

CONSPECTUS
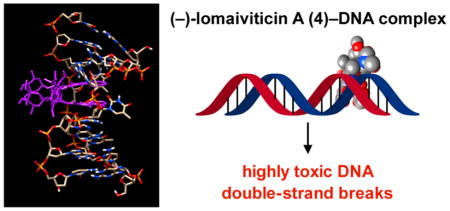
(–)-Lomaiviticin A (4) is a complex C2-symmetric bacterial metabolite that contains two diazofluorene functional groups. The diazofluorene consists of naphthoquinone, cyclo-pentadiene, and diazo substituents fused through a σ- and π-bonding network. Additionally, (–)-lomaiviticin A (4) is a potent cytotoxin, with half-maximal inhibitory potency (IC50) values in the low nanomolar range against many cancer cell lines. Because of limitations in supply, its mechanism of action had remained a “black box” since its isolation in the early 2000s. In this Account, I describe how studies directed toward the total synthesis of (–)-lomaiviticin A (4) provided a platform to elucidate the emergent properties of this metabolite and thereby connect chemical reactivity with cellular phenotype. We first developed a convergent strategy to prepare the diazofluorene (9 + 10 → 13). We then adapted this chemistry to the synthesis of lomaiviticin aglycon (21/22) and the natural monomeric diazofluorene (–)-kinamycin F (3). The key step in the lomaiviticin aglycon (21/22) synthesis involved the stereoselective oxidative coupling of two monomeric diazofluorenes (2 × 18→ 20) to establish the cojoining carbon–carbon bond of the target. As the absolute stereochemistry of the aglycon and carbohydrate residues of (–)-lomaiviticin A (4) were unknown, we developed a semisynthetic route to the metabolite that proceeds in one step and 42% yield by diazo transfer to the more abundant isolate (–)-lomaiviticin C (6). This allowed us to complete the stereochemical assignment of (–)-lomaiviticin A (4) and provided a renewable source of material. Using this material, we established that the remarkable cytotoxic effects of (–)-lomaiviticin A (4) derive from the induction of highly toxic double-strand breaks (DSBs) in DNA. At the molecular level, 1,7-nucleophilic additions to each electrophilic diazofluorene trigger homolytic decomposition pathways that produce sp2 radicals at the carbon atoms of each diazo group. These radicals abstract hydrogen atoms from the deoxyribose of DNA, a process known to initiate strand cleavage. NMR spectroscopy and molecular mechanics simulations were used to elucidate the mode of DNA binding. These studies showed that both diazofluorenes of (–)-lomaiviticin A (4) penetrate into the duplex. This mode of non-covalent binding places each diazo carbon atom in close proximity to each DNA strand. Throughout these studies, isolates containing one diazofluorene, such as (–)-lomaiviticin C (6) and (–)-kinamycin C (2), were used as controls. Consistent with our mechanistic model, these compounds do not induce DSBs in DNA and are several orders of magnitude less potent. Reactivity studies suggest that (–)-lomaiviticin A (4) is more electrophilic than simple monomeric diazofluorenes. We attribute this to through-space delocalization of the developing negative charge in the transition state for 1,7-addition. Consistent with this mechanism of action, (–)-lomaiviticin A (4) displays selective low-picomolar potencies toward DNA DSB repair-deficient cell types. The emergent properties of (–)-lomaiviticin A (4) derive from the specific arrangement of diazo, naphthoquinone, cyclopentadiene, and ketone functional groups. These functional groups work together to yield, essentially, a masked vinyl radical that can be exposed under biological conditions. Furthermore, the rotational symmetry of the metabolite, deriving from dimerization, allows it to interact with the antiparallel symmetry of DNA and affect cleavage of the duplex.
Graphical Abstract
INTRODUCTION
“Emergence” is the phenomenon by which novel properties arise from a given level of hierarchical complexity. Although atoms interacting in molecules, which in turn interact with each other, is the very definition of both emergence and of chemistry,1 emergence per se is not a concept that is typically discussed (or taught) by chemists. For example, chemists agree that benzene is aromatic and that this property is not possessed by the six carbon and hydrogen atoms that constitute it. Chemical ligands bind biological targets with specificity due to their particular arrangement of atoms, while fragments or individual atoms of the same ligands have little or no target affinity. Similarly, emergent properties cannot be understood by a reductionist approach, and they are difficult or impossible to predict because of the diversity of molecular architectures that are accessible from a small number of fragments. It could be argued that emergence is an obvious element of organic chemistry. However, our work to elucidate the mechanism of action of (–)-lomaiviticin (4) (Figure 1), a C2-symmetric dimeric cytotoxin, exemplifies (in my mind) an instance where we would not have understood the most important aspects of the chemical system without thinking carefully about emergence per se. (–)-Lomaiviticin A (4) and the related monomeric isolates known as the kinamycins contain diazotetrahydrobenzo[b] fluorene (diazofluorene) functional groups. These groups comprise redox-active naphthoquinone, cyclopentadiene, and diazo residues fused in a σ- and π-bonding network. I will describe the manner in which lomaiviticin’s complex properties are more than the sum of its functional group parts and the way in which conceptualizing the system using the idea of emergent properties helped us see this system clearly and may help in other similar cases. Thinking carefully about how we generate chemical insight in these complex systems advances our understanding of structure, bonding, and reactivity and, in the case of (–)-lomaiviticin A (4) and other bioactive molecules, has obvious potential benefits to pharmaceutical development. The extent to which the work described herein could potentially ever be automated is an interesting question.
Figure 1.

Structures of (–)-kinamycins A, C, and F (1–3, respectively) and (–)-lomaiviticins A–E (4–8, respectively).
The kinamycins were first isolated in the 1970s by Ōmura and co-workers.2 Their absolute stereochemistry was established by X-ray crystallography. For nearly 25 years the kinamycins were believed to possess a cyanamide in place of the diazo substituent; the circuitous path to the correct structures has been reviewed.3 (–)-Lomaiviticins A (4) and B (5) were discovered by George Ellestad (Wyeth), Chris Ireland (University of Utah), and co-workers.4 The absolute stereochemistries of the aglycon residues of 4 and 5 were assigned by analogy to the kinamycins, but the stereochemistry of the carbohydrates was not determined. In 2012 we reported the isolation and structure determination of (–)-lomaiviticins C–E (6–8) and the relative and absolute stereochemical assignments of (–)-lomaiviticins A (4) and B (5).5 Shortly thereafter, Moore and co-workers reported the isolation and some key elements of the biosynthesis of 4–8.6 Other elements of lomaiviticin biosynthesis, including insights into the origin of the diazo group, have been elucidated.7 Kinamycin biosynthesis8 and our synthetic work and important contributions from other laboratories3,9 have been reviewed.
(–)-Lomaiviticin A (4) is a potent cytotoxin, with half-maximal inhibitory potency (IC50) values in the nanomolar–picomolar range against many cancer cell lines. It was noted that (–)-lomaiviticin A (4) damaged DNA,4 but no details of this study were disclosed. Kinamycins possess IC50 values in the hundreds of nanomolar range.
OVERVIEW OF THE SYNTHETIC STRATEGY
Scheme 1 shows the strategy that we developed to access the diazofluorene. A key step, finding precedent in the synthesis of diazocyclopentadiene by von Doering in 1953,10 involves diazo transfer to hydroxyfulvene 12 to install the dinitrogen substituent. Although unknown at the time, diazofluorenes 13 are converted to hydroxyfulvenes 12 under nucleophilic or reductive conditions. The transformation of 12 to 13 completes a redox cycle between these two states.
Scheme 1.

Convergent Assembly of the Diazofluorene
We adapted this chemistry to the synthesis of (–)-kinamycin F (3)11 and the chain and ring isomers of lomaiviticin aglycon (21 and 22, respectively).12,11b (–)-Kinamycin F (3) was prepared in seven steps from ketone 15 and juglone 16 (Scheme 2A). We pursued the oxidative coupling of two monomeric diazofluorenes to access lomaiviticin aglycon (21/22, Scheme 2B). In exploratory experiments we found that the oxidative coupling of enoxysilanes derived from monomeric diazofluorenes was relatively efficient and general. The primary obstacle to the synthesis of 21/22 was diastereocontrol. The lomaiviticins possess the “syn,syn” stereorelationship with respect to the cojoining bond and the adjacent tertiary carbon–oxygen bonds (blue bonds in 4–8; Figure 1). This arrangement calls for bond formation to the more hindered face of each monomer in the coupling.
Scheme 2.

(A) Synthesis of (–)-Kinamycin F (3); (B) Synthesis of the Chain and Ring Isomers of Lomaiviticin Aglycon (21 and 22, Respectively)
In early experiments we prepared the unnatural anti,anti coupling product (not shown), but efforts to invert the stereochemistry were unsuccessful. The bridging bond is hindered, and the nucleophile and base sensitivity of the diazofluorene constrained the scope of viable experimental conditions. Accordingly, we focused on attaining substrate control. We found that the enoxysilane derived from exo-mesityl diazofluorene 18 underwent oxidative coupling with manganese tris(hexafluoroacetylacetonate) (19) in benzene to provide the syn,syn product 20 with 1.5–3:1 selectivity versus the anti,anti product. The C1-symmetric syn,anti product was not formed under these conditions. This suggests, surprisingly, that the transition state giving rise to the syn,anti-product is higher in energy than that giving rise to the syn,syn diastereomer. In dichloromethane the anti,anti dimer was formed exclusively. The stereoselectivity may derive in part from tight ion pairing after electron transfer, which drives bond formation to the concave face of the oxidized substrate (see the Scheme 2B inset). The basis for stereoselectivity in the incoming (unoxidized) monomer is less apparent. The coupling product 20 was deprotected under acidic conditions; neutralization at low temperature provided the aglycon chain isomer 21, which cyclized to the ring isomer 22.
Isolation of (–)-Lomaiviticin C (6) and Semisynthesis of (–)-Lomaiviticin A (4)
Our attention then turned to synthesis of (–)-lomaiviticin A (4). As noted, the absolute stereochemistries of the carbohydrates were unknown.4 The presence of D- and L-oleandrose in nature and the unique nature of the aminosugar made it impossible to advance a plausible assignment. We sought to obtain natural (–)-lomaiviticin A (4) but learned through conversations with Dr. Jeff Janso (Pfizer), who was part of the Wyeth natural products group, that the natural material was depleted. Dr. Janso granted us permission to request the producing strain from the USDA. This change in direction proved more enabling than a small sample of (–)-lomaiviticin A (4) itself, as it ultimately provided a renewable source of material.
We were looking for (and identified) (–)-lomaiviticin A (4) in the bacterial cultures, but our attention was drawn to a more abundant metabolite, which we named (–)-lomaiviticin C (6) (Scheme 3).5,6 (–)-Lomaiviticin C (6) is identical to (–)-lomaiviticin A (4) save for the conversion of one diazofluorene to a hydroxyfulvene. The L configuration of the carbohydrates was elucidated by acidic hydrolysis, isolation, and comparison to standards.13 Using ROESY analysis and the absolute stereochemistry of the aminosugar, we deduced the aglycon configuration, thereby completing the stereochemical assignment. Treatment of (–)-lomaiviticin C (6) with trifyl azide and trifluoroacetic acid provided semisynthetic (–)-lomaiviticin A (4), which was indistinguishable from a fully natural sample. Thus, this transformation allowed us to relay our stereochemical assignments from (–)-lomaiviticin C (6) to (–)-lomaiviticin A (4) and provided a renewable source of (–)-lomaiviticin A (4) [yields of (–)-lomaiviticin C (6) are up to 120-fold higher, and it is stable toward storage]. We believe that (–)-lomaiviticin C (6) is produced from (–)-lomaiviticin A (4) during the fermentation. This degradation may constitute a fortuitous detoxification pathway for the producing strain, as (–)-lomaiviticin C (6) is several orders of magnitude less cytotoxic. We also identified the mono- and dimethylated derivatives (–)-lomaiviticin D (7) and E (8) (Figure 1).
Scheme 3.

Method To Produce Semisynthetic (–)-Lomaiviticin A (4); Pairs of Colored Balls in (–)-Lomaiviticin C (6) Denote ROESY Interactions Used To Elucidate the Absolute Stereochemistry of the Aglycon Residue
THE DIMERIC DIAZOFLUORENE IS REQUIRED FOR POTENT CYTOTOXICITY
Earlier we attempted to deduce structure–function relationships from monomeric diazofluorenes, but extrapolating these results to (–)-lomaiviticin A (4) seemed tenuous. We now know that there is no reason to expect any monomeric diazofluorene to approach the potency of (–)-lomaiviticin A (4). Table 1 shows that (–)-lomaiviticin A (4) possesses IC50 values in the picomolar–nanomolar range against the K562, LNCaP, HCT-116, and HeLa cancer cell lines. (–)-Lomaiviticin C (6) is several orders of magnitude less potent. Also worthy of note is the surprising determination that (–)-kin-amycin C (2) is more potent than (–)-lomaiviticin C (6) in three of these four cell lines. While the additional structural elements of (–)-lomaiviticin C (6) may influence properties not captured in this assay, from the perspective of cell viability these are insufficient to provide a potent cytotoxin. These results directed our attention toward the dimeric diazofluorene of (–)-lomaiviticin A (4).
Table 1.
IC50 Values (in nM) of (–)-Lomaiviticin A (4), (–)-Lomaiviticin C (6), and (–)-Kinamycin C (2) against the K562, LNCaP, HCT-116, and HeLa Cell Lines
| compound | K562 | LNCaP | HCT-116 | HeLa |
|---|---|---|---|---|
| (–)-lomaiviticin A (4) | 0.12 | 0.31 | 0.034 | 4.5 |
| (–)-lomaiviticin C (6) | 470 | 330 | 220 | 590 |
| (–)-kinamycin C (2) | 72 | 120 | 270 | 520 |
| relative potency 4:6 | 4750 | 1060 | 5120 | 131 |
(–)-Lomaiviticin A (4) Induces Double-Strand Breaks in DNA in Vitro through a Single Binding Event
(–)-Lomaiviticin A (4) was discovered during an effort to identify enediynes from a strain of Salinispora symbiotic with a marine ascidian.4 The molecule was isolated by activity-guided fractionation using the biochemical prophage induction assay (BIA), a sensitive method for the detection of DNA-damaging agents.14 It was noted that “lomaiviticin A cleaved double-stranded DNA under reducing conditions”, but these studies were never disclosed.4
We evaluated the activity of (–)-lomaiviticin A (4) in an in vitro plasmid DNA damage assay15 employing the 4361 base pair (bp) supercoiled resistance plasmid pBR322. The induction of DNA single-strand breaks (SSBs) and double-strand breaks (DSBs) results in relaxation (unwinding) and linearization, respectively, of the supercoiled plasmid. The plasmid, relaxed, and linearized DNA can be separated by electrophoresis and visualized. This assay does not require radiolabeled material and is easily modulated to gain insight into the mechanism of DNA damage.
Figure 2A shows that incubating pBR322 (38 μM in base pairs) with (–)-lomaiviticin A (4) (2.0 μM) and dithiothreitol (DTT) (0.5 mM) at 37 °C for 16 h resulted in consumption of the supercoiled plasmid and a large proportion of linearized DNA.16 Nicking and linearization was also observed using 0.5 μM (–)-lomaiviticin A (4), although some unmodified plasmid remained. SSBs predominated in the absence of DTT.
Figure 2.

Analysis of in vitro DNA damage by (–)-lomaiviticin A (4). (A) (–)-Lomaiviticin A (4) induces DNA DSBs in vitro. (B) Freifelder–Trumbo analysis of DNA damage by (–)-lomaiviticin A (4). Over a 70-fold concentration range, the ratio of SSBs to DSBs induced by (–)-lomaiviticin A (4) was invariant (5.3 ± 0.6):1, indicating that DNA cleavage occurs through a single binding event (“first-order mechanism”). See ref 16 for conditions.
DNA damage by the kinamycins is mediated by reactive oxygen species (ROS), hydrogen peroxide, and/or iron.3 Surprisingly, control experiments indicated that DNA damage by (–)-lomaiviticin A (4) is independent of these factors. This is consistent with the studies outlined below, which support nucleophilic addition to (–)-lomaiviticin A (4) as a step preceding DNA cleavage. The less electrophilic diazofluorene of the kinamycins may render alternative pathways, such as ROS production by redox cycling of the naphthoquinone, more favorable. Other classes of quinonoid natural products, such as the anthracyclines, generate ROS by redox cycling.17
A Freifelder–Trumbo analysis18 was used to determine whether DNA DSBs induced by (–)-lomaiviticin A (4) derive from a single binding event (“first-order mechanism”; Figure 2B) or from an accumulation of closely spaced SSBs (“second-order mechanism”). This assay has been used to assess the nature of DNA DSBs induced by bleomycin.18c The fractions of relaxed and linearized DNA that remained after treatment with (–)-lomaiviticin A (4) were quantified, and the numbers of SSBs and DSBs per DNA molecule were determined.18 Consistent with a “first-order mechanism”, the ratio of SSBs to DSBs was constant (5.3 ± 0.6):1 over a 70-fold concentration range and lower than that expected if DSBs arose from an accumulation of closely spaced SSBs (red series). DNA DSBs were not observed in parallel experiments using 1 mM (–)-lomaiviticin C (6) or (–)-kinamycin C (2) in the presence or absence of DTT. These experiments provide the outlines of a mechanistic model wherein the potency of (–)-lomaiviticin A (4) derives from the induction of highly toxic19 DNA DSBs. Metabolites containing one diazofluorene do not induce DSBs and are orders of magnitude less potent.
(–)-Lomaiviticin A (4) Activates the DNA Damage Response
We sought to determine whether the DNA cleavage activity of (–)-lomaiviticin A (4) is recapitulated in tissue culture. Pulsed-field gel electrophoresis (PFGE)20 and the neutral comet unwinding assay21 are related methods for detecting DNA DSBs. They are based on the selective migration of DNA fragments during electrophoresis (undamaged DNA is immobile). The comet assay is more sensitive and was used in our studies.16,22 Following exposure, the cells were embedded in agarose on slides, lysed under neutral conditions, and subjected to electrophoresis. We visualized the DNA by staining with SYBR Green. The “head of the comet” consists of a circular spot of undamaged DNA, while the “tail” comprises DNA fragments. The amount of DNA damage can be expressed quantitatively as the tail moment, defined as the product of the tail length and the fraction of fluorescence in the tail.
We employed K562 leukemia cells since this cell line was sensitive to monomeric diazofluorenes. Figure 3A shows that extensive DNA damage was observed after cells were exposed to 5 or 50 nM (–)-lomaiviticin A (4) for 30 min. Figure 3B shows that 5 nM (–)-lomaiviticin A (4) was comparable to 40 Gy of ionizing radiation (IR) or 100 μM hydrogen peroxide. (–)-Lomaiviticin C (6) and (–)-kinamycin C (2) did not damage DNA at concentrations of 300 nM.
Figure 3.

Analysis of DNA damage by (–)-lomaiviticin A (4), (–)-lomaiviticin C (6), and (–)-kinamycin C (2) using the neutral comet unwinding assay: (A) fluorescence images; (B) tail moments. See refs 16 and 22 for conditions.
The comet assay is a widely used molecular biology method, but the interpretation of the results is not free from debate.23,21b To corroborate our findings, we probed for activation of the DNA damage response (DDR), a signaling network that detects DNA damage and rapidly repairs it. DNA DSBs are resolved by the nonhomologous end joining (NHEJ)24 or homologous recombination (HR)25 repair pathways. We interrogated the DDR factors phospho-SER139-H2AX (γH2AX)26 and p53 binding protein 1 (53BP1).27 γH2AX is a phosphorylated form of the histone protein H2AX that is generated for a megabase region surrounding a DSB. It amplifies the DDR signal and localizes NHEJ and HR repair proteins around the lesion.26 53BP1 is a nuclear protein that translocates to the site of cleavage, activates checkpoint signaling, and initiates repair.27 Both γH2AX and 53BP1 form foci that can be visualized. Figure 4 shows that γH2AX and 53BP1 foci were observed following incubation of K562 cells with 0.5 nM (–)-lomaiviticin A (4) for 4 h.16,22 Treatment with 300 nM (–)-lomaiviticin C (6) or (–)-kinamycin C (2) did not generate similar responses. Ataxia telangiectasia mutated (ATM) kinase28 and DNA-dependent protein kinase (DNA-PK)29 are mediators of H2AX phosphorylation in response to DSBs. Pretreatment with inhibitors of ATM or DNA-PK abolished the γH2AX response,22 further linking this phenotype to DSB repair. Parallel results were obtained in HeLa cells.16 These assays provided conclusive evidence that (–)-lomaiviticin A (4) induces DNA DSBs and activates the DDR at low-nanomolar concentrations.
Figure 4.

Immunofluorescence imaging of γH2AX and 53BP1 foci in K562 cells treated with (–)-lomaiviticin A (4), (–)-lomaiviticin C (6), or (–)-kinamycin C (2). See refs 16 and 22 for conditions.
(–)-Lomaiviticin A (4) Cleaves DNA by a Hydrogen Atom Abstraction Pathway
These data establish DNA DSBs as the likely biological basis for the potency of (–)-lomaiviticin A (4) but provide little insight into the molecular mechanism of DNA cleavage. The relative potencies of (–)-lomaiviticin A (4) and (–)-lomaiviticin C (6) provided evidence that hydrodediazotization diminishes the activity (Table 1 and Figure 5A). Earlier mechanism of action studies employing synthetic diazofluorenes (e.g., 25 and 26) or the kinamycins implicated ROS, covalent adducts 27, vinyl radicals 28, and addition to hydroxyfulvene 29 or acylfulvene 30 electrophiles as underlying sources of cytotoxicity (Figure 5B).3
Figure 5.

(A) Hydroxyfulvenes 23 are less potent than the corresponding diazofluorene 24 (left). Model systems 25 and 26 were studied in the first experiments implicating vinyl radicals in diazofluorene toxicity (right). (B) Intermediates suggested by prior mechanism of action studies3 to form from the kinamycins and/or the lomaiviticins. Pathways for the generation of 28, 29, and 30 directly from the diazofluorene or from the addition product 27 were proposed in prior studies.3
Our data supported vinyl radicals (as first proposed by Dmitrienko30 and Feldman31 on the basis of studies of 25 and 26) as the most significant for cytotoxic effects. As discussed above, the plasmid-damaging activity of (–)-lomaiviticin A (4) was independent of ROS, and covalent adducts were excluded on the basis of semipreparative DNA cleavage experiments in which >90% mass recovery was attained (vide infra). The decreased potency of (–)-lomaiviticin C (6) relative to (–)-lomaiviticin A (4) (Table 1; a similar trend was observed for pairs of synthetic monomeric hydroxyfulvenes–diazofluorenes) excluded hydroxyfulvenes and the acylfulvenes that form from them.32
Isotope labeling experiments were conducted to probe for the production of vinyl radicals 4· and 6· in the presence of DNA (Scheme 4). We removed all exchangeable protons in calf thymus (CT) DNA and DTT by repeated lyophilization from D2O and incubated a mixture of DTT-d4 (1 equiv), CT DNA-dn, and (–)-lomaiviticin A (4) for 48 h at 37 °C in D2O. We isolated (–)-lomaiviticin C (6) and the double reduction product 31 in 34% and 37% yield containing 67% and 39% hydrogen atom incorporation at the vinylic positions, respectively. In the absence of DTT-d4, (–)-lomaiviticin C (6) and the double reduction product 31 were isolated in 81% and 18% yield and contained 88% and 60% hydrogen atom incorporation at the vinylic positions, respectively. A two-electron process would involve N–D or O–D bond cleavage, resulting in deuteration of the vinylic position. The preponderance of protiation implicates removal of a non-exchangeable hydrogen atom by the vinyl radicals 4· and 6·, most likely from the deoxyribose backbone, a process that is known to initiate strand breaks.33
Scheme 4.

Pathway for the Hydrodediazotization of (–)-Lomaiviticin A (4) and (–)-Lomaiviticin C (6)
Remarkably, the hydrodediazotization of (–)-lomaiviticin A (4) to form (–)-lomaiviticin C (6) is several orders of magnitude faster than the transformation of (–)-lomaiviticin C (6) to the double reduction product 31.34 Thus, addition of N-acetyl-L-cysteine methyl ester and triethylamine (40 equiv each) to a solution of (–)-lomaiviticin A (4) in methanol-d4 at 5 °C resulted in instantaneous conversion to (–)-lomaiviticin C (6) (97%). This solution slowly converted to the double reduction product 31 with a half-life of 5 h at 5 °C (87% yield of 31 based on 4). In this experiment, the reduction products contained >95% deuterium atom incorporation at the vinylic positions. Isotope labeling experiments revealed that the vinylic C–H/D bond in both products derived from C–H/D bond cleavage in methanol.
Figure 6A shows diazosulfide 32, which formed instantaneously in 81% yield at –50 °C when benzylthiol was used as the nucleophile. Upon warming to –20 °C, diazosulfide 32 underwent decomposition to give (–)-lomaiviticin C (6) with a half-life of 49 min, demonstrating it as a kinetically competent intermediate in this transformation.
Figure 6.

(A) Structure of diazosulfide 32. (B) Minimized structure of (–)-lomaiviticin A (4) in water [B3LYP/6-31G(d)].
Our data allow us to construct a mechanistic pathway to describe DNA cleavage by (–)-lomaiviticin A (4), as summarized in Scheme 4. 1,7-Nucleophilic addition of thiol to a diazofluorene of (–)-lomaiviticin A (4) forms a diazosulfide intermediate (e.g., 32; Figure 6A) that decomposes to give vinyl radical 4·. The couplings of arenediazonium ions with thiolates to generate diazosulfides and their decomposition to form aryl radicals are well-known.35 Although the diazofluorene is formally not an arenediazonium ion, Dmitrienko30 had shown that this functional group displays enhanced (diazonium-like) electrophilicity. Hydrogen atom abstraction from the deoxy-ribose backbone initiates SSB formation with production of (–)-lomaiviticin C (6). In a parallel but slower transformation, vinyl radical 6· is generated and induces scission of the complementary strand (with production of 31) leading to the DSB. Figure 2B shows that the ratio of SSBs to DSBs was (5.3 ± 0.6):1 over a 70-fold concentration range.16 This could be explained by competitive dissociation of (–)-lomaiviticin C (6) from the duplex before the second cleavage occurs. The antitumor agent bleomycin cleaves DNA by the stepwise introduction of SSBs,36 and the ratio of SSBs to DSBs produced by bleomycin is in the range of 3.3–6.0:1 (depending on the assay conditions).18b,37 This too has been rationalized by invoking competitive dissociation of bleomycin before the second strand break. We speculate that the enhanced reactivity of (–)-lomaiviticin A (4) may be due to delocalization of the developing negative charge in the transition state for nucleophilic addition into the adjacent diazofluorene. In the DFT-minimized structure, the diazofluorenes of (–)-lomaiviti-cin A (4) are within 4 Å (Figure 6B). Deprotonation of (–)-lomaiviticin C (6) by excess base in solution may further reduce its electrophilicity.
(–)-Lomaiviticin A (4) Binds DNA by Penetration of Both Diazofluorenes into the Duplex
We initiated studies to determine the nature of the interaction of (–)-lomaiviticin A (4) with DNA. Table 2 presents the results of fluorescence intercalator displacement assays,38 which revealed that (–)-lomaiviticin A (4) binds alternating AT regions preferentially.39 The concentration of drug at which 50% of the fluorescent intercalator is displaced (DC50) using the palindromic duplex d(CGCATATGCG)2 was 850 nM, and the ratio of duplex to ligand (rdl) was 1.0, indicating a single binding site.
Table 2.
DC50 and rdl Values for (–)-Lomaiviticin A (4)a
| entry | oligonucleotide duplexes | DC50 (per base pair) | rdl |
|---|---|---|---|
| 1 |
|
18.3 ± 1.4 μM | 0.09 |
| 2 |
|
2.70 ± 0.38 μM | 0.2 |
| 3 |
|
2.14 ± 0.18 μM | 0.5 |
| 4 |
|
1.87 ± 0.14 μM | 0.5 |
| 5 |
|
0.85 ± 0.05 μM | 1.0 |
For the conditions, see ref 39.
We selected the 8 bp duplex d(GCTATAGC)2 for study because its 1H NMR resonances had been assigned.40 A 1:1 complex was obtained when equimolar amounts of the duplex and (–)-lomaiviticin A (4) (1.1 μM each) were combined in phosphate-buffered D2O. Three aspects of the complex were immediately obvious (Figure 7). First, binding lifts the degeneracy of (–)-lomaiviticin A (4) and (fortuitously) resolves the aromatic C–H resonances. Each was shifted upfield by 0.47–1.76 ppm, which is indicative of an intercalative-like interaction.41 Second, the pairs of aromatic C–H resonances could be assigned by COSY, which in turn allowed us to identify an interdiazofluorene NOE, indicating that these residues were in proximity. Third, the other positionally equivalent sites within (–)-lomaiviticin A (4) were within 0.20 ppm of each other, suggesting pseudo-C2-symmetric binding. Collectively these observations suggest that (–)-lomaiviticin A (4) adopts a conformation wherein the diazofluorene residues are in approximately parallel planes. Additionally, both of these residues appear to penetrate the duplex, which was unexpected.
Figure 7.

Key elements in the binding of (–)-lomaiviticin A (4) to the duplex d(GCTATAGC)2.
An NOE walk established A4/T13 and T5/A12 as the binding site. The N-H protons of T5 and T13 resonated as broad singlets in the range of 11.2–11.7 ppm, which is indicative of disruption of base pairing. Base pairing at the duplex ends remained, as evidenced by minimal perturbations of the NOE contacts and nominal changes in chemical shifts.
Figure 8A–C shows the structural model that was generated using AMBER.42 The structure shows that (–)-lomaiviticin A (4) approaches from the minor groove. Both diazofluorene residues penetrate the duplex, leading to significant distortion (Figure 8C). Binding flips A4, T5, and T13 out of the duplex. The T5 and T13 exchangeable protons lost hydrogen-bonding character and were observed as broad peaks in a δ range typical of free thymine (11.2–11.7 ppm), indicative of complete disruption of A4/T13 and T5/A12 base pairing. Base flipping has been observed for non-covalent (e.g., actinomycin)43 and covalent (e.g., trioxacarcin A)44 DNA binders. It is likely that this mechanism is responsible for the sequence preferences of (–)-lomaiviticin A (4). The enthalpic penalty associated with disruption (flipping) of A–T pairs is less than that of G–C pairs because of reduced hydrogen-bonding and base-stacking interactions.45 There are several interactions that may compensate for this distortion of the duplex and render the overall binding favorable. The aminosugar residues are within 4.3 Å of the nearest phosphate group, and it is likely that they stabilize the complex through an electrostatic interaction. Additionally, a π contact between A12 and the bottom diazofluorene (as oriented in Figure 8A, distance = 3.3 Å) may further stabilize the complex. Finally, there appears to be an edge–face interaction between T13 and the top diazofluorene (as oriented in Figure 8A, distance = 2.9 Å).
Figure 8.

Structure of (–)-lomaiviticin A (4) bound to d-(GCTATAGC)2. (–)-Lomaiviticin A (4) is shown in purple. The diazo nitrogen atoms are shown as yellow spheres. (A) Front view. (B) Side view. (C) Ribbon view showing the distortion of the backbone.
This binding is compatible with the molecular mechanism of DNA cleavage outlined in Scheme 4. A6 H5′ and A6 H4′ are located 4.2 and 4.3 Å, respectively, from the nearest diazo carbon while A14 H1′ and T13 H4′ are located 4.2 and 4.9 Å, respectively, from the alternate diazo carbon atom. Comparison to literature data suggests that these distances are compatible with the stepwise induction of DNA SSBs by hydrogen atom abstraction. Bleomycin undergoes a translocation of 15–18 Å between strand breaks.46
The interdiazofluorene distance is compressed (by 0.3 Å) upon binding. This compression may increase the stereo-electronic activation outlined in Figure 6B, leading to heightened reactivity on binding. Thus, this constitutes another example of shape-dependent catalysis, which was beautifully elucidated in the context of DNA-mediated activation of CC-1065 and duocarmycin by Boger and co-workers.47 Finally, the bound conformation of (–)-lomaiviticin A (4) is only 0.40 kcal/mol higher in energy than the free structure, suggesting substantial preorganization. Nonbonded interactions between the oleandrose residues may prevent rotation of the diazofluorenes away from each other (Figure 9).
Figure 9.

The oleandrose residues of (–)-lomaiviticin A (4) engage in nonbonded interactions as the diazofluorenes are rotated away from each other.
Translational Development of (–)-Lomaiviticin A (4)
Knowledge of the mechanism of action of (–)-lomaiviticin A (4) enables rational approaches to translational development. Advances in genome sequencing indicate that many cancers are driven by mutations in DNA repair pathways.48 These mutations promote tumorigenesis but also provide a therapeutic opportunity because they can sensitize tumors to DNA-damaging agents.49 We hypothesized that DNA DSB repair-deficient cell lines may be sensitized toward (–)-lomaiviticin A (4). In collaboration with the Glazer laboratory (Yale School of Medicine), we evaluated (–)-lomaiviticin A (4) against paired cell lines that are proficient or deficient in single DNA DSB repair factors but otherwise isogenic.22 Breast cancer type 2, early onset (BRCA2)-deficient and phosphatase and tensin homologue deleted on chromosome ten (PTEN)-deficient cell lines were sensitized toward (–)-lomaiviticin A (4) with LC50 values of 1.5–2.0 pM. PTEN50 and BRCA251 are involved in DNA DSB repair, and mutations in BRCA251 and PTEN52 are widespread in cancers. The paired BRCA2-proficient cell line was 12-fold less sensitive to 4 pM (–)-lomaiviticin A (4), while the paired PTEN-proficient line was fully viable at this concentration. Sensitivity to (–)-lomaiviticin (4) conferred by deficiencies in ATM- and X-ray repair cross complementing 5 (KU80) are also consistent with the involvement of DNA damage and DSB formation. We also found that the ataxia telangiectasia and Rad3-related protein (ATR) inhibitor VE-82153 synergizes with (–)-lomai-viticin A (4).54 Exposure of K562 cells to either 10 μM VE-821 or 500 pM (–)-lomaiviticin A (4) alone for up to 48 h was nonlethal. However, when treated with 10 μM VE-821 + 500 pM (–)-lomaiviticin A (4), the cells suffered 81% and 94% killing after 24 and 48 h, respectively. Collectively these results suggest many exciting directions for translational development, including systemic application of (–)-lomaiviticin A (4) and VE-821 to treat DNA DSB repair-proficient tumor types and evaluation of (–)-lomaiviticin A (4) against BRCA2- and PTEN-deficient tumors.
CONCLUSION
Through a combination of chemical, in vitro, and tissue culture studies, we have been able to elucidate the emergent properties of (–)-lomaiviticin A (4). Understanding these properties in turn has allowed us connect its potent cytotoxicity with its structure and chemical reactivity. The key property of the diazofluorene is its ability to serve as a vinyl radical precursor that can be unmasked under biological conditions. This property can be considered emergent because it derives from the specific spatial configuration of the diazo, cyclopentadiene, naphthoquinone, and D-ring ketone. Individually or in different spatial configurations, these functional groups would not give rise to this behavior. Additionally, the combination of aminosugar residues and the aromatic diazofluorenes leads to the emergence of a high-affinity ligand for DNA. Finally, the unique mode of C2-symmetric dimerization gives (–)-lomaiviticin A (4) an element of rotational symmetry that allows it to interact with the antiparallel symmetry of DNA. In this way, each radical precursor is positioned in close proximity and in the correct orientation to C–H bonds of the deoxyribose backbones of opposing strands. Ultimately this mode of interaction brings about hydrogen atom abstraction and DNA cleavage. The absence of this symmetry in (–)-lomaiviticin C (6) and (–)-kinamycin C (2) removes this property, leading to agents that are ultimately much less potent. Moreover, the absence of aminosugar residues in (–)-kinamycin C (2) reduces the affinity of this molecule for DNA. These remarkable properties of (–)-lomaiviticin A (4) are reminiscent of other DNA-damaging agents, such as the enediynes,55 bleomycins,36 and duocarmyins,56 which also possess emergent properties deriving from specific combinations of functional groups.
Though we have made progress in the study of (–)-lomaiviticin A (4), several important issues remain unresolved. First, as noted above, we observed hydro-dediazotization of (–)-lomaiviticin A (4) in the presence of DNA but in the absence of added thiol nucleophile. This raises the intriguing possibility that DNA itself may be reactive toward the first diazo group of (–)-lomaiviticin A (4). Alternatively, other mechanisms such as electron transfer to the diazofluorene may predominate in the absence of a strong nucleophile. Further study is required to elucidate this point. In addition, much remains to be learned about the structure–function relationships surrounding other regions of the molecule. For example, if the aminosugar simply provides electrostatic stabilization to the DNA–(–)-lomaiviticin A (4) complex, simpler substituents may be equally effective. Finally, the total chemical synthesis of (–)-lomaiviticin A (4) remains an unsolved problem that we continue to investigate.
Acknowledgments
I am grateful to the many co-workers and collaborators who contributed to the work described in this Account and whose names are included in the references. I am also immeasurably indebted to those scientists who pioneered the study of natural product–DNA recognition and damage and the use of synthesis to elucidate the emergent properties of these fascinating metabolites. I thank Professor Alison M. Sweeney (The University of Pennsylvania) for helpful comments on the manuscript. Financial support from the National Institutes of Health (R01 GM-090000) is gratefully acknowledged.
Biography
Seth B. Herzon completed his undergraduate degree at Temple University and his graduate studies at Harvard University under the guidance of Professor Andrew G. Myers. He conducted postdoctoral work with Professor John F. Hartwig at the University of Illinois at Urbana–Champaign. Since 2008 he has been at Yale University, currently as a Professor of Chemistry and Pharmacology and a member of the Yale Comprehensive Cancer Center. His research focuses on studies of DNA-damaging natural products, human microbiota metabolites, and antibiotic development.
Footnotes
The author declares no competing financial interest.
References
- 1.Luisi PL. Emergence in chemistry: Chemistry as the embodiment of emergence. Found Chem. 2002;4:183–200. [Google Scholar]
- 2.(a) Ito S, Matsuya T, Ōmura S, Otani M, Nakagawa A, Takeshima H, Iwai Y, Ohtani M, Hata T. A new antibiotic, kinamycin. J Antibiot. 1970;23:315–317. doi: 10.7164/antibiotics.23.315. [DOI] [PubMed] [Google Scholar]; (b) Hata T, Ōmura S, Iwai Y, Nakagawa A, Otani M, Ito S, Matsuya T. A new antibiotic, kinamycin: Fermentation, isolation, purification and properties. J Antibiot. 1971;24:353–359. doi: 10.7164/antibiotics.24.353. [DOI] [PubMed] [Google Scholar]; (c) Ōmura S, Nakagawa A, Yamada H, Hata T, Furusaki A, Watanabe T. Structure of kinamycin c, and the structural relation among kinamycin a, b, c, and d. Chem Pharm Bull. 1971;19:2428–2430. [Google Scholar]; d) Furusaki A, Matsui M, Watanabe T, Ōmura S, Nakagawa A, Hata T. Crystal and molecular structure of kinamycin c p-bromobenzoate. Isr J Chem. 1972;10:173–187. [Google Scholar]; (e) Ōmura S, Nakagawa A, Yamada H, Hata T, Furusaki A, Watanabe T. Structures and biological properties of kinamycin a, b, c, and d. Chem Pharm Bull. 1973;21:931–940. doi: 10.1248/cpb.21.931. [DOI] [PubMed] [Google Scholar]; (f) Cone MC, Seaton PJ, Halley KA, Gould SJ. New products related to kinamycin from streptomyces murayamaensis. I. Taxonomy, production, isolation and biological properties. J Antibiot. 1989;42:179–188. doi: 10.7164/antibiotics.42.179. [DOI] [PubMed] [Google Scholar]; (g) Seaton PJ, Gould SJ. New products related to kinamycin fromstreptomyces murayamaensis Ii. Structures of pre-kinamycin ketoanhydrokinamycin and kinamycins e and f. J Antibiot. 1989;42:189–197. doi: 10.7164/antibiotics.42.189. [DOI] [PubMed] [Google Scholar]
- 3.Herzon SB, Woo CM. The diazofluorene antitumor antibiotics: Structural elucidation, biosynthetic, synthetic, and chemical biological studies. Nat Prod Rep. 2012;29:87–118. doi: 10.1039/c1np00052g. [DOI] [PubMed] [Google Scholar]
- 4.He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. Lomaiviticins A and B, potent antitumor antibiotics fromMicromonospora lomaivitiensis. J Am Chem Soc. 2001;123:5362–5363. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- 5.Woo CM, Beizer NE, Janso JE, Herzon SB. Isolation of lomaiviticins c–e. Transformation of lomaiviticin c to lomaiviticin a, complete structure elucidation of lomaiviticin a, and structure–activity analyses. J Am Chem Soc. 2012;134:15285–15288. doi: 10.1021/ja3074984. [DOI] [PubMed] [Google Scholar]
- 6.Kersten RD, Lane AL, Nett M, Richter TKS, Duggan BM, Dorrestein PC, Moore BS. Bioactivity-guided genome mining reveals the lomaiviticin biosynthetic gene cluster in salinispora tropica. ChemBioChem. 2013;14:955–962. doi: 10.1002/cbic.201300147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Janso JE, Haltli BA, Eustaquio AS, Kulowski K, Waldman AJ, Zha L, Nakamura H, Bernan VS, He H, Carter GT, Koehn FE, Balskus EP. Discovery of the lomaiviticin biosynthetic gene cluster in salinispora pacifica. Tetrahedron. 2014;70:4156–4164. doi: 10.1016/j.tet.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang P, Hong GJ, Wilson MR, Balskus EP. Production of stealthin c involves an s–n-type smiles rearrangement. J Am Chem Soc. 2017;139:2864–2867. doi: 10.1021/jacs.6b10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gould SJ. Biosynthesis of the kinamycins. Chem Rev. 1997;97:2499–2510. doi: 10.1021/cr9600215. [DOI] [PubMed] [Google Scholar]
- 9.(a) Marco-Contelles J, Molina MT. Naturally occurring diazo compounds: The kinamycins. Curr Org Chem. 2003;7:1433–1442. [Google Scholar]; (b) Arya DP. Diazo and diazonium DNA cleavage agents: Studies on model systems and natural product mechanisms of action. Top Heterocycl Chem. 2006;2:129–152. [Google Scholar]; (c) Nawrat CC, Moody CJ. Natural products containing a diazo group. Nat Prod Rep. 2011;28:1426–1444. doi: 10.1039/c1np00031d. [DOI] [PubMed] [Google Scholar]; (d) Herzon SB. The kinamycins. In: Li JJ, Corey EJ, editors. Total Synthesis of Natural Products: At the Frontiers of Organic Chemistry. Springer; Berlin: 2012. pp. 39–65. [Google Scholar]
- 10.Doering WvE, DePuy CH. Diazocyclopentadiene1. J Am Chem Soc. 1953;75:5955–5957. [Google Scholar]
- 11.(a) Woo CM, Lu L, Gholap SL, Smith DR, Herzon SB. Development of a convergent entry to the diazofluorene antitumor antibiotics: Enantioselective synthesis of kinamycin f. J Am Chem Soc. 2010;132:2540–2541. doi: 10.1021/ja910769j. [DOI] [PubMed] [Google Scholar]; (b) Woo CM, Gholap SL, Lu L, Kaneko M, Li Z, Ravikumar PC, Herzon SB. Development of enantioselective synthetic routes to (–)-kinamycin f and (–)-lomaiviticin aglycon. J Am Chem Soc. 2012;134:17262–17273. doi: 10.1021/ja307497h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herzon SB, Lu L, Woo CM, Gholap SL. 11-step enantioselective synthesis of (–)-lomaiviticin aglycon. J Am Chem Soc. 2011;133:7260–7263. doi: 10.1021/ja200034b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gholap SL, Woo CM, Ravikumar PC, Herzon SB. Synthesis of the fully glycosylated cyclohexenone core of lomaiviticin a. Org Lett. 2009;11:4322–4325. doi: 10.1021/ol901710b. [DOI] [PubMed] [Google Scholar]
- 14.Elespuru RK, White RJ. Biochemical prophage induction assay: A rapid test for antitumor agents that interact with DNA. Cancer Res. 1983;43:2819–2830. [PubMed] [Google Scholar]
- 15.Burden DA, Froelich-Ammon SJ, Osheroff N. Topoisomerase ii-mediated cleavage of plasmid DNA. Methods in molecular biology. 2000;95:283–289. doi: 10.1385/1-59259-057-8:283. [DOI] [PubMed] [Google Scholar]
- 16.Colis LC, Woo CM, Hegan DC, Li Z, Glazer PM, Herzon SB. The cytotoxicity of (–)-lomaiviticin a arises from induction of double-strand breaks in DNA. Nat Chem. 2014;6:504–510. doi: 10.1038/nchem.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaudiano G, Koch TH. Redox chemistry of anthracycline antitumor drugs and use of captodative radicals as tools for its elucidation and control. Chem Res Toxicol. 1991;4:2–16. doi: 10.1021/tx00019a001. [DOI] [PubMed] [Google Scholar]
- 18.(a) Freifelder D, Trumbo B. Matching of single-strand breaks to form double-strand breaks in DNA. Biopolymers. 1969;7:681–693. [Google Scholar]; (b) Povirk LF, Wübker W, Köhnlein W, Hutchinson F. DNA double-strand breaks and alkali-labile bonds produced by bleomycin. Nucleic Acids Res. 1977;4:3573–3580. doi: 10.1093/nar/4.10.3573. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boger DL, Honda T, Menezes RF, Colletti SL. Total synthesis of bleomycin a2 and related agents. 3. Synthesis and comparative evaluation of deglycobleomycin a2,epideglycobleomycin a2, deglycobleomycin a1, and desacetamido-, descarboxamido-, desmethyl-, and desimidazolyldegly-cobleomycin a2. J Am Chem Soc. 1994;116:5631–5646. [Google Scholar]
- 19.Aparicio T, Baer R, Gautier J. DNA double-strand break repair pathway choice and cancer. DNA Repair. 2014;19:169–175. doi: 10.1016/j.dnarep.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joshi N, Grant SG. DNA double-strand break damage and repair assessed by pulsed-field gel electrophoresis. Methods Mol Biol. 2004;291:121–129. doi: 10.1385/1-59259-840-4:121. [DOI] [PubMed] [Google Scholar]
- 21.(a) Olive PL, Banath JP. The comet assay: A method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]; (b) Forchhammer L, Johansson C, Loft S, Möller L, Godschalk RWL, Langie SAS, Jones GDD, Kwok RWL, Collins AR, Azqueta A, Phillips DH, Sozeri O, Stępnik M, Palus J, Vogel U, Wallin H, Routledge MN, Handforth C, Allione A, Matullo G, Teixeira JP, Costa S, Riso P, Porrini M, Møller P. Variation in the measurement of DNA damage by comet assay measured by the ecvag† inter-laboratory validation trial. Mutagenesis. 2010;25:113–123. doi: 10.1093/mutage/gep048. [DOI] [PubMed] [Google Scholar]
- 22.Colis LC, Hegan DC, Kaneko M, Glazer PM, Herzon SB. Mechanism of action studies of lomaiviticin a and the monomeric lomaiviticin aglycon. Selective and potent activity toward DNA double-strand break repair-deficient cell lines. J Am Chem Soc. 2015;137:5741–5747. doi: 10.1021/ja513117p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins AR, Oscoz AA, Brunborg G, Gaivão I, Giovannelli L, Kruszewski M, Smith CC, Štětina R. The comet assay: Topical issues. Mutagenesis. 2008;23:143–151. doi: 10.1093/mutage/gem051. [DOI] [PubMed] [Google Scholar]
- 24.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sung P, Klein H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 26.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. Γh2ax and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panier S, Boulton SJ. Double-strand break repair: 53bp1 comes into focus. Nat Rev Mol Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 28.Kastan MB, Lim DS. The many substrates and functions of atm. Nat Rev Mol Cell Biol. 2000;1:179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 29.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. Atm and DNA-pk function redundantly to phosphorylate h2ax after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 30.Laufer RS, Dmitrienko GI. Diazo group electrophilicity in kinamycins and lomaiviticin a: Potential insights into the molecular mechanism of antibacterial and antitumor activity. J Am Chem Soc. 2002;124:1854–1855. doi: 10.1021/ja0167809. [DOI] [PubMed] [Google Scholar]
- 31.Feldman KS, Eastman KJ. Studies on the mechanism of action of prekinamycin, a member of the diazoparaquinone family of natural products: Evidence for both sp2 radical and orthoquinonemethide intermediates. J Am Chem Soc. 2006;128:12562–12573. doi: 10.1021/ja0642616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mulcahy SP, Woo CM, Ding WD, Ellestad GA, Herzon SB. Characterization of a reductively-activated elimination pathway relevant to the biological chemistry of the kinamycins and lomaiviticins. Chem Sci. 2012;3:1070–1074. [Google Scholar]
- 33.Burrows CJ, Muller JG. Oxidative nucleobase modifications leading to strand scission. Chem Rev. 1998;98:1109–1152. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 34.Xue M, Herzon SB. Mechanism of nucleophilic activation of (–)-lomaiviticin a. J Am Chem Soc. 2016;138:15559–15562. doi: 10.1021/jacs.6b09657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abeywickrema AN, Beckwith ALJ. Mechanistic and kinetic studies of the thiodediazoniation reaction. J Am Chem Soc. 1986;108:8227–8229. [Google Scholar]
- 36.(a) Stubbe J, Kozarich JW, Wu W, Vanderwall DE. Bleomycins: A structural model for specificity, binding, and double strand cleavage. Acc Chem Res. 1996;29:322–330. [Google Scholar]; (b) Boger DL, Cai H. Bleomycin: Synthetic and mechanistic studies. Angew Chem, Int Ed. 1999;38:448–476. doi: 10.1002/(SICI)1521-3773(19990215)38:4<448::AID-ANIE448>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]; (c) Chen J, Stubbe J. Bleomycins: Towards better therapeutics. Nat Rev Cancer. 2005;5:102–112. doi: 10.1038/nrc1547. [DOI] [PubMed] [Google Scholar]; (d) Hecht SM. Anticancer Agents from Natural Products. 2. CRC Press; Boca Raton, FL: 2011. Bleomycin group antitumor agents; pp. 451–478. [Google Scholar]
- 37.Absalon MJ, Kozarich JW, Stubbe J. Sequence specific double-strand cleavage of DNA by febleomycin. 1. The detection of sequence-specific double-strand breaks using hairpin oligonucleotides. Biochemistry. 1995;34:2065–2075. doi: 10.1021/bi00006a029. [DOI] [PubMed] [Google Scholar]
- 38.(a) Boger DL, Lee JK. Development of a solution-phase synthesis of minor groove binding bis-intercalators based on triostin a suitable for combinatorial synthesis. J Org Chem. 2000;65:5996–6000. doi: 10.1021/jo000382r. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Fink BE, Brunette SR, Tse WC, Hedrick MP. A simple, high-resolution method for establishing DNA binding affinity and sequence selectivity. J Am Chem Soc. 2001;123:5878–5891. doi: 10.1021/ja010041a. [DOI] [PubMed] [Google Scholar]; (c) Tse WC, Boger DL. A fluorescent intercalator displacement assay for establishing DNA binding selectivity and affinity. Acc Chem Res. 2004;37:61–69. doi: 10.1021/ar030113y. [DOI] [PubMed] [Google Scholar]
- 39.Woo CM, Li Z, Paulson EK, Herzon SB. Structural basis for DNA cleavage by the potent antiproliferative agent (–)-lomaiviticin a. Proc Natl Acad Sci U S A. 2016;113:2851–2856. doi: 10.1073/pnas.1519846113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishimura T, Okobira T, Kelly AM, Shimada N, Takeda Y, Sakurai K. DNA binding of tilorone: 1h nmr and calorimetric studies of the intercalation. Biochemistry. 2007;46:8156–8163. doi: 10.1021/bi602402m. [DOI] [PubMed] [Google Scholar]
- 41.Tse WC, Boger DL. Sequence-selective DNA recognition: Natural products and nature’s lessons. Chem Biol. 2004;11:1607–1617. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 42.Cheatham TE, Case DA. Twenty-five years of nucleic acid simulations. Biopolymers. 2013;99:969–977. doi: 10.1002/bip.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chou SH, Chin KH, Chen FM. Looped out and perpendicular: Deformation of watson–crick base pair associated with actinomycin d binding. Proc Natl Acad Sci U S A. 2002;99:6625–6630. doi: 10.1073/pnas.102580399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pfoh R, Laatsch H, Sheldrick GM. Crystal structure of trioxacarcin a covalently bound to DNA. Nucleic Acids Res. 2008;36:3508–3514. doi: 10.1093/nar/gkn245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yakovchuk P, Protozanova E, Frank-Kamenetskii MD. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 2006;34:564–574. doi: 10.1093/nar/gkj454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Absalon MJ, Wu W, Kozarich JW, Stubbe J. Sequence-specific double-strand cleavage of DNA by febleomycin. 2. Mechanism and dynamics. Biochemistry. 1995;34:2076–2086. doi: 10.1021/bi00006a030. [DOI] [PubMed] [Google Scholar]
- 47.(a) Boger DL, Garbaccio RM. Catalysis of the cc-1065 and duocarmycin DNA alkylation reaction: DNA binding induced conformational change in the agent results in activation. Bioorg Med Chem. 1997;5:263–276. doi: 10.1016/s0968-0896(96)00238-6. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Garbaccio RM. Shape-dependent catalysis: Insights into the source of catalysis for the cc-1065 and duocarmycin DNA alkylation reaction. Acc Chem Res. 1999;32:1043–1052. [Google Scholar]; (c) Wolkenberg SE, Boger DL. Mechanisms of in situ activation for DNA-targeting antitumor agents. Chem Rev. 2002;102:2477–2496. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- 48.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 49.Hosoya N, Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014;105:370–388. doi: 10.1111/cas.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bassi C, Ho J, Srikumar T, Dowling RJO, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Nuclear pten controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341:395–399. doi: 10.1126/science.1236188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roy R, Chun J, Powell SN. Brca1 and brca2: Different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hopkins BD, Hodakoski C, Barrows D, Mense SM, Parsons RE. Pten function: The long and the short of it. Trends Biochem Sci. 2014;39:183–190. doi: 10.1016/j.tibs.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM, Pollard JR. Selective killing of atm- or p53-deficient cancer cells through inhibition of atr. Nat Chem Biol. 2011;7:428–430. doi: 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]
- 54.Colis LC, Herzon SB. Synergistic potentiation of (–)-lomaiviticin a cytotoxicity by the atr inhibitor ve-821. Bioorg Med Chem Lett. 2016;26:3122–3126. doi: 10.1016/j.bmcl.2016.04.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borders DB, Doyle TW. Enediyne Antibiotics as Antitumor Agents. Marcel Dekker; New York: 1995. [Google Scholar]
- 56.Tichenor MS, Boger DL. Yatakemycin: Total synthesis, DNA alkylation, and biological properties. Nat Prod Rep. 2008;25:220–226. doi: 10.1039/b705665f. [DOI] [PubMed] [Google Scholar]