

Abstract
Peroxisomal biogenesis disorders due to PEX gene defects are classified into many subgroups, of which Zellweger spectrum disorders (ZSDs) represent the major subgroup. The ZSDs are clinical and biochemical disorders divided into three phenotypes: neonatal, adolescence, or adult. Clinical presentations vary with severity of the condition. Metabolic abnormalities occur due to functional peroxisomal defects that could be detected in blood and urine. No cure or definitive management exists to date; only supportive and palliative measures are applied to prevent worse sequelae. We experienced a case of oxalate renal stones in a patient with ZSD. This patient had hyperoxaluria and hyperglycolic aciduria with clinically associated clues that correlate with urinary oxalate load. Urinary oxalate and glycolate excretion were assessed. Radiological workup revealed renal involvement with urolithiasis and nephrocalcinosis. Urinalysis and ultrasonography for stones and hyperoxaluria should be used to screen patients with ZSD for early intervention to prevent renal damage.
Keywords: Peroxisome biogenesis disorder, Zellweger spectrum disorder, Zellweger syndrome
Introduction
Zellweger spectrum disorders (ZSDs) are a heterogeneous group of autosomal recessive disorders characterized by a defect in peroxisome formation and mutations in 1 of 13 PEX genes.[1] Patients with ZSD accumulate very long chain fatty acids (VLCFAs), phytanic and pristanic acid, C27-bile acid intermediates, and pipecolic acid in plasma.[2] The core features are liver dysfunction, developmental delay, and other neurological abnormalities, adrenocortical dysfunction, and hearing and visual abnormalities due to microangiopathy of the brain, retina, and cochlea.
Case Report
A 19-month-old male infant with Zellweger syndrome was diagnosed by partial molecular testing VLCFA. He was born full term through spontaneous vaginal delivery after an uneventful pregnancy to nonconsanguineous parents as a second child. At the age of 6 months, his family noticed small gravels in the diaper. Thereafter, workup was done, and he was diagnosed with left renal stones through abdominal ultrasonography. He was followed up regularly by a pediatric nephrologist. He did not have a history of any documented urinary tract infection but had chronic constipation. The patient had a family history of nephrolithiasis in his first-degree relative (brother) who was diagnosed and died of the same condition at the age of 1 year. He was referred to our center at the age of 15 months, when he developed recurrent mild pain at the flanks while urinating (i.e., renal colic) and exhibited blood in the urine (gross hematuria). The pain lasted for a short period, which was not associated with vomiting.
The patient was admitted and evaluated by a multidisciplinary team, including a pediatric nephrologist, hematologist, geneticist, and pediatric anesthesiologist in addition to the pediatric urology team, and a complete workup was performed. He appeared emaciated, dysmorphic craniofacial features with a high forehead and broad nasal bridge and global muscle wasting and hypotonia.
Metabolic workup of renal stone was performed. Urine organic acid analysis showed hyperoxaluria. Urine analysis showed microscopic hematuria (urinary red blood cell, 3700/hpf), no white blood cells, and negative nitrates. Urine examination revealed calcium oxalate crystals. Spot urine test showed urine citrate, 103 mg/24 h; urine calcium, 1.4 mmol/L; urine PO4, 3 mmol/24 h; urine uric acid, 541 mg/24 h; urine creatinine, 390 mmol/24 h; and urine oxalate, 0.12 mg/24 h. Plasma VLCFA levels were markedly elevated. His coagulation profile was prolonged: prothrombin time 22.2 s (11.5–16.5), partial thromboplastin time 52.5 (26–39), and INR 1.87 (0.8–1.3). Mixing study of the blood showed low factor VII: 21 (40–170) and XI 56 (75–155), high factor VIII 290 (50–200) with normal other factors: II 68 (40–170), V 108 (40–120), IV 76 (50–150), X79 (40–120), and XII 80 (70–145) U/mL.
Renal ultrasonography showed multiple left renal stones with grade I hydronephrosis (SFU grading system) with normal right kidney. Computed tomography [Figure 1] showed multiple variable left renal stones, with the largest stone seen in the pelvis, which measured 8 mm × 10 mm in longitudinal diameter. The other stones filled the lower calyx, with the largest stone measuring 4 mm × 10 mm.
Figure 1.
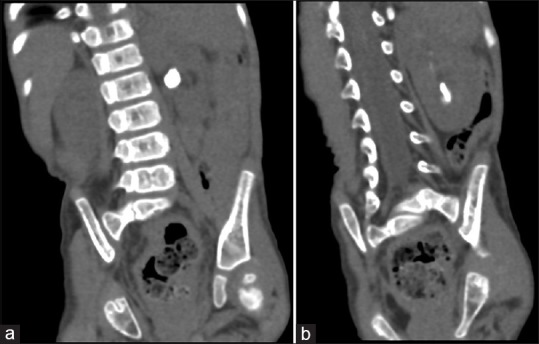
Abdominal computed tomogram (a) stone in the renal pelvis, (b) stone in the lower calyx
The treatment for kidney stones was supportive. From the pediatric urology point of view, the best treatment option for him was left percutaneous nephrolithotomy or multiple sessions of extracorporeal shock wave lithotripsy and insertion of a double JJ stent. The patient was generally hypotonic as well as exhibited coagulopathy and deficiency of factors VII and XI. Life expectancy of patients with this syndrome was determined.
His parents were counseled and explained different treatment modalities with their complications in addition to classification of the patient as high risk due to his general condition, with a high risk of bleeding and general anesthesia. After extensive discussion, his family decided not to perform any surgical intervention and to undergo conservative follow-up particularly that the patient's brother died because of the same condition. We explained that they should observe for any change in voiding habit, unexplained fever, colic, irritability, or reduced feeding. They were discharged with clinic follow-up in the nephrology, genetics, urology, and general pediatric department. Written informed consent was obtained from patient family.
Discussion
The incidence of Zellweger spectrum disorder is 1 in 50,000 newborns.[3] Neonatal–infantile, childhood, and adolescent–adult (late) presentations have direct impact on prognosis. Children with a severe phenotype (neonatal–infantile presentation with severe clinical symptoms) have a poor prognosis and usually die within the 1st year of life. Patients who present in childhood or adolescence usually have a better prognosis but can develop progressive liver disease.[4]
Hyperoxaluria was an obvious laboratory finding in our patient, as well as elevated glycolate levels in the urine. Compared with patients with primary hyperoxaluria, patients with ZSD have higher levels of glycolate and oxalate in the urine. As shown in our case, intermittent diarrhea was a notable clinical sign in combination with urinary oxalate levels.[5] We excluded any Vitamin C supplementation or any diseases that increased the enteric absorption of oxalates, where hyperoxaluria seen in patients with malabsorption syndromes due to high enteric oxalate uptake. Enteric hyperoxaluria and oxalate urolithiasis in patients with ileal resection are caused by intestinal hyperabsorption of oxalate.[6,7] Cessation of glycolate metabolism may result from corrupted import, which occurs due to the absence of physiological allotment of Glyoxylate aminotransferase (AGT) inside the peroxisome. This results in elevated glycolate levels despite apparent normal AGT activity in patients with ZSD. Not all glycolate will be converted to glycine by AGT in patients with ZSD but will instead be converted to oxalate by cytosolic enzymatic system, such as lactate dehydrogenases.[8] The disease severity is linked to the presence of hyperoxaluria because a positive correlation was found between oxalates level in the urine and disease severity, which is expressed by the compound developmental score. We recommend that any patient known to have ZSD should be screened for urine oxalate levels through periodic urine analysis. In cases of patients with maldevelopment and poor bladder control, small urine collections should be performed instead of 24-h urine sample, which is more recommended due to potential sample errors.[9] In cases of hyperoxaluria, we should perform repeated analysis with supplementation of oral citrate to prevent calcium oxalate stone formation and nephrocalcinosis. Our target is to improve the patient's quality of life through proper early supportive intervention due to lack of definitive and curative lines of management of such cases.[10]
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Waterham HR, Ebberink MS. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta. 2012;1822:1430–41. doi: 10.1016/j.bbadis.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Gould S, Raymond G, Valle D. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001. The peroxisome biogenesis disorders; pp. 131–42. [Google Scholar]
- 3.Steinberg SJ, Snowden A, Braverman NE, Chen L, Watkins PA, Clayton PT, et al. A PEX10 defect in a patient with no detectable defect in peroxisome assembly or metabolism in cultured fibroblasts. J Inherit Metab Dis. 2009;32:109–19. doi: 10.1007/s10545-008-0969-8. [DOI] [PubMed] [Google Scholar]
- 4.Berendse K, Engelen M, Linthorst GE, van Trotsenburg AS, Poll-The BT. High prevalence of primary adrenal insufficiency in zellweger spectrum disorders. Orphanet J Rare Dis. 2014;9:133. doi: 10.1186/s13023-014-0133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nancy E. Braverman, Gerald V. Raymond, William B. Rizzo, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016;117:313–21. doi: 10.1016/j.ymgme.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emmett M, Guirl MJ, Santa Ana CA, Porter JL, Neimark S, Hofmann AF, et al. Conjugated bile acid replacement therapy reduces urinary oxalate excretion in short bowel syndrome. Am J Kidney Dis. 2003;41:230–7. doi: 10.1053/ajkd.2003.50012. [DOI] [PubMed] [Google Scholar]
- 7.Traxer O, Huet B, Poindexter J, Pak CY, Pearle MS. Effect of ascorbic acid consumption on urinary stone risk factors. J Urol. 2003;170:397–401. doi: 10.1097/01.ju.0000076001.21606.53. [DOI] [PubMed] [Google Scholar]
- 8.Ikeda M, Kanouchi H, Minatogawa Y. Characterization of peroxisomal targeting signals on alanine: Glyoxylate aminotransferase. Biol Pharm Bull. 2008;31:131–4. doi: 10.1248/bpb.31.131. [DOI] [PubMed] [Google Scholar]
- 9.Simons J. Phenotypic variability in fraternal twins with pex1 mutations: Zellweger syndrome with discordant clinical phenotype. Hered Genet. 2013;2:213–7. [Google Scholar]
- 10.Danpure CJ, Rumsby G. Molecular aetiology of primary hyperoxaluria and its implications for clinical management. Expert Rev Mol Med. 2004;6:1–6. doi: 10.1017/S1462399404007203. [DOI] [PubMed] [Google Scholar]