

ABSTRACT
Invasive aspergillosis (IA) remains the primary cause of morbidity and mortality in chronic granulomatous disease (CGD) patients, often due to infection by Aspergillus species refractory to antifungals. This motivates the search for alternative treatments, including immunotherapy. We investigated the effect of exogenous type I interferon (IFN) activation on the outcome of IA caused by three Aspergillus species, A. fumigatus, A. nidulans, and A. tanneri, in CGD mice. The animals were treated with poly(I):poly(C) carboxymethyl cellulose poly-l-lysine (PICLC), a mimetic of double-stranded RNA, 24 h preinfection and postinfection. The survival rates and lung fungal burdens were markedly improved by PICLC immunotherapy in animals infected with any one of the three Aspergillus species. While protection from IA was remarkable, PICLC induction of type I IFN in the lungs surged 24 h posttreatment and returned to baseline levels by 48 h, suggesting that PICLC altered early events in protection against IA. Immunophenotyping of recruited leukocytes and histopathological examination of tissue sections showed that PICLC induced similar cellular infiltrates as those in untreated-infected mice, in both cases dominated by monocytic cells and neutrophils. However, the PICLC immunotherapy resulted in a marked earlier recruitment of the leukocytes. Unlike with conidia, infection with A. nidulans germlings reduced the protective effect of PICLC immunotherapy. Additionally, antibody depletion of neutrophils totally reversed the protection, suggesting that neutrophils are crucial for PICLC-mediated protection. Together, these data show that prophylactic PICLC immunotherapy prerecruits these cells, enabling them to attack the conidia and thus resulting in a profound protection from IA.
KEYWORDS: Aspergillus fumigatus, Aspergillus nidulans, Aspergillus tanneri, poly(I:C) (PICLC), chronic granulomatous disease (CGD), conidia, neutrophils, phagocyte recruitment, type I IFN
IMPORTANCE
Patients with chronic granulomatous disease (CGD) are highly susceptible to invasive aspergillosis (IA). While Aspergillus fumigatus is the most-studied Aspergillus species, CGD patients often suffer IA caused by A. nidulans, A. tanneri, and other rare species. These non-fumigatus Aspergillus species are more resistant to antifungal drugs and cause higher fatality rates than A. fumigatus. Therefore, alternative therapies are needed to protect CGD patients. We report an effective immunotherapy of mice infected with three Aspergillus species via PICLC dosing. While protection from IA was long lasting, PICLC induction of type I IFN surged but quickly returned to baseline levels, suggesting that PICLC was altering early events in IA. Interestingly, we found responding immune cells to be similar between PICLC-treated and untreated-infected mice. However, PICLC immunotherapy resulted in an earlier recruitment of the leukocytes and suppressed fungal growth. This study highlights the value of type I IFN induction in CGD patients.
INTRODUCTION
Invasive aspergillosis (IA) is one of the most serious fungal diseases that immunocompromised patients encounter throughout the world (1, 2). The annual global burden of IA is estimated to be more than 300,000 with a high fatality rate (30 to 95%) (3). The patient populations at the highest risk for IA are those with prolonged neutropenia from intensive myeloablative chemotherapy and those with primary immune deficiency such as chronic granulomatous disease (CGD) (4, 5). CGD is a rare genetic disorder of the NADPH-oxidase system, in which phagocytes are defective in generating the microbicidal reactive oxygen species (ROS) and their metabolites (4), which are required for host defense against a variety of pathogens. As a result, CGD patients are susceptible to recurrent, life-threatening bacterial and fungal infections (1, 5, 6). The primary etiologic agent of IA in both neutropenic and CGD patient populations is Aspergillus fumigatus, a species generally susceptible to triazoles and amphotericin B, which are the recommended antifungals for first-line therapy. It is noteworthy that occurrences of infection caused by azole-resistant A. fumigatus mutants have been increasingly reported (7). In contrast to patients with secondary immunosuppression, CGD patients are also presented with less well characterized pathogenic Aspergillus species such as A. nidulans (8), A. pseudoviridinutans (9), A. tanneri (10), A. udagawae (11), and A. calidoustus (12). These species are almost exclusively isolated from CGD patients, and all are more resistant to currently available antifungals than A. fumigatus (S. Seyedmousavi, M. S. Lionakis, M. Parta, S. W. Peterson, K. J. Kwon-Chung, submitted for publication). Generally, infections in CGD patients caused by these species are chronic and require higher doses of antifungals or longer durations of therapy or result in total therapy failure (8, 10, 11). The predilection of these non-fumigatus Aspergillus species for CGD patients is not yet understood.
Despite the availability of antifungal prophylaxis for CGD patients (13), infections by Aspergillus species are nonetheless the leading cause of mortality in these patients (14, 15). Thus, there is a clear need for alternative therapeutic modalities, especially to combat infections by species refractory to currently available antifungal therapy. One such strategy is immunotherapy, which aims to enhance or restore antifungal immunity in patients (16, 17). One such antifungal immunotherapy in CGD is type II interferon (gamma interferon [IFN-γ]) administration, which promotes a protective phenotype against aspergillosis (18, 19). While IFN-γ administration is generally beneficial, recent CGD cases of A. tanneri proved fatal even after combined IFN-γ and antifungal treatment (10), suggesting that alternative immunotherapies are needed.
Induction of type I IFN is another immune therapy strategy studied for eventual use against cancer and/or infectious disease. The role of type I IFN is well documented in host defense against viral infection (20), but it has also been demonstrated to affect the outcome of infections with bacteria (21), parasites (22, 23), and fungi (24–27). The type I IFN family is a multigene cytokine family that encodes 13 partially homologous IFN-α subtypes in humans (14 in mice), a single IFN-β, and several less well defined single-gene products (IFN-ε, IFN-τ, IFN-κ, IFN-ω, IFN-δ, and IFN-ζ) (25, 26). During fungal infection, type I IFN-mediated signaling exerted a protective role against Candida albicans and Cryptococcus neoformans in mice (24, 27, 28), while it had a deleterious effect against Histoplasma capsulatum (29) and Candida glabrata (30). Poly(I:C) and its stabilized derivative [Hiltonol; poly(I:C) condensed with poly-l-lysine and carboxymethyl cellulose (PICLC)] are synthetic mimics of the viral double-stranded RNA (dsRNA) designed to stimulate prolonged and high-level production of type I IFNs (31–34). PICLC is currently being investigated in phase II clinical studies in patients with metastatic solid tumors: sarcomas, melanoma, squamous cell skin cancer, and head and neck cancers (clinicaltrials.gov, NCT01984892) (35).
In the current study, we investigated the effect of an elevated type I IFN environment induced by PICLC immunotherapy in murine aspergillosis caused by A. fumigatus and two non-fumigatus Aspergillus species, A. tanneri and A. nidulans, uniquely associated with especially problematic IA in CGD patients (8, 10). PICLC immunotherapy protected mice from all three species by reducing the growth of fungi in the lung. Analysis of immune cells in the lung indicated that PICLC immunotherapy recruited significantly higher numbers of neutrophils and other myeloid leukocytes earlier than in the untreated control mice.
RESULTS
Immunotherapy with PICLC protects CGD mice against A. fumigatus infection.
Since exogenous induction of type I IFN has shown positive results for the experimental treatment of cancer (35) and some infectious diseases (24), we resolved to investigate the efficacy of exogenous induction of type I IFN by PICLC in the setting of IA caused by Aspergillus species which are refractory to antifungal treatment in CGD patients. PICLC immunotherapy significantly improved survival compared to the nontreated controls in mice infected with A. fumigatus (Fig. 1A) and resulted in significant suppression of the fungal burden as measured by lung fungal DNA load throughout the experimental period (Fig. 1B). In histopathology sections taken 3 days postinfection, small lesions were visible throughout the lung in both groups of infected animals. However, fewer infectious foci were observed in the animals which received PICLC immunotherapy (Fig. 1C).
FIG 1 .

PICLC protects CGD mice against A. fumigatus infection. CGD mice were treated with either PICLC (15 μg PICLC in 30 μl PBS/mouse) or PBS via pharyngeal aspiration 24 h pre- and post-infection with A. fumigatus (5 × 103 conidia/mouse). (A) The survival of mice (n = 6) was monitored for 15 days. (B) At 3, 6, and 15 days postinfection, lungs (n = 12) were harvested and fungal burdens were estimated by measuring fungal DNA by qPCR. *, P ≤ 0.05. (C) In H&E-stained histology sections taken at 3 days postinfection, fewer infectious foci were observed in the animals which received PICLC immunotherapy. Bar, 2 mm.
PICLC immunotherapy protects CGD mice against A. nidulans infection.
To expand these results to non-fumigatus Aspergillus species, identical PICLC immunotherapy was tested in A. nidulans infection. PICLC immunotherapy was also protective in this model, as 80% of the mice in the control group (phosphate-buffered saline [PBS] treated) succumbed to IA within 28 days postinfection, whereas no deaths were observed in the PICLC immunotherapy group during the same period (Fig. 2A). Fungal DNA loads were substantially higher in the lungs of the control group mice than the PICLC-dosed mice, suggesting that growth of A. nidulans in host tissue was hampered by PICLC immunotherapy (Fig. 2B). While fewer lung infectious foci were observed 3 days postinfection in PICLC immunotherapy mice than in control mice, individual foci were similarly sized and contained similar germinated fungal elements (Fig. 2C and D, top row). By 15 days postinfection, foci had matured into granulomas in both PICLC immunotherapy and control mice, but fungal elements were noticeably smaller and fewer in lung granulomas of PICLC immunotherapy mice than in infected-untreated controls (Fig. 2D, bottom row).
FIG 2 .

PICLC protects CGD mice against A. nidulans infection. CGD mice were treated with either PICLC (15 μg PICLC in 30 μl PBS/mouse) or PBS via pharyngeal aspiration 24 h pre- and post-infection with A. nidulans (5 × 103 conidia/mouse). (A) The survival of mice (n = 6) was monitored for 28 days. (B) Lungs from infected mice were taken at 3 and 15 days postinfection (n = 8), and fungal burdens were estimated by measuring fungal DNA by qPCR. *, P ≤ 0.05. (C) Montaged 25× images of H&E-stained sections taken 3 days postinfection showing size and frequency of infectious foci in PICLC immunotherapy and control mice. Bar, 2 mm. (D) Higher-magnification images of infectious foci at 3 days postinfection (pi) (top row) and granuloma at 15 days postinfection (bottom row). Left columns, H&E-stained sections; right columns, GMS-stained sections. Bars, 100 µm (H&E images) and 50 µm (GMS images).
PICLC immunotherapy protects CGD mice against A. tanneri infection.
A. tanneri is a novel species noted for its high baseline resistance to antifungal drugs (10), isolated from two fatal cases of IA in CGD patients. While infection kinetics were different between A. tanneri and the two abovementioned Aspergillus species, PICLC immunotherapy was nonetheless protective against A. tanneri. As shown in Fig. 3A, all mice in the control group succumbed to IA in 90 days, whereas 100% of the mice dosed with PICLC were still alive at 90 days postinfection. Fungal DNA loads were also substantially higher in the lungs of the control group mice than in the PICLC-treated mice (Fig. 3B). Histology confirmed the presence of granulomas at 30 days postinfection in the lungs of control group mice, but fewer and smaller lesions were observed in the PICLC-dosed mice (Fig. 3C). Thus, PICLC immunotherapy was protective against multiple Aspergillus species.
FIG 3 .

PICLC protects CGD mice against A. tanneri infection. CGD mice were treated with either PICLC (15 μg PICLC in 30 μl PBS/mouse) or PBS via pharyngeal aspiration 24 hours pre- and post-infection with A. tanneri (5 × 103 conidia/mouse). (A) The survival of mice (n = 6) was monitored for 90 days. (B) Fungal DNA loads as measured by qPCR in the lungs of PICLC-treated and control mice (n = 8) at 15 and 30 days postinfection. *, P ≤ 0.05. (C) Histology confirmed the presence of granulomas at 30 days postinfection in the lungs of control group mice, but fewer lesions were observed in the PICLC-dosed mice. Bar, 2 mm.
Type I IFN is briefly induced by PICLC in the lungs of CGD mice.
To confirm that PICLC was able to induce type I IFN in CGD mice, IFN protein was measured following a single dose of PICLC. As shown in Fig. 4, there was significant expression (P value < 0.05) of IFN-α and IFN-β from 6 to 30 hours postinjection, which then returned to near baseline by 48 h postinjection. These data indicate that CGD mice mount a type I IFN response to PICLC exposure and that this response lasts for approximately 2 days following injection.
FIG 4 .

PICLC dosing results in a robust but temporary induction of type I IFN in the lungs of CGD mice. Uninfected CGD mouse lung homogenate was analyzed for IFN-α (A) and IFN-β (B) at the indicated time points following a single PICLC aspiration. There was significant expression of IFN-α and IFN-β from 6 to 48 hours postinjection (*, P ≤ 0.05).
PICLC immunotherapy induces significant leukocyte recruitment in the lungs.
Since the IFN response to PICLC lasts approximately 2 days following injection (Fig. 4) coupled with effective protection from IA (Figs. 1 to 3), we hypothesized that PICLC immunotherapy results in some immunologic change that impacts the early events of Aspergillus infections. To characterize the effects of PICLC on these early immune events, leukocytes were isolated from the lungs of A. nidulans-infected or control mice with or without PICLC dosing at the time of infection (0 days postinfection) or 1 and 3 days postinfection. Full leukocyte recruitment into the lungs was monitored utilizing a fluorescein isothiocyanate (FITC)-conjugated CD45-specific antibody injected into the mice intravenously 3 to 5 min prior to euthanasia. This technique marks vascular cells with FITC by flow cytometry. In samples taken 24 h after a single dose of PICLC and before infection (day 0), a significant influx of immune cells was observed compared to control samples (Fig. 5A, 0 days postinfection). Histopathological analysis of similarly dosed samples shows that these recruited leukocytes are especially notable as infiltrates accumulating near airways (Fig. 6A and C) and as newly recruited cells near endothelium (Fig. 6B). It should be noted that this time point represents the condition of the lungs at the time of infection in the PICLC immunotherapy-receiving animals. Immunophenotyping of the lung leukocyte populations showed that PICLC results in the recruitment of several immune cell types. Monocyte-derived myeloid cells and neutrophils dominated this infiltrate, but increased numbers of blood monocytes, NK cells, and plasmacytoid dendritic cells were also observed in response to PICLC (Fig. 5B).
FIG 5 .

Immunophenotyping of leukocytic infiltrates. Immunophenotyping of single-cell suspensions derived from A. nidulans-infected and PICLC-dosed or control lungs from CGD mice isolated 0, 1, and 3 days postinfection. Consistent with the previous PICLC dosing schedule, mice analyzed on days 0 and 1 received one dose of PICLC, while mice harvested on day 3 received the second PICLC dose. (A) Total CD45-positive (immune) cells in the lungs. (B) Calculated total cells per lung for various immune cell populations (y axis): alveolar macrophages (AM), blood monocytes (mono), monocyte-derived myeloid cells (MDMC), classical dendritic cells (cDC), plasmacytoid dendritic cells (pDC), neutrophils (polymorphonuclear leukocytes [PMN]), eosinophils (EOS), natural killer cells (NK), B cells, CD4 T cells, and CD8 T cells. Note that all the measured cells are parenchymal, CD45-FITC negative, except for the blood monocytes, which are CD45-FITC positive. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. (C) Representative flow plot panels gated on neutrophils depicting parenchymal staining of cells on the y axis and forward scatter (FSC) on the x axis. Data displayed are from one representative experiment from two total experiments. Indicated statistical comparisons are between PBS and PICLC conditions and corresponding time points.
FIG 6 .
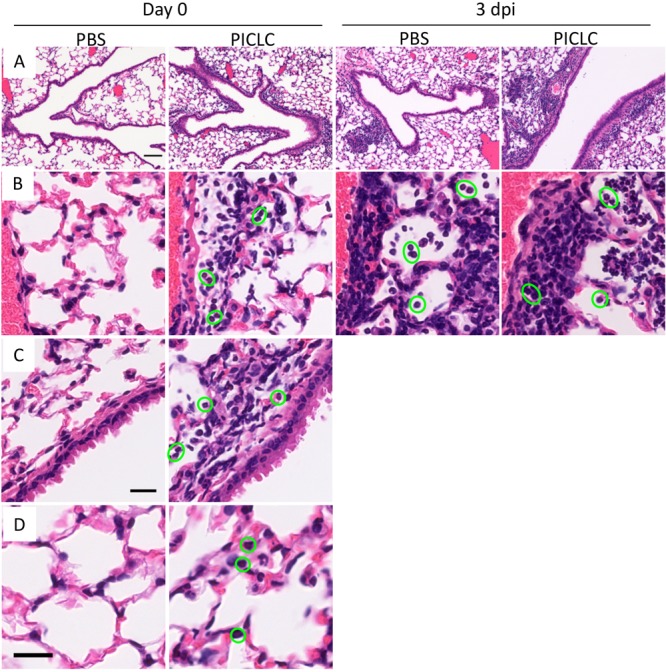
PICLC dosing recruits immune cells to lung airways and alveoli but does not seem to alter the composition of early infectious foci. Hematoxylin-and-eosin-stained lung sections harvested from PICLC-dosed or control CGD mice (day 0) or PICLC-dosed or control mice infected with A. nidulans (day 3 postinfection [dpi]). Images in row A were acquired using an ×100 magnification, whereas all other images were acquired at ×400. (A) Lung airways and recruited leukocytes (visible at this magnification as dark areas). (B) Perivascular lung areas depicting immune cell extravasation. (C) Airway epithelia and surrounding tissue with recruited immune cells. (D) Lung alveoli. Note that in rows B, C, and D neutrophils are highlighted with green circles. Bars, 100 µm (A) and 20 µm (C and D) (bar in row C also applies to row B).
Interestingly, the numbers of recruited CD45+ leukocytes and of most individual immune cell types were converging in the lungs of infected mice either with or without PICLC immunotherapy at 1 and 3 days postinfection (Fig. 5A and B). This is consistent with similar periendothelial accumulation of recruited cells (Fig. 6B) in the two mouse groups. Thus, A. nidulans infection resulted in a robust recruitment of immune cells between 24 and 72 hours postinfection in the mice not receiving immunotherapy, but these mice were unable to control the fungus and eventually succumbed to the infection (Fig. 2). While PICLC-dosed mice recruit essentially the same types of cells, the timing of the recruitment was earlier and correlated with protection (Fig. 2). Importantly, Aspergillus species go through a gradual morphological change in this period from dormant spores (conidia) to developing hyphae. Thus, we hypothesized that this early recruitment allows immune cells to act on conidia, which are more susceptible to the antifungal activities mustered by CGD hosts (36).
Protection by PICLC immunotherapy is significantly diminished in CGD mice infected with germinated conidia.
To test the specificity of PICLC immunotherapy for the conidial stage of IA, we compared the differences between CGD mice infected with resting conidia and with conidia germinated for 5 h (germlings) of A. nidulans (Fig. 7A). We hypothesized that infection with germlings would bypass whatever susceptibility exists in the conidial stage and that early prerecruited leukocytes would be less efficient in eradicating germlings, rendering PICLC immunotherapy less protective. In Fig. 7B, CGD mice infected with the germinated A. nidulans conidia were only partially protected by PICLC immunotherapy, whereas mice infected with resting conidia were completely protected. This indicates that a significant portion of PICLC immunotherapeutic protection is attributable to the ability of prerecruited immune cells to suppress conidia.
FIG 7 .
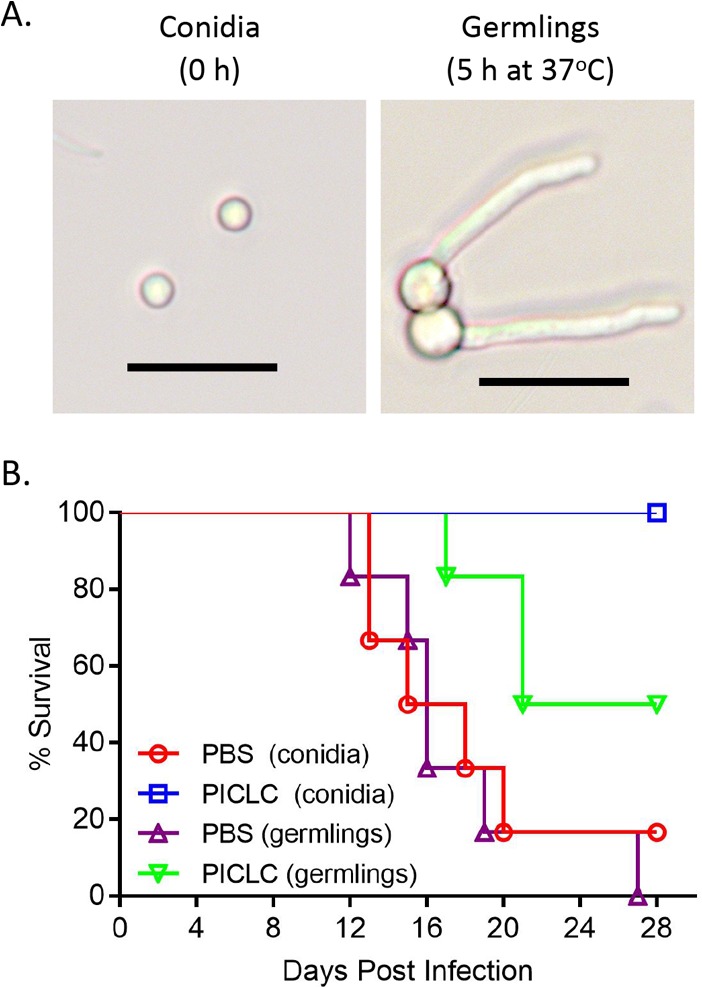
Infection with germlings compromises PICLC protection. (A) A. nidulans germlings were generated by incubating fresh conidia for 5 h in RPMI medium at 37°C. Germination was monitored by estimation of germ tube length by light microscopy. Bars, 10 µm. (B) CGD mice were left untreated or PICLC dosed as described above and then infected with resting conidia or pregerminated cells. Survival of infected mice was monitored for 28 days.
Neutrophils are required for PICLC immunotherapy-mediated protection from IA.
We observed significantly more neutrophils in PICLC immunotherapy mice than in control mice at experimental day 0 (after receiving one dose of PICLC but before infection), but no difference was detected between the treated and untreated infected mice at days 1 and 3 postinfection (Fig. 5B). Infiltration of neutrophils was also noted by histopathology, gathering on the parenchymal side of the endothelium, near airways, and in the alveoli of PICLC-dosed mice at day 0 (Fig. 6C and D, green-circled cells). As neutrophils have a well-established role in IA infection and were highly recruited by PICLC, we probed the role of neutrophils in PICLC-mediated protection utilizing monoclonal antibody (MAb) depletion. The 1A8 monoclonal antibody, administered to the mice prior to infection, efficiently depleted neutrophils, resulting in a 90% reduction in recruited neutrophils 1 day postinfection (Fig. 8A). This depletion of neutrophils resulted in significantly higher day 3 fungal burdens than in those mice receiving PICLC immunotherapy alone (Fig. 8B). These data suggest that neutrophils in the lung may play a protective role in PICLC immunotherapy.
FIG 8 .

Depletion of neutrophils with a monoclonal antibody reverses PICLC protection. CGD mice were dosed with PICLC and infected with A. nidulans as described above. One group of mice was treated with 1A8 monoclonal antibody to deplete neutrophils prior to PICLC dosing and infection. (A) At day 1 postinfection, cells were isolated from mouse lungs and neutrophils were enumerated by flow cytometry. n = 2 mice per group. (B) Three days postinfection, lungs were harvested and fungal load was measured using qPCR. n = 4 mice per group. Data from one representative experiment from two total experiments are displayed. *, P ≤ 0.05; ns, not significant.
DISCUSSION
PICLC immunotherapy was effective in reducing mortality, pathology, and fungal loads in the CGD model of murine IA for all three Aspergillus species tested. Prophylactic PICLC immunotherapy recruited leukocytes so that immune cells were present in the lungs in significant numbers and localized specifically in the alveoli preceding the arrival of the infectious conidia. However, a similar set of leukocytes was recruited in mice infected with Aspergillus alone with a delayed kinetics compared to PICLC immunotherapy. While we have not excluded the importance of other types of immune cells, we observed neutrophils to be the critical cells for mediating this protection based on their presence in the alveoli subsequent to PICLC dosing and the loss of protection upon depletion of these cells. Several studies have demonstrated the critical role that neutrophils play in host defense against Aspergillus (reviewed in references 37 to 41). Our data support the hypothesis that PICLC immunotherapy induces very early recruitment of neutrophils, which destroy a significant fraction of the fungus before it can fully differentiate into hyphae.
We suggest that the earliest stages of Aspergillus infection are a window of opportunity for CGD hosts where efficient induction of immunity can limit infection because our results also indicated that the in vitro-differentiated germlings were more refractory to PICLC immunotherapy than conidia (Fig. 7B). While CGD neutrophils are defective in hyphal killing due to their deficiency in phagosomal oxidase activity, recent studies have shown that these cells do retain killing activity against Aspergillus conidia via nonoxidative pathways specifically involving CD11b/CD18 integrin and phosphatidylinositol 3-kinase (PI3K)- and lactoferrin-mediated iron depletion (36, 40, 42). However, the Aspergillus factors which contribute to this conidial susceptibility to nonoxidative killing remain uncharacterized. Additionally, the most susceptible stage of Aspergillus may not be conidia but rather the early stages of conidium germination, because lactoferrin-mediated iron depletion arrests the growth of Aspergillus at the germling stage but not at the initiation of germination (42). Ultimately, the factors that result in the susceptibility of Aspergillus conidia to nonoxidative killing remain an exciting avenue of future study, especially since manipulation of any putative susceptibility factors may itself become a possible therapeutic strategy for IA.
We consider our results as having important implications for Aspergillus pathobiology. Following deposition of Aspergillus conidia into the lung, a fraction of these conidia are destroyed by the relatively numerous resident alveolar macrophages (AMs) (39, 43, 44) and the relatively scarce neutrophils that are resident in the lungs or are recruited in the first few hours of infection (36, 38, 42). However, some conidia escape these initial defenses (Fig. 9, i and ii). These conidia swell and germinate in response to environmental cues in the first 5 to 12 h of an infection (Fig. 9B). In humans and mice with normal immune function, neutrophils kill hyphal Aspergillus through ROS production (36, 40), which stifles the infection subclinically (Fig. 9, iv). Neutrophils from CGD patients and CGD model mice, however, are unable to kill germinated Aspergillus (36, 40, 42). This allows the progressive growth of fully germinated vegetative hyphae (Fig. 9, v), which is associated with morbidity. PICLC prophylaxis alters this trajectory in CGD mice as immunotherapy recruits the immune cells, especially neutrophils, into alveoli, which intercept a significant fraction of conidia (Fig. 9, iii). The few remaining conidia in PICLC immunotherapy lungs do seem to germinate normally and generate similar infectious foci (Fig. 2C) but at a greatly reduced frequency per animal (Fig. 1C and 2C), eventually resulting in reduced overall fungal loads (Fig. 9, vi) and a significantly better survival outcome of the PICLC-treated mice.
FIG 9 .

Model of early immune response against Aspergillus in CGD versus normal lungs. Illustrative model of the early pulmonary pathobiology of Aspergillus infection showing very early stages, just after conidia enter the alveoli (A), and later following Aspergillus germination and leukocyte recruitment (B). Several alveoli are depicted, with each alveolus representing a condition examined in this work. In non-CGD lungs, a fraction of conidia are killed by alveolar macrophages, leaving some conidia to germinate (i), but upon recruitment of fully fungicidal neutrophils, the infection is contained in a reactive oxygen species-dependent manner (iv). In CGD lungs, initial stages proceed similarly (ii). These infectious foci recruit many neutrophils (v), but due to their deficiency in ROS production, these cells cannot contain the infection and contain many hyphae. PICLC prophylaxis alters the initial stages in CGD lungs in that the prerecruited neutrophils drastically reduce the initial infectious load at the conidial stage (iii). The few fungal elements that do survive and germinate in PICLC-treated CGD lungs do seem to develop into infectious foci (vi), but the overall infection is nonlethal.
The mechanisms and shortcomings of IA immunotherapy elucidated in this study have important implications for the conception and development of future IA-targeted immunotherapies. These data and others (36, 39, 40, 42) clearly separate early IA into two major immunologic stages roughly corresponding to the dominant fungal morphology. We suggest that effective immunotherapy for IA will require enhancing immunity during one, or both, of these stages, probably by addressing the immunologic deficiencies of each stage. During the initial stage of infection, Aspergillus conidia are killed via nonoxidative killing by macrophages and neutrophils in both healthy and CGD patients (36). A clear limitation of immunity during this stage is the relatively small population of phagocytes present in unperturbed lungs (Figs. 5 and 6). Therefore, putative immunotherapeutic approaches which aim to act on this first stage need to result in the enhanced recruitment of neutrophils and perhaps other phagocytes capable of killing conidia. Our data predict that significant numbers of leukocytes need to be present within the first few hours of infection to be efficacious, so successful strategies which target this stage will probably require prophylactic dosing, such as demonstrated here by PICLC. Once the Aspergillus germinates into full hyphae, generation of reactive oxygen is required to kill the mold. So, while there are plenty of local neutrophils at this stage, CGD neutrophils are incapable of fungal control. This suggests that an immunomodulatory strategy that targets this later stage would need to enhance hyphal killing but not necessarily improve recruitment. An example of immunotherapy aimed at this later stage is recombinant IFN-γ treatment (described clinically in reference 18), which improves activity of neutrophils against Aspergillus hyphae in vitro (45). While the evidence presented in this study strongly supports PICLC immunotherapy acting during the conidial stage, we cannot rule out some additional PICLC effects which act later at the hyphal stage. In fact, while histological analysis of infected lungs early after infection shows similarly composed infectious foci with and without PICLC immunotherapy (Fig. 2C), later time points show markedly less fungal material in granuloma cores from PICLC-dosed mice than from untreated groups (Fig. 2D). Furthermore, infection with germlings only partially reversed PICLC-mediated protection (Fig. 7). Together, these results suggest that PICLC immunotherapy induces some additional antihyphal activity in CGD mice, perhaps similar to that of IFN-γ (45). Type I IFN has been shown to positively influence several potentially relevant neutrophil activities in other models such as direct cytotoxicity (46), neutrophil extracellular trap formation (47, 48), and maximal neutrophil differentiation allowing full release of cytotoxic factors (47), reinforcing the plausibility of PICLC immunotherapy having some activity during the hyphal stage. Future studies are therefore necessary to test whether any PICLC-mediated antihyphal activities exist and whether they show efficacy against IA in vivo.
Overall, while the data presented here provide some insight into the generalized parameters for IA immunotherapies, we believe that the efficacy shown here by PICLC merits further development as a possible clinical prophylactic agent for patients at high risk of IA. While prophylactic regimens can be burdensome, several prophylactic strategies are standard practice in certain immunocompromised populations, including CGD patients who are at risk for IA. PICLC immunotherapy may also have some distinct advantages over existing antifungal strategies (summarized in reference 13). Prophylactic antifungal chemotherapy can be effective against susceptible organisms, but resistant organisms are unaffected, and despite the common prophylactic regimens, a significant clinical IA burden remains (14, 15). Because immune therapies, such as PICLC, function by entirely different mechanisms, they can be effective even against antifungal-resistant organisms. As proof of this principle, here we show the efficacy of PICLC immunotherapy against two such azole-resistant species, A. nidulans and A. tanneri (Figs. 2 and 3). Furthermore, PICLC may be less costly than recombinant IFN-γ (13), as polymer compounds are typically less expensive to manufacture than recombinant proteins. In some situations, type I IFN can lead to less immunopathology than IFN-γ (49). Relatedly, long-term PICLC dosing was well tolerated in mice (24). Finally, we showed that PICLC immunotherapy is effective in the mouse A. tanneri IA model, suggesting that this treatment may be useful in preventing IA caused by Aspergillus species which are refractory to IFN-γ treatment (10). Thus, while it is too early to speculate about the efficacy of PICLC immunotherapy compared to other prophylactic regimens, the data presented here demonstrate clear efficacy in the CGD model of IA and suggest further study of this promising immunotherapy.
MATERIALS AND METHODS
Fungal strains.
Three Aspergillus species isolated from IA cases were used for in vitro and in vivo studies, including Aspergillus fumigatus B-5233 (50) and A. nidulans M24 and A. tanneri NIH 1004 (10), all three strains being isolated from CGD patients. The isolates were stored in 10% glycerol at −80°C and were revived on malt extract agar (MEA) for 5 to 7 days at 37°C. All isolates were freshly cultured on MEA for 5 to 7 days at 37°C before preparation of the inoculum.
Animals and husbandry.
The gp91phox− CGD mice (51) (7- to 11-week-old males) from Jackson Laboratories, USA, were housed under standard conditions, with drink and feed supplied ad libitum. To evaluate the efficacy of PICLC immunotherapy, animals were randomized into groups of 22 (6 mice for survival analysis, 8 mice for the determination of fungal burdens in the lung using quantitative real-time PCR [qPCR], and 8 mice for histopathological analysis). In addition, CGD mice were randomized in groups of 3, 5, and 5 mice to quantify interferon, immunophenotyping of leukocytic infiltrates, and depletion of neutrophils with a monoclonal antibody. All PICLC immunotherapy experiments for each strain were performed using two independent replicates. To avoid selection bias, all studies were blind, in that animals were preassigned to survival, qPCR, and histopathology groups at the time of infection.
Animal infection model.
CGD mice were infected with 30 µl freshly harvested Aspergillus conidial suspension (5 × 103 CFU/mouse) by the pharyngeal aspiration technique, as described previously (52). Before performing the experiment, the conidial suspension was filtered through sterile 30-µm cell strainers (MACS Smart strainers; Miltenyi Biotec, Auburn, CA) to remove any hyphae and conidial clumps, and the number of conidia was counted in a hemacytometer. The inoculum size was determined from hemacytometer counts and conidial viability. After the inoculum was adjusted to the required concentration, the conidial suspension was stored overnight at 4°C until use. In all survival studies, experienced individuals who were blind to the animal treatment monitored the infected mice at least twice daily.
Histopathological analysis.
For some experiments lungs were isolated from randomly chosen surviving animals for histopathological analysis. Preparation of histopathological sections and staining with hematoxylin and eosin (H&E) and Gomori’s methenamine-silver (GMS) were performed by Histoserv Inc., Germantown, MD. Color microscopy images were acquired using a Zeiss Axio Observer inverted microscope and Zeiss Zen microscope software. All images were acquired using identical illumination and camera settings for each magnification setting. Images displayed here were identically scaled, cropped, and resized. Image acquisition and postprocessing used Zen software (Carl Zeiss Microscopy, Jena, Germany).
PICLC formulation and dosing.
The animals were treated with PICLC [poly(I:C) condensed with poly-l-lysine and carboxymethyl cellulose], a stabilized synthetic analogue of viral dsRNA via pharyngeal aspiration 24 h preinfection and postinfection. The commercial formulation of PICLC (Hiltonol) was supplied by Oncovir Inc., Washington, DC. Working-concentration drug solutions (15 μg PICLC in 30 μl PBS/mouse) were prepared each day of administration. The control mice received PBS.
Determination of fungal burden in lung.
On the indicated days postchallenge, lungs were isolated and DNA was extracted from lungs using the Fast DNA Spin kit (MP Biomedicals, Santa Ana, CA) according to the manufacturer’s protocol as described previously (10). The concentration of total DNA isolated from lung tissue was measured by the NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Aspergillus loads were determined by real-time qPCR using primers and probe (6-carboxyfluorescein [FAM] labeled) targeting the 28S-ITS2 region of the ribosomal subunit gene of each Aspergillus species, as described previously (53). The primer and probe sequences used in the current study are shown in Table S1 in the supplemental material. The qPCRs were run with 250 ng of lung DNA as the template. To determine the Aspergillus load in each organ sample (picograms per nanogram of total DNA isolated), the fungal DNA concentration was calculated from a standard curve derived from an 8-fold dilution series of the genomic DNA of each Aspergillus species.
Primer and probe sequences used for qPCR assays. Download TABLE S1, DOC file, 0.04 MB (42KB, doc) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Quantification of interferon in naive CGD mice.
Lungs of uninfected CGD mice were isolated 6, 24, 30, and 48 h after a single dose of PICLC or PBS, and then each mouse lung was homogenized in 1 ml of lysis buffer (0.5% Tween 20, 1 mM EDTA, 1 mM benzamidine, 0.1 mM benzethonium chloride, and 0.1 mM phenylmethylsulfonyl fluoride [PMSF]) using a probe homogenizer (TH; Omni Inc., Kennesaw, GA). Cleared (centrifuged at 7,000 × g for 5 min) homogenate was analyzed for total protein concentration (RC/DC protein assay kit; Bio-Rad, Hercules, CA) as well as IFN-α and IFN-β concentration using VeriKine mouse alpha interferon and beta interferon enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN).
In vivo efficacy of PICLC against A. nidulans hyphae.
To assess the impact of PICLC treatment on A. nidulans germlings versus conidia, the conidia were germinated for 5 h at 37°C in RPMI 1640 medium containing l-glutamine but without phenol red (Gibco, Gaithersburg, MD). Prepared in this manner, Aspergillus conidia swell and germinate to form 6- to 8-µm-long germlings (Fig. 7A). These germlings were then inoculated into mice via pharyngeal aspiration identically to the conidia described above.
Vascular staining, lung dissection, and isolation of immune cells.
Mice were intravenously injected with FITC-conjugated anti-CD45 antibody (BioLegend, San Diego, CA) 3 to 5 min before euthanasia to mark immune cells still present in the vasculature. Following humane euthanasia, mouse lungs were isolated and suspensions of single cells were obtained by enzymatic and mechanical disruption using the gentleMACS dissociator system (Miltenyi Biotec, Auburn, CA) as previously described (54). Live cells were stained using acridine orange-propidium iodine and counted using a Cellometer K2 cytometer (Nexcelom, Lawrence, MA).
Flow cytometry.
Following isolation and enumeration, 1 × 106 live cells from single-cell suspensions were preblocked in mouse Fc block (BD Biosciences, USA), and then extracellular antigens were stained with SiglecF-BV421, B220-BV510, major histocompatibility complex class II (MHC-II)-BV510, CD11c-BV605, Ly6C-BV711, CD11b-BV785, PDCA1-phycoerythrin (PE), Ly6G-allophycocyanin (APC), and CD45-APC-R700 for myeloid cells and granulocytes. Other samples were stained with B220-BV510, CD8-BV605, CD49b-BV711, NK1.1-BV785, CD3e-peridinin chlorophyll protein (PerCP)-Cy5.5, CD4-PE, CD19-PE-Cy7, and CD45-APC-R700 for lymphocytes. Samples were then stained for viability using fixable viability dye eFluor780 (eBioscience, Thermo Fisher Scientific, Waltham, MA), rinsed, and then fixed. Myeloid panels were permeabilized with the FoxP3/transcription factor staining buffer kit (eBioscience, Thermo Fisher Scientific, Waltham, MA) and then stained using CD68-PerCP-Cy5.5 for the intracellular antigen. Analysis of stained cells was performed with a BD Fortessa flow cytometer using FACSDiva software (BD Biosciences, USA). Compensation settings were acquired using OneComp beads (eBioscience, Thermo Fisher Scientific, Waltham, MA) for the antibody channels and ArC amine-reactive beads (Molecular Probes, Eugene, OR) for the viability stain. Analysis of flow cytometry data utilized FlowJo software (BD Biosciences, USA). The gating strategy for flow cytometry analysis is shown in Fig. S1.
Demonstration of gating strategy for immunophenotyping of lung cells for myeloid populations. Pictured are populations in an uninfected CGD mouse. Initial gates eliminating doublets and debris are not shown. Vascular versus parenchymal gating using intravenously injected antibody staining was performed for all cell types but is demonstrated only for monocytic cells and neutrophils for simplicity. Download FIG S1, PDF file, 0.5 MB (536.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
In vivo depletion of cell subsets.
Neutrophils were depleted using anti-Ly6G monoclonal antibody (MAb) 1A8 (Bio X Cell, West Lebanon, NH). Antibodies were administered to groups of 5 CGD mice by intraperitoneal injection on days −6, −4, and −1 preinfection and day +1 postinfection. Injections comprised 500 μg per mouse in 200 µl. Depletion efficiency was monitored using flow cytometry as described above.
Ethics statement.
The Institutional Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases approved all animal studies (approval no. LCIM-5E). Studies were performed in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (55).
Statistical analysis.
All data analyses were performed using GraphPad Prism, version 7 (GraphPad Software, San Diego, CA). Mortality data were analyzed by the log rank test. Student’s t test and two-way analysis of variance (ANOVA) followed by multiple-comparison test were used to define whether there are significant differences between indicated samples. Statistical significance for comparisons was defined as a P value of ≤0.05 (two-tailed), which was reported as follows: nonsignificant (NS), P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
ACKNOWLEDGMENT
This work was supported by a research fund from the intramural program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Citation Seyedmousavi S, Davis MJ, Sugui JA, Pinkhasov T, Moyer S, Salazar AM, Chang YC, Kwon-Chung KJ. 2018. Exogenous stimulation of type I interferon protects mice with chronic granulomatous disease from aspergillosis through early recruitment of host-protective neutrophils into the lung. mBio 9:e00422-18. https://doi.org/10.1128/mBio.00422-18.
REFERENCES
- 1.Segal BH, Romani LR. 2009. Invasive aspergillosis in chronic granulomatous disease. Med Mycol 47(Suppl 1):S282–S290. doi: 10.1080/13693780902736620. [DOI] [PubMed] [Google Scholar]
- 2.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 3.Bongomin F, Gago S, Oladele RO, Denning DW. 2017. Global and multi-national prevalence of fungal diseases—estimate precision. J Fungi 3:57. doi: 10.3390/jof3040057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. 2000. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine 79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Segal BH, Holland SM. 2000. Primary phagocytic disorders of childhood. Pediatr Clin North Am 47:1311–1338. doi: 10.1016/S0031-3955(05)70273-X. [DOI] [PubMed] [Google Scholar]
- 6.Almyroudis NG, Holland SM, Segal BH. 2005. Invasive aspergillosis in primary immunodeficiencies. Med Mycol 43(Suppl 1):S247–S259. doi: 10.1080/13693780400025203. [DOI] [PubMed] [Google Scholar]
- 7.Seyedmousavi S, Verweij PE. 2015. Azole resistance in Aspergillus fumigatus: mechanisms, route of resistance selection and clinical implications, p 403–421. In Gotte M, Berghuis A, Matlashewski G, Wainberg MA, Sheppard D (ed), Handbook of antimicrobial resistance. Springer, New York, NY. doi: 10.1007/978-1-4939-0694-9_22. [DOI] [Google Scholar]
- 8.Segal BH, DeCarlo ES, Kwon-Chung KJ, Malech HL, Gallin JI, Holland SM. 1998. Aspergillus nidulans infection in chronic granulomatous disease. Medicine 77:345–354. doi: 10.1097/00005792-199809000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Vinh DC, Shea YR, Jones PA, Freeman AF, Zelazny A, Holland SM. 2009. Chronic invasive aspergillosis caused by Aspergillus viridinutans. Emerg Infect Dis 15:1292–1294. doi: 10.3201/eid1508.090251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugui JA, Peterson SW, Clark LP, Nardone G, Folio L, Riedlinger G, Zerbe CS, Shea Y, Henderson CM, Zelazny AM, Holland SM, Kwon-Chung KJ. 2012. Aspergillus tanneri sp. nov., a new pathogen that causes invasive disease refractory to antifungal therapy. J Clin Microbiol 50:3309–3317. doi: 10.1128/JCM.01509-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vinh DC, Shea YR, Sugui JA, Parrilla-Castellar ER, Freeman AF, Campbell JW, Pittaluga S, Jones PA, Zelazny A, Kleiner D, Kwon-Chung KJ, Holland SM. 2009. Invasive aspergillosis due to Neosartorya udagawae. Clin Infect Dis 49:102–111. doi: 10.1086/599345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hubka V, Kubatova A, Mallatova N, Sedlacek P, Melichar J, Skorepova M, Mencl K, Lyskova P, Sramkova B, Chudickova M, Hamal P, Kolarik M. 2012. Rare and new etiological agents revealed among 178 clinical Aspergillus strains obtained from Czech patients and characterized by molecular sequencing. Med Mycol 50:601–610. doi: 10.3109/13693786.2012.667578. [DOI] [PubMed] [Google Scholar]
- 13.Gallin JI, Alling DW, Malech HL, Wesley R, Koziol D, Marciano B, Eisenstein EM, Turner ML, DeCarlo ES, Starling JM, Holland SM. 2003. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl J Med 348:2416–2422. doi: 10.1056/NEJMoa021931. [DOI] [PubMed] [Google Scholar]
- 14.King J, Henriet S, Warris A. 2016. Aspergillosis in chronic granulomatous disease. J Fungi 2:15. doi: 10.3390/jof2020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beauté J, Obenga G, Le Mignot L, Mahlaoui N, Bougnoux ME, Mouy R, Gougerot-Pocidalo MA, Barlogis V, Suarez F, Lanternier F, Hermine O, Lecuit M, Blanche S, Fischer A, Lortholary O. 2011. Epidemiology and outcome of invasive fungal diseases in patients with chronic granulomatous disease: a multicenter study in France. Pediatr Infect Dis J 30:57–62. doi: 10.1097/INF.0b013e3181f13b23. [DOI] [PubMed] [Google Scholar]
- 16.Lehrnbecher T, Kalkum M, Champer J, Tramsen L, Schmidt S, Klingebiel T. 2013. Immunotherapy in invasive fungal infection—focus on invasive aspergillosis. Curr Pharm Des 19:3689–3712. doi: 10.2174/1381612811319200010. [DOI] [PubMed] [Google Scholar]
- 17.Carvalho A, Cunha C, Bistoni F, Romani L. 2012. Immunotherapy of aspergillosis. Clin Microbiol Infect 18:120–125. doi: 10.1111/j.1469-0691.2011.03681.x. [DOI] [PubMed] [Google Scholar]
- 18.The International Chronic Granulomatous Disease Cooperative Study Group 1991. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N Engl J Med 324:509–516. doi: 10.1056/NEJM199102213240801. [DOI] [PubMed] [Google Scholar]
- 19.Delsing CE, Gresnigt MS, Leentjens J, Preijers F, Frager FA, Kox M, Monneret G, Venet F, Bleeker-Rovers CP, van de Veerdonk FL, Pickkers P, Pachot A, Kullberg BJ, Netea MG. 2014. Interferon-gamma as adjunctive immunotherapy for invasive fungal infections: a case series. BMC Infect Dis 14:166. doi: 10.1186/1471-2334-14-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Versteeg GA, García-Sastre A. 2010. Viral tricks to grid-lock the type I interferon system. Curr Opin Microbiol 13:508–516. doi: 10.1016/j.mib.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macri G, Ruggeri A, Leanderson T, Teti G. 2007. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol 178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- 22.Une C, Andersson J, Orn A. 2003. Role of IFN-alpha/beta and IL-12 in the activation of natural killer cells and interferon-gamma production during experimental infection with Trypanosoma cruzi. Clin Exp Immunol 134:195–201. doi: 10.1046/j.1365-2249.2003.02294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diefenbach A, Schindler H, Donhauser N, Lorenz E, Laskay T, MacMicking J, Röllinghoff M, Gresser I, Bogdan C. 1998. Type 1 interferon (IFNalpha/beta) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity 8:77–87. doi: 10.1016/S1074-7613(00)80460-4. [DOI] [PubMed] [Google Scholar]
- 24.Sionov E, Mayer-Barber KD, Chang YC, Kauffman KD, Eckhaus MA, Salazar AM, Barber DL, Kwon-Chung KJ. 2015. Type I IFN induction via poly-ICLC protects mice against cryptococcosis. PLoS Pathog 11:e1005040. doi: 10.1371/journal.ppat.1005040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. 2015. Type I interferons in infectious disease. Nat Rev Immunol 15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pestka S, Krause CD, Walter MR. 2004. Interferons, interferon-like cytokines, and their receptors. Immunol Rev 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 27.del Fresno C, Soulat D, Roth S, Blazek K, Udalova I, Sancho D, Ruland J, Ardavín C. 2013. Interferon-beta production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 38:1176–1186. doi: 10.1016/j.immuni.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 28.Biondo C, Midiri A, Gambuzza M, Gerace E, Falduto M, Galbo R, Bellantoni A, Beninati C, Teti G, Leanderson T, Mancuso G. 2008. IFN-alpha/beta signaling is required for polarization of cytokine responses toward a protective type 1 pattern during experimental cryptococcosis. J Immunol 181:566–573. doi: 10.4049/jimmunol.181.1.566. [DOI] [PubMed] [Google Scholar]
- 29.Inglis DO, Berkes CA, Hocking Murray DR, Sil A. 2010. Conidia but not yeast cells of the fungal pathogen Histoplasma capsulatum trigger a type I interferon innate immune response in murine macrophages. Infect Immun 78:3871–3882. doi: 10.1128/IAI.00204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bourgeois C, Majer O, Frohner IE, Lesiak-Markowicz I, Hildering K-S, Glaser W, Stockinger S, Decker T, Akira S, Muller M, Kuchler K. 2011. Conventional dendritic cells mount a type I IFN response against Candida spp. requiring novel phagosomal TLR7-mediated IFN-beta signaling. J Immunol 186:3104–3112. doi: 10.4049/jimmunol.1002599. [DOI] [PubMed] [Google Scholar]
- 31.Levy HB, Baer G, Baron S, Buckler CE, Gibbs CJ, Iadarola MJ, London WT, Rice J. 1975. A modified polyriboinosinic-polyribocytidylic acid complex that induces interferon in primates. J Infect Dis 132:434–439. doi: 10.1093/infdis/132.4.434. [DOI] [PubMed] [Google Scholar]
- 32.Levy HB, Riley FL, Lvovsky E, Stephen EE. 1981. Interferon induction in primates by stabilized polyriboinosinic acid-polyribocytidylic acid: effect of component size. Infect Immun 34:416–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ammi R, De Waele J, Willemen Y, Van Brussel I, Schrijvers DM, Lion E, Smits EL. 2015. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharm Ther 146:120–131. doi: 10.1016/j.pharmthera.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 34.Cheever MA. 2008. Twelve immunotherapy drugs that could cure cancers. Immunol Rev 222:357–368. doi: 10.1111/j.1600-065X.2008.00604.x. [DOI] [PubMed] [Google Scholar]
- 35.Salazar AM, Erlich RB, Mark A, Bhardwaj N, Herberman RB. 2014. Therapeutic in situ autovaccination against solid cancers with intratumoral poly-ICLC: case report, hypothesis, and clinical trial. Cancer Immunol Res 2:720–724. doi: 10.1158/2326-6066.CIR-14-0024. [DOI] [PubMed] [Google Scholar]
- 36.Gazendam RP, van Hamme JL, Tool ATJ, Hoogenboezem M, van den Berg JM, Prins JM, Vitkov L, van de Veerdonk FL, van den Berg TK, Roos D, Kuijpers TW. 2016. Human neutrophils use different mechanisms to kill Aspergillus fumigatus conidia and hyphae: evidence from phagocyte defects. J Immunol 196:1272–1283. doi: 10.4049/jimmunol.1501811. [DOI] [PubMed] [Google Scholar]
- 37.Park SJ, Mehrad B. 2009. Innate immunity to Aspergillus species. Clin Microbiol Rev 22:535–551. doi: 10.1128/CMR.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feldmesser M. 2006. Role of neutrophils in invasive aspergillosis. Infect Immun 74:6514–6516. doi: 10.1128/IAI.01551-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knox BP, Huttenlocher A, Keller NP. 2017. Real-time visualization of immune cell clearance of Aspergillus fumigatus spores and hyphae. Fungal Genet Biol 105:52–54. doi: 10.1016/j.fgb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Margalit A, Kavanagh K. 2015. The innate immune response to Aspergillus fumigatus at the alveolar surface. FEMS Microbiol Rev 39:670–687. doi: 10.1093/femsre/fuv018. [DOI] [PubMed] [Google Scholar]
- 41.Bonnett CR, Cornish EJ, Harmsen AG, Burritt JB. 2006. Early neutrophil recruitment and aggregation in the murine lung inhibit germination of Aspergillus fumigatus conidia. Infect Immun 74:6528–6539. doi: 10.1128/IAI.00909-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zarember KA, Sugui JA, Chang YC, Kwon-Chung KJ, Gallin JI. 2007. Human polymorphonuclear leukocytes inhibit Aspergillus fumigatus conidial growth by lactoferrin-mediated iron depletion. J Immunol 178:6367–6373. doi: 10.4049/jimmunol.178.10.6367. [DOI] [PubMed] [Google Scholar]
- 43.Ibrahim-Granet O, Philippe B, Boleti H, Boisvieux-Ulrich E, Grenet D, Stern M, Latge JP. 2003. Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infect Immun 71:891–903. doi: 10.1128/IAI.71.2.891-903.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waldorf AR, Levitz SM, Diamond RD. 1984. In vivo bronchoalveolar macrophage defense against Rhizopus oryzae and Aspergillus fumigatus. J Infect Dis 150:752–760. doi: 10.1093/infdis/150.5.752. [DOI] [PubMed] [Google Scholar]
- 45.Ahlin A, Elinder G, Palmblad J. 1997. Dose-dependent enhancements by interferon-gamma on functional responses of neutrophils from chronic granulomatous disease patients. Blood 89:3396–3401. [PubMed] [Google Scholar]
- 46.Otten MA, Rudolph E, Dechant M, Tuk CW, Reijmers RM, Beelen RHJ, van de Winkel JGJ, van Egmond M. 2005. Immature neutrophils mediate tumor cell killing via IgA but not IgG Fc receptors. J Immunol 174:5472–5480. doi: 10.4049/jimmunol.174.9.5472. [DOI] [PubMed] [Google Scholar]
- 47.Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, Dummer R, Simon HU, Yousefi S. 2004. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem 279:44123–44132. doi: 10.1074/jbc.M405883200. [DOI] [PubMed] [Google Scholar]
- 48.Andzinski L, Kasnitz N, Stahnke S, Wu CF, Gereke M, von Köckritz-Blickwede M, Schilling B, Brandau S, Weiss S, Jablonska J. 2016. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer 138:1982–1993. doi: 10.1002/ijc.29945. [DOI] [PubMed] [Google Scholar]
- 49.Moreira-Teixeira L, Sousa J, McNab FW, Torrado E, Cardoso F, Machado H, Castro F, Cardoso V, Gaifem J, Wu X, Appelberg R, Castro AG, O’Garra A, Saraiva M. 2016. Type I IFN inhibits alternative macrophage activation during Mycobacterium tuberculosis infection and leads to enhanced protection in the absence of IFN-gamma signaling. J Immunol 197:4714–4726. doi: 10.4049/jimmunol.1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sugui JA, Peterson SW, Figat A, Hansen B, Samson RA, Mellado E, Cuenca-Estrella M, Kwon-Chung KJ. 2014. Genetic relatedness versus biological compatibility between Aspergillus fumigatus and related species. J Clin Microbiol 52:3707–3721. doi: 10.1128/JCM.01704-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pollock JD, Williams DA, Gifford MAC, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. 1995. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 52.Rao GVS, Tinkle S, Weissman D, Antonini J, Kashon M, Salmen R, Battelli L, Willard P, Hubbs A, Hoover M. 2003. Efficacy of a technique for exposing the mouse lung to particles aspirated from the pharynx. J Toxicol Environ Health A 66:1441–1452. doi: 10.1080/15287390306417. [DOI] [PubMed] [Google Scholar]
- 53.Seyedmousavi S, Melchers WJG, Bakkers J, Verweij PE, Mouton JW. 2012. Targeting 28S rDNA-gene of Aspergillus fumigatus for pharmacokinetics and pharmacodynamics (PK/PD) studies of voriconazole in a non-neutropenic murine model of invasive aspergillosis. Mycoses 55:125–126. [Google Scholar]
- 54.Davis MJ, Eastman AJ, Qiu Y, Gregorka B, Kozel TR, Osterholzer JJ, Curtis JL, Swanson JA, Olszewski MA. 2015. Cryptococcus neoformans-induced macrophage lysosome damage crucially contributes to fungal virulence. J Immunol 194:2219–2231. doi: 10.4049/jimmunol.1402376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals 2011. Guide for the care and use of laboratory animals, 8th edition National Academies Press, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer and probe sequences used for qPCR assays. Download TABLE S1, DOC file, 0.04 MB (42KB, doc) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Demonstration of gating strategy for immunophenotyping of lung cells for myeloid populations. Pictured are populations in an uninfected CGD mouse. Initial gates eliminating doublets and debris are not shown. Vascular versus parenchymal gating using intravenously injected antibody staining was performed for all cell types but is demonstrated only for monocytic cells and neutrophils for simplicity. Download FIG S1, PDF file, 0.5 MB (536.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.