

Abstract
Ryanodine receptors (RyRs) are calcium release channels expressed in the sarco-endoplasmic reticula of many cell types including cardiac and skeletal muscle cells. In recent years, Ca2+ leak through RyRs has been implicated as a major contributor for development of diseases including heart failure, muscle myopathies, Alzheimer’s disease and diabetes, making it an important therapeutic target. Recent mammalian RyR1 cryo-EM structures of multiple functional states have clarified long-standing questions including the architecture of the transmembrane pore and cytoplasmic domains, location and architecture of the channel gate, ligand binding sites and the gating mechanism. As we advance toward complete models of RyRs this new information enables determination of domain-domain interfaces and the location and structural effects of disease-causing RyR mutations.
Keywords: ryanodine receptors, calcium channel, cryo-EM, structure, excitation-contraction coupling
RyRs are Ca2+ channels required for muscle contraction
Calcium (Ca2+) release from intracellular stores is an important step in many signaling pathways (see glossary) including cardiac and skeletal muscle excitation-contraction coupling (EC coupling, see glossary). Upon plasma membrane depolarization, Voltage dependent Ca2+ channels (dihydropyridine receptors, DHPR also known as CaV1.1) are activated, followed by activation of the sarcoplasmic reticulum (SR) ryanodine (see glossary) receptors (RyRs) that release intracellular Ca2+ necessary for muscle contraction (Figure 1). Distinct genes encode the three different mammalian RyR isoforms and share ~70% sequence homology [2]. The type 1 isoform (RyR1) is expressed in skeletal muscle, while cardiac muscle contains predominately RyR2. RyR1, RyR2, and RyR3 are all distributed in different parts of the brain as well as in many other cell types [3]. RyRs are expressed in all striated muscles from Caenorhabditis elegans to humans. Inositol 1,4,5-triphosphate receptors (IP3Rs) are the other major type of calcium release channels; they have high structural and functional homology to RyRs and are also located on the ER of many cell types.
Figure 1. Ca2+ handling components in skeletal muscle Excitation-contraction coupling.
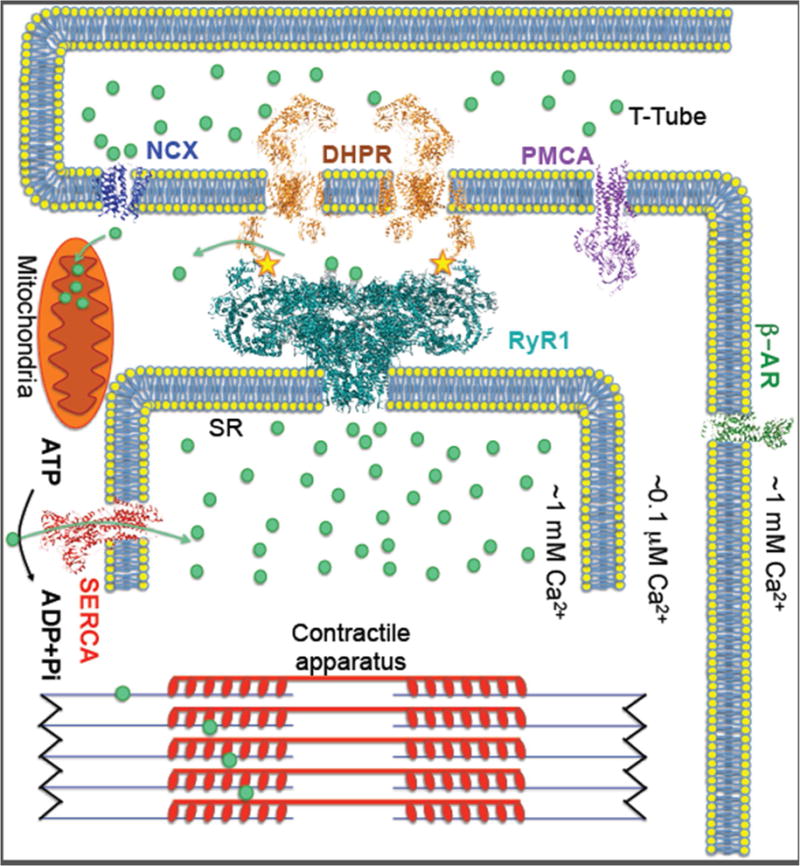
Upon plasma membrane depolarization DHPR (dihydropyridine receptor) mechanically activates RyR1 to release SR Ca2+, which triggers muscle contraction. The Ca2+ ATPase SERCA1a pumps Ca2+ back into the SR and PMCA (plasma membrane Ca2+ ATPase) pump out the Ca2+ from cytoplasm causing muscle relaxation. The RyR1 complex includes: calstabin1, which stabilizes the closed state of the channel, kinases and phosphatases that modulate the channel through phosphorylation (yellow stars marks the phosphorylation site), and calmodulin. RyR1 can be activated by stress pathways via β-adrenergic receptors (β-AR) resulting in increased Ca2+ release and enhanced muscle performance. SR Ca2+ release may also modulate mitochondrial function, particularly during states of chronic stress (e.g. muscular dystrophy), resulting in generation of reactive oxygen species that can oxidize RyR1 and deplete calstabin1 from the channel, rendering the channels leaky. Leaky RyR1 channels were shown to be a contributing factor to diseases including muscle myopathies, Duchenne muscular dystrophy and cancer.
RyRs are the largest known ion channels. It is composed of four identical ~565 kDa protomers that form a single central transmembrane pore and a large cytoplasmic assembly that bridges the gap between the SR and the transverse (T) tubules; specialized invaginations of the plasma membrane that penetrate deep into the muscle fiber [4]. The size and the relative stability of the detergent-purified protein in aqueous solution made RyR1 an ideal target for one of the earliest single particle negative-stain studies [5] and later, electron cryo-microscopy (cryo-EM) [6] reconstruction (see glossary) studies of a purified membrane protein. Since then Terrence Wagenknecht’s group and others published multiple cryo-EM structures that identified the locations of many of the channel domains using insertion mutations and antibodies to identify the location of specific domains [7]. In 2005 two RyR1 structures in the closed state were published suggesting the location of membrane-spanning α-helices and providing the first hints to the pore architecture [8, 9]. Structural homology with the IP3Rs was suggested in a subnanometer resolution cryo-EM structure of the RyR1 [10]. Samso et al. were the first to show large conformational changes in the cytoplasmic assembly between the RyR1 open and closed states [11]. However, all of these structures were obtained at relatively low resolution.
In early 2015 RyR1 joined the cryo-EM resolution-revolution [12]. In one of the major scientific breakthroughs of that year [13], 3D structures were obtained for the mammalian RyR1 [14–16] and the mammalian IP3R1 [17] in the closed state (PDB: 4UWA, 3J8E AND 3J8H) at resolutions ranging from 3.8 to 6.1 Å. Although a clear picture of the channel pore region has emerged, the precise mechanism for the channel activation remained vague until the open and the ‘primed’ states of RyR1 [18–20] and RyR2 [21] were solved. Together, these ~85% complete structures immensely improved our understanding of the fundamental physiology of intracellular Ca2+ release in muscle EC-coupling and in many other cellular contexts. The new high-resolution RyR1 structures provided the location, structure and mechanism of the channel gate, the composition of the channel pore, the nature of the open and closed states of the channel, the location of ligand binding sites, and provide a plausible mechanism for the channel activation. These structures provide a firm foundation to advance our molecular understanding of the role of Ca2+ release in many Ca2+ signaling pathways and in the mechanism of important diseases such as muscle myopathies [22] Duchenne muscular dystrophy [23], malignant hyperthermia (MH) [24] and cancer [25] (associated with RyR1), heart failure [26], catecholaminergic polymorphic ventricular tachycardia (CPVT) [27], and as recently demonstrated, Alzheimer’s disease [28], and diabetes [29] (associated with RyR2).
RyR molecular architecture
Earlier cryo-EM structure studies predicted the RyR1 protomer boundaries [10] as well as many of the RyR1 functional domains and their locations within the overall cryo-EM density (reviewed in detail by Hwang et al. [7]). A few RyR1 domains could be expressed, folded, crystallized and then docked into the cryo-EM maps [30–33]. For the most part, these predictions have been confirmed by the recent high-resolution structures with few discrepancies between the models.
RyR1 structure can be divided into three main regions: the N-terminal ‘shell’, the core solenoid (also described as the central domain [15]) and the transmembrane (TM) domain. Discrepancies in domains and sequence assignments can be found between the three 2015 RyR1 structures (e.g. the order of the SPRY domains in Zalk et al. [14] and the direction of the α-solenoid repeats in Efremov et al. [16]) but the puzzle is getting clearer with each added structure (Figure 2). The availability of high-resolution crystal structures of RyR1 fragments not only greatly improved domain assignment and sequence registration in the full-length RyRs models but were also invaluable in correcting the cryo-EM maps allowing for an accurate calibration of the pixel size by real-space correlation with the crystal structures [20]. Significant differences in local resolution between the different domains can be observed in all RyRs cryo-EM maps; from ~3 Å at the channel core, where clear side chains were de-novo assigned, dropping to >7 Å at the RY3&4 domain and in the channel periphery where sequence registration and connectivity are still ambiguous.
Figure 2. RyR1 domain assignment.
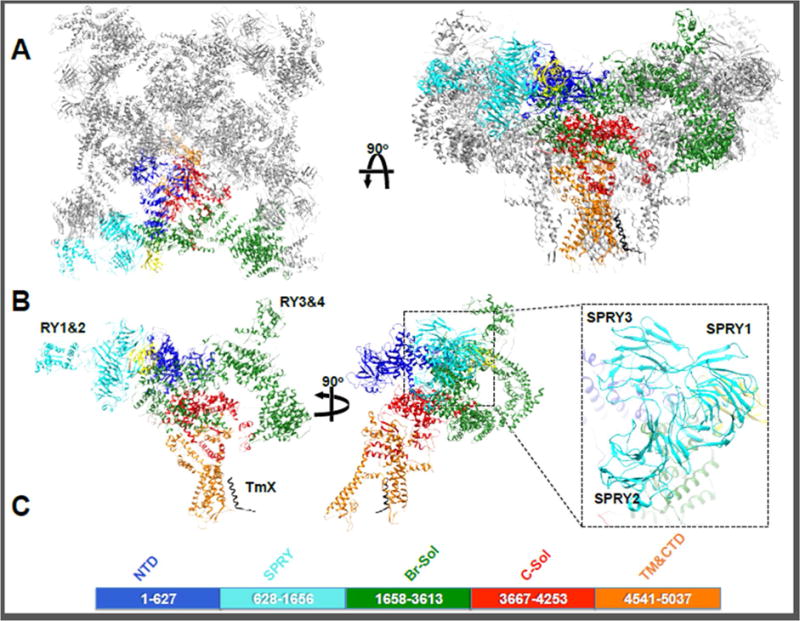
A RyR1 tetramer viewed from the cytosol (left) and from the plane of the membrane. One protomer of the tetramer is shown in colors. B - The RyR1 protomer can be divided into 5 main domains: The NTD is depicted in blue, the three SPRY domains and RYR1&2 in cyan, the Bridging solenoid including Ry3&4 in green, the core solenoid in red and the pore domain in orange. The modulatory subunit calstabin is shown in yellow. A newly identified transmembrane helix TmX is shown in black. The orientation of the 3 SPRY domains in the inset and the location of RY1&2 and RY3&4 are indicated. C – Indicates the sequence boundaries of the 5 main domains.
The cytosolic shell
The cytosolic shell is the is the largest assembly, composed of the N-terminal domains NTD-A (amino acids 1–208), NTD-B (209–392) and N-terminal solenoid (Nsol, 393–627), the 3 SPRY (628–1656) domains, the two RYR (RY1&2, 850–1054 and RY3&4, 2735–2938) domains and the large (~35% of the sequence) Junctional and bridging solenoids (Jsol and BrB, also known as the ‘handle’ domain, 1657–2144 and 2145–3613 respectively). The cytoplasmic assembly also serves as the scaffold for RyR modulatory proteins including calstabin1 (FKBP12), calmodulin, kinases including PKA and CamKII (in RyR2) and their anchoring proteins, phosphatases including PP1 and PP2A (in RyR2) and their anchoring proteins and the phosphodiesterase PDE4D3 [34–36]. Built on a scaffold of 37 α-solenoid repeats this massive cytoplasmic assembly allosterically controls channel gating (the pore opening and closing) and these modulators can regulate channel activation (change open probability) by regulating conformational changes in the pore and surrounding the transmembrane segments [20].
The NTD β-trefoil forms the central cytosolic vestibule containing a ‘hot spot’ for disease causing mutations located at the interfaces between the four protomers [33] (Figure 3). Furthermore, the NTD forms an inter-domain interaction with another mutation hot spot cluster on the bridging solenoid, between NTD-B and BrB of the adjacent protomer (figure 3 insets), highlighting its important role in allosterically modulating the pore gating by stabilizing these inter-domain contacts to keep the ‘upward’ conformation and stabilize the closed state [20, 37]. Intriguingly, this NTD-B-BrB interaction is in close proximity to DP4 (DPc10 in RyR2) (Figure 3, pink), a short RyR1 peptide (Leu2442-Pro2477 in RyR1 and Gly2460-Pro2495 in RyR2) on BrB where 3 MH (in RyR1) and a CPVT (in RyR2) associated mutations are located. This peptide was shown to increase RyR1 channel open probability and mean open time by disrupting inter-domain interactions that stabilize the channel closed state [38, 39]. In RyR2 it increases SR Ca2+ leak that causes arrhythmias that characterize CPVT [40].
Figure 3. Clusters of mutation hot spots.
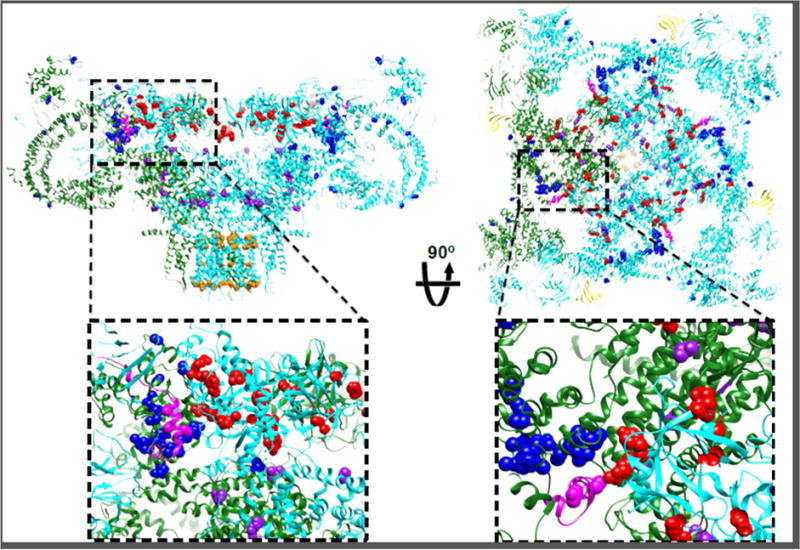
Disease-causing RyR1 mutations (http://www.uniprot.org/uniprot/P21817) on the NTD (red spheres), bridging solenoid (blue spheres), core solenoid (purple spheres) and on the transmembrane domain (orange spheres), viewed from the side (left) and from the cytosol (right). Insets show the domain-domain interface between the NTD and the bridging solenoid at the ‘zipping’ point where DP4 (magenta) is located. On the right inset the interaction between NTD-A (cyan) and NTD-B of the adjacent protomer (green) also shows mutations clusters on both sides of this domain-domain interface.
Crystal structures of SPRY1 and SPRY2 [30, 31] were docked within a cluster of three SPRY domains. Discrepancy in the order of the three SPRY domains locations in the cluster is now settled with agreement that SPRY1 forms part of the calstabin1 binding site; SPRY2, previously proposed to form the DHPR binding site [41], is buried underneath SPRY3; and SPRY3 forms contact sites with the bridging solenoid of the adjacent protomer (figure 2 inset).
The enormous bridging solenoid is curled counterclockwise (as viewed from the cytosol) under the SPRY domains of the adjacent protomer. The bridging solenoid is a dynamic part of the channel and exhibits distinct conformations in all functional states. Identification of disease causing mutations reveals that they are clustered at inter-domain interfaces highlighting the importance of these sequences in channel function (figure 3).
The two RYR repeats [32, 42] were docked to the ‘clamp’ region (RY1&2) and to the top of the cytosolic assembly between helices 19 and 20 of the bridging solenoid (Ry3&4, the PKA phosphorylation domain). Given its location and that phosphorylation of this site increases open probability, addition of a phosphate group could promote the downward conformation of the cytosolic shell to exert its effect, possibly by modifying interdomain interactions or by modifying protein-protein interactions with plasma membrane proteins like the DHPR.
The core solenoid
The core solenoid is the rigid complex connecting the cytosolic shell with the channel pore. The core solenoid of one protomer forms contact sites with NTD-B of the same protomer and NTD-A of the adjacent one. An EF-hand pair (residues 4072–4135) interacts with the S2-S3 helical bundle of the adjacent protomer upon channel priming and the ‘thumb and forefinger’ (TaF) domain (also known as the ‘U motif’) engulfs the CTD. The core solenoid, jointly with the CTD, harbors the binding site for Ca2+, caffeine and nucleotides and mediates the transmission of information between the cytoplasmic assembly and the pore region upon channel activators binding (figure 4).
Figure 4. RyR1 gate latch.
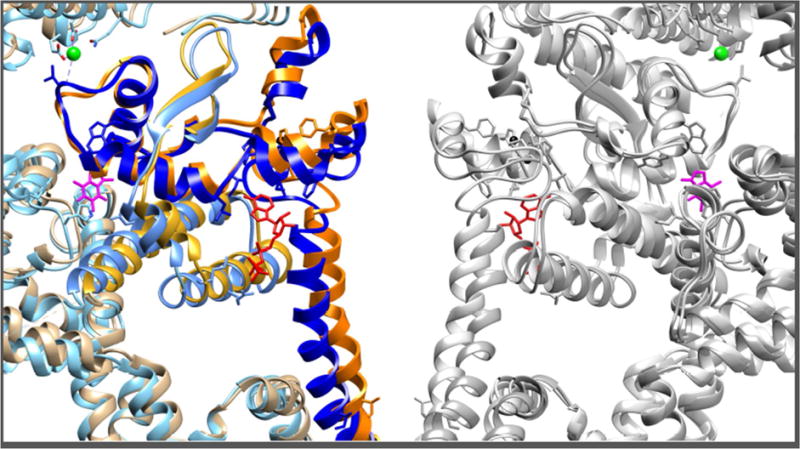
The CTD forms intimate interactions with the core solenoid TaF domain. Upon Ca2+ and ATP binding, the conformational changes in the cytoplasmic shell are coupled to the bowing of the pore S6 helix and the dilation of the pore aperture (Ile4937). Dashed arrows represent the apparent direction of movements from closed to open states. Ca2+, ATP and Caffeine, all RyR1 activators, bind at different sites of the CTD-core solenoid inter-domain interfaces. Here one protomer is shown in orange (closed state) and blue (open state).
The transmembrane domain
The RyRs membrane spanning architecture resembles that of the 6 transmembrane (6TM, see glossary) cation-channels superfamily with the characteristic domain-swap organization: The pore-forming segments S5 and S6 are connected to the S1–S4 segments via a juxtamembrane S4–S5 linker forming a pinwheel-like structure. The ion path is formed by S6, and similar to the selectivity filter in sodium and potassium channels, a short pore helix and an extended segment form the narrow path at the luminal side of the channel pore. However, consistent with the fact that RyR1 is not highly selective (the conductance for K and Na is ~5 times that for Ca2+ and the channel is only 11:1 selective for cations over anions), the RyR1 P-loop does not seem to form the potassium channels signature structure that removes the ion hydration shell when it enters the channel path [14].
Eight negatively charged residues on each protomer at the selectivity filter and many more on the luminal loops of the transmembrane domain form a negatively charged ‘bowl’ that penetrates ~1/3 of the way through the membrane [14]. These negative charges most probably serve to concentrate Ca2+ close to the pore mouth contributing to the high conductance of RyR1 for Ca2+ (~100 pS). Seven acidic residues forming negatively charged ‘rings’ on the cytosolic side of the aperture on S6 (S6c) were shown to serve a similar function [43]. The pore is composed of carboxylic groups from Asp4899 and Glu4900 and carbonyl groups from Ala4893, Gly4894 and Gly4895 (in RyR1) resembling features of other Ca2+ conducting channels such as TRPV1 [44], CavAB [45] and the IP3R1 [17].
The ‘bundle-crossing’ surrounded by the amphipathic S4–S5 linkers forms the pore aperture with a single gate at Ile4937 in RyR1 and Ile4868 in RyR2 that comprises the narrowest part of the conduction pathway. Helices S1–S4 form a pseudo voltage sensor domain (pVSD) of similar size and architecture to the voltage sensors in other 6TM channels but lacking all but one of the positive charges that define these voltage sensors.
Three unique features in the RyR1 transmembrane region distinguish it from other 6TM channels: 1) a long negatively charged and disordered luminal S1–S2 loop. 2) The S2–S3 helical bundle (also named VSC), a conserved (71% identity between human RyR1 and human RyR2) and unique motif to RyRs, it is not present in any other known 6TM cation channel, including the closely related IP3R1 channels. The location on the pVSD suggests a role in ‘sensing’ conformational changes in the cytosolic EF-hand domain located in close proximity (figure 5) that may modulate channel gating. An intriguing conserved proline-rich domain is located within the S2–S3 domain of RyR1 (but not in RyR2) suggesting a RyR1-specific protein-binding site for an as yet unidentified SH3 domain containing protein. 3) S6c - the long cytosolic extension of S6, ending at the CTD zinc-fingers, directly controls the channel gating as it connects the pore aperture with the CTD, where Ca2+, ATP and caffeine are bound. This long cytosolic extension of S6 is also found in RyR2 and IP3R1 where S6c extends all the way through the NTD vestibule and terminates at the IP3 binding domain [17].
Figure 5. RyR1 moving parts.
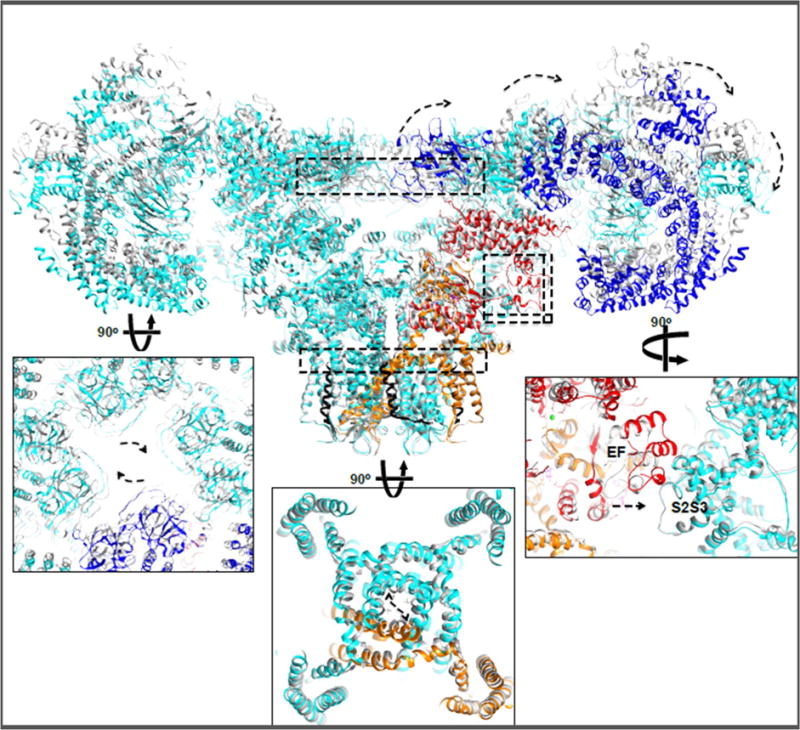
Upon Ca2+ and ATP binding dilated channel pore aperture can be observed coupled to exo-rotation of the cytoplasmic shell and clockwise translation of the NTD cytoplasmic vestibule. Dashed arrows represent the apparent direction of movements from closed to open states. Similar conformational changes were observed in in the presence of PCB95 or ruthenium red. Curiously, an ~8 Å translation of the EF-hand pair towards the S2–S3 helical bundle is observed also in the RyR1 ‘primed’ state. Insets show enlarged views of the structures at the indicated dashed rectangles.
A 7th helix
An intriguing transmembrane density located close to the pVSD and S5 of the adjacent protomer was identified in the Zalk et al. structure [14]. This density was confirmed to be a protein helix identified in some but not all of the structures described by des Georges et al. [20], suggesting it is a RyR1 helix that is a part of an unstable TM hairpin. Interestingly, a similar transmembrane density was identified in the IP3R1 cryo-EM map (EMDB_6369), modeled as a loop between TM1 and TM2 (PDB_3JAV). A plausible explanation for this helix is a predicted hairpin N-terminal to S1 [46], between Glu4253 and Phe4540. Based on its position, this hairpin may serve a modulatory role similar to the 2TM beta-subunit of the large-conductance, voltage- and Ca2+-gated potassium (BK) channels [47], but further studies are necessary to resolve the role of this TM segment.
The RyR1 C-terminal domain is located at the cytosolic end of S6c engulfed by the core solenoid TaF domain. A zinc ion, coordinated by cysteines 4958 and 4961 and histidines 4978 and 4983 [15], rigidifies the hinge between S6c and the CTD. The essential role of the CTD in controlling the RyR1 channel gate became obvious when the binding sites for three RyR1 major activators (Ca2+, caffeine and ATP [48–50]) were all found surrounding the CTD (Figure 4). Multiple RyR1 cryo-EM structures in the presence of different combinations of these RyR activators yielded difference maps that enabled identification of the binding sites for Ca2+, ATP and caffeine [20]. The CTD, engulfed by the core solenoid, transmits the conformational changes from the cytosolic assembly to the pore aperture when channel activators are bound (figure 4).
The detailed descriptions of the Ca2+ release channels architecture have immensely improved our understanding of Ca2+ release channels. That said, these structures reveals very little on the channels gating mechanism. Ideas on the biology of the channels and how activators such as Ca2+ and ATP affect the channel gating and how plasma membrane depolarization can allosterically affect the channel pore could only be achieved by comparing multiple conformations of the channels.
RyR1 Gating: the moving parts
Simultaneous binding of Ca2+ along with ATP (an essential ligand for channel activation [50]) and caffeine, or with ryanodine, yielded detailed RyR1 structures in the open states [20]. Two other RyR1 structures in the Ca2+ activated state stabilized with Ca2+ and ruthenium red [18] or Ca2+ and PCB95 [19], and a structure of RyR2 with Ca2+ and PCB95 [21], described global conformational changes involved in the channel gating. RyR1 structures in des Georges et al. [20] described a Ca2+ bound (closed) state (3.8 Å), Ca2+/ATP/Caffeine-bound open states (4.4 Å), Ca2+-free states (4.5 Å) and a ryanodine-bound state, providing detailed description of the RyR1 gating mechanism. Of note is the Wei et al. cryo-EM map (EMD-8074) that indeed shows Ca2+ density in the same site as the des Georges et al. maps but describe a constricted aperture state [52].
Tilting of the CTD and the TaF domains toward the plane of the SR membrane results in bowing of S6c and dilation of the pore aperture. Rigidifying the interface between the CTD and regions of the core solenoid by Ca2+ (a.a. Glu3893, Glu3967, Thr5001), ATP (Lys4211, Ly4214 and Arg4215 interacts with Pi groups and Leu4985 and Phe4959 with the adenosine) and caffeine (a.a.Trp4716) allows for the tight control of the cytosolic assembly over this complex gate latch (figure 4).
Morphing between the Ca2+-free state and the Ca2+/ATP/Caffeine open state (CAC), as well as between the CAC 3D classes (open vs. closed) reveals that the transition from a closed to open pore involves conformational changes in multiple regions (figure 4 and 5); 1) The cytosolic shell undergoes the largest conformational change in both the ligand-bound and in the ligand-free states of the channel [19, 20]. Upon gating the large solenoid repeat domain ‘splits’ into two rigid bodies and bends at a flexible joint between the junctional solenoid (a.a. 1657-2144) and the bridging solenoid (a.a. 2145-3613). 2) Bending of S6c and the rotation of Ile4937, located at the pore aperture, results in an increase of the aperture diameter from ~2 Å to ~10 Å, enough to let a hydrated Ca2+ ion (~8.5 Å) through the pore. The bowing of S6 is also accompanied by displacements of the S4–S5 linker. A small aperture dilation (~2 Å) was observed in the presence of 10 mM Ca2+ [16] where the channel is presumably in its Ca2+-inhibited state [48] consistent with a non-conducting or closed state.
Interactions that are broken and formed in the transition between closed and open states in the pore include the S6c salt bridge between E4942, R4944 and E4948 on the adjacent S6c. In the open state the E4942-R4944 interaction breaks and R4944 now form salt bridge with D4938 while E4942 interacts with E4948, both may stabilize the open conformation.
In the presence of RyR1 activators the degree of exo-rotation of the cytoplasmic shell correlates with the degree of the S6 bowing and dilation of the pore aperture. In addition, a RyR1 cryo-EM data set collected in the presence of ryanodine (locked at a unitary open state) exhibited a homogeneous particles population where all 3D classes are in the open state and in the downward shell conformation. These two observations suggest a bidirectional mechanical coupling between the shell motion and the dilation of the pore. In the absence of Ca2+ or ATP this exo-rotation of the RyR1 cytoplasmic shell is uncoupled from the pore dilation [20].
Additions of Ca2+ alone or ATP without Ca2+ involve conformational changes within the activation module that do not cause dilation of the pore aperture. These conformations, described as a “primed state”, are distinct from the ligand-free states; most noticeable is the EF-hand pair interaction with the adjacent protomer S2S3 helical bundle while these domains are separated in the ligand-free state (figure 5). Of importance is the observation that the RyR1 EF-hand pair appears to be in its apo conformation in all states, including the Ca2+ bound states, and is unlikely to play a Ca2+ sensing role in these conditions in RyR1 [53] nor, as been recently described, in RyR2 [54].
Other than the described conformational changes, and quite unexpectedly, few changes are observed in the structure of pVSD during channel gating (figure 5). Naturally, the characteristic charges on S4 that are sensing the changes in membrane potential in voltage-gated channels are missing in RyRs and in IP3Rs.
The RyR1 and IP3R1 share conserved architecture in the NTD, the core solenoid the transmembrane domain and the CTD [16, 17]. Structural similarities in the NTD and in the interfaces between sub-domains of the NTD suggested similar functional role for inter-domain interfaces in the channel activation [51, 55]. R.M.S.D. (Root Mean Square Deviation, the average distance between atoms in superimposed proteins) calculated for 77 atom pairs in the transmembrane domains and the CTDs is 1.135 Å suggesting very high structural and functional similarity in the pore-forming domains of the two proteins.
While we do not have an IP3R structure in its open state two main differences between RyRs and IP3Rs structures should be pointed and are most likely closely related to two different tactics for a similar gating mechanism. One is the lack of the large bridging solenoid in IP3R, and the second is the IP3R-unique long extension of the CTD all the way through the cytosolic vestibule to the NTD where the IP3 binding domain is located [17]. We anticipate that IP3 binding will serve similar role to the RyR1 shell movement in bending the IP3R1 LNK domain (homologous to the RyR1 CTD) following the bowing of S6 to open the pore aperture.
To conclude, the apparent coupling mechanism between the movement of the cytosolic shell and the opening of the pore aperture is now the basis for the allosteric mechanism by which the DHPR on the plasma membrane can activate the release of Ca2+ from the SR stores. The detailed description can now explain the activating role of Ca2+ ions, ATP and caffeine as well as provide an explanation for the effect of disease causing mutations in the interfaces between the moving domains.
Concluding remarks and outstanding questions
While the recent RyRs structures have answered some of the long-standing questions in the field, many remained unanswered questions (see also outstanding questions box). We expect the fast evolving cryo-EM technologies to keep playing a major role in supplying the data to complete the following gaps in our knowledge:
Outstanding Questions.
What is the role of Ca2+ in activation and inhibition of the channel? What is the inhibitory mechanism of Magnesium?
How does DHPR activate the channel?
What is the molecular mechanism of coupled gating?
What is the location and mechanism of disease-causing mutations?
What is the regulatory mechanism of the RyR-associated proteins?
How do post-translational modifications such as phosphorylation, oxidation and nitrosylation regulate the channel function?
What are the binding sites and mechanisms of drugs targeted to RyRs?
Completion of the model
Gaps and ambiguity still exist in large parts of all RyR1 and RyR2 models. These gaps are located mostly where low local resolution in the maps impedes reliable model building and prediction of location and orientation of key residues including disease-causing mutations. Given the known rigidity of these domains and the recent advances in cryo-EM software development we expect these gaps will be bridged and the model will be completed in the very near future.
The role of Ca2+
All open states whose structure has been solved required activating concentrations of Ca2+. In planar lipid bilayers RyR1 exhibits a bell-shaped [Ca2+] response curve with low open probability (Po) at ~100nM Ca2+, maximal Po at ~100 μM Ca2+ and low Po at the mM Ca2+ range [48]. This behavior suggests the presence of at least two cytosolic Ca2+-binding sites: a high affinity (μM) activation site and a low affinity (mM) inhibitory site. While Ca2+ is required for the channel activation of the solubilized protein, Ca2+ entry is not required for RyR1 activation in the skeletal muscle [56, 57].
DHPR
In skeletal muscle, EC-coupling depends on the physical interaction between DHPR and RyR1 to translate plasma membrane depolarization into Ca2+ release from the SR stores located more than 200 Å away [58, 59] (scheme in figure 1). While we know that eliminating the ability of the DHPR to conduct Ca2+ has little or no effect on EC-coupling [56], one can now think of the DHPR as the ‘puppeteer’ controlling the RyR1 cytoplasmic shell and the channel gate, pulling upwards to close the channel [60] and ‘letting go’ to open it. However, the DHPR components that are necessary for the protein-protein interaction with RyR1 are still not clear. Multiple domains have been associated with the DHPR-RyR1 complex including the α1s II-III loop [61, 62], the β1a-subunit [63], STAC3 [64–66] and junctophilins [67]. A step forward in our understanding of this interaction came from the recent cryo-EM structure of DHPR by Wu at al. [68]. Unfortunately, the role played by the II-III loop (residues 670–797) is still vague as it is unresolved in the current model, most likely due to its inherent unstructured nature [69]. Place et al. [70] have recently described RyR1 cleavage by calpain3 upon intense exercise. Calpain cleaves RyR1 N-terminally to residues in SPRY3 (1383–1400) causing EC-uncoupling. A RyR1-DHPR complex structure is the most anticipated missing link in our understanding of EC-coupling.
Coupled gating
In the SR membrane RyRs channels form checkerboard-like arrays of 10–100s of channels [71], with each channel forming contact sites with four other channels, one in each corner. In the terminal cisternae only every other RyR1 is apposed to a plasma membrane VDCC that activates it [4]. This means 50% of the RyR1 channels are not directly activated by DHPR [4]. Those channels are most likely activated either by the Ca2+ released from the DHPR-activated channels (Ca2+ induced Ca2+ release) or directly via coupled gating, where the gating of two or more channels are coordinated and dependent upon the presence of calstabin1 [72, 73]. There is still uncertainty regarding the contact sites between RyRs in the array. Freeze-fracture experiments [4] and 2D crystallization in lipid membranes [74] suggest that RyR1 forms ‘R-arrays’ with the contact sites interface is formed by RY1&2 and the solenoid from the adjacent protomer. Recent work using purified RyR2 suggested that interactions between RyRs in solution occurs at the RY1&2 and calstabin2-SPRY1 interface [75]. High-resolution cryo-electron tomography and sub-tomogram averaging [76] of RyR arrays might resolve this missing link.
RyR-associated proteins and drugs
Ca2+ leak through RyRs due to chronic post-translational modifications including phosphorylation, oxidation and nitrosylation has been implicated as contributing to diseases including heart failure [26], muscle fatigue, Duchenne muscular dystrophy [23], Alzheimer’s disease [28, 77, 78], cancer [25] and diabetes [29]. Our capability to detect RyR1-bound small molecules set the ground for the design and better understanding of the mechanism of novel therapeutics that target leaky RyRs including the Ca2+-stabilizing benzothiazepine derivatives known as Rycals.
Trends.
Ca2+ ions are pivotal to signaling pathways involving extra cellular and intracellular Ca2+ stores and protein transporters including ion channels and ATP dependent Ca2+ pumps.
Recent advances in cryo-electron microscopy detectors, software and specimen preparation have revolutionized structural biology allowing the solution of near-atomic resolution structures of large protein complexes including the Ca2+ release channels ryanodine receptors (RyRs) and IP3 receptors.
Structures of RyR1 at multiple functional states helped identify ligand-binding sites and unravel the channel’s gating mechanism.
Acknowledgments
We would like to thank the members of the laboratories of Filippo Mancia, Wayne A. Hendrickson and Joachim Frank, in particular Oliver Clarke and Amedee Des Georges for their invaluable insights on the structure determination of RyR1. This project has received funding from National Institutes of Health (R01AR060037 and R01HL061503), the Fondation Leducq, Servier, to A.R.M., the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No 708572 to R.Z. A.R.M. is a consultant to, and owns shares in, ARMGO Pharma, Inc., a biotech company targeting RyR channels for therapeutic purposes.
Glossary
- Cryo-EM single particle reconstruction
The process of calculating a 3D density map from 2D electron microscope projections of particles in vitrified ice. In order to obtain the 3D information different slices of the Fourier space, calculated from real space 2D projections at different orientations, have to be gathered to sample the entire information. 3D image classification can then be used to identify distinct conformations within the particles in a data set
- Excitation-contraction coupling
the signaling pathway whereby muscle membrane depolarization activates RyRs to release the massive intracellular Ca2+ that is required to trigger the muscle contractile apparatus. In skeletal muscle DHPR mechanically activate RyR1 via protein-protein interactions. In cardiac muscle, the small flow of Ca2+ through DHPR (CaV1.2) activates the RyR2 in a process termed ‘Ca2+ induced Ca2+ release’. Other components involved in the process are depicted in figure 1 and thoroughly reviewed elsewhere [1]
- Ryanodine
a plant alkaloid that binds to the open state of RyRs with high affinity (nM) and specificity. At low concentrations, ryanodine locks the channel in a long lasting open state with a characteristic ~half the conduction for Ca2+. At high (mM) concentrations ryanodine will block the channel
- Ca2+ signaling
Ca2+ ions serve as second messengers in signaling pathways in almost all eukaryotic cells. Ca2+ signaling is typically triggered by its influx (from outside the cell) or its release (from intracellular stores) followed by a short-term spike in its cytosolic concentration
- Six transmembrane cation channel superfamily
A family of transmembrane Ion channels with a typical pinwheel-like structure (domain swap) where the ion path is formed by four sets (tetramer) of segments (S) 5 and 6. A pore loop between S5 and S6 usually form the selectivity filter and located at the ion entrance to the pore while the S5 and S6 form the narrow part of the gate at the exit of the ion path. S1–S4 form the voltage sensor in voltage gated channels where the positively charged amino acid residues on S4 move through the membrane in response to membrane depolarization to activate the channel gate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rebbeck RT, et al. Skeletal muscle excitation-contraction coupling: who are the dancing partners? The international journal of biochemistry & cell biology. 2014;48:28–38. doi: 10.1016/j.biocel.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Rossi D, Sorrentino V. Molecular genetics of ryanodine receptors Ca2+- release channels. Cell calcium. 2002;32:307–319. doi: 10.1016/s0143416002001987. [DOI] [PubMed] [Google Scholar]
- 3.Giannini G, et al. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. The Journal of cell biology. 1995;128:893–904. doi: 10.1083/jcb.128.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Block BA, et al. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. The Journal of cell biology. 1988;107:2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saito A, et al. Ultrastructure of the calcium release channel of sarcoplasmic reticulum. The Journal of cell biology. 1988;107:211–219. doi: 10.1083/jcb.107.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radermacher M, et al. Cryo-electron microscopy and three-dimensional reconstruction of the calcium release channel/ryanodine receptor from skeletal muscle. The Journal of cell biology. 1994;127:411–423. doi: 10.1083/jcb.127.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang JH, et al. Mapping domains and mutations on the skeletal muscle ryanodine receptor channel. Trends in molecular medicine. 2012;18:644–657. doi: 10.1016/j.molmed.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Ludtke SJ, et al. The pore structure of the closed RyR1 channel. Structure (Camb) 2005;13:1203–1211. doi: 10.1016/j.str.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samso M, et al. Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nature structural & molecular biology. 2005;12:539–544. doi: 10.1038/nsmb938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serysheva II, et al. Subnanometer-resolution electron cryomicroscopy-based domain models for the cytoplasmic region of skeletal muscle RyR channel. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9610–9615. doi: 10.1073/pnas.0803189105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samso M, et al. Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS biology. 2009;7:e85. doi: 10.1371/journal.pbio.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai XC, et al. How cryo-EM is revolutionizing structural biology. Trends in biochemical sciences. 2015;40:49–57. doi: 10.1016/j.tibs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Adler EM. 2015: Signaling Breakthroughs of the Year. Science signaling. 2016;9:eg1. doi: 10.1126/scisignal.aad9795. [DOI] [PubMed] [Google Scholar]
- 14.Zalk R, et al. Structure of a mammalian ryanodine receptor. Nature. 2014 doi: 10.1038/nature13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan Z, et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature. 2014 doi: 10.1038/nature14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Efremov RG, et al. Architecture and conformational switch mechanism of the ryanodine receptor. Nature. 2014 doi: 10.1038/nature13916. [DOI] [PubMed] [Google Scholar]
- 17.Fan G, et al. Gating machinery of InsPR channels revealed by electron cryomicroscopy. Nature. 2015 doi: 10.1038/nature15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei R, et al. Structural insights into Ca(2+)-activated long-range allosteric channel gating of RyR1. Cell research. 2016;26:977–994. doi: 10.1038/cr.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai XC, et al. The Central domain of RyR1 is the transducer for long-range allosteric gating of channel opening. Cell research. 2016;26:995–1006. doi: 10.1038/cr.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.des Georges A, et al. Structural Basis for Gating and Activation of RyR1. Cell. 2016;167:145–157 e117. doi: 10.1016/j.cell.2016.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng W, et al. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science. 2016 doi: 10.1126/science.aah5324. [DOI] [PubMed] [Google Scholar]
- 22.Mathews KD, Moore SA. Multiminicore myopathy, central core disease, malignant hyperthermia susceptibility, and RYR1 mutations: one disease with many faces? Arch Neurol. 2004;61:27–29. doi: 10.1001/archneur.61.1.27. [DOI] [PubMed] [Google Scholar]
- 23.Bellinger AM, et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nature medicine. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keating KE, et al. Detection of a novel RYR1 mutation in four malignant hyperthermia pedigrees. Human molecular genetics. 1994;3:1855–1858. doi: 10.1093/hmg/3.10.1855. [DOI] [PubMed] [Google Scholar]
- 25.Waning DL, et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nature medicine. 2015 doi: 10.1038/nm.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wehrens XH, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304:292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 27.Priori SG, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 28.Oules B, et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:11820–11834. doi: 10.1523/JNEUROSCI.0875-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santulli G, et al. Calcium release channel RyR2 regulates insulin release and glucose homeostasis. The Journal of clinical investigation. 2015 doi: 10.1172/JCI84937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuchi Z, et al. Crystal structures of ryanodine receptor SPRY1 and tandem-repeat domains reveal a critical FKBP12 binding determinant. Nature communications. 2015;6:7947. doi: 10.1038/ncomms8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lau K, Van Petegem F. Crystal structures of wild type and disease mutant forms of the ryanodine receptor SPRY2 domain. Nature communications. 2014;5:5397. doi: 10.1038/ncomms6397. [DOI] [PubMed] [Google Scholar]
- 32.Yuchi Z, et al. Disease mutations in the ryanodine receptor central region: crystal structures of a phosphorylation hot spot domain. Structure. 2012;20:1201–1211. doi: 10.1016/j.str.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 33.Tung CC, et al. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010;468:585–588. doi: 10.1038/nature09471. [DOI] [PubMed] [Google Scholar]
- 34.Reiken S, et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. The Journal of cell biology. 2003;160:919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marx SO, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 36.Lehnart SE, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kimlicka L, et al. Disease mutations in the ryanodine receptor N-terminal region couple to a mobile intersubunit interface. Nature communications. 2013;4:1506. doi: 10.1038/ncomms2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto T, et al. Postulated role of interdomain interaction within the ryanodine receptor in Ca(2+) channel regulation. The Journal of biological chemistry. 2000;275:11618–11625. doi: 10.1074/jbc.275.16.11618. [DOI] [PubMed] [Google Scholar]
- 39.Shtifman A, et al. Interdomain interactions within ryanodine receptors regulate Ca2+ spark frequency in skeletal muscle. The Journal of general physiology. 2002;119:15–32. doi: 10.1085/jgp.119.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Z, et al. The RyR2 central domain peptide DPc10 lowers the threshold for spontaneous Ca2+ release in permeabilized cardiomyocytes. Cardiovascular research. 2006;70:475–485. doi: 10.1016/j.cardiores.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 41.Peralvarez-Marin A, et al. 3D Mapping of the SPRY2 domain of ryanodine receptor 1 by single-particle cryo-EM. PloS one. 2011;6:e25813. doi: 10.1371/journal.pone.0025813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma P, et al. Structural determination of the phosphorylation domain of the ryanodine receptor. The FEBS journal. 2012;279:3952–3964. doi: 10.1111/j.1742-4658.2012.08755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu L, et al. Two rings of negative charges in the cytosolic vestibule of type-1 ryanodine receptor modulate ion fluxes. Biophysical journal. 2006;90:443–453. doi: 10.1529/biophysj.105.072538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao M, et al. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang L, et al. Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature. 2014;505:56–61. doi: 10.1038/nature12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Du GG, et al. Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1) Proceedings of the National Academy of Sciences of the United States of America. 2002;99:16725–16730. doi: 10.1073/pnas.012688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu G, et al. Positions of the cytoplasmic end of BK alpha S0 helix relative to S1–S6 and of beta1 TM1 and TM2 relative to S0–S6. The Journal of general physiology. 2015;145:185–199. doi: 10.1085/jgp.201411337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bezprozvanny I, et al. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 49.Rousseau E, et al. Activation of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum by caffeine and related compounds. Archives of biochemistry and biophysics. 1988;267:75–86. doi: 10.1016/0003-9861(88)90010-0. [DOI] [PubMed] [Google Scholar]
- 50.Dutka TL, Lamb GD. Effect of low cytoplasmic [ATP] on excitation-contraction coupling in fast-twitch muscle fibres of the rat. The Journal of physiology. 2004;560:451–468. doi: 10.1113/jphysiol.2004.069112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seo MD, et al. Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature. 2012;483:108–112. doi: 10.1038/nature10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clarke OB, Hendrickson WA. Structures of the colossal RyR1 calcium release channel. Current opinion in structural biology. 2016;39:144–152. doi: 10.1016/j.sbi.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fessenden JD, et al. Mutational analysis of putative calcium binding motifs within the skeletal ryanodine receptor isoform, RyR1. The Journal of biological chemistry. 2004;279:53028–53035. doi: 10.1074/jbc.M411136200. [DOI] [PubMed] [Google Scholar]
- 54.Guo W, et al. The EF-hand Ca2+ Binding Domain Is Not Required for Cytosolic Ca2+ Activation of the Cardiac Ryanodine Receptor. The Journal of biological chemistry. 2016;291:2150–2160. doi: 10.1074/jbc.M115.693325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mak DD, Foskett JK. Ryanodine receptor resolution revolution: Implications for InsP3 receptors? Cell calcium. 2017;61:53–56. doi: 10.1016/j.ceca.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dirksen RT, Beam KG. Role of calcium permeation in dihydropyridine receptor function. Insights into channel gating and excitation-contraction coupling. The Journal of general physiology. 1999;114:393–403. doi: 10.1085/jgp.114.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Armstrong CM, et al. Twitches in the presence of ethylene glycol bis(− aminoethyl ether)-N,N′-tetracetic acid. Biochimica et biophysica acta. 1972;267:605–608. doi: 10.1016/0005-2728(72)90194-6. [DOI] [PubMed] [Google Scholar]
- 58.Tanabe T, et al. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 1988;336:134–139. doi: 10.1038/336134a0. [DOI] [PubMed] [Google Scholar]
- 59.Nakai J, et al. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- 60.Lacampagne A, et al. Two mechanisms for termination of individual Ca2+ sparks in skeletal muscle. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7823–7828. doi: 10.1073/pnas.97.14.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu X, et al. Activation of the skeletal muscle calcium release channel by a cytoplasmic loop of the dihydropyridine receptor. The Journal of biological chemistry. 1994;269:6511–6516. [PubMed] [Google Scholar]
- 62.Tanabe T, et al. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 1990;346:567–569. doi: 10.1038/346567a0. [DOI] [PubMed] [Google Scholar]
- 63.Szpyt J, et al. Three-dimensional localization of the alpha and beta subunits and of the II-III loop in the skeletal muscle L-type Ca2+ channel. The Journal of biological chemistry. 2012;287:43853–43861. doi: 10.1074/jbc.M112.419283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nelson BR, et al. Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11881–11886. doi: 10.1073/pnas.1310571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horstick EJ, et al. Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nature communications. 2013;4:1952. doi: 10.1038/ncomms2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reinholt BM, et al. Stac3 is a novel regulator of skeletal muscle development in mice. PloS one. 2013;8:e62760. doi: 10.1371/journal.pone.0062760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Golini L, et al. Junctophilin 1 and 2 proteins interact with the L-type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. The Journal of biological chemistry. 2011;286:43717–43725. doi: 10.1074/jbc.M111.292755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu J, et al. Structure of the voltage-gated calcium channel Cav1.1 complex. Science. 2015;350:aad2395. doi: 10.1126/science.aad2395. [DOI] [PubMed] [Google Scholar]
- 69.Cui Y, et al. A dihydropyridine receptor alpha1s loop region critical for skeletal muscle contraction is intrinsically unstructured and binds to a SPRY domain of the type 1 ryanodine receptor. The international journal of biochemistry & cell biology. 2009;41:677–686. doi: 10.1016/j.biocel.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 70.Place N, et al. Ryanodine receptor fragmentation and sarcoplasmic reticulum Ca2+ leak after one session of high-intensity interval exercise. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:15492–15497. doi: 10.1073/pnas.1507176112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Franzini-Armstrong C, et al. Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophysical journal. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marx SO, et al. Coupled gating between cardiac calcium release channels (ryanodine receptors) Circulation research. 2001;88:1151–1158. doi: 10.1161/hh1101.091268. [DOI] [PubMed] [Google Scholar]
- 73.Marx SO, et al. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors) Science. 1998;281:818–821. doi: 10.1126/science.281.5378.818. [DOI] [PubMed] [Google Scholar]
- 74.Yin CC, et al. Physical coupling between ryanodine receptor-calcium release channels. Journal of molecular biology. 2005;349:538–546. doi: 10.1016/j.jmb.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 75.Cabra V, et al. Ultrastructural Analysis of Self-Associated RyR2s. Biophysical journal. 2016;110:2651–2662. doi: 10.1016/j.bpj.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hagen WJ, et al. Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. Journal of structural biology. 2016 doi: 10.1016/j.jsb.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peng J, et al. Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neuroscience letters. 2012;516:274–279. doi: 10.1016/j.neulet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chakroborty S, et al. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer’s disease. PloS one. 2012;7:e52056. doi: 10.1371/journal.pone.0052056. [DOI] [PMC free article] [PubMed] [Google Scholar]