

Summary
This study reports the complete genome sequence of chimpanzee herpesvirus (ChHV), an alphaherpesvirus isolated from a chimpanzee. Although closely related to human herpes simplex virus type 2 (HSV2), the level of sequence diversity confirms that ChHV is sufficiently distinct to be considered a member of a different virus species rather than a variant strain of HSV2. Phylogenetic comparison with other simplexviruses at several levels supports the hypothesis that HSV2 and ChHV co-evolved with their respective human and chimpanzee hosts and raises questions regarding the evolutionary origins of HSV1.
The alphaherpesviruses are exquisitely adapted to their natural host species and phylogenetic analyses suggest they have co-evolved with their hosts [3]. Limited phylogenetic analysis of an alphaherpesvirus (ChHV) isolated from a chimpanzee placed it within a hominid virus clade as expected based on host phylogeny [2]. However, ChHV and HSV2 are more closely related to each other than either is to HSV1. The questions that these initial phylogenetic analyses raised concerning the origins of the three hominid viruses warranted further analysis of the ChHV isolate. Here, we report and analyze the complete sequence of the ChHV genome.
ChHV strain 105640 was originally isolated in 2004 from a chimpanzee (Pan troglodytes) during an outbreak of oral lesions in a captive colony and was propagated in Vero cells as described [2]. Viral DNA was purified from infected cells on NaI gradients, sequencing was performed on a Genome Sequencer 20 (Roche) using the manufacturer’s Shotgun Sequencing protocol, gaps were closed using a conventional shotgun library, and the sequence was assembled and analyzed as described previously [5]. The genomic termini were mapped by homology with HSV2, since the sequences were highly similar. Protein sequence comparisons were performed using an in-house perl script implementing the Needleman-Wunsch global alignment algorithm, and phylogenetic trees were constructed with Mega 5 [4].
The genome of ChHV (GenBank accession JQ360576) is 153,158 bp long with an overall G + C content of 68.1 %. The organization of genes and repeat regions of the ChHV genome is orthologous with that of HSV2 (Fig. 1), and the overall genome sequence identity at the DNA level is 92 % and 79 % with HSV2 and HSV1, respectively. The close sequence similarity between ChHV and HSV2 extends along the entire genome, with the greatest divergence being in non-coding regions of the inverted repeats.
Fig. 1.
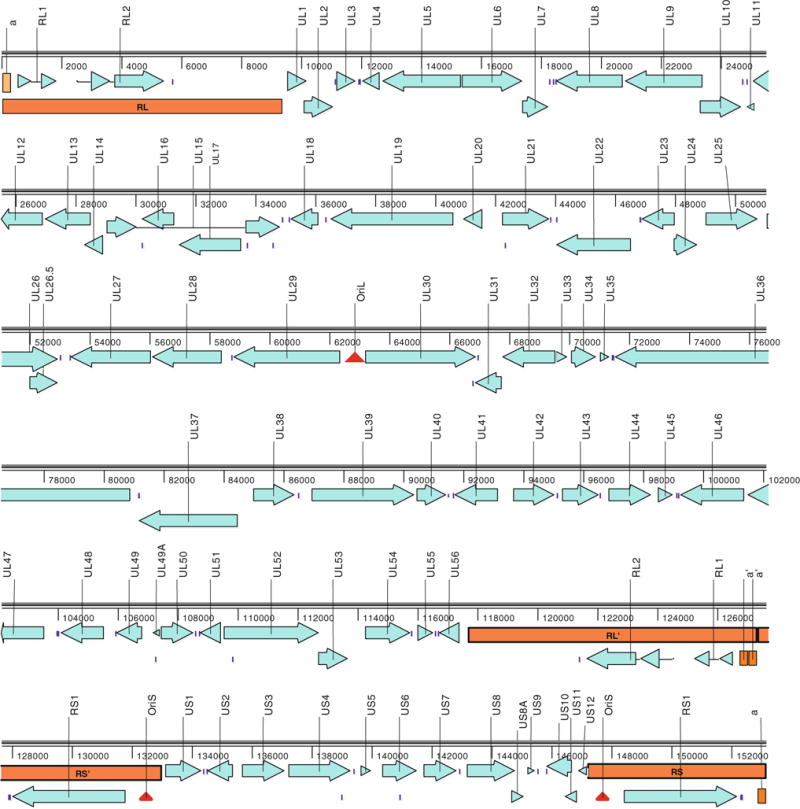
Genetic map of the ChHV genome. ORFs are indicated in aquamarine, inverted repeat regions in orange, origins of replication as red triangles, and polyadenylation signals as small blue bars (color figure online)
Phylogenetic trees constructed by whole genome alignment of the other six completely sequenced primate simplexviruses (not shown) confirmed that HSV2 is more closely related to ChHV than to HSV1. All three viruses do, however, group together in a hominid virus clade, distinct from the cercopithecine monkey virus clade and the more distant platyrrhine monkey virus SQHV (squirrel monkey herpesvirus 1). Like HSV1 and HSV2, ChHV has asymmetric short-region origin of replication (OriS) sequences and an RL1 gene with similarity to the mammalian GADD45 protein, whereas the cercopithecine simplexviruses have symmetrical OriS sequences and lack an RL1 gene homologue [3].
All open reading frames (ORFs) of HSV2 are conserved in ChHV, with an average sequence identity at the amino acid (AA) level of 91 %. The average AA sequence identity level between ChHV and HSV1 is 76 %. ORFs RL1 (γ34.5), RL2 (ICP0), US5 (gJ), US11 (RNA-binding tegument protein) and US12 (ICP47) are significantly less conserved than other genes; the AA sequence identity between these ChHV and HSV2 proteins is 56.4 %, 80.1 %, 62.1 %, 82.9 % and 66.4 %, respectively. The ChHV RL1 gene stop codon lies further downstream as compared to HSV2 (and HSV1), adding 80 AAs that do not show similarity to any known protein. This region of the ChHV and HSV2 strain G genomes was cloned and re-sequenced, and these results were confirmed. There are two putative introns in the RL2 gene of both HSV2 and ChHV, while there are three introns in HSV1. US4 (glycoprotein G) is full length in HSV2, ChHV and the cercopithecine simplexviruses, while it lacks the soluble domain in HSV1.
The remarkable lack of unique features that differentiate ChHV from HSV2 raises the question of whether ChHV is merely an HSV2 variant that infected a captive chimpanzee. To address this possibility, the thymidine kinase (TK; UL23) gene sequence of ChHV was aligned with TK gene sequences of 186 strains of HSV1 and 114 strains of HSV2 (from GenBank), and the average evolutionary distances were calculated using the maximum composite likelihood method with standard errors estimated by bootstrap analysis [4]. The ChHV-HSV2 TK distance (0.041 ± 0.011 base substitutions/site) is significantly higher (p < 0.05) than distances within strains of HSV1 (0.011 ± 0.003) or HSV2 (0.007 ± 0.002). Similar analysis of 63 HSV2 glycoprotein G gene (US4) sequences showed a distance of 0.003 ± 0.001 substitutions/site among HSV2 strains and 0.088 ± 0.008 between HSV2 strains and ChHV. These results support the conclusion that ChHV is not a variant strain of HSV2 [2]. Previous serological cross-reactivity data [1, 2] also indicated that HSV2 and ChHV were closely related but distinct viruses, and that other captive chimpanzees harbored a virus antigenically similar to ChHV.
Phylogenetic analyses [2] based on estimated times of divergence of host species supported the hypothesis that ChHV and other primate simplexviruses coevolved with their hosts, with the sole exception of HSV1. We plotted pairwise distances obtained by aligning the DNA polymerase gene (UL30) of HSV1, HSV2, ChHV, simian B virus (BV), herpesvirus Papio 2 (HVP2) and SQHV, versus the pairwise distances between the mitochondrial genomes of their host species (Homo sapiens, Pan troglodytes, Macaca mulatta, Papio hamadryas and Saimiri sciureus). As shown in Fig. 2 (triangles), pairwise distances align on a credible straight line (R2 = 0.90). The distance of the ChHV/HSV2 pair from the Homo//Pan pair (point a) and other points on the graph are compatible with coevolution of ChHV and HSV2 with their respective chimpanzee and human hosts. HSV1 lies considerably off of the “coevolution straight line” (open circle), but it falls directly on it (solid circle) if the mitochondrial genome of Pongo abelii (orangutan) is used as the HSV1 host species. The point representing the distance of ChHV/HSV1 vs. Pan/Pongo mitochondrial DNA also falls close to the straight line (gray circle). Similar results, with points defining a straight line (R2 = 0.96), were obtained by aligning whole genome sequences of simplexviruses (not shown). It is possible that HSV1 and HSV2 evolved independently in different epidemiological niches within the human host. However, these results suggest that HSV1 may have been acquired at some point in human evolution from another primate host, possibly related to modern Pongidae, while HSV2 is the original human simplexvirus.
Fig. 2.
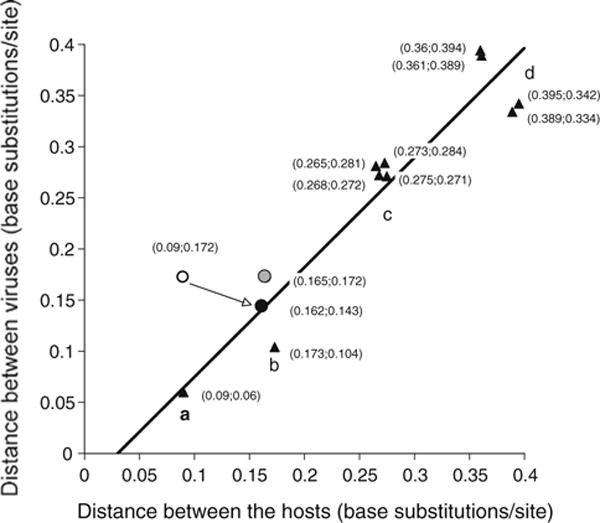
Correlation of evolutionary distance between primate simplexviruses and their host species. The nucleotide sequences of UL30 ORF of HSV2, ChHV, HVP2, BV and SQHV were aligned, and pairwise distances were plotted against pairwise distances of aligned mitochondrial genomes of the putative hosts in these combinations (triangles): a ChHV/HSV2 vs. Pan/Homo; b HVP2/BV vs. Papio/Macaca; c BV/other viruses vs. Macaca/other hosts; d SQHV/other viruses vs. Saimiri/other hosts. The regression line has a slope of 1.07 and an R2 value of 0.90. The open circle represents the pairs HSV1/ChHV vs. Homo/Pan, the solid circle represents the pairs HSV1/HSV2 vs. Pongo/Homo, and the gray circle represents the pairs ChHV/HSV1 vs. Pan/Pongo (color figure online)
In conclusion, the lack of distinctive features between ChHV and HSV2 is remarkable. Molecular and phylogenetic analyses support the notion that ChHV is not a variant strain of HSV2, but rather that ChHV and HSV2 coevolved with their Pan and Homo hosts. Based on these findings, we propose that ChHV be considered for formal designation as a member of a new species, “Panine herpesvirus 3”.
Acknowledgments
This work was supported in part by funding from the Public Health Agency of Canada and the US Public Health Service (P40 RR012317).
Contributor Information
Alberto Severini, Department of Medical Microbiology and Infectious Diseases, University of Manitoba, Winnipeg, MB, Canada; National Microbiology Laboratory, Public Health Agency of Canada, 1015 Arlington Street, Winnipeg, MB R3E 3R2, Canada.
Shaun D. Tyler, National Microbiology Laboratory, Public Health Agency of Canada, 1015 Arlington Street, Winnipeg, MB R3E 3R2, Canada
Geoffrey A. Peters, National Microbiology Laboratory, Public Health Agency of Canada, 1015 Arlington Street, Winnipeg, MB R3E 3R2, Canada
Darla Black, Department of Veterinary Pathobiology, Center for Veterinary Health Sciences, Oklahoma State University, Stillwater, OK, USA.
R. Eberle, Department of Veterinary Pathobiology, Center for Veterinary Health Sciences, Oklahoma State University, Stillwater, OK, USA
References
- 1.Eberle R, Hilliard JK. Serological evidence for variation in the incidence of herpesvirus infections in different species of apes. J Clinl Microbiol. 1989;27:1357–1366. doi: 10.1128/jcm.27.6.1357-1366.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luebcke E, Dubovi E, Black DH, Ohsawa K, Eberle R. Isolation and characterization of a chimpanzee alphaherpesvirus. J Gen Virol. 2006;87:11–19. doi: 10.1099/vir.0.81606-0. [DOI] [PubMed] [Google Scholar]
- 3.McGeoch DJ, Rixon FJ, Davison AJ. Topics in herpesvirus genomics and evolution. Virus Res. 2006;117:90–104. doi: 10.1016/j.virusres.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tyler S, Severini A, Black D, Walker M, Eberle R. Structure and sequence of the saimiriine herpesvirus 1 genome. Virology. 2011;410:181–191. doi: 10.1016/j.virol.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]