

 This report details the first organocatalytic method for nitrenoid transfer and its application to intermolecular, site-selective C(sp3)–H amination.
This report details the first organocatalytic method for nitrenoid transfer and its application to intermolecular, site-selective C(sp3)–H amination.
Abstract
This report details the first organocatalytic method for nitrenoid transfer and its application to intermolecular, site-selective C(sp3)–H amination. The method utilizes a trifluoromethyl iminium salt as the catalyst, iminoiodinanes as the nitrogen source, and substrate as the limiting reagent. Activated, benzylic, and aliphatic substrates can all be selectively functionalized in yields up to 87%. A mechanistic proposal for the observed reactivity supported by experimental evidence invokes the intermediacy of a diaziridinium salt or related organic nitrenoid, species that have not been previously explored for the purpose of C–H amination. Finally, examples of late-stage functionalization of complex molecules highlight the selectivity and potential utility of this catalytic method in synthesis.
Introduction
The discovery of new methods for the direct amination of C–H bonds remains a topic of broad interest due to the prevalence of nitrogen in biologically active natural products and pharmaceuticals.1 Among the most captivating and potentially transformative problems of current interest in this area is chemo- and site-selective intermolecular C(sp3)–H amination in the absence of directing groups.2 When the substrate is used as the limiting reagent, such transformations can enable both the direct synthesis of valuable amines from inexpensive precursors and the late-stage functionalization of complex molecules. Although the importance of developing catalytic methods having these features has been appreciated for some time, only recently have initial solutions emerged, which primarily rely on the use of transition metal complexes to promote nitrenoid transfer, either as catalysts or enzyme cofactors (Scheme 1).3,4 Considerable challenges remain to achieving the goal of general methods for intermolecular C–H amination that demonstrate site selectivity when applied to complex substrates.
Scheme 1. Catalytic approaches to intermolecular C(sp3)–H amination via nitrenoid transfer.

Despite the noted advantages of organocatalysis over transition metal or enzymatic catalysis, including potentially complementary reactivity as well as lower cost, toxicity, and impact on the environment,5 to date no organocatalytic approach to nitrenoid transfer in a C–H amination reaction has been reported.6–8 In contrast to nitrogen transfer, organocatalysis of C–H hydroxylation, which typically harnesses the oxygen-transfer capability of three-membered diheterocyclic oxidants such as dioxiranes, oxaziridines, and oxaziridinium salts, is well established.9 We envisioned that nitrogen transfer from these or related species might be achieved and harnessed in a catalytic cycle, leading to a new organocatalytic platform for site-selective C–H amination distinct from previous metal-based approaches. We report herein the application of iminium salt catalysts to the catalysis of selective C–H amination (Scheme 1). The intermediacy of a diaziridinium salt or related species is proposed to account for the observed reactivity, which offers a novel catalytic approach to nitrenoid transfer.
Results and discussion
Previous explorations of nitrogen transfer from oxaziridines and related species have demonstrated reactivity towards olefin aziridination10 and with strong organometallic nucleophiles,11 but no examples of C–H insertion have been reported. In contrast, the use of iminium salt catalysts offered a different opportunity (Scheme 2a). We have previously demonstrated that trifluoromethyl iminium salts can catalyse site-selective aliphatic C–H hydroxylation by aqueous hydrogen peroxide via an oxaziridinium salt.9b We hypothesized that an appropriate iminium salt might similarly catalyse C–H amination by a nitrenoid precursor, potentially involving the intermediacy of a diaziridinium salt or related organic nitrenoid. In principle, the cationic nature of the catalyst should render the nitrogen to be transferred more electrophilic than in the oxaziridine case. Aside from a single report by Lambert on the isolation and unambiguous characterization of an NH-diaziridinium salt,12 very little is known about their reactivity. In that case, evaluation of the nitrogen transfer ability of the diaziridinium was hampered by a propensity for decomposition via ring expansion (Scheme 2b). Given the role of the nitrogen lone pair in facilitating this rearrangement, we favoured a strategy for catalytic nitrogen transfer that might proceed via a diaziridinium or related nitrenoid with a less available nitrogen lone pair.
Scheme 2. Design of the organocatalytic amination reaction.

Consequently, we began our investigations by screening common tosyl nitrenoid precursors as reagents for amination of tetralin catalysed by iminium salt 5a. The use of one equivalent of the iminoiodinane PhINTs in the presence of 20 mol% catalyst led to selective benzylic amination at room temperature using dichloromethane as the solvent (Table 1). The use of two equivalents proved optimal; conversion was diminished when three equivalents were used and no overoxidation products were observed.13 In all cases, yield is limited by conversion; along with the product 4 unreacted tetralin is the only other species observed in more than trace amounts. Initial mechanistic information revealed by control experiments is consistent with the requirement of the iminium species for catalysis of amination. For example, neither tetrafluoroboric acid (which could plausibly be formed in situ by the reaction of trace amounts of water with the catalyst) nor the tetrafluoroborate counterion itself contribute substantially to overall conversion (Table 1, entries 10 & 11). In addition, yields are also dependent on catalyst structure; e.g. iminium salt 5b, which we have previously shown to be suboptimal for oxaziridinium-mediated catalytic hydroxylation,9a gives substantially reduced yield of amination products (Table 1, entry 4).14 Electronic modification of preferred catalyst 5a (Table 1, entries 5–9) resulted in comparable or diminished performance. As such, 5a was selected for further evaluation.
Table 1. Optimization of iminium salt-catalyzed amination a .

| |||
| Entry | Catalyst | Conditions | Yield b |
| 1 | 5a (R1 = Me, R2 = H, R3 = H) | 1 equiv. PhINTs | 14% (20%) |
| 2 | 5a | As shown | 64% (78%) |
| 3 | 5a | 3 equiv. PhINTs | 32% (40%) |
| 4 | 5b (R1 = H, R2 = H, R3 = H) | As shown | 14% (24%) |
| 5 | 5c (R1 = Me, R2 = Cl, R3 = H) | As shown | 46% (52%) |
| 6 | 5d (R1 = Me, R2 = Br, R3 = H) | As shown | 36% (43%) |
| 7 | 5e (R1 = Me, R2 = H, R3 = OMe) | As shown | 62% (78%) |
| 8 | 5f (R1 = Me, R2 = H, R3 = Br) | As shown | 44% (50%) |
| 9 | 5g (R1 = Me, R2 = H, R3 = Cl) | As shown | 36% (46%) |
| 10 | HBF4·Et2O | As shown | 6% (12%) |
| 11 | NaBF4 | As shown | 0% (0%) |
| 12 | — | No catalyst | 0% (0%) |
aReaction conditions unless otherwise noted: 3 (0.5 mmol), PhINTs (1.0 mmol), catalyst (20 mol%), anhydrous CH2Cl2 (2 mL), under N2.
bIsolated yield. Uncorrected GC conversion in parentheses.
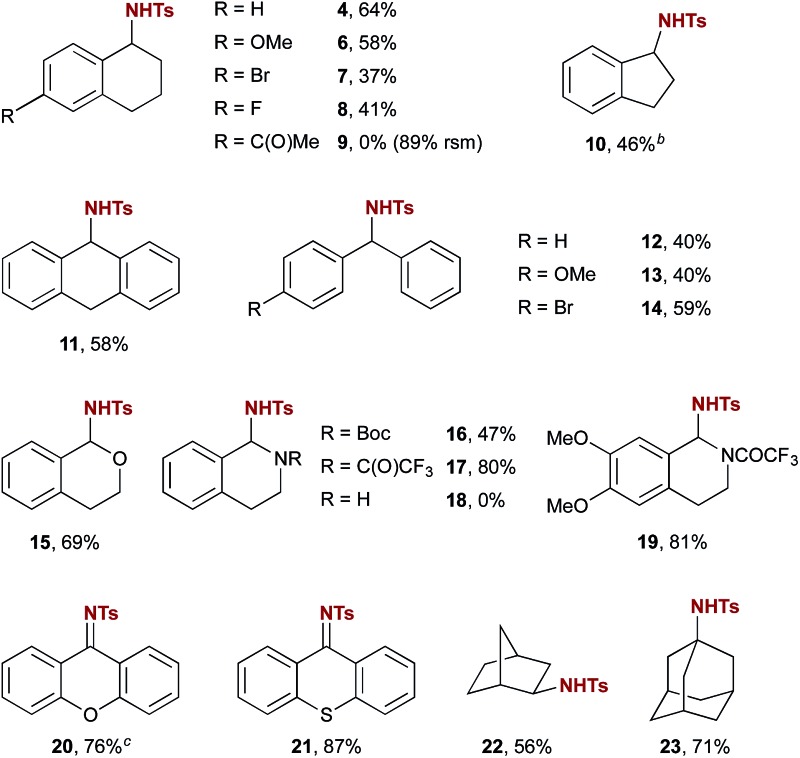
Exploration of the substrate scope of this new catalytic amination method revealed applicability to several different classes of substrates. As shown in Table 2, amination of cyclic benzylic substrates and doubly benzylic acyclic substrates generally proceeded smoothly (e.g. products 4–14).15 One exception, 6-acetyl tetralin, provided no observed products of amination (e.g. product 9). The high yield of recovered starting material (89%) suggests that ketones are compatible with the reaction conditions, but that they may decrease reactivity in this case through an electron-withdrawing effect. In the case of indane, product 10 was isolated as an inseparable mixture of the sulfonamide shown and the corresponding imine in a 6 : 1 ratio. Resubjecting the product mixture to the reaction conditions led to further imine formation with concomitant consumption of 10, confirming that the imine arises from overoxidation of the initial sulfonamide product. Substrates with multiple differentiated benzylic positions wherein one is adjacent to a heteroatom (ethers, amides, and carbamates) showed a strong preference for amination at the more activated position (e.g. products 15–17 and 19). The secondary amine 1,2,3,4-tetrahydroisoquinoline promoted a rapid decomposition of PhINTs; no products of amination were observed. The capability of 5a to catalyse amination adjacent to carbamates and amides provides further evidence that imine formation in the case of 10 likely arises from a second amination of the initial product followed by elimination of tosylamide. Substrates with a strong driving force favouring this overoxidation, such as xanthene and thioxanthene, gave selectively products of C–H imination (20 & 21) and are among the highest yielding substrates investigated.
Table 2. Scope of the reaction: benzylic and aliphatic substrates a .

|
|
aReaction conditions unless otherwise noted: substrate (0.5 mmol), PhINTs (1.0 mmol), 5a (0.1 mmol), anhydrous CH2Cl2 (2 mL), under N2.
bIsolated as an inseparable 6 : 1 mixture of 10 with the corresponding imine.
cIsolated as an inseparable 5.9 : 1 mixture of 20 with the corresponding amine.
The capability of this novel organocatalytic method also extends to aliphatic C–H amination (22 & 23). Notably, amination of norbornane was highly diastereoselective; only the exo product 22 was observed. In all cases amination was highly site-selective; the products shown are the only products observed in more than trace amounts with unreacted substrate making up the balance of the material. A feature of this reaction characteristic of selective intermolecular C–H functionalization reactions is a high dependence of yield on slight variations in substrate structure. In the event, yields varied from low (36%) to high (87%). Although general trends are difficult to discern, substrates that were converted in the highest yields generally benefited from substantial stereoelectronic weakening of the C–H bond of interest (e.g.15–17, 19 and 23).
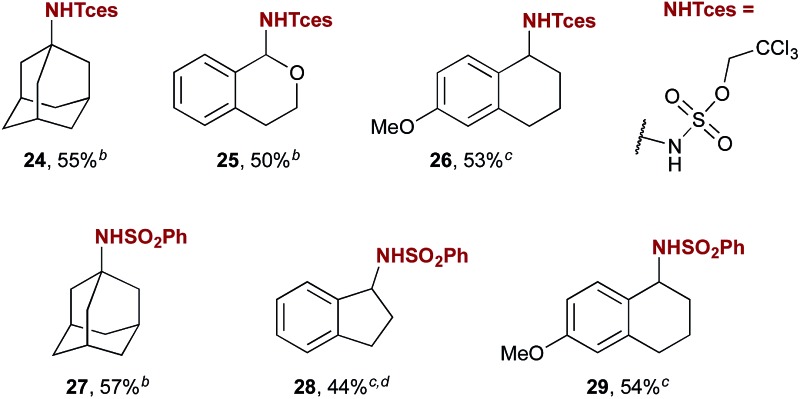
Flexibility in protecting group choice is a feature of this method that increases its potential utility in synthesis, especially given the relatively harsh conditions required for tosyl deprotection that are inherent in the use of the commonly employed PhINTs.3f An investigation of the use of alternative iminoiodinanes revealed that the Tces protecting group, which can be cleaved under milder reductive conditions,16 is compatible with amination catalysed by 5a. Yields for activated, benzylic, and aliphatic substrates are only modestly lower when PhINTces is used (Table 3, products 24–26). The iminoiodinane derived from benzenesulfonamide is also well tolerated (e.g. products 27–29), however those derived from relatively electron poor sulfonamides such as nosyl are not. Overall, this initial investigation of iminoiodinane scope reveals that protecting group choice can be partially tuned to synthetic need.
Table 3. Scope of the reaction: alternative iminoiodinanes a .

|
|
aReaction conditions unless otherwise noted: substrate (1 equiv.), PhINR (2 equiv.), 5a (0.2 equiv.), anhydrous CH2Cl2 (0.25 M), under N2.
bReaction performed on 0.1 mmol scale.
cReaction performed on 0.25 mmol scale.
dIsolated as an inseparable mixture of 28 with the corresponding imine.
Despite recent advances in transition metal catalysed intermolecular C–H amination, few methods are available that have been demonstrated to be applicable to the site-selective late-stage amination of complex molecules.2a Examination of iminium salt-catalyzed C–H amination for this purpose revealed promising initial findings (Fig. 1). For example, amination of ambroxide gave product 30, albeit with modest diastereoselectivity, demonstrating a high degree of site selectivity and the capability of 5a to catalyse amination of non-benzylic ethereal C–H bonds. A dehydroabietylamine17 derivative was selectively aminated at the benzylic position, providing 31 as a 1 : 1 mixture of diastereomers. In order to evaluate whether the final diastereomeric mixture for these substrates arises from the initial amination step or is thermodynamically controlled by a subsequent substitution reaction, we performed a crossover experiment by including 4-methoxybenzenesulfonamide in the reaction mixture, using isochroman as a test substrate (Scheme 3). No crossover was observed, suggesting that stereoselectivity is set in the C–H amination step. Of relevance to the reaction mechanism, this experiment also rules out tosylamide liberated by decomposition of PhINTs as the source of nitrogen in the initial amination step.
Fig. 1. Products of late-stage site-selective amination catalysed by 5a.

Scheme 3. Sulfonamide crossover experiment.

In order to further probe the capabilities of the iminium catalyst we evaluated its performance in an aziridination reaction of trans-stilbene (Scheme 4). Reaction under the standard catalytic conditions but at reduced temperature (4 °C) provided trans-aziridine 35 in 51% yield.18 In comparison, C–H amination is slow at this reduced temperature (trace conversion for a selection of substrates tested). Overall, the observed aziridination capability of catalyst 5a under these unoptimized conditions is consistent with formation of a species capable of nitrenoid transfer.
Scheme 4. Aziridination of trans-stilbene (unoptimized results).

Kinetic isotope effect (KIE) and Hammett analysis experiments were performed in order to further assess the mechanism and transition state of amination. The KIE observed for an intermolecular competition experiment involving isochroman (kH/kD = 2.5) is consistent with C–H cleavage being the rate-determining step (Scheme 5). In comparison to transition metal-catalyzed nitrenoid insertion reactions, the magnitude of this KIE is small and most in line with that observed for Rh2(OAc)4-catalyzed intramolecular amination (kH/kD = 2.6), which is proposed to proceed via concerted nitrenoid C–H insertion.18 Larger KIEs (generally >5) have been observed for nitrenoid insertion reactions thought to proceed through stepwise mechanisms.3f The observed KIE is also similar to that observed for oxygen insertion by TFDO (kH/kD = 2.2 for tertiary hydroxylation),19 for which concerted insertion and radical rebound mechanisms have been proposed.20,21 Hammett studies involving 6-substituted tetralin derivatives suggest an increase in positive charge at the benzylic carbon during the transition state, giving a ρ value of –2.5 for a plot of log(kAr/kPh) vs. σp.22 The previously discussed lack of reactivity of 6-acetyl tetralin and the high degree of diastereoselectivity observed for the exo C–H bond of norbornane (where heterolytic bond cleavage can be stabilized by hyperconjugation) are consistent with this observation. A tighter correlation with σp (R2 = 0.98) than with σ+ (R2 = 0.91) was observed, consistent with a limited role for resonance stabilization of cationic charge. No linear relationship was observed for σ˙. Overall, both the Hammett and KIE studies provide evidence against free radical intermediates or substantial homolytic character of C–H bond cleavage at the site of amination during the transition state.
Scheme 5. Kinetic isotope effect determination.

In an attempt to identify catalyst-derived species formed from the reaction of PhINTs with catalyst 5a, we performed a reaction under the standard conditions but in the absence of any substrate and analysed the crude mixture by LCMS using electrospray ionization to minimize fragmentation. This reaction resulted in a complex mixture of products. Importantly, we did observe a mass consistent with the parent ion peak of diaziridinium 37 along with major fragments that were also consistent with its formation (Scheme 6).23,24 Despite this evidence and the support it provides for the mechanistic hypothesis that guided the design of this method, the involvement of a diaziridinium is still speculative and will ultimately require more detailed investigations into the nature of any nitrenoid generated during the course of the reaction.
Scheme 6. LCMS evidence in support of a postulated diaziridinium intermediate.

Conclusions
In summary, an effective process for iminium salt-catalyzed C–H amination has been detailed. The use of iminoiodinanes as the nitrogen source allows for amination of a variety of substrates at room temperature, and the site selectivity observed for amination of complex substrates reveals potential utility for late-stage diversification. Mechanistic evidence supports the hypothesis that a rarely encountered diaziridinium salt could be the active nitrogen transfer agent, adding to the novelty of the method. To our knowledge, this is the first example of organocatalysis of nitrenoid transfer in any context, and presents an alternative to the use of catalysts containing rare or toxic metals. More broadly, this work combined with our earlier efforts establishes iminium salt catalysis as a general platform for both nitrogen and oxygen transfer in a C–H functionalization process. Future studies are aimed at expanding this platform to address additional challenges in selective C–H functionalization.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
Financial support for this work from the National Institutes of Health (R01 GM124092) and the University of Virginia (generous startup funds) is gratefully acknowledged.
Footnotes
†Electronic supplementary information (ESI) available: Experimental procedures, characterization data for all new products, copies of NMR spectra. See DOI: 10.1039/c7sc03968a
References
- For selected recent reviews see: ; (a) Yamaguchi J., Yamaguchi A. D., Itami K. Angew. Chem., Int. Ed. 2012;51:8960. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; (b) Du Bois J. Org. Process Res. Dev. 2011;15:758. doi: 10.1021/op200046v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vitaku E., Smith D. T., Njardarson J. T. J. Med. Chem. 2014;57:10257. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- (a) Park Y., Kim Y., Chang S. Chem. Rev. 2017;117:9247. doi: 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]; (b) Roizen J. L., Harvey M. E., Du Bois J. Acc. Chem. Res. 2012;45:911. doi: 10.1021/ar200318q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Alderson J. M., Corbin J. R., Schomaker J. M. Acc. Chem. Res. 2017;50:2147. doi: 10.1021/acs.accounts.7b00178. [DOI] [PubMed] [Google Scholar]
- (a) Prier C. K., Zhang R. K., Buller A. R., Brinkmann-Chen S., Arnold F. H. Nat. Chem. 2017;9:629. doi: 10.1038/nchem.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dolan N. S., Scamp R. J., Yang T., Berry J. F., Schomaker J. M. J. Am. Chem. Soc. 2016;138:14658. doi: 10.1021/jacs.6b07981. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Alderson J. M., Phelps A. M., Scamp R. J., Dolan N. S., Schomaker J. M. J. Am. Chem. Soc. 2014;136:16720. doi: 10.1021/ja5094309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Buendia J., Grelier G., Darses B., Jarvis A. G., Taran F., Dauban P. Angew. Chem., Int. Ed. 2016;55:7530. doi: 10.1002/anie.201602022. [DOI] [PubMed] [Google Scholar]; (e) Bess E. N., DeLuca R. J., Tindall D. J., Oderinde M. S., Roizen J. L., Du Bois J., Sigman M. S. J. Am. Chem. Soc. 2014;136:5783. doi: 10.1021/ja5015508. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Roizen J. L., Zalatan D. N., Du Bois J. Angew. Chem., Int. Ed. 2013;52:11343. doi: 10.1002/anie.201304238. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Liu Y., Guan X., Wong E. L.-M., Liu P., Huang J.-S., Che C.-M. J. Am. Chem. Soc. 2013;135:7194. doi: 10.1021/ja3122526. [DOI] [PubMed] [Google Scholar]; (h) Michaudel Q., Thevenet D., Baran P. S. J. Am. Chem. Soc. 2012;134:2547. doi: 10.1021/ja212020b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Gephart III R. T., Warren T. H. Organometallics. 2012;31:7728. [Google Scholar]; (j) Lescot C., Darses B., Collet F., Retailleau P., Dauban P. J. Org. Chem. 2012;77:7232. doi: 10.1021/jo301563j. [DOI] [PubMed] [Google Scholar]; (k) Liang C., Collet F., Robert-Peillard F., Müller P., Dodd R. H., Dauban P. J. Am. Chem. Soc. 2008;130:343. doi: 10.1021/ja076519d. [DOI] [PubMed] [Google Scholar]; (l) Fiori K. W., Du Bois J. J. Am. Chem. Soc. 2007;129:562. doi: 10.1021/ja0650450. [DOI] [PubMed] [Google Scholar]; (m) Liang C., Robert-Peillard F., Fruit C., Müller P., Dodd R. H., Dauban P. Angew. Chem., Int. Ed. 2006;45:4641. doi: 10.1002/anie.200601248. [DOI] [PubMed] [Google Scholar]; (n) Yu X.-Q., Huang J.-S., Zhou X.-G., Che C.-M. Org. Lett. 2000;2:2233. doi: 10.1021/ol000107r. [DOI] [PubMed] [Google Scholar]
- Site-selective catalytic azidation can be a complementary approach, see: ; (a) Karimov R. R., Sharma A., Hartwig J. F. ACS Cent. Sci. 2016;2:715. doi: 10.1021/acscentsci.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang X., Bergsten T. M., Groves J. T. J. Am. Chem. Soc. 2015;137:5300. doi: 10.1021/jacs.5b01983. [DOI] [PubMed] [Google Scholar]; (c) Sharma A., Hartwig J. F. Nature. 2015;517:600. doi: 10.1038/nature14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan D. W. C. Nature. 2008;455:304. doi: 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]
- Qin Y., Zhu L., Luo S. Chem. Rev. 2017;117:9433. doi: 10.1021/acs.chemrev.6b00657. [DOI] [PubMed] [Google Scholar]
- For an alternative organocatalytic approach to that described here, relying on intermolecular trapping of carbocation or carbon-centered radical intermediates, see: ; (a) Pandey G., Laha R. Angew. Chem., Int. Ed. 2015;54:14875. doi: 10.1002/anie.201506990. [DOI] [PubMed] [Google Scholar]; (b) Amaoka Y., Kamijo S., Hoshikawa T., Inoue M. J. J. Org. Chem. 2012;77:9959. doi: 10.1021/jo301840e. [DOI] [PubMed] [Google Scholar]
- For an alternative transition metal-free approach to intermolecular amination see: Fan R., Li W., Pu D., Zhang L., Org. Lett., 2009, 11 , 1425 . [DOI] [PubMed] [Google Scholar]
- (a) Shuler W. G., Johnson S. L., Hilinski M. K. Org. Lett. 2017;19:4790. doi: 10.1021/acs.orglett.7b02178. [DOI] [PubMed] [Google Scholar]; (b) Wang D., Shuler W. G., Pierce C. J., Hilinski M. K. Org. Lett. 2016;18:3826. doi: 10.1021/acs.orglett.6b01832. [DOI] [PubMed] [Google Scholar]; (c) Pierce C. J., Hilinski M. K. Org. Lett. 2014;16:6504. doi: 10.1021/ol503410e. [DOI] [PubMed] [Google Scholar]; (d) Adams A. M., Du Bois J. Chem. Sci. 2014;5:656. [Google Scholar]; (e) Litvinas N. D., Brodsky B. H., Du Bois J. Angew. Chem., Int. Ed. 2009;48:4513. doi: 10.1002/anie.200901353. [DOI] [PubMed] [Google Scholar]; (f) Brodsky B. H., Du Bois J. J. Am. Chem. Soc. 2005;127:15391. doi: 10.1021/ja055549i. [DOI] [PubMed] [Google Scholar]
- (a) Andreae S., Schmitz D. Synthesis. 1991:327. [Google Scholar]; (b) For an example of aziridination by a diaziridine acting as a nucleophile, see: Ishihara H., Hori K., Sugihara H., Ito Y. N., Katsuki T., Helv. Chim. Acta, 2002, 85 , 4272 . [Google Scholar]
- (a) Zhou Z., Ma Z., Behnke N. E., Gao H., Kürti L. J. Am. Chem. Soc. 2017;139:115. doi: 10.1021/jacs.6b12712. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao H., Zhou Z., Kwon D.-H., Coombs J., Jones S., Behnke N. E., Ess D. H., Kürti L. Nat. Chem. 2017;9:681. doi: 10.1038/nchem.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J. M., Lambert T. H. Tetrahedron. 2014;70:4111. [Google Scholar]
- Portionwise addition of PhINTs offered no improvement and in most cases substantially diminished yields
- An additional control reaction revealed that the imine derived from loss of the N-methyl group of 5a, which could also plausibly be formed under the reaction conditions, does not catalyse amination of tetralin (0% yield)
- Attempted aminations of n-alkylbenzenes, such as ethylbenzene and n-nonylbenzene, led to no more than trace conversion
- (a) Guthikonda K., Du Bois J. J. Am. Chem. Soc. 2002;124:13672. doi: 10.1021/ja028253a. [DOI] [PubMed] [Google Scholar]; (b) Barrett A. G. M., Betts M. J., Fenwick A. J. Org. Chem. 1985;50:169. [Google Scholar]
- Kuzu O. F., Gowda R., Sharma A., Robertson G. P. Mol. Cancer Ther. 2014;13:1690. doi: 10.1158/1535-7163.MCT-13-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziridine 35 was isolated as an inseparable mixture of 35 with the N-tosyl imine derived from 2,2-diphenylacetaldehyde (10 : 1 favouring 35). A control experiment confirmed that this side product arises from decomposition of 35 under the reaction conditions
- Harvey M. E., Musaev D. G., Du Bois J. J. Am. Chem. Soc. 2011;133:17207. doi: 10.1021/ja203576p. [DOI] [PubMed] [Google Scholar]
- Curci R., d'Accolti L., Fusco C. Acc. Chem. Res. 2006;39:1. doi: 10.1021/ar050163y. [DOI] [PubMed] [Google Scholar]
- Zou L., Paton R. S., Eschenmoser A., Newhouse T. R., Baran P. S., Houk K. N. J. Org. Chem. 2013;78:4037. doi: 10.1021/jo400350v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the ESI for details, including experimental procedure and data analysis
- For computational support of the possibility of direct nitrogen transfer from a diaziridinium, see: Washington I., Houk K. N., Armstrong A., J. Org. Chem., 2003, 68 , 6497 . [DOI] [PubMed] [Google Scholar]
- See the ESI for details
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.