

Practical Implications
Consider adult-onset Alexander disease in the differential diagnosis of possible behavioral-variant frontotemporal dementia.
Alexander disease is a clinically heterogeneous condition associated with glial fibrillary acidic protein (GFAP) gene mutations initially described in infants, but juvenile and adult forms exist. Adult-onset Alexander disease (AOAD) has an insidious onset of symptoms localized largely to brainstem, but may also include cognitive dysfunction.
Though only a minority of patients with AOAD are reported with neurobehavioral dysfunction,1 formal neuropsychological testing in adults has not been reported. Here we present the neurobehavioral findings of 3 siblings with genetically proven AOAD.
The affected family was ascertained through the University of British Columbia Hospital Clinic for Alzheimer Disease and Related Disorders. Research ethics protocols for the study were approved and informed consent was obtained.
Clinical and genealogic histories and neurologic examinations were performed for all 3 siblings. In addition, 2 of the siblings each underwent 2 standard neuropsychological examinations, along with the State Trait Anxiety Inventory, Beck Depression Inventory, and the Behavioral Rating Scale to quantify behavioral impairment.
Neuroimaging was performed with GE Medical Systems (Chicago, IL) Signa HDxt 1.5T scanner.
Blood samples were collected from affected siblings and unaffected mother. Genomic DNA was extracted using standard protocols. Sibling 1 had whole exome sequencing performed, and selected candidate mutations were validated through Sanger sequencing as previously described.2
The siblings' father developed dementia, bulbar signs, and spastic quadriplegia, and died at age 67. Their paternal aunt and grandfather developed progressive parkinsonism in midlife.
All affected siblings had MRI signs highly suggestive of AOAD (figure e-1 at Neurology.org/cp). Whole exome sequencing found sibling 1 to be heterozygous for a missense substitution in GFAP p.N386S (c.1157A>G; NM_002055.4). Sanger sequencing of DNA samples from siblings 2 and 3 and their unaffected mother confirmed that this mutation segregates with disease.
Table 1 summarizes neurologic findings. Sibling 1's initial presentation was described previously.3 Later difficulties with memory, multitasking, judgment, apathy, empathy, compulsive behaviors. and insight emerged. While there was no social or occupational impairment at initial assessment, follow-up revealed clear functional deterioration, meeting criteria of dementia due to AOAD.
Table 1.
Neurologic findings
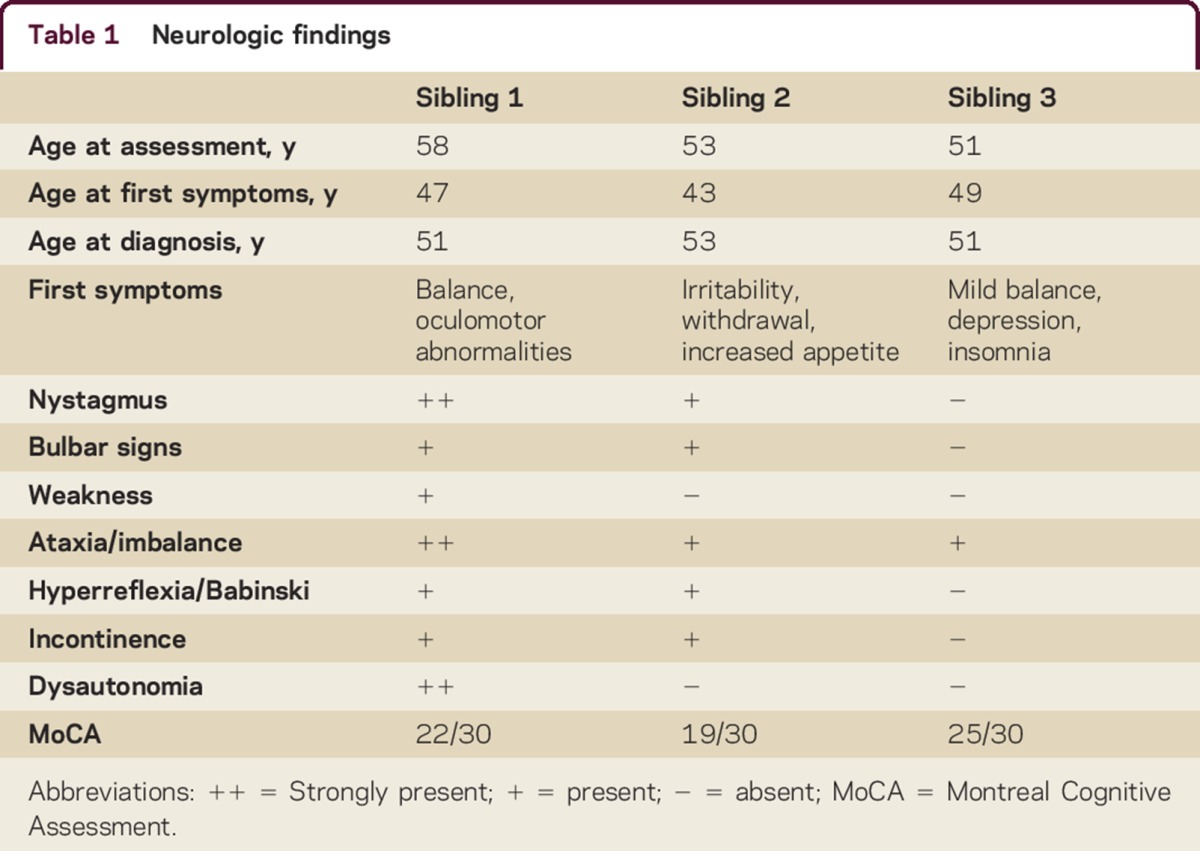
Sibling 2 was evaluated at age 53. Earlier he was diagnosed with depression, with symptoms of irritability and social withdrawal leading to loss of employment. Lacking insight, he progressively became apathetic and impulsive, gaining 40 pounds from uncontrolled appetite. With cognitive and functional impairment, he met criteria for dementia due to AOAD.
Sibling 3 developed imbalance at age 49. She reported insomnia and depression at initial assessment. She met criteria for mild cognitive impairment, with inefficiencies in attention, planning, concentration, and verbal memory retrieval, without reported functional impairment. Observed behavior included inflexibility and reduced insight and affective range. She declined neuropsychological evaluation.
Siblings 1 and 2 underwent 2 neuropsychological examinations 13 months apart. Table 2 summarizes results. Sibling 1's scores fell within the average range and Sibling 2's in the borderline range of general ability and overall intellectual function, adjusted for age and education.
Table 2.
Neuropsychological findings
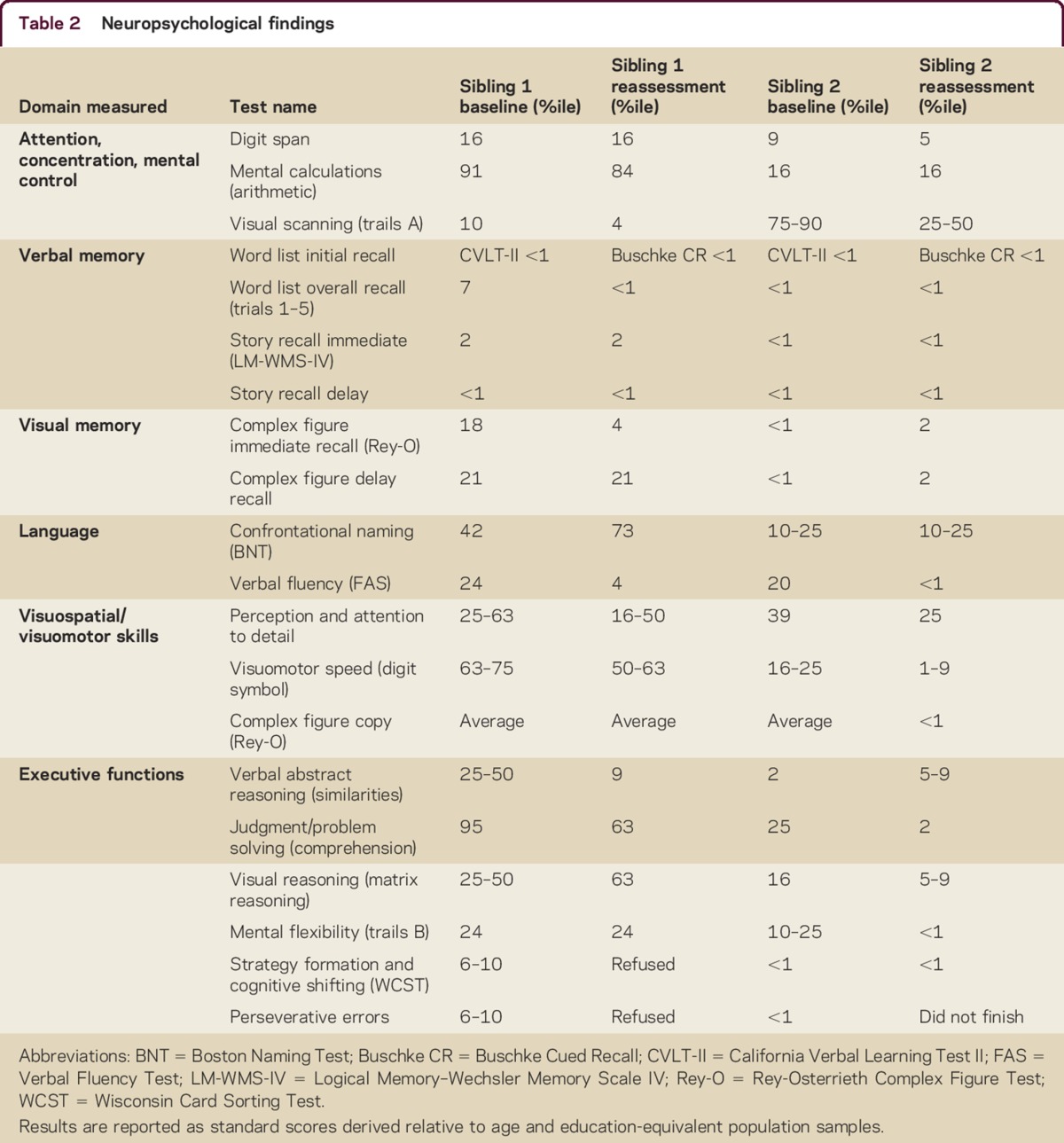
Behaviorally, both showed impairments in insight, affective range, expressive deficit, and impulsivity. On testing, sibling 1 developed rule violations. Sibling 2 demonstrated problems with effort, perseverance, and latency of response. Neither endorsed symptoms of depression or anxiety.
Both siblings showed deficits in attention and executive function domains, with declining abilities between assessments. In verbal memory/learning, both siblings showed poor initial learning, free recall, and retrieval, with sibling 2 performing worse. For both, visual memory was less markedly impaired and visuospatial/visuomotor and language skills were relatively preserved.
DISCUSSION
We present a neurobehavioral assessment of 3 siblings with genetically confirmed AOAD. Their cognitive profile includes progressive decline in attention, executive function, and memory. Behavioral changes include reduced insight and affective range, expressive deficits, and impulsivity. Their neurobehavioral profile reflects frontotemporal dysfunction, meeting criteria4 for possible behavioral-variant frontotemporal dementia (bvFTD).
The predominant neurobehavioral findings in our patients are consistent with other neurocognitive reports in noninfantile Alexander disease.5,6 GFAP p.N386S was previously described presenting only with transient diplopia,7 suggesting that other factors possibly modify phenotypic expression, and neurobehavioral symptoms in AOAD may be underrecognized unless they are scrutinized in detail.
Structural imaging in this family did not indicate frontotemporal changes: neurobehavioral alterations are likely driven by functional network impairment.
We detail the neurobehavioral profiles of 3 siblings with AOAD, characterized as including both frontotemporal behavioral and cognitive impairments, in the context of brainstem symptoms. AOAD should be on the differential when assessing patients with possible bvFTD. Neurobehavioral symptoms in AOAD may be underrecognized. Further study is needed to better characterize phenotype of neurocognitive impairment in AOAD and investigation of network abnormalities with functional imaging is warranted to investigate the mechanism of frontotemporal dysfunction.
AUTHOR CONTRIBUTIONS
M.L. Lichtenstein: study concept and design, acquisition of data, analysis and interpretation of data, drafting, revising, and final approval of manuscript. E. Dwosh: acquisition of data, analysis and interpretation of data, revising and final approval of manuscript. A.R. Chowdhury: acquisition of data, revising and final approval of manuscript. M.J. Farrer: acquisition of data, analysis and interpretation of data, revising and final approval of manuscript. M.B. McKenzie: acquisition of data, analysis and interpretation of data, revising and final approval of manuscript. I. Guella: acquisition of data, analysis and interpretation of data, revising and final approval of manuscript. D.M. Evans: acquisition of data, analysis and interpretation of data, revising and final approval of manuscript. H.B. Nygaard: analysis and interpretation of data, revising and final approval of manuscript. J.R. Shewchuk: analysis and interpretation of data, revising and final approval of manuscript. S. Hayden: acquisition of data, analysis and interpretation of data, revising and final approval of manuscript. J.J.S. Barton: acquisition of data, analysis and interpretation of data, revising and final approval of manuscript. H.H. Feldman: study concept and design, acquisition of data, analysis and interpretation of data, revising and final approval of manuscript.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the family for their ongoing support of research endeavors.
Footnotes
Supplemental data at Neurology.org/cp
STUDY FUNDING
Genetic analyses were supported by the Canada Excellence Research Chairs program and Leading Edge Endowment Funds, provided by the Province of British Columbia, LifeLabs, and Genome BC for the Dr. Donald Rix BC Leadership Chair.
DISCLOSURES
M.L. Lichtenstein receives research support from Association for Frontotemporal Degeneration. E. Dwosh receives a stipend from UBC Department of Medical Genetics for conference-related travel expenses. A.R. Chowdhury has received funding for travel or speaker honoraria from Novartis and Eisai. Her spouse serves on scientific advisory boards for Astellas, MSD, Pfizer, Mundipharma, GlaxoSmithKline, Bayer, and Novartis; serves on speakers' bureaus for Astellas, Pfizer, Novartis, MSD, Janssen, and Bristol-Myers Squibb; and receives research support from Sanofi. M.J. Farrer has served on scientific advisory boards for Michael J. Fox Foundation, Parkinson's Society Canada, EURAC, and Parkinson's UK; serves on the editorial board of Parkinsonism & Related Disorders; is author on patents re: Lrrk2 gene and mutations; is founder and CSO of and holds stock/stock options in Neurocode Labs Inc.; receives research support from the Canadian Federal Government (CERC, CFI, CIHR) and Cunhill Foundation; and receives royalty payments from Lundbeck Inc. and Merck for Lrrk2 mouse models and from Isis Pharma for SNCA mouse model. M.B. McKenzie and I. Guella report no disclosures. D.M. Evans is employed by and holds stocks/stock options in Neurocode Labs. H.B. Nygaard serves as an Associate Editor for Neuroscience Letters and receives research support from National Institutes of Health/National Center for Advancing Translational Sciences, Weston Brain Institute, and Michael Smith Foundation for Health Research. J.R. Shewchuk holds stock/stock options in SolAeroMed. S. Hayden reports no disclosures. J.J.S. Barton serves on a scientific advisory board for Vycor Medical Corporation; has received honoraria as a speaker for the LAUNCH program of Serono Inc; serves as an executive for Medicologic Inc.; serves on the editorial boards of Cortex and PLoS One; receives publishing royalties from UptoDate; receives research support from the National Institute of Mental Health, UBC Academic Enhancement grant; BC Network for Aging Research, Canadian Institutes of Health Research, and Natural Sciences and Engineering Research Council; and has participated in medicolegal cases. H.H. Feldman reports consultative services through service agreements with University of British Columbia and University of California, San Diego, to Eli Lilly, Tau Rx, and Merck; has served as a member of a Data Safety Monitoring Board for Eisai Pharmaceuticals and Diagnostic Monitoring Committee for Genentech/Banner Health; has received publishing royalties from Atlas of Alzheimer's Disease (Informa Health, 2007); is author on a patent re: Detecting and treating dementia; has served as a member of a research policy committee for the Alzheimer's Society of Canada and the Scientific Advisory Board of the Tau Consortium; serves on the Research Executive Committee of the Canadian Consortium of Neurodegeneration and Aging funded by the Canadian Institutes of Health Research; and is Director of the Alzheimer Disease Cooperative Study funded through the National Institute on Aging, which has undertaken clinical trials with sponsors including AC Immune and QR Pharma. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
REFERENCES
- 1.Balbi P, Salvini S, Fundaro C, et al. The clinical spectrum of late-onset Alexander disease: a systematic literature review. J Neurol 2010;257:1955–1962. [DOI] [PubMed] [Google Scholar]
- 2.Steele JC, Guella I, Szu-Tu C, et al. Defining neurodegeneration on Guam by targeted genomic sequencing. Ann Neurol 2015;77:458–468. [DOI] [PubMed] [Google Scholar]
- 3.Pfeffer G, Abegg M, Vertinsky AT, Ceccherini I, Caroli F, Barton JJ. The ocular motor features of adult-onset alexander disease: a case and review of the literature. J Neuroophthalmol 2011;31:155–159. [DOI] [PubMed] [Google Scholar]
- 4.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Restrepo J, Bernardin L, Hammeke T. Neurocognitive decline in Alexander disease. Clin Neuropsychol 2011;25:1266–1277. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida T, Sasayama H, Mizuta I, et al. Glial fibrillary acidic protein mutations in adult-onset Alexander disease: clinical features observed in 12 Japanese patients. Acta Neurol Scand 2011;124:104–108. [DOI] [PubMed] [Google Scholar]
- 7.Sugiyama A, Sawai S, Ito S, et al. Incidental diagnosis of an asymptomatic adult-onset Alexander disease by brain magnetic resonance imaging for preoperative evaluation. J Neurol Sci 2015;354:131–132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.