

Abstract
The neuronal mitochondrial metabolite N-acetylaspartate (NAA) is decreased in the multiple sclerosis (MS) brain. NAA is synthesized in neurons by the enzyme N-acetyltransferase-8-like (NAT8L) and broken down in oligodendrocytes by aspartoacylase (ASPA) into acetate and aspartate. We have hypothesized that NAA links the metabolism of axons with oligodendrocytes to support myelination. To test this hypothesis, we performed lipidomic analyses using liquid chromatography–tandem mass spectrometry (LC–MS/MS) and high-performance thin-layer chromatography (HPTLC) to identify changes in myelin lipid composition in postmortem MS brains and in NAT8L knockout (NAT8L−/−) mice which do not synthesize NAA. We found reduced levels of sphingomyelin in MS normal appearing white matter that mirrored decreased levels of NAA. We also discovered decreases in the amounts of sphingomyelin and sulfatide lipids in the brains of NAT8L−/− mice compared to controls. Metabolomic analysis of primary cultures of oligodendrocytes treated with NAA revealed increased levels of α-ketoglutarate, which has been reported to regulate histone demethylase activity. Consistent with this, NAA treatment resulted in alterations in the levels of histone H3 methylation, including H3K4me3, H3K9me2, and H3K9me3. The H3K4me3 histone mark regulates cellular energetics, metabolism, and growth, while H3K9me3 has been linked to alterations in transcriptional repression in developing oligodendrocytes. We also noted the NAA treatment was associated with increases in the expression of genes involved in sulfatide and sphingomyelin synthesis in cultured oligodendrocytes. This is the first report demonstrating that neuronal-derived NAA can signal to the oligodendrocyte nucleus. These data suggest that neuronal-derived NAA signals through epigenetic mechanisms in oligodendrocytes to support or maintain myelination.
Keywords: Multiple sclerosis, Oligodendrocytes, Myelin lipids, Mass spectrometry, N-acetylaspartate, Histone methylation
Introduction
Multiple sclerosis (MS) is an inflammatory, demyelinating disease of the central nervous system (CNS) that destroys myelin, oligodendrocytes, and neurons, resulting in conduction abnormalities and neurological disability (Noseworthy et al. 2000). MS has been traditionally considered a white matter disease, but cortical pathology including extensive gray matter lesions and cortical atrophy has been reported (Bo et al. 2006; Fisher et al. 2008). The mechanisms responsible for cortical pathology are not clear, but we and others have reported mitochondrial damage in MS cortex. Mitochondrial defects including decreased expression of mitochondrial electron transport chain subunit genes in neurons and cytochrome oxidase inhibition have been reported in normal appearing gray matter (NAGM) in MS (Dutta et al. 2006; Pandit et al. 2009; Broadwater et al. 2011; Campbell et al. 2011; Witte et al. 2013). It has also been demonstrated that the neuronal metabolite NAA is decreased in the MS brain (Gonen et al. 2000; Inglese et al. 2004) and that NAA synthesis is regulated by electron transport chain activity (Li et al. 2013), suggesting a dysfunction of neuronal metabolism in MS (Cader et al. 2007). In vivo measurements of NAA and brain volume by magnetic resonance spectroscopy (MRS) and imaging (MRI) in MS patients have shown that there are considerable decreases in NAA,, which precede neuronal atrophy and these decreases in NAA are reversible in remitting phases of disease (Ge et al. 2004; Khan et al. 2005). Therefore, decreased NAA is not merely a marker of neuronal loss, but also an indication of a dysfunction of neuronal metabolism prior to neuronal degeneration.
N-acetyl aspartate is synthesized by the enzyme N-acetyltransferase-8-like (NAT8L) in neurons and is catabolized by aspartoacylase (ASPA) in oligodendrocytes; thus, it has been suggested that NAA mediates cross talk between neurons and oligodendrocytes. However, the function of NAA in oligodendrocytes and the signaling mechanisms involved have not been clearly defined. It is known that NAA is broken down by cytosolic ASPA into acetate and aspartate in oligodendrocytes and radiolabelling studies have demonstrated that the resulting acetate can be converted to acetyl-CoA and incorporated into myelin lipids (Mehta and Namboodiri 1995; Chakraborty et al. 2001; Ledeen et al. 2006). The inability to break down NAA as a result of defective ASPA in Canavan disease in humans or loss of ASPA in mice results in hypomyelination as well as changes in differentiation of oligodendrocytes (Madhavarao et al. 2005; Kumar et al. 2009; Mattan et al. 2010), indicating that NAA catabolism in oligodendrocytes is necessary for proper myelination.
Recently, the enzyme which synthesizes NAA in neurons, NAT8L, has been knocked out in mice, and surprisingly early analyses of these mice by transmission electron microscopy (TEM) suggest that they appear to myelinate normally even though NAA has been shown to be absent (Guo et al. 2015). However, at 2 months g-ratios were found to be decreased in NAT8L−/− mice, indicating thicker myelin in the corpus callosum (Guo et al. 2015). Changes in myelin lipid composition have not been analyzed in these mice. In the present study, we have performed lipidomic analysis by liquid chromatography–tandem mass spectrometry (LC−MS/MS) and high-performance thin-layer chromatography (HPTLC) to identify changes in myelin lipid composition that are correlated with decreased NAA levels in postmortem MS and control brains and in NAT8L knockout (NAT8L−/−) mice. We have also performed cell culture experiments to delineate signaling pathways mediated by NAA in oligodendrocytes which may impact their development and myelination potential.
Methods
Isolation of myelin lipids
MS and control postmortem frozen brain tissue was obtained from the Rocky Mountain MS Center and the Brain and Spinal Cord Resource Center at UCLA under institutional review board guidelines. MS and control tissue were matched for brain region, age, and postmortem interval as closely as possible. Myelin was isolated from normal appearing white matter (NAWM) from nine postmortem MS and nine control brains by sucrose density gradient according to Norton and Poduslo (1973). Myelin was also isolated from the brains of three C57Bl/6 male mice and three NAT8L−/− male mice. All protocols involving animals were conducted in accordance with institutional animal care guidelines approved at either UC Davis or Kent State University. NAT8L−/− brains were a kind gift from Dr. David Pleasure. The NAT8L−/− mice are constitutive knockouts generated at the UC Davis KOMP Repository (Project ID VG11213). C57Bl/6 control mice were obtained from Jackson Labs (Bar Harbor, ME). All mice were one year old. In brief, either postmortem tissue (0.5–1 g) or whole mouse brains were homogenized in 20 ml of 0.30 M sucrose solution and layered over 18 ml of 0.83 M sucrose solution followed by ultracentrifugation at 75,000 g, 4 °C for 30 min. Crude myelin was collected from the interface of two sucrose solutions, resuspended in 20 ml of Tris–HCl buffer, and centrifuged at 75,000 g, 4 °C for 15 min. The pellet was resuspended again and centrifuged at 12,000×g (15 min.), and the cloudy supernatant removed. To obtain the purified myelin, the pellet was resuspended in 0.30 M sucrose solution. The suspension was then layered over 0.83 M sucrose solution in ultracentrifuge tubes, and the whole process repeated again. Myelin was dissolved in 50 mM Tris–HCl and 2 mM of EDTA, pH 7.4, and the protein concentration was determined from an aliquot by Bradford method. This determination was used to be sure that we isolated lipid from equal amount of myelin from each sample. Myelin lipids were then isolated by an adjusted Bligh and Dyer (1959) procedure. We add the internal lipid standard (heptadecanoyl ethanolamide) to the myelin before lipid isolation. One part of purified myelin was mixed with 3.4 parts of solvent [chloroform/methanol/1 M HCl (10:23:1)] and kept on ice for 15 min. It was mixed with one part of 0.1 M HCl and 1 part of chloroform for phase separation and centrifuged at 3500 rpm, 0 °C for 5 min. The chloroform phase was collected, mixed with one part of buffer (0.1 M NaCl, 0.1 M EDTA, 0.05 M Tris pH 8.2), followed by centrifugation at 3500 rpm, 0 °C for 5 min. The chloroform phase were then collected again and mixed with 1/15 part of isopropanol followed by evaporation of solvent in a roto-evaporator. Lipids were dissolved in methanol/chloroform/water (v: v: v, 45:45:10) mixture containing 5 mM ammonium acetate for LC–MS/MS and in chloroform/methanol (2:1) for HPTLC.
Lipidomic analysis of myelin lipid composition
A direct-infusion mass spectrometric analysis (MS/MSALL) method was applied for analysis of the lipophilic metabolites. LC/MS-grade water and methanol were purchased from Fisher Scientific (Fair Lawn, NJ). Without chromatographic pre-separation, metabolites obtained in the organic phase after extraction were resuspended in a methanol/chloroform/water (v: v: v, 45:45:10) mixture containing 5 mM ammonium acetate (HPLC, ≥99.0 % Sigma-Aldrich BioRegent, St. Louis, MO) and injected into the mass spectrometry with a flow rate of 7 μL min−1. The ion source nebulizer gas (GS1) used was set at 14 psi, the heater gas (GS2) at 15 psi, and the curtain gas (CUR) at 25 psi.
A mass range of 200–1200 Da was scanned followed by 1000 individual MS/MS experiments. In the survey TOF MS scan, 80.0 V DP (declustering potential) and 10.0 V CE (collision energy) was selected for positive charged ions, and −80.0 V DP and −10.0 V CE for negative ions. With a 3000-ms accumulation time, ions with mass range starting at 200.050 Da and finishing with 1200.049 Da were detected. The product ion information-dependent acquisition (IDA) scan selected candidate ions step by step and split precursor ions detected by the survey TOF MS scan into 1000 steps with 1.001 Da window width, generating MS/MS spectra for each precursor ions within 300 ms. Precursors were then fragmented with ±30.0 to 50.0 V collision energy spread under positive or negative mode, respectively. Shotgun lipidomic data were processed using LipidView software (AB Sciex) with mass tolerance of 0.05 Da, min % intensity = 0.1 %, and S/N ≥ 3, and lipids levels were normalized based on the level of internal lipid standard (heptadecanoyl ethanolamide). A Student’s t test was performed to determine statistical significance of changes in myelin lipid levels between MS and control cortical samples and between NAT8L−/− and control (C57Bl/6) mice. A representative LC–MS/MS chromatogram showing the distribution of different lipids from myelin lipid isolated from an MS brain is shown in Supplementary Fig. 1.
HPTLC
Lipid was extracted from equal amounts of myelin and standardized to protein concentration in the protein fraction. For high-performance thin-layer chromatography (HPTLC), the dry lipids were dissolved in chloroform/methanol solution (2:1) in 37 °C water bath for 15 min. The HPTLC plate was dried under vacuum at 100 °C to activate the plate, and samples and standard mixture were spotted on the plate. The mobile phase was chloroform/methanol/water (65:25:4). Lipids were visualized by dipping the plates with 3 % cupric acetate in 8 % phosphoric acid followed by heating at 130 °C for 30 min (Fewster et al. 1969). The plates were scanned immediately, and densitometry was performed with ImageJ software. The average levels of sulfatides and galactocerebrosides were determined in myelin isolated from nine MS and nine control samples and from three wild-type (WT) and three NAT8L−/− mouse brains. Data are shown as the relative concentration of lipids per mg protein from two separate experiments. Statistical significance of changes in myelin lipids was determined with a Student’s t test with p < 0.05 considered significant.
Transmission electron microscopy (TEM)
To detect ultrastructural changes in myelin, we performed TEM of corpus callosum in one WT and one NAT8L−/− mouse, both 7 months old. Mice were deeply anesthetized by intraperitoneal injection of ketamine (150 mg kg−1) and xylazine (16 mg kg−1). Mice were then briefly perfused with 500 units of heparin in 0.1 M phosphate followed sequentially with freshly prepared 4 % paraformaldehyde then 3 % gluteraldehyde in 0.1 M phosphate buffer. Coronal frontal cortical slices (1–2 mm in thickness, between bregma coordinates 0.26 and −0.82) were trimmed to contain the corpus callosum, then post-fixed with 2 % osmium tetroxide, and embedded in Epon 812 resin. Thin sections (70 nm) were stained with uranyl acetate and lead citrate. To quantify axonal/myelin g-ratios and intraperiod myelin thickness, typically, the number of axons and their g-ratios were counted in 25 × 0.25 μm2 sequential micrographs in order to sample the cross-sectional area of corpus callosum in its virtual entirety.
Measuring NAA by HPLC
Levels of NAA were measured in postmortem MS and control brain tissue and in brains of WT and NAT8L ± mice by high-performance liquid chromatography (HPLC) as previously described (Li et al. 2013). NAA was measured in 50 mg cortical gray matter tissue adjacent to the NAWM analyzed for myelin lipid composition by mass spectrometry. A Whatman partisil 10 SAX anion-exchange column (4.6 mm × 250 mm) was used in an Agilent 1100 Series HPLC Value System. NAA concentrations were determined in triplicate. Pearson’s correlation analyses were performed to identify lipid changes in postmortem samples that were correlated with changes in NAA concentration. Correlations were considered significant if p < 0.05.
Primary oligodendrocyte cultures
Oligodendrocytes were harvested from the cerebrum of Sprague–Dawley 16-day rat embryos (E16), and tissue from all fetuses was pooled for culture preparation. The cortices were mechanically dissociated through a 140-μm-pore-sized mesh in 10 % FCS in EMEM without any antibiotics. Cells were further dissociated by trituration with a fire-polished pipette approximately 20 times and then suspended in 10 % FCS/EMEM and sieved through 70-μm-pore-sized mesh (×2). The number of viable cells was determined by using trypan blue exclusion in a hemocytometer. The cells were maintained in EMEM media supplement with 10 % fetal bovine serum (FBS) for 3 weeks. The first week, cells were seeded on poly-L-lysine-coated 100-mm dishes. After 7 days, cells were transferred to 100-mm uncoated culture dishes using 0.05 % trypsin in PBS. After another 7 days, cells were passaged again and cultured in 10 % FCS/EMEM at a density of 3 × 106 cells per plate for 2 days. Media were changed from FCS/EMEM to DMEM supplemented with 10 μg mL−1 insulin, 0.5 μg mL−1 transferrin 100 μg mL−1 BSA, 60 ng mL−1 progesterone, 16 μg mL−1 putrescine, 40 μg mL−1 sodium selenite, 60 ng mL−1 N-acetylcysteine, 5 μM forskolin and 10 ng mL−1 PDGF and cultured for an additional 2 days. Differentiation was initiated by removing PDGF and supplementing with 30 ng mL−1 T3, 40 ng mL−1 T4, and 10 ng mL−1 neurotrophin-3 (NT-3). For NAA treatment, cells were treated with a final concentration of either 2 μM or 20 μM NAA in the media for 7 days during differentiation. pH and cell number were monitored and found not to be changed with NAA treatment. Some cells were cultured on coverslips and immunostained with an antibody to CNPase and a fluorescently labelled secondary antibody. Images were acquired with a Fluoview 1000 confocal microscope.
Metabolite extraction and HILIC–MS
Tricarboxylic acid (TCA) cycle intermediates and metabolites were analyzed by hydrophilic interaction liquid chromatography–mass spectrometry (HILIC–MS) from primary cultures of oligodendrocytes treated with NAA for 7 days. The extraction method utilized a modified form of the Bligh and Dyer (1959) extraction. Cells were lysed by the addition of 180 μL HPLC-grade water and 20 μL methanol and removed from tissue culture dishes by gentle scraping. The cell suspensions were subjected to three cycles of freezing in liquid nitrogen, followed by thawing, and sonication to release cellular metabolites. To each sample, 750 μL 1:2 (v: v) chloroform/methanol and 125 μL chloroform were added, and samples were vortexed thoroughly followed by the addition of 250 μL of water. After incubation at −20 °C for 1 h, samples were centrifuged at 1000×g for 10 min at 4 °C to yield a two-phase system: aqueous layer on top, organic layer below, with a protein disk interphase. The aqueous phase, containing hydrophilic chemicals, and the organic phase, containing lipophilic chemicals, were collected into 1.5-mL tubes separately. All the extracted samples were dried in a CentriVap Concentrator (LABCONCO, Kansas, MO) at room temperature and then preserved at −80 °C until resuspension and analysis. Protein pellets were used to normalize extracted metabolites quantities based on protein concentration with a bicinchoninic acid (BCA) assay.
Chromatographic separation for TCA cycle intermediates was carried out on a Micro200 LC (Eksigent, Redwood, CA) equipped with a HILIC column (Luna 3μ NH2 100Å, 150 mm × 1.0 mm, Phenomenex, Torrance, CA). The hydrophilic metabolites were resuspended in 200 μL of 35 % acetonitrile (aqueous solution) purchased from Fisher Scientific (Fair Lawn, NJ, USA) and 5 μL of the sample was injected into the column. Solvents for the liquid chromatography consist of a mobile phase A of water and a mobile phase B of acetonitrile, each with the addition of 5 mM ammonium acetate and 5 mM ammonium hydroxide (LC/MS, ≥25 % in H2O, Sigma-Aldrich, BioRegent, St. Louis, MO). The flow rate was 30 μL min−1. The gradient consisted of the following linear changes in mobile phase B in time: 0 min 100 %, 0.5 min 98 %, 1 min 95 %, 5 min 80 %, 10 min 4.7 %, 17 min 0 %, 17.1 min 100 %, 23 min 100 %.
Samples were analyzed on a 5600+ TripleTOF® Mass Spectrometry Spectrometer (AB SCIEX, Framingham, MA, USA) and processed with IDA. The ion source nebulizer gas (GS1) used was set at 15 psi, heater gas (GS2) was 20 psi, and the curtain gas (CUR) was 25 psi. In the IDA experiment, a TOF MS scan is selected as survey scan for the mass range of 60–1000 Da. This survey scan utilized a 250-ms accumulation time for precursor ions acquisition. In the positive mode, +5000 V ion spray voltage was used and a +100 V declustering potential (DP) was selected to increase the detection of precursor ions. The background threshold for candidate ion selection was set to 10 counts s−1 to eliminate peaks with a low signal-to-noise (S/N) ratio and abundance. Fragmentation data were subsequently collected by using a collision energy spread (CES) of +(25–40) V. Samples analyzed in negative mode used the same GS1, GS2 and CUR in IDA criteria. A −4500 V ion spray voltage was used, and a −100 V declustering potential (DP) was selected for better precursor ion detection. Fragmentation data are shown in Supplementary Fig. 2 and were collected by using a collision energy spread (CES) of −(40–25) V. Metabolomic data for measurements of TCA cycle intermediates were obtained from at least two separate experiments. Statistical significance of changes in metabolite concentrations between control and NAA-treated cells was determined with a Student’s t test, p < 0.05 considered significant.
α-Ketoglutarate quantification
The levels of α-ketoglutarate were determined by select reaction monitoring (MRM) using 5600+ TripleToF® Mass Spectrometry Spectrometer. α-Ketoglutaric acid was purchased from Sigma-Aldrich (BioRegent, St. Louis, MO), and standards were diluted in series concentrations. A tuned condition was selected for quantification: GS1 was 25 psi, GS2 was 20 psi, CUR was 25 psi, and source temperature was 300 °C. The −4500 V ion spray voltage was used as well as −60 V DP and −10 CE. MS/MS spectrum was generated by using product scan. Five microliters of cell extraction sample was injected in RPLC column (Kinetex 5 μ EVO C18 100 Å, 21 cm × 15 mm, Phenomenex, Torrance, CA) and processed a 20-min isocratic (50:50 water/acetonitrile with 5 mM ammonium hydroxide) at 40 μL min−1 flow rate. The standard curve and MS/MS spectrum are shown in Supplementary Fig. 3.
Western blotting
Control and NAA-treated oligodendrocyte cultures and subcortical white matter isolated from three WT and three NAT8L∓ mice (1 year old) were homogenized with lysis buffer (Tris–HCl, EDTA, NaCl, PMSF, SDS, and protease inhibitor cocktail) followed by centrifugation at 1000×g for 10 min at 4 °C to remove the debris. Nuclear, cytoplasmic, and mitochondrial fractions were isolated by using differential centrifugation. Protein was separated on 15 % SDS-polyacrylamide gels and transferred to nitrocellulose membranes. To determine the level of H3K4me3/H3K9me3/H3K9me2/acetylated H3 over H3 blots were incubated with their respective primary antibody overnight. H3, H3K4Me3, and H3K9me2 were purchased from Abcam (Cambridge, MA), H3K9me3 from Mybiosource (San Diego, CA), and acetylated H3 from Millipore (Temecula, CA). To detect ASPA and myelin basic protein (MBP) expression, Western blotting was also performed with a primary antibody to ASPA (Thermo Scientific, Waltham, MA) and MBP (Millipore, Temecula, CA). Levels of ASPA and MBP were normalized to GAPDH (Millipore, Billerica, MA). After washing with TBST, blots were incubated in the corresponding HRP-conjugated secondary antibody. The immunoreactive bands were visualized by luminol reagent under a fluorescent imaging camera. The integrated density of the bands was measured with ImageJ software from three separate experiments. Levels of H3K4me3/H3K9me3/H3K9me2/acetylated H3 were normalized to histone H3.
QRT-PCR
Relative expression of mRNA for genes involved in oligodendrocyte differentiation including transcription factor 7-like 2 (TCF7l2) and oligodendrocyte transcription factor (olig2) and lipid synthesis including sphingomyelin synthase (Sgms1), cerebroside sulfotransferase (Ugt8), and galactosyl transferase (Gal3st1) was quantitated by QRT-PCR. Briefly, RNA was isolated from control and NAA-treated oligodendrocyte cultures. First, cells were scraped and homogenized in TRIzol (Invitrogen, Carlsbad, CA) followed by addition of chloroform for phase separation. Next, the mixture was centrifuged at 10,000×g for 10 min at 4 °C. The aqueous layer containing RNA was removed and RNA precipitated by isopropanol prior to washing with 70 % ethanol. QRT-PCR was performed with Brilliant III Ultra-Fast SYBR Green (Agilent Technologies, Santa Clara, CA) and gene-specific primers spanning an intron.
Results
Myelin lipid composition is altered in postmortem MS cortex and in NAT8L knockout mice
In order to understand the relationship between NAA and myelination in MS, we performed shotgun lipidomics (LC−MS/MS) to identify changes in myelin lipid composition in NAWM in MS cortex and measured NAA concentrations by HPLC in the same tissue blocks. We analyzed nine MS and nine control cortical tissue blocks. Seven parietal and two motor cortex samples were analyzed for MS and controls. Table 1 lists donor and tissue characteristics. The average age for controls was 69 ± 16.8 years and for MS donors was 61 ± 11.9 years. Average PMI for controls was 12.4 h, and for MS samples, it was 9.8 h. A representative LC−MS/MS chromatogram showing the distribution of different lipids from myelin lipid isolated from an MS brain is shown in Supplementary Fig. 1. Our lipidomics analysis of all eighteen samples revealed a significant decrease in sphingomyelin (SM) in MS myelin compared to controls (Fig. 1a). Sphingomyelin content was reduced by 28 % in MS myelin. We also determined relative levels of sulfatides, important constituents of myelin by HPTLC, as we could not efficiently detect sulfides with our current shotgun lipidomics method. A representative HPTLC plate showing the migration of lipids from MS and control samples is shown in Fig. 1b. Lipid standards including galactocerebrosides (GalC) and sulfatides were run in the first lane. The mobility of the sulfatide standard was slightly reduced compared to the sulfatides in the myelin lipid samples due to the absence of cholesterol and other lipids. Quantitation for sulfatides in MS and control samples is shown in Fig. 1c and shows that sulfatides are also decreased in MS samples by 30 %.
Table 1.
Postmortem cortical samples for lipidomics
| Sample ID | Sex | Age (years) | PMI (h) | Region |
|---|---|---|---|---|
| C1 | M | 80 | 12 | Parietal |
| C2 | F | 83 | 17.6 | Parietal |
| C3 | M | 82 | 14 | Parietal |
| C4 | M | 52 | 16 | Parietal |
| C5 | F | 75 | 15.4 | Motor |
| C6 | M | 58 | 9 | Parietal |
| C7 | F | 35 | 9.3 | Motor |
| C8 | M | 80 | 9.5 | Parietal |
| C9 | F | 76 | 9 | Parietal |
| MS1 | M | 69 | 9 | Parietal |
| MS2 | F | 62 | 6.5 | Parietal |
| MS3 | F | 64 | 7.8 | Parietal |
| MS4 | F | 79 | 3 | Motor |
| MS5 | F | 36 | 3 | Parietal |
| MS6 | M | 57 | 6.5 | Motor |
| MS7 | M | 67 | 5.5 | Parietal |
| MS8 | F | 53 | 3 | Parietal |
| MS9 | F | 63 | 23 | Parietal |
Fig. 1.
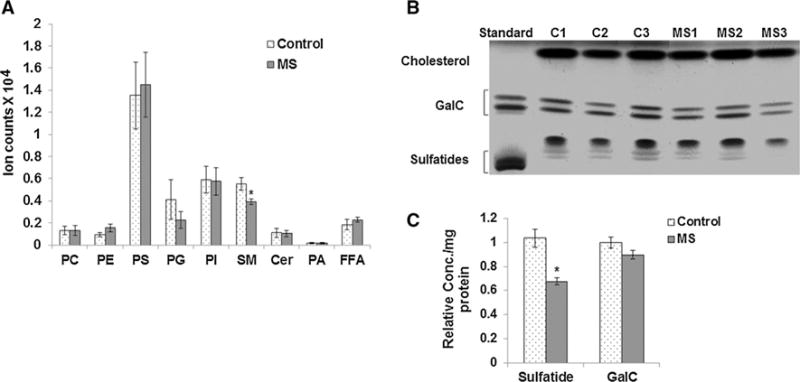
Lipidomic analysis of myelin lipids isolated from MS and control NAWM by LC–MS/MS and HPTLC. a Samples were analyzed by LC–MS/MS to obtain quantitative and qualitative information on multiple lipid classes including phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidylserine (PS), and phospatidic acid (PA), sphingomyelin (SM), ceramide (Cer), phosphatidylinositol (PI), and free fatty acids (FFA). Lipidomics data obtained through LC−MS/MS from NAWM from 9 MS and 9 control samples show a significant decrease in sphingomyelin (SM) lipids in MS myelin. b Representative HPTLC image shows bands for cholesterol, galactocerebrosides (GalC), and sulfatides visualized by copper acetate spray and heating. Lipid standards for GalC and sulfatide were run in the first lane. The mobility of the sulfatide standard is altered slightly compared to the myelin lipids isolated from the brain tissue due to the absence of cholesterol and other lipids. c Quantitation shows that average sulfatide levels were found to be significantly reduced in MS (n = 9) compared to control samples (n = 9). Error bars represent SEM. *p ≤ 0.05
NAA concentrations were also measured by HPLC in gray matter adjacent to the white matter analyzed for myelin lipid content. Representative HPLC chromatograms for a control (top panel) and an MS (bottom panel) sample show that NAA elutes at 5.1 min (Fig. 2a). Quantitation from all eighteen tissue blocks shows that average NAA levels are decreased to a similar magnitude as sphingomyelin and sulfatides in MS samples (Fig. 2b). A Pearson’s correlation test revealed a significant correlation (r = 0.55; p = 0.04, for two-tailed t test) between decreased neuronal NAA levels and relative levels of sphingomyelin (Fig. 2c). The correlation between NAA and sulfatides in our postmortem samples did not reach statistical significance. Sphingomyelin levels were not correlated with age or PMI. Sphingomyelin can be broken down into ceramide under inflammatory conditions, but no increase in ceramide was observed either in myelin (Fig. 1) or in total cellular lipids (data not shown), suggesting that maintenance of sphingomyelin constituents in myelin is defective (Quarles et al. 2006).
Fig. 2.
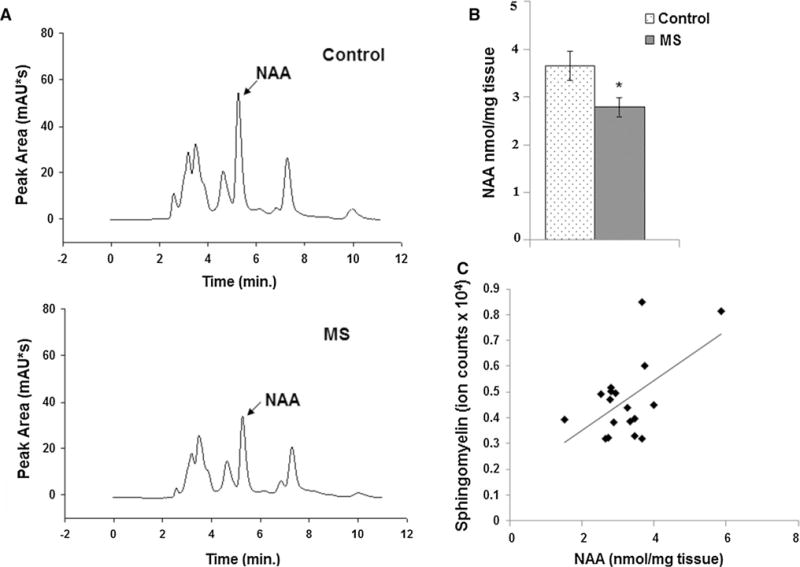
The neuronal mitochondrial metabolite NAA is correlated with levels of sphingomyelin. a Representative HPLC chromatogram of NAA measurement from cortical gray matter tissue blocks from control and MS postmortem brain shows the NAA peak eluting at 5.1 min. b Quantitation shows that average NAA concentration is decreased in MS samples (n = 9) compared to controls (n = 9). c Pearson’s correlation test shows a significant correlation between decreased neuronal NAA levels and relative levels of sphingomyelin (r = 0.55; p = 0.04). *p ≤ 0.05
Our lipidomics data from postmortem NAWM showed that reduced sphingomyelin was correlated with NAA in MS brains. These data suggest a relationship between NAA levels and myelin lipid composition. To test directly whether depletion of NAA causes changes in myelin lipids, we analyzed brains from 1-year-old NAT8L−/− mice by mass spectrometry and HPTLC. The enzyme NAT8L synthesizes NAA, and it has been shown that NAA is not synthesized in NAT8L−/− mice (Guo et al. 2015). Analyzing myelin lipids from the brains of these mice, we found significant decreases in sphingomyelin (SM), phosphatidylcholine (PC), phosphatidylserine (PS), and phosphatidic acid (PA) by LC−MS/MS (Fig. 3a) and sulfatides by HPTLC (Fig. 3b, c).
Fig. 3.
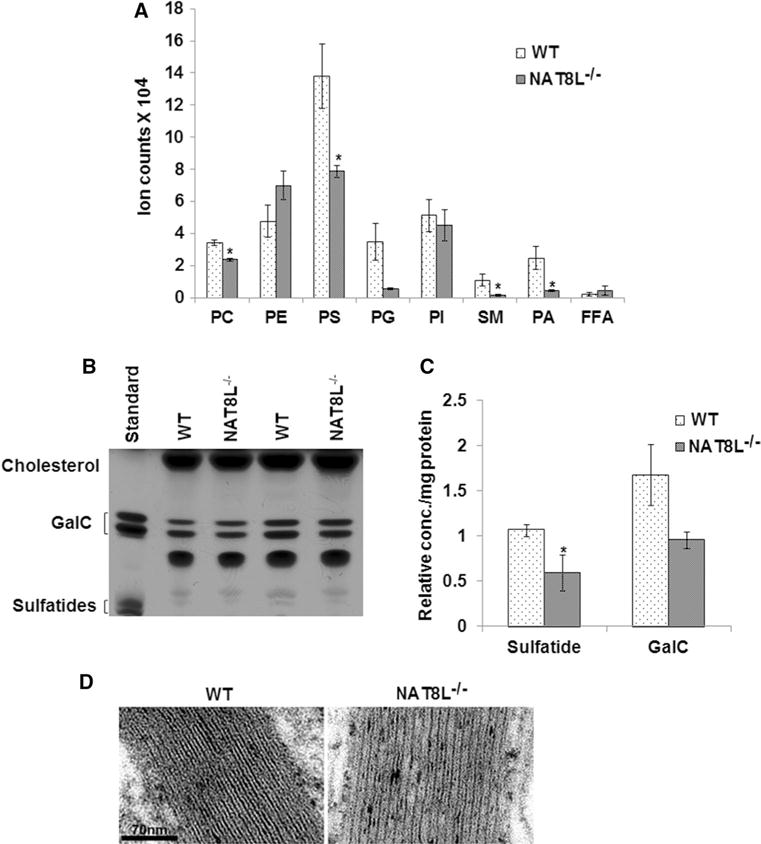
NAT8L−/− mice exhibit changes in myelin lipids and in myelin structure. a Lipidomics data of myelin lipids isolated from 1-year-old NAT8L−/− and wild-type C57Bl/6 control (WT) mouse brains show significant decrease in sphingomyelin (SM), phosphatidylcholine (PC), phosphatidylserine (PS), and phosphatidic acid (PA) in NAT8L−/− brains compared to controls. b Representative HPTLC shows bands for the myelin lipids cholesterol, galactocerebrosides (GalC), and sulfatides visualized by copper acetate spray and heating. Lipid standards for GalC and sulfatide were run in the first lane. c After densitometry, sulfatide levels were found to be significantly reduced in myelin isolated from NAT8L−/− mouse brains (n = 3) compared to WT (n = 3). d High-power EM images show less uranyl acetate/lead citrate counterstaining in NAT8L−/− corpus callosum myelin as compared to WT. The intraperiod distances were measured from high-powered images and were found to be significantly increased in the NAT8L−/− mouse (9.50 ± 0.56 nm and 10.95 ± 0.6 in WT and NAT8L−/− mice, respectively), indicating less compact myelin as a result of NAA depletion. Error bars represent SEM. *p ≤ 0.05
We next examined myelin structure by TEM in one 7-month-old WT and one 7-month-old NAT8L−/− mouse. The g-ratios, determined in corpus callosum, were not significantly changed in the NAT8L knockout (0.76 ± 0.06 for WT and 0.75 ± 0.04 for the NAT8L−/−; mean ± SD, p 0.12, Student’s t test). However, we did find that osmication and uranyl acetate/lead citrate counterstaining is less than normal in the NAT8L−/− mouse, indicating semiquantitatively that there are both less membrane lipids in the intraperiod lines and less proteins in the less condensed interperiod lines (Fig. 3d). We measured the intraperiod lines from high-powered images, and the intraperiod distances are 9.50 ± 0.56 nm (mean ± SEM, n = 8 measurements) and 10.95 ± 0.6 (n = 8, p < 0.0002, one-tailed t test) in WT and NAT8L−/− mice, respectively. These data suggest that the NAT8L−/− animals must have fewer myelin sheaths that are less compacted to account for identical g-ratios.
NAA modulates levels of TCA metabolites in primary cultures of oligodendrocytes
Our lipidomic analysis shows that NAA depletion does have an effect on the lipid constituents of myelin. To begin to understand how NAA may be signaling in oligodendrocytes to change myelin lipid composition, we treated rat primary oligodendrocyte cultures with NAA. To determine that our cultures were expressing oligodendrocyte-specific markers, we performed immunostaining for the oligodendrocyte-specific marker CNPase and Western blotting for ASPA (Fig. 4). These data demonstrate that our cultured oligodendrocytes exhibit the morphology of oligodendrocytes and also express oligodendrocyte-specific markers including ASPA which is necessary to catabolize NAA into acetate and aspartate. These cells responded to NAA in culture by increasing ASPA and myelin basic protein (MBP) expression (Fig. 4b). Acetate and aspartate resulting from break down of NAA by ASPA can enter the TCA cycle. Acetate can be converted into acetyl-CoA and incorporated into fatty acids and enter mitochondria and the TCA cycle after oxidation of fatty acids. Aspartate can also enter the TCA cycle after conversion to oxaloacetate by the oxidoreductase aspartate dehydrogenase. To determine whether these products of NAA catabolism are entering the TCA cycle, we measured levels of TCA cycle intermediates by HILIC–MS in primary cultures of oligodendrocytes treated with NAA (2 μM and 20 μM concentration). We found increased aspartate, which we expected since NAA is broken down into acetate and aspartate by ASPA in oligodendrocytes. We also found that NAA treatment increased levels of TCA cycle intermediates in a dose-dependent manner at the beginning of the cycle including citrate/isocitrate and α-ketoglutarate, but metabolites at the end of the cycle including malate and fumarate were not significantly changed (Figs. 4, 5).
Fig. 4.
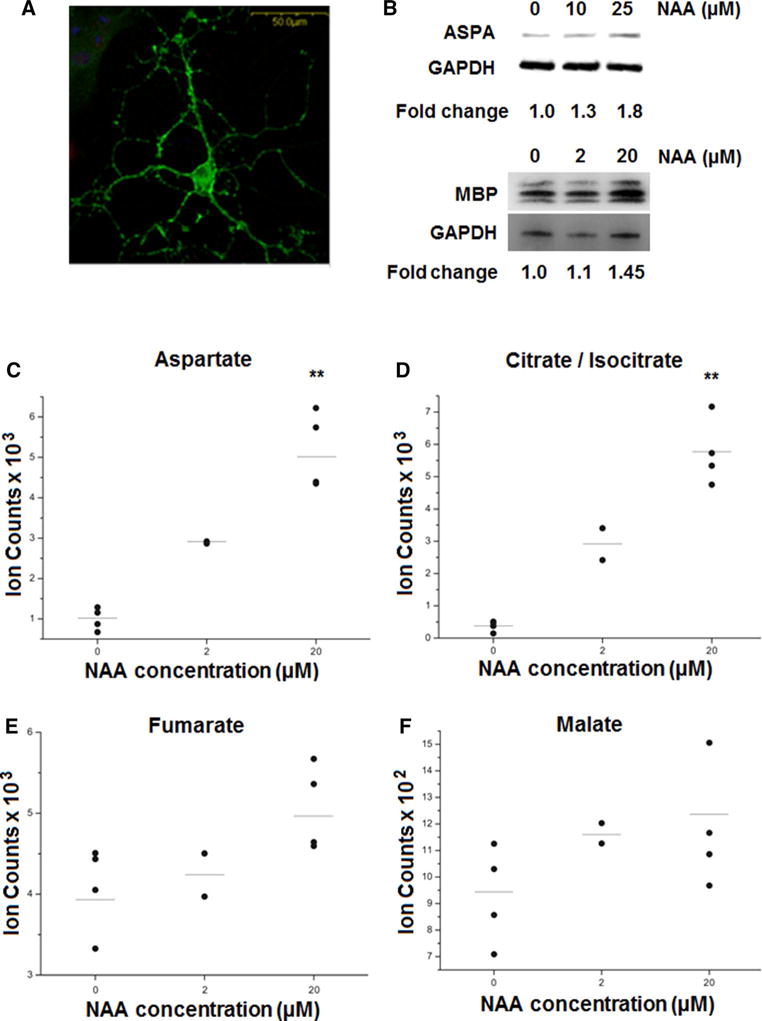
Metabolites related to intermediary metabolism are increased in primary cultures of oligodendrocytes treated with NAA. a Confocal image shows a cultured oligodendrocyte immunostained for CNPase. Scale bar represent 500 μm. b Western blot shows that treatment of cultured oligodendrocytes with NAA increases ASPA and MBP levels. Fold changes of ASPA and MBP normalized to GAPDH are shown. In c–f metabolites were measured in NAA-treated oligodendrocytes by HILIC–mass spectrometry. NAA treatment increased the levels of aspartate (c) and citrate/isocitrate (d) but not fumarate (e) or malate (f) in primary cultures of oligodendrocytes untreated and treated with 2 and 20 μM NAA. Error bars represent SEM. **p ≤ 0.01
Fig. 5.
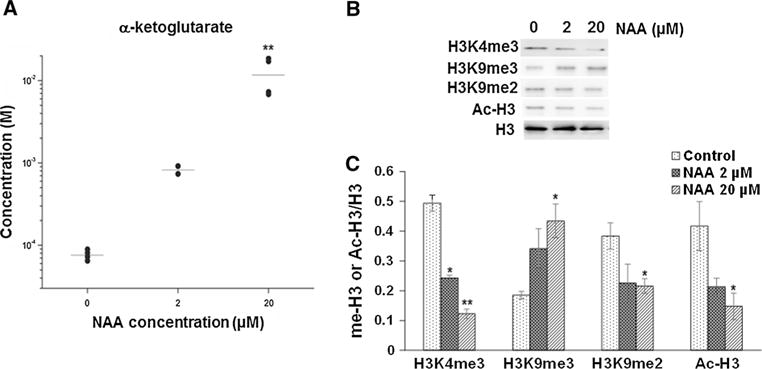
The concentration of a-ketoglutarate and levels of histone H3 methylation are regulated by NAA. a The concentration of α-ketoglutarate was measured by HILIC–mass spectrometry in control cultured oligodendrocytes, cultures treated with 2 and 20 μM NAA. b Representative Western blots for H3K4me3, H3K9me3, H3K9me2, acetylated histone H3 (Ac-H3), and histone H3 from control oligodendrocyte primary cultures and cultures treated with either 2 or 20 μM NAA. c Bar diagram showing the densitometry analysis for H3K4me3/H3K9me3/H3K9me2/Ac-H3 normalized to H3 levels. Error bars represent SEM. *p ≤ 0.05, **p ≤ 0.01
NAA regulates histone methylation patterns and gene expression related to oligodendrocyte differentiation and myelin lipid synthesis
Changes in levels of TCA cycle intermediates have been shown to regulate gene expression through epigenetic mechanisms (Salminen et al. 2014). The TCA cycle intermediate α-ketoglutarate is of particular importance because it is a cofactor required for activity of histone demethylases. Increases in α-ketoglutarate have been shown to activate histone demethylases and lead to decreases in histone H3 methylation. To determine whether NAA could be changing the balance of TCA cycle intermediates and histone H3 methylation in oligodendrocytes, we measured levels of H3K4me3, H3K9me3, and H3K9me2 by Western blotting. Figure 5 shows that NAA decreases levels of H3K4me3 and H3K9me2 and increases H3K9me3 in oligodendrocytes in a dose-dependent manner. Addition of 2 and 20 μM NAA decreased H3K4me3 in oligodendrocyte cultures by 50 and 74 %, while increasing H3K9me3 by 75 and 125 %, respectively. While NAA may be activating histone demethylases and leading to reduced H3K4me3 and H3K9me2 by increasing α-ketoglutarate, other pathways and enzymes must also be involved since H3K9me3 methylation is elevated. We also measured levels of histone H3 acetylation in NAA-treated oligodendrocyte cultures since histone deacetylation is required for OPCs to differentiate into mature oligodendrocytes (Shen et al. 2005). We found that NAA treatment decreased levels of acetylated H3 (Fig. 5), suggesting that NAA signaling is involved in oligodendrocyte maturation and or myelination. Changes in H3K4me3 as a result of NAA signaling were confirmed in 1–year-old NAT8L+/− mice which exhibits reduced levels of NAA (Fig. 6). Consistent with our cell culture data showing that increased NAA catabolism in oligodendrocytes results in reduced H3K4me3, these data show that depleting NAA in mice results in increased H3K4me3.
Fig. 6.
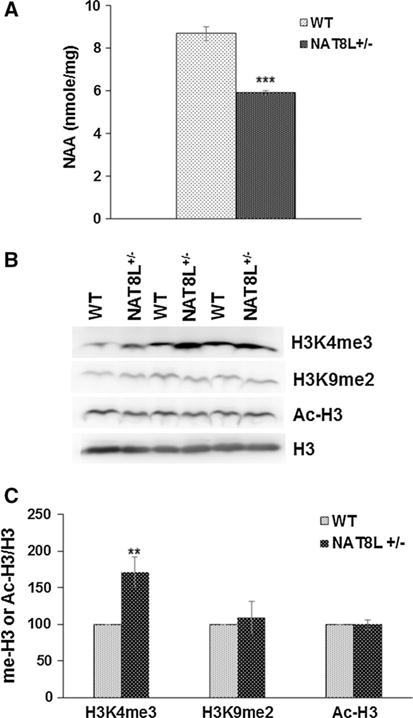
Reductions in NAA in vivo result in increased levels of H3K4me3. a Bar diagram showing NAA concentration is decreased in NAT8L+/− mice as compared to WT mice (n = 3). b Representative Western blots for H3K4me3, H3K9me2, acetylated histone H3 (Ac-H3), and histone H3 from white matter of 1-year-old WT and NAT8L+/− mice. c Bar diagram showing the densitometry analysis for H3K4me3/H3K9me2/Ac-H3 normalized to H3 levels. Error bars represent SEM. **p ≤ 0.01, ***p ≤ 0.001
The potential that changes in methylation and acetylation of histones might affect the expression of specific genes related to oligodendrocyte differentiation and myelin lipid synthesis was also examined. After treating with NAA, we found an increase in mRNA expression for genes related to oligodendrocyte differentiation (TCF7l2, Olig2) and lipid synthesis (Ugt8, Sgms1). NAA treatment (20 μM) increased TCF7l2 and Olig2 mRNA by approximately 30 and 300 % and Ugt8 and Sgms1 by 50 and 55 %, respectively (Fig. 7).
Fig. 7.
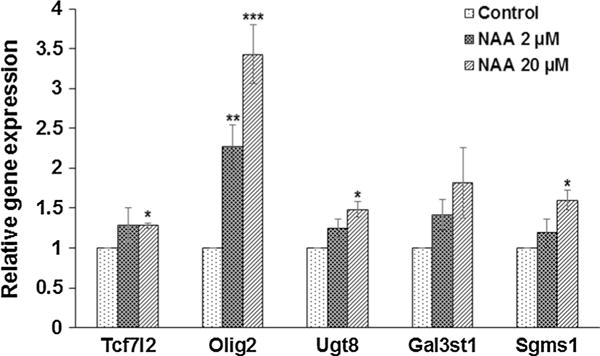
NAA increases mRNA expression of genes related to oligodendrocyte differentiation and myelin lipid synthesis. Bar diagrams showing the differential expression of TCF7l2, Olig2, Ugt8, Gal3st1, and Sgms1 in primary culture of oligodendrocyte treated with 0, 2, and 20 μM of NAA. Data were normalized with respect to β-actin. Relative gene expression data are shown relative to controls, which are adjusted to 1. Error bars represent SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001
Discussion
NAA is a neuronal mitochondrial metabolite that has provided a noninvasive marker of neuronal health and viability for decades; however, its function has remained elusive. NAA synthesis is an energy-dependent process and has been shown to be decreased in neurodegenerative conditions and in traumatic brain injury both of which involve tissue damage, neuronal loss, and mitochondrial impairment (Signoretti et al. 2001). In MS, levels of NAA have been found to be reduced and to be strongly correlated with disability (Bjartmar et al. 2000; De Stefano et al. 2001; Mathiesen et al. 2006). While many studies have documented changes in NAA in neurodegenerative diseases and in MS, the role of NAA is not entirely clear. It has been shown that NAA catabolism by ASPA in oligodendrocytes can provide acetyl groups for myelin lipid synthesis (Moffett et al. 2011). In humans, a mutation in ASPA results in Canavan disease which is marked by dysmyelination and leukodystrophy (Matalon et al. 1988; Kaul et al. 1993). Mice with deletions or mutations of the ASPA gene exhibit similar pathologies including white matter vacuolation and impaired lipid synthesis (Matalon et al. 2000; Madhavarao et al. 2005). These findings led many to speculate that acetyl-CoA derived from NAA breakdown in oligodendrocytes was required for myelination. Recently, the NAT8L enzyme which synthesizes NAA in neurons was knocked out in mice. Surprisingly, these mice did appear to myelinate normally during development (Guo et al. 2015). Further insights into the mechanisms of Canavan disease were apparent when ASPA-deficient mice (ASPAnur7/nur7) were bred with NAT8L−/− mice which do not synthesize NAA. The NAT8L deletion rescued the ASPAnur7/nur7 leukodystrophy and demonstrated that a toxic buildup of NAA rather than an inability to break down NAA was likely involved in Canavan disease pathology.
While excess NAA has been shown to result in myelin defects and leukodystrophy, our data suggest that reductions in NAA may also be deleterious and impact myelination. We found decreases in sphingomyelin and sulfatides in myelin lipids isolated from NAWM in MS samples compared to controls. These observations are supported by other studies which have analyzed postmortem white matter in MS and reported lipid abnormalities and changes in lipid content including sphingolipids and sulfatides (Alling et al. 1971; Marbois et al. 2000; Laule et al. 2013). The fact that these decreased sphingomyelin levels were correlated with NAA levels suggested that defects in neuronal metabolism in MS and reduced NAA may be involved in changes to myelin lipid composition. Our lipidomics data and TEM data obtained from NAT8L−/− mouse brains directly demonstrate a link between NAA and myelin. These data show that even though NAT8L−/− mice appear to myelinate normally during development, aged NAT8L−/− mice have abnormalities in myelin lipid composition consistent with changes observed in MS postmortem NAWM. Our data are supported by a study showing that the inability to catabolize NAA results in myelin lipid changes which can occur at even earlier stages (Traka et al. 2008). Myelin lipid content was analyzed by TLC in mice with a mutation in the ASPA gene (ASPAnur7/nur7) at postnatal day 21. These mice were found to have reduced levels of galactocerebrosides and PE.
We found that depletion of NAA in NAT8L−/− mice leads to decreased levels of several important myelin lipids including sphingomyelin and sulfatides. Changes in sphingolipid content can lead to changes in myelin structure. A study that analyzed postmortem NAWM by electrospray ionization tandem mass spectrometry reported a decrease in sphingolipids and an increase in phospholipid content in MS myelin (Wheeler et al. 2008). After performing biophysical modeling the authors concluded that this change in lipid content would result in myelin decompaction. This event would be consistent with the structural changes we observed in an aged NAT8L−/− mouse. We found that while the g-ratio was not changed in corpus callosum in a 7-month-old NAT8L−/− mouse, the intraperiod distance was increased, suggesting that myelin is decompacted in these animals. Changes in myelin lipid content have also been shown to impact myelin function. Mice with a deletion of the UDP-galactose/ceramide galactosyltransferase (CGT) gene which is required for synthesis of galactocerebrosides and sulfatides showed defects in axonal conduction and developed tremors and ataxia (Coetzee et al. 1996). Our data demonstrate that NAA does play a role in determining myelin lipid composition and suggest that decreased NAA in MS may contribute to changes in myelin function and/or stability.
While others have suggested that NAA catabolism in oligodendrocytes may support myelination by providing a supply of acetyl-CoA for synthesis of myelin lipids, our data suggest that a separate mechanism may be involved. We have delineated a novel pathway mediating cross talk between neuronal/axonal mitochondria and the oligodendrocyte nucleus (Fig. 8). This pathway appears to act on differentiating oligodendrocytes. The neuronal mitochondrial metabolite NAA may regulate myelin lipid composition by signaling to the oligodendrocyte nucleus, altering histone methylation patterns and gene expression. In support of this mechanism we have shown that exposure to NAA can change levels of TCA cycle intermediates in oligodendrocytes. Changes in products of intermediary metabolism have been shown to regulate epigenetic modifications in the nucleus. These epigenetic modifications include methylation of histones and DNA and link mitochondrial energetics to changes in chromatin conformation and gene expression programs (Gut and Verdin 2013; Salminen et al. 2014). Our data suggest that the acetyl groups and aspartate generated from NAA catabolism can potentially modulate TCA cycle activity. Changes in α-ketoglutarate are of particular interest, since this TCA cycle intermediate has been shown to regulate epigenetic modifications to histones in the nucleus by activating histone demethylases. These enzymes are a family of 2-oxoglutarate (α-ketoglutarate)-dependent dioxygenases (2-OGDO) which oxidize C-H bonds (Vissers et al. 2014).
Fig. 8.
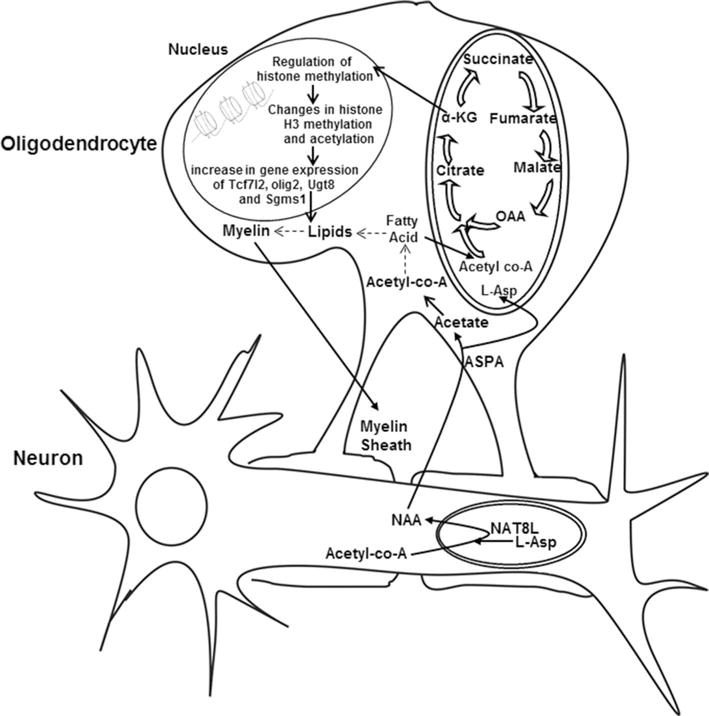
Schematic shows the relationship between neuronal and oligodendrocyte metabolism. Potential signaling pathways mediated by NAA in oligodendrocytes. NAA is synthesized from aspartate and acetyl-CoA in neuronal mitochondria by the enzyme NAT8L. It is catabolized in oligodendrocytes by ASPA into aspartate and acetate. The resulting acetate can be converted to acetyl-CoA in the cytoplasm which is incorporated into myelin lipids (pathway shown by dotted lines). However, our data suggest the existence of a novel signaling pathway. In this pathway (shown by bold arrows), the products of NAA catabolism enter the TCA cycle and signal to the nucleus of oligodendrocytes. Acetate can be converted to acetyl-CoA and incorporated into lipids which can be oxidized to generate acetyl-CoA in mitochondria which then can enter the TCA cycle. Aspartate (L-Asp) can be converted to oxaloacetate by aspartate dehydrogenase and can also enter the TCA cycle. These products then alter levels of TCA cycle intermediates including α-ketoglutarate (α-KG). TCA cycle intermediates and metabolites measured including aspartate, citrate/isocitrate, α-KG, succinate, fumarate, and malate are shown. Increased α-KG can regulate histone demethylases resulting in changes in methylation of histone H3 which can then lead to changes in chromatin conformation and gene expression
In addition to changes in TCA cycle intermediates, we also found that histone H3 methylation patterns were changed in NAA-treated oligodendrocytes. Both H3K4me3 and H3K9me3 histone marks were altered in NAA-treated cells and changes in H3K4me3 were confirmed in subcortical white matter of NAT8L+/− mice. These histone marks have been shown to be involved in cellular differentiation programs. H3K4me3 marks promoters of actively transcribed genes and reductions in H3K4me3 result in slowed metabolism and growth, and increased differentiation (Shyh-Chang et al. 2013). The repressive histone H3 methyl mark H3K9me3 has been shown to repress inappropriate gene expression programs and is increased during oligodendrocyte differentiation (Liu et al. 2015). Our data showing that NAA decreases H3K4me3 and increases H3K9me3 suggest that NAA mediates signals to the oligodendrocyte nucleus, changing chromatin structure and gene expression programs that may support myelination. This idea is supported by our gene expression data, showing that NAA regulates TCF7l2 and olig2, transcription factors involved in oligodendrocyte differentiation (Hammond et al. 2015; Wegener et al. 2015), as well as genes involved in synthesis of the myelin lipids sphingomyelin and sulfatides. Histone methylation and deacetylation patterns necessary for proper cell identity can be lost in aging oligodendrocytes (Shen et al. 2008). Our data suggest that exposure to NAA may provide a mechanism to prevent this loss of epigenetic cell memory.
In conclusion, our findings suggest that the NAT8L−/− mouse model may provide insight into the changes which occur in oligodendrocytes and in myelin in MS. These mice at first appeared to myelinate normally; however, our data show that there are changes in levels of H3K4me3 and myelin lipid content in these mice as they age. Similarly, in MS myelination occurs normally during development, but over time is disrupted. We found changes in myelin lipids in NAWM consistent with those observed in NAT8L−/− mice. Whether the mitochondrial abnormalities reported in MS are primary or secondary to inflammation and the disease process remains to be determined. In either case, our data suggest that neuronal mitochondrial dysfunction in MS and decreased NAA can mediate signals to the nucleus in oligodendrocytes by changing epigenetic modifications to histones. Ultimately, decreased NAA synthesis in neurons and catabolism in oligodendrocytes may leave axons more vulnerable to demyelination due to changes in gene expression in oligodendrocytes and myelin lipid composition.
Supplementary Material
Acknowledgments
We would like to thank the Rocky Mountain MS Center who is funded by the National Multiple Sclerosis Society and the Human Brain and Spinal Fluid Resource Center at UCLA for MS and control tissue. This research was funded in part by a grant from the College of Arts and Sciences at Kent State University (JM).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00221-016-4789-z) contains supplementary material, which is available to authorized users.
References
- Alling C, Vanier MT, Svennerholm L. Lipid alterations in apparently normal white matter in multiple sclerosis. Brain Res. 1971;35:325–336. doi: 10.1016/0006-8993(71)90478-1. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Kidd G, Mörk S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl-aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48:893–901. [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Bo L, Geurts JJ, Mork SJ, van der Valk P. Grey matter pathology in multiple sclerosis. Acta Neurol Scand Suppl. 2006;183:48–50. doi: 10.1111/j.1600-0404.2006.00615.x. [DOI] [PubMed] [Google Scholar]
- Broadwater L, Pandit A, Azzam S, Clements R, Vadnal J, Yong VW, Freeman EJ, Gregory RB, McDonough J. Analysis of the mitochondrial proteome in multiple sclerosis cortex. Biochim Biophys Acta. 2011;1812:630–641. doi: 10.1016/j.bbadis.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cader S, Johansen-Berg H, Wylezinska M, Palace J, Behrens TE, Smith S, Matthews PM. Discordant white matter N-acetylasparate and diffusion MRI measures suggest that chronic metabolic dysfunction contributes to axonal pathology in multiple sclerosis. Neuroimage. 2007;36(1):19–27. doi: 10.1016/j.neuroimage.2007.02.036. [DOI] [PubMed] [Google Scholar]
- Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, Lassmann H, Turnbull DM, Mahad DJ. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol. 2011;69(3):481–492. doi: 10.1002/ana.22109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty G, Mekala P, Yahya D, Wu G, Ledeen RW. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: evidence for myelin-associated aspartoacylase. J Neurochem. 2001;78(4):736–745. doi: 10.1046/j.1471-4159.2001.00456.x. [DOI] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Suzuki K, Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86(2):209–219. doi: 10.1016/s0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Narayanan S, Francis GS, Arnaoutelis R, Tartaglia MC, Antel JP, Matthews PM, Arnold DL. Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol. 2001;58(1):65–70. doi: 10.1001/archneur.58.1.65. [DOI] [PubMed] [Google Scholar]
- Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, Gudz T, Macklin WB, Lewis DA, Fox RJ, Rudick R, Mirnics K, Trapp BD. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59:478–489. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- Fewster ME, Burns BJ, Mead JF. Quantitative densitometric thin-layer chromatography of lipids using copper acetate reagent. J Chromatogr. 1969;43(1):120–126. doi: 10.1016/s0021-9673(00)99173-8. [DOI] [PubMed] [Google Scholar]
- Fisher E, Lee JC, Nakamura K, Rudick RA. Gray matter atrophy in multiple sclerosis: a longitudinal study. Ann Neurol. 2008;64(3):255–265. doi: 10.1002/ana.21436. [DOI] [PubMed] [Google Scholar]
- Ge Y, Gonen O, Inglese M, Babb JS, Markowitz CE, Grossman RI. Neuronal cell injury precedes brain atrophy in multiple sclerosis. Neurology. 2004;62(4):624–627. doi: 10.1212/wnl.62.4.624. [DOI] [PubMed] [Google Scholar]
- Gonen O, Catalaa I, Babb JS, Ge Y, Mannon LJ, Kolson DL, Grossman RI. Total brain N-acetylaspartate: a new measure of disease load in MS. Neurology. 2000;54(1):15–19. doi: 10.1212/wnl.54.1.15. [DOI] [PubMed] [Google Scholar]
- Guo F, Bannerman P, Mills Ko E, Miers L, Xu J, Burns T, Li S, Freeman E, McDonough J, Pleasure D. Ablation of N-acetyl-L-aspartate synthesis prevents leukodystrophy in a murine model of Canavan disease. Ann Neurol. 2015;77(5):884–888. doi: 10.1002/ana.24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502(7472):489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- Hammond E, Lang J, Maeda Y, Pleasure D, Angus-Hill M, Xu J, Horiuchi M, Deng W, Guo F. The Wnt effector transcription factor 7-like 2 positively regulates oligodendrocyte differentiation in a manner independent of Wnt/β-catenin signaling. J Neurosci. 2015;35(12):5007–5022. doi: 10.1523/JNEUROSCI.4787-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese M, Ge Y, Filippi M, Falini A, Grossman RI, Gonen O. Indirect evidence for early widespread gray matter involvement in relapsing-remitting multiple sclerosis. Neuroimage. 2004;21:1825–1829. doi: 10.1016/j.neuroimage.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Kaul R, Gao GP, Balamurugan K, Matalon R. Cloning of the human aspartoacylase cDNA and a common missense mutation in Canavan disease. Nat Genet. 1993;5(2):118–123. doi: 10.1038/ng1093-118. [DOI] [PubMed] [Google Scholar]
- Khan O, Shen Y, Caon C, Bao F, Ching W, Reznar M, Buccheister A, Hu J, Latif Z, Tselis A, Lisak R. Axonal metabolic recovery and potential neuroprotective effect of glatiramer acetate in relapsing-remitting multiple sclerosis. Mult Scler. 2005;11(6):646–651. doi: 10.1191/1352458505ms1234oa. [DOI] [PubMed] [Google Scholar]
- Kumar S, Biancotti JC, Matalon R, de Vellis J. Lack of aspartoacylase activity disrupts survival and differentiation of neural progenitors and oligodendrocytes in a mouse model of Canavan disease. J Neurosci Res. 2009;87:3415–3427. doi: 10.1002/jnr.22233. [DOI] [PubMed] [Google Scholar]
- Laule C, Pavlova V, Leung E, Zhao G, MacKay AL, Kozlowski P, Traboulsee AL, Li DK, Moore GR. Diffusely abnormal white matter in multiple sclerosis: further histologic studies provide evidence for a primary lipid abnormality with neurodegeneration. J Neuropathol Exp Neurol. 2013;72(1):42–52. doi: 10.1097/NEN.0b013e31827bced3. [DOI] [PubMed] [Google Scholar]
- Ledeen RW, Wang J, Wu G, Lu ZH, Chakraborty G, Meyenhofer M, Tyring SK, Matalon R. Physiological role of N-acetylaspartate: contribution to myelinogenesis. Adv Exp Med Biol. 2006;576:131–143. doi: 10.1007/0-387-30172-0_9. discussion 361–363. [DOI] [PubMed] [Google Scholar]
- Li S, Clements R, Sulak M, Gregory R, Freeman E, McDonough J. Decreased NAA in gray matter is correlated with decreased availability of acetate in white matter in postmortem multiple sclerosis cortex. Neurochem Res. 2013;38(11):2385–2396. doi: 10.1007/s11064-013-1151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Magri L, Zhang F, Marsh NO, Albrecht S, Huynh JL, Kaur J, Kuhlmann T, Zhang W, Slesinger PA, Casaccia P. Chromatin landscape defined by repressive histone methylation during oligodendrocyte differentiation. J Neurosci. 2015;35(1):352–365. doi: 10.1523/JNEUROSCI.2606-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Arun P, Moffett JR, Szucs S, Surendran S, Matalon R, Garbern J, Hristova D, Johnson A, Jiang W, Namboodiri MA. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan’s disease. Proc Natl Acad Sci USA. 2005;102(14):5221–5226. doi: 10.1073/pnas.0409184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbois BN, Faull KF, Fluharty AL, Raval-Fernandes S, Rome LH. Analysis of sulfatide from rat cerebellum and multiple sclerosis white matter by negative ion electrospray mass spectrometry. Biochim Biophys Acta. 2000;1484(1):59–70. doi: 10.1016/s1388-1981(99)00201-2. [DOI] [PubMed] [Google Scholar]
- Matalon R, Michals K, Sebesta D, Deanching M, Gashkoff P, Casanova J. Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease. Am J Med Genet. 1988;29:463–471. doi: 10.1002/ajmg.1320290234. [DOI] [PubMed] [Google Scholar]
- Matalon R, Rady PL, Platt KA, Skinner HB, Quast MJ, Campbell GA, Matalon K, Ceci JD, Tyring SK, Nehls M, Surendran S, Wei J, Ezell EL, Szucs S. Knock-out mouse for Canavan disease: a model for gene transfer to the central nervous system. J Genet Med. 2000;2(3):165–175. doi: 10.1002/(SICI)1521-2254(200005/06)2:3<165::AID-JGM107>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Mathiesen HK, Jonsson A, Tscherning T, Hanson LG, Andresen J, Blinkenberg M, Paulson OB, Sorensen PS. Correlation of global N-acetyl aspartate with cognitive impairment in multiple sclerosis. Arch Neurol. 2006;63(4):533–536. doi: 10.1001/archneur.63.4.533. [DOI] [PubMed] [Google Scholar]
- Mattan NS, Ghiani CA, Lloyd M, Matalon R, Bok D, Casaccia P, de Vellis J. Aspartoacylase deficiency affects early postnatal development of oligodendrocytes and myelination. Neurobiol Dis. 2010;40(2):432–443. doi: 10.1016/j.nbd.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta V, Namboodiri MA. N-Acetylaspartate as an acetyl source in the nervous system. Brain Res Mol Brain Res. 1995;31(1–2):151–157. doi: 10.1016/0169-328x(95)00044-s. [DOI] [PubMed] [Google Scholar]
- Moffett JR, Arun P, Ariyannur PS, Garbern JY, Jacobowitz DM, Namboodiri AM. Extensive aspartoacylase expression in the rat central nervous system. Glia. 2011;59(10):1414–1434. doi: 10.1002/glia.21186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE. Myelination in rat brain: method of myelin isolation. J Neurochem. 1973;21(4):749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- Pandit A, Vadnal J, Houston S, Freeman E, McDonough J. Impaired regulation of electron transport chain subunit genes by nuclear respiratory factor 2 in multiple sclerosis. J Neurol Sci. 2009;279:14–20. doi: 10.1016/j.jns.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Quarles RH, Macklin WB, Morell P. Myelin formation, structure and biochemistry. In: Siegel GJ, Agranoff BW, Albers RW, et al., editors. Basic neurochemistry: molecular, cellular and medical aspects. 6th. Lippincott-Raven; Philadelphia: 2006. pp. 51–71. [Google Scholar]
- Salminen A, Kauppinen A, Hiltunen M, Kaarniranta K. Krebs cycle intermediates regulate DNA and histone methylation: epigenetic impact on the aging process. Ageing Res Rev. 2014;16:45–65. doi: 10.1016/j.arr.2014.05.004. [DOI] [PubMed] [Google Scholar]
- Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J Cell Biol. 2005;169(4):577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Liu A, Li J, Wolubah C, Casaccia-Bonnefil P. Epigenetic memory loss in aging oligodendrocytes in the corpus callosum. Neurobiol Aging. 2008;3:452–463. doi: 10.1016/j.neurobiolaging.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, Asara JM, Daley GQ, Cantley LC. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339:222–226. doi: 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signoretti S, Marmarou A, Tavazzi B, Lazzarino G, Beaumont A, Vagnozzi R. N-Acetylaspartate reduction as a measure of injury severity and mitochondrial dysfunction following diffuse traumatic brain injury. J Neurotrauma. 2001;18(10):977–991. doi: 10.1089/08977150152693683. [DOI] [PubMed] [Google Scholar]
- Traka M, Wollmann RL, Cerda SR, Dugas J, Barres BA, Popko B. Nur7 is a nonsense mutation in the mouse aspartoacylase gene that causes spongy degeneration of the CNS. J Neurosci. 2008;28(45):11537–11549. doi: 10.1523/JNEUROSCI.1490-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers MC, Kuiper C, Dachs GU. Regulation of the 2-oxoglutarate-dependent dioxygenases and implications for cancer. Biochem Soc Trans. 2014;42(4):945–951. doi: 10.1042/BST20140118. [DOI] [PubMed] [Google Scholar]
- Wegener A, Deboux C, Bachelin C, Frah M, Kerninon C, Seilhean D, Weider M, Wegner M, Nait-Oumesmar B. Gain of Olig2 function in oligodendrocyte progenitors promotes remyelination. Brain. 2015;138(Pt 1):120–135. doi: 10.1093/brain/awu375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler D, Bandaru VV, Calabresi PA, Nath A, Haughey NJ. A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain. 2008;131(Pt 11):3092–3102. doi: 10.1093/brain/awn190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte ME, Nijland PG, Drexhage JA, Gerritsen W, Geerts D, van Het Hof B, Reijerkerk A, de Vries HE, van der Valk P, van Horssen J. Reduced expression of PGC-1α partly underlies mitochondrial changes and correlates with neuronal loss in multiple sclerosis cortex. Acta Neuropathol. 2013;125(2):231–243. doi: 10.1007/s00401-012-1052-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.