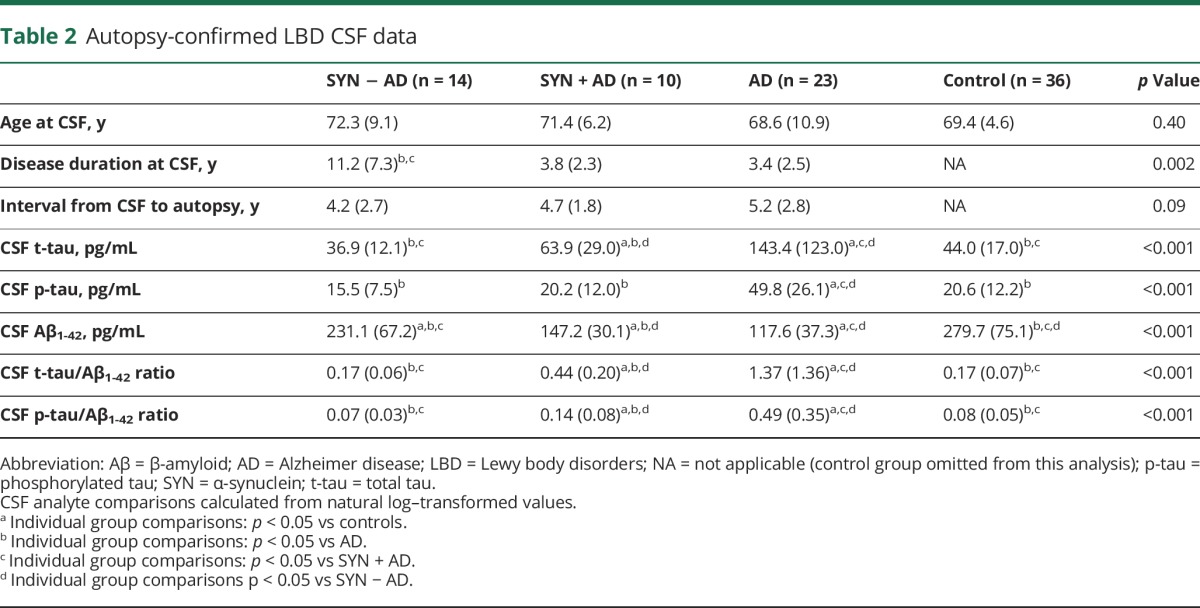
Figure 1. CSF biomarkers in neuropathologic groups of LBD.

Graphs depict individual data points for natural log transformed values CSF (A) t-tau, (B) p-tau, (C) Aβ1-42, (D) t-tau/Aβ1-42 ratio, and (E) p-tau/Aβ1-42 ratio for the normal control (green), SYN − AD (blue), SYN + AD (red), and AD (orange) groups. Bars represent median and interquartile range. Bar denotes p < 0.05. *p < 0.01, **p < 0.001 difference between groups. Aβ = β-amyloid; AD = Alzheimer disease; p-tau = phosphorylated tau; SYN = α-synuclein; t-tau = total tau.
The SYN − AD group had lower levels of CSF Aβ1-42 compared to controls (t = 2.3, df = 48, p < 0.05) (figure 1), and 4 of the 6 patients with SYN pathology and no AD copathology had levels of CSF Aβ1-42 below the mean (table 2) of our control group (range 136–274 pg/mL).
To test the direct relationship between CSF analytes and corresponding neuropathologic substrates, we used univariate linear regression (figure 2 and table e-4, links.lww.com/WNL/A257). We found a mild association between increasing CSF t-tau and global cerebral tau (R2 = 0.15, β = 0.3, p = 0.04) and with amyloid plaque scores (R2 = 0.16, β = 0.17, p = 0.05). We also found lower CSF Aβ1-42 to be moderately associated with increasing global cerebral tau (R2 = 0.43, β = −0.31, p < 0.001), amyloid plaque (R2 = 0.49, β = −0.20, p < 0.001), and SYN scores (R2 = 0.31, β = −0.20, p = 0.004). To test the association of CSF Aβ1-42 with SYN pathology independently from Aβ pathology, we adjusted for global cerebral amyloid plaque score and found a significant independent association with the global cerebral SYN score (β = −0.11, p < 0.05). Furthermore, 3 of the 4 patients with SYN − AD with no cerebral amyloid pathology and low CSF Aβ1-42 had a neocortical distribution of SYN pathology and a global cerebral pathology score ≥1.5. We did not find an association of CSF Aβ1-42 with the global subcortical SYN scores (R2 = 0.08, β = −0.1, p = 0.2), but we did find an association with an average of all total SYN cortical and subcortical regions (R2 = 0.3, β = −0.2, p = 0.009).
Figure 2. Relationship between CSF biomarkers and postmortem pathology.

Scatterplots depict individual patient data of CSF analyte levels plotted against global cerebral pathology scores coded for neuropathologic diagnosis. Fitted lines and R2 values derived from linear regression models predicting CSF analytes. *p < 0.05, **p ≤ 0.01. Aβ = β-amyloid; AD = Alzheimer disease; p-tau = phosphorylated tau; SYN = α-synuclein.
The t-tau/Aβ1-42 ratio was also significantly associated with increasing global cerebral tau (R2 = 0.47, β = 0.63, p < 0.001), amyloid plaque (R2 = 0.46, β = 0.36, p < 0.001), and SYN scores (R2 = 0.27, β = 0.36, p = 0.01). We did not find an association of CSF p-tau with these pathologies or CSF t-tau with global cerebral SYN scores (all p > 0.1). We did not find a significant association of any demographic feature with CSF t-tau, p-tau, or t-tau/Aβ1-42 ratio (data not shown), while CSF Aβ1-42 had significant univariate associations with years from disease onset to CSF collection and clinical diagnosis (table e-3, links.lww.com/WNL/A257). Covariate-adjusted models for CSF Aβ1-42 yielded results similar to the univariate models above (table e-3).
We performed ROC analyses to assess the preliminary evidence of the predictive value of CSF AD biomarkers for both underlying concomitant AD pathology and SYN neocortical stage in LBD (figure 3). We found the highest diagnostic AUC value for t-tau/Aβ1-42 ratio >0.30 (AUC 0.92, 95% CI 0.67–1.0, p < 0.001), with 90% sensitivity and 100% specificity for SYN + AD at this cut point. To avoid overfitting, we also examined a previously validated diagnostic threshold of t-tau/Aβ1-42 ratio of 0.34 to predict AD pathology,15 which also had high diagnostic accuracy (AUC 0.85, 95% CI 0.67–1.0, p = 0.004) with 70% sensitivity and 100% specificity for SYN + AD. Finally, we examined the t-tau/Aβ1-42 ratio to predict any level of AD (i.e., low, medium, or high level) with SYN pathology compared to SYN with no AD copathology and found high diagnostic accuracy (AUC 0.87, 95% CI 0.72–1.0, p = 0.008) with a lower optimal cut point of 0.17 (sensitivity 78%, specificity 83%).
Figure 3. Diagnostic accuracy of CSF biomarkers for AD pathology and neocortical LBD stage in LBD.

Receiver operating curves for diagnostic accuracy of CSF biomarkers to predict (A) SYN + AD pathology or (B) neocortical LBD stage pathology. Tables list optimal cut point with sensitivity, specificity, AUC, and 95% CI derived from a random sampling procedure with 1,000 bootstrap samples. Aβ = β-amyloid; AD = Alzheimer disease; AUC = area under the curve; CI = confidence interval; LBD = Lew body disorders; p-tau = phosphorylated tau; SYN = α-synuclein; t-tau = total tau.
We found CSF Aβ1-42 to have the highest predictive value for a neocortical stage of SYN pathology (AUC 0.76, 95% CI 0.54–0.94) with 77% sensitivity and 82% specificity using a cut point of 185 pg/mL. We did not find significant predictive value of CSF t-tau, p-tau, or p-tau/Aβ1-42 ratio for neocortical SYN stage (AUC 0.47–0.67, p > 0.1 for all).
Discussion
Here, we provide tissue validation for AD CSF biomarkers in a relatively large and well-characterized autopsy-confirmed LBD cohort. Previous CSF studies in LBD focus largely on clinical samples without autopsy confirmation, which significantly limits the interpretation because of the poor clinical diagnostic accuracy of LBD phenotypes16,17 and high frequency of mixed pathologies across the clinical spectrum of LBD.1–3,5,7 Furthermore, the few previous CSF studies in LBD that include autopsy samples18–21 examined small numbers of patients, used only categorical measures of AD pathology, and did not examine SYN pathology. We found both a robust difference in AD CSF biomarker levels between SYN + AD and SYN − AD categorical neuropathologic groups (figure 1) and direct associations of these antemortem measurements with continuous measures of postmortem AD pathology (figure 2). Furthermore, we found high diagnostic accuracy of the CSF t-tau/Aβ1-42 ratio to distinguish patients with LBD with SYN + AD pathology from those with low/no AD copathology (figure 3). Finally, we found an association of antemortem CSF Aβ1-42 and t-tau/Aβ1-42 ratio with severity of postmortem cerebral SYN pathology (figure 2), which was independent of severity of cerebral amyloidosis, and high diagnostic accuracy to predict neocortical stage SYN pathology (figure 3). These data have important implications for clinical care and therapeutic trials in LBD.
AD and SYN pathology commonly coexist in LBD. Several modalities of evidence, including autopsy,1–5,7 genetic,22 and neuroimaging studies,23–25 highlight the detrimental effects of AD copathology on cognition and prognosis in LBD. Furthermore, increasing cerebral AD pathology is often associated with higher cerebral SYN pathology in LBD.1–3,5–7 These clinical studies mirror the growing cell26,27 and animal model28 data that support a hypothesis of distinct strains29 of pathogenic SYN that spread throughout the CNS, along with varying degrees of AD-associated tauopathy.30 Thus, it is imperative for clinical care and clinical trials in LBD to accurately detect the subset of patients with AD copathology because these patients appear to have a divergent natural history and possible altered response to therapeutics compared to those with relatively “pure” synuclienopathy.7 Our data highlight the potential prognostic use of AD CSF biomarkers in LBD to identify patients with AD copathology who are at risk for rapid decline. Previous work in AD has found a similar direct association of AD CSF biomarkers with both postmortem AD pathology31 and in vivo molecular imaging of AD pathology.32 Furthermore, AD CSF biomarkers are currently used in AD clinical trials to track target engagement for tau- and amyloid-directed therapies. While AD-targeted therapies are currently understudied in LBD, our data suggest that CSF t-tau/Aβ1-42 and Aβ1-42 levels may be used in a similar manner in future LBD clinical trials targeting AD copathology.
Our cohort included patients with clinical PD, PDD, and DLB. CSF biomarker studies in living patients with LBD largely find groupwise differences in AD CSF biomarkers between these clinical LBD phenotypes (see elsewhere for comprehensive review9). Postmortem3,5 and emerging in vivo tau and amyloid molecular imaging studies24,25 report similar findings of variable but largely increasing levels of AD copathology across the spectrum of PD, PDD, and DLB. Despite these groupwise differences, we and others previously found that no clear pathologic or genetic substrate could clearly substantiate the clinical distinction of PDD and DLB.2,33 Furthermore, it is recommended that studies examining the underlying biology of synucleinopathies include the full spectrum of LBD.34 Despite this strong rationale for our study design, it is possible that clinical phenotype could have influenced our results; however, when we adjusted for clinical diagnosis, we still found a significant association of CSF Aβ1-42 with postmortem global cerebral pathologies (table e-3, links.lww.com/WNL/A257), and clinical diagnosis did not appear to influence the other CSF biomarkers. Further study is needed in larger groups of patients identified during prodromal presymptomatic period before the onset of clinical symptoms35,36 and followed up to autopsy to fully elucidate the associations of CSF biomarkers across clinical phenotypes.
We did not find an association of CSF p-tau with postmortem pathology or a difference in CSF p-tau in our LBD pathology groups compared to normal controls (figure 1) in this dataset. Tau is hyperphosphorylated in AD-associated neurofibrillary pathology, and CSF levels of both t-tau and p-tau in AD reflect the severity of postmortem tau pathology,31 likely through release of pathogenic tau protein into the CSF from degenerating ghost tangles. In contrast, CSF t-tau can also be elevated in a range of nonneurodegenerative insults to the CNS and may reflect nonspecific neuronal damage. Some data suggest that CSF tau biomarkers may be influenced by SYN pathology in a manner that is distinct from aging and AD,9 but the exact nature of this interaction is currently unclear. Indeed, we find that AD without SYN pathology had altered CSF biomarkers compared to SYN + AD (figure 1), despite similar plaque and tangle stages (table 1 and table e-1, links.lww.com/WNL/A257). Our CSF measurement uses an immunoassay that does not allow direct assessment of phosphorylation at each individual peptide, and we examined only 1 phospho-epitope, so it is possible that analytic factors could also contribute to this negative finding.
We found a moderate association of antemortem CSF Aβ1-42 and the CSF t-tau/Aβ1-42 ratio with postmortem global cerebral synuclein scores. Experimental model data suggest both an interaction between Aβ and SYN fibrils to promote synapse loss37 and SYN pathology38 and synergistic interactions between tau and SYN polymerization.26,27 Furthermore, low baseline CSF Aβ1-42 has been linked to greater cognitive decline in PD39 and DLB,40 and cerebral SYN pathology is one of the strongest correlates of dementia in PD.1 Thus, our findings reinforce the prognostic association of CSF Aβ1-42 and t-tau/Aβ1-42 ratio in LBD and provide a link between antemortem CSF AD biomarkers and postmortem cerebral SYN pathology. Postmortem findings in LBD find strong correlations of all 3 pathologies,1–3,5 and it is possible that the association between CSF Aβ1-42 and SYN pathology could be influenced by cerebral amyloidosis. However, when we included global cerebral amyloid plaque score, we found an independent association with SYN pathology. Furthermore, patients with SYN − AD had lower CSF Aβ1-42 compared to normal controls (figure 1), and the majority of this subset of patients with SYN − AD with no cerebral amyloid pathology and low CSF Aβ1-42 had significant neocortical SYN pathology, suggesting that cerebral SYN pathology alone may influence CSF Aβ1-42 levels. However, we cannot rule out very early amyloid pathologic processes that are not detectable with standard staining techniques in these patients. Our findings of high diagnostic accuracy of CSF Aβ1-42 to predict neocortical synucleinopathy in the context of the clinical LBD spectrum are important. CSF analysis is a relatively low-cost biomarker approach for LBD, and neuroimaging techniques cannot currently detect cortical synuclein pathology. Furthermore, CSF SYN assays are yet to be fully optimized, and values may be influenced by a range of confounding factors.9 Our findings of an association of CSF Aβ1-42 with global cerebral SYN scores and total SYN scores, but not subcortical SYN scores, suggest that low CSF Aβ1-42 is associated with increased progression of SYN pathology from the brainstem to cortical regions. While we cannot determine the topographic distribution of SYN pathology at the time of CSF collection, the association with a widespread neocortical pattern of SYN pathology at end-stage disease suggests that AD CSF biomarkers could lead to early detection of patients with LBD with AD copathology at greater risk for progression of cerebral SYN pathology.
There are additional limitations of our work to consider. Despite the rarity of autopsy-confirmed samples with antemortem CSF and the relative size of our cohort, we cannot fully assess potential clinical variables that may influence CSF analyte levels. Furthermore, while we used a bootstrapping procedure for ROC curves, these data provide proof of concept for the detection of AD copathology in LBD, and the absolute diagnostic cut points found here (figure 3) require replication in future larger autopsy-confirmed datasets. Finally, our data are retrospective and from a tertiary academic center, which may limit generalizability to the general population.
We find predictive value for AD CSF biomarkers in LBD for both AD and SYN pathology. Future work with tissue validation of CSF biomarkers from a larger group of individuals followed up prospectively with serial molecular imaging and clinical assessments will be important to further characterize the dynamic changes in tau, amyloid, and SYN pathology across the LBD spectrum toward the goal of personalized molecular therapies.
Acknowledgment
The authors thank the patients and their families who made this study possible.
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- AUC
area under the curve
- CI
confidence interval
- DLB
dementia with Lewy bodies
- LBD
Lewy body disorders
- PD
Parkinson disease
- PDD
Parkinson disease with dementia
- p-tau
phosphorylated tau
- ROC
receiver operating curve
- SYN
α-synuclein
- t-tau
total tau
Footnotes
Editorial, page 537
Author contributions
D.J.I.: drafting/revising manuscript, study concept/design, analysis, interpretation of data, statistical analysis, obtained funding. S.X.X.: statistical supervision, revising manuscript. D.C. and N.N.: acquisition of data, revising manuscript. R.S.A. and C.T.M.: study concept/design, revising manuscript. E.B.L.: acquisition of data, revising manuscript. D.A.W.: acquisition of data, revising manuscript, obtained funding. D.W.: study concept/design, acquisition of data, revising manuscript. A.C.-P., J.E.D., M.S., A.S., H.I.H., and L.M.S.: acquisition of data, revising manuscript. M.G. and J.Q.T.: study concept/design, acquisition of data, revising manuscript, obtained funding.
Study funding
Funding provided by the National Institute on Aging (AG010124), National Institute of Neurological Disorders and Stroke (NS088341, NS053488), and National Center for Advancing Translational Sciences of the NIH under award TL1TR001880. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosure
D. Irwin reports travel stipend for academic research meeting from GE Healthcare. S. Xie receives research funding from US NIH and serves as a consultant to Roche on 2 cancer clinical trials. D. Coughlin, N. Nevler, R. Akhtar, C. McMillan, and E. Lee, report no disclosures relevant to the manuscript. D. Wolk received grant funding from Merck, Biogen, Avid Radiopharmaceuticals and Eli Lilly and personal fees from GE Healthcare, Merck, and Janssen. D. Weintraub has received research funding or support from Michael J. Fox Foundation for Parkinson's Research, NIH (National Institute for Neurological Disorders and Stroke), Novartis Pharmaceuticals, Department of Veterans Affairs, Avid Radiopharmaceuticals, Alzheimer's Disease Cooperative Study, and International Parkinson and Movement Disorder Society; honoraria for consultancy from Acadia, Biogen, Biotie (Acorda), Bracket, Clintrex LLC, Eisai Inc, Eli Lilly, Lundbeck, Takeda, UCB, and the CHDI Foundation; license fee payments from the University of Pennsylvania for QUIP and QUIP-RS; royalties from Wolters Kluwer; and fees for legal consultation for lawsuits related to medication prescribing in patients with PD. A. Chen-Plotkin reports no disclosures relevant to the manuscript. J. Duda has received research funding or support from Michael J. Fox Foundation for Parkinson's Research, NIH, and Department of Veterans Affairs. M. Spindler, A. Siderowf, H. Hurtig, L. Shaw, M. Grossman, and J. Trojanowski report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 2012;72:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irwin DJ, Grossman M, Weintraub D, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 2017;16:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol 2008;115:427–436. [DOI] [PubMed] [Google Scholar]
- 4.Jellinger KA, Seppi K, Wenning GK, Poewe W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson's disease. J Neural Transm (Vienna) 2002;109:329–339. [DOI] [PubMed] [Google Scholar]
- 5.Compta Y, Parkkinen L, O'Sullivan SS, et al. Lewy- and Alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain 2011;134:1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halliday G, Hely M, Reid W, Morris J. The progression of pathology in longitudinally followed patients with Parkinson's disease. Acta Neuropathol 2008;115:409–415. [DOI] [PubMed] [Google Scholar]
- 7.Irwin DJ, Lee VM, Trojanowski JQ. Parkinson's disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 2013;14:626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mollenhauer B, Parnetti L, Rektorova I, et al. Biological confounders for the values of cerebrospinal fluid proteins in Parkinson's disease and related disorders. J Neurochem 2016;139(suppl 1):290–317. [DOI] [PubMed] [Google Scholar]
- 10.Xie SX, Baek Y, Grossman M, et al. Building an integrated neurodegenerative disease database at an academic health center. Alzheimers Dement 2011;7:e84–e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toledo JB, Van Deerlin VM, Lee EB, et al. A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement 2014;10:477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montine TJ, Monsell SE, Beach TG, et al. Multisite assessment of NIA-AA guidelines for the neuropathologic evaluation of Alzheimer's disease. Alzheimers Dement 2016;12:164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raftery AE. Bayesian model selection in social research. Sociol Methodol 1995;25:111–163. [Google Scholar]
- 15.Irwin DJ, McMillan CT, Toledo JB, et al. Comparison of cerebrospinal fluid levels of tau and Abeta 1-42 in Alzheimer disease and frontotemporal degeneration using 2 analytical platforms. Arch Neurol 2012;69:1018–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adler CH, Beach TG, Hentz JG, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology 2014;83:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson PT, Jicha GA, Kryscio RJ, et al. Low sensitivity in clinical diagnoses of dementia with Lewy bodies. J Neurol 2010;257:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Doring F, Trenkwalder C, Schlossmacher MG. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol 2011;10:230–240. [DOI] [PubMed] [Google Scholar]
- 19.Slaets S, Le Bastard N, Theuns J, et al. Amyloid pathology influences abeta1-42 cerebrospinal fluid levels in dementia with Lewy bodies. J Alzheimers Dis 2013;35:137–146. [DOI] [PubMed] [Google Scholar]
- 20.Brunnstrom H, Hansson O, Zetterberg H, Londos E, Englund E. Correlations of CSF tau and amyloid levels with Alzheimer pathology in neuropathologically verified dementia with Lewy bodies. Int J Geriatr Psychiatry 2013;28:738–744. [DOI] [PubMed] [Google Scholar]
- 21.Toledo JB, Brettschneider J, Grossman M, et al. CSF biomarkers cutoffs: the importance of coincident neuropathological diseases. Acta Neuropathol 2012;124:23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuang D, Leverenz JB, Lopez OL, et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 2013;70:223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kantarci K, Lowe VJ, Boeve BF, et al. AV-1451 tau and beta-amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann Neurol 2017;81:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomperts SN, Locascio JJ, Makaretz SJ, et al. Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol 2016;73:1334–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomperts SN, Locascio JJ, Marquie M, et al. Brain amyloid and cognition in Lewy body diseases. Movement Disord 2012;27:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo JL, Covell DJ, Daniels JP, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 2013;154:103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo JL, Lee VM. Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett 2013;587:717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luk KC, Kehm V, Carroll J, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012;338:949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Covell DJ, Robinson JL, Akhtar RS, et al. Novel conformation-selective alpha-synuclein antibodies raised against different in vitro fibril forms show distinct patterns of Lewy pathology in Parkinson's disease. Neuropathol Appl Neurobiol 2017;43:604–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 2014;20:130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tapiola T, Alafuzoff I, Herukka SK, et al. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol 2009;66:382–389. [DOI] [PubMed] [Google Scholar]
- 32.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol 2006;59:512–519. [DOI] [PubMed] [Google Scholar]
- 33.Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 2006;67:1931–1934. [DOI] [PubMed] [Google Scholar]
- 34.Lippa CF, Duda JE, Grossman M, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 2007;68:812–819. [DOI] [PubMed] [Google Scholar]
- 35.Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson's disease. Mov Disord 2015;30:1600–1611. [DOI] [PubMed] [Google Scholar]
- 36.McKeith I, Taylor JP, Thomas A, Donaghy P, Kane J. Revisiting DLB diagnosis: a consideration of prodromal DLB and of the diagnostic overlap with Alzheimer disease. J Geriatr Psychiatry Neurol 2016;29:249–253. [DOI] [PubMed] [Google Scholar]
- 37.Bachhuber T, Katzmarski N, McCarter JF, et al. Inhibition of amyloid-beta plaque formation by alpha-synuclein. Nat Med 2015;21:802–807. [DOI] [PubMed] [Google Scholar]
- 38.Masliah E, Rockenstein E, Veinbergs I, et al. β-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci USA 2001;98:12245–12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siderowf A, Xie SX, Hurtig H, et al. CSF amyloid {beta} 1-42 predicts cognitive decline in Parkinson disease. Neurology 2010;75:1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemstra AW, de Beer MH, Teunissen CE, et al. Concomitant AD pathology affects clinical manifestation and survival in dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2017;88:113–118. [DOI] [PubMed] [Google Scholar]