

Abstract
The current global obesity pandemic is clearly linked to both the increasing prevalence of and preference for foods high in calories, specifically fat and sucrose, and declining levels of daily physical activity. A less commonly discussed possible explanation is that risk of obesity begins in utero as a result of developmental plasticity during early life. This idea fits into the broader Developmental Origins of Health and Disease hypothesis, which holds that stressful in utero exposure manifests as disease in adulthood. In this review, we highlight several studies that have revealed the role of epigenetics in multigenerational transmission of developmentally programmed obesity and associated cardiometabolic disease.
Obesity and Disease
The worldwide prevalence of obesity more than doubled between 1980 and 2014, and 41 million children under the age of five were overweight or obese in 20141. Globally, obesity and overweight are associated with more deaths than undernourishment, and World Health Organization reports have shown that obese and overweight people outnumber underweight people in all corners of the planet except in some parts of sub-Saharan Africa and Asia1. This shocking increase in obesity has led to rises in the incidence of cardiovascular disease (CVD), metabolic syndrome, musculoskeletal diseases, and some cancers. Although several large-scale genome-wide studies have led to identification of over 50 genetic loci thought to be associated with obesity-related phenotypes, these loci have miniscule effects on obesity susceptibility and account for a fraction of the total variance2. Several studies, however, have produced compelling data to suggest that the increasing incidence of cardiometabolic disease reflects adverse conditions during the pre-and perinatal period.
Developmental Plasticity and the Developmental Origins of Health and Disease Hypothesis
In human development, the first 1000 days after fertilization is a critical stage during which developmental plasticity is possible. After this period, very few organ systems and tissues have the ability to adapt and respond to physiologic challenges. The Developmental Origins of Health and Diseases (DOHAD) hypothesis proposes that adverse events during this period predispose offspring to poor health in later life3.
The original evidence to support the DOHAD hypothesis arose from retrospective epidemiologic studies that revealed a strong association between low birth weight and metabolic disease such as CVD, insulin resistance, and type 2 diabetes in adulthood3-5. These initial studies were validated in other cohorts and populations, including the offspring of survivors of the Dutch famine in 1944, in which those exposed to famine during the intrauterine period had increased rates of obesity, diabetes, and CVD as adults 6,7. Subsequent epidemiological studies reported similar findings in those who were exposed to overnutrition during pregnancy. Some of these offspring were born large for gestational age, whereas others were born small and underwent catch-up growth. The individuals at both ends of this spectrum experienced permanent physiologic and metabolic effects that predisposed them to CVD in their later years. Thus, the long-term effects of birthweight reflect a U-shaped curve in which both extremes are detrimental in later life.8,9
Epigenetics in DOHaD
Over the last 10 years, DOHAD researchers have begun to focus on identifying the mechanisms responsible for associations between in utero exposures and adult disease outcomes. Such work has revealed roles for several regulatory systems, organs, and tissues in developmental programing. These include the hormones insulin and leptin10, the hypothalamic-pituitary-adrenal axis11, adipose tissue signaling and metabolism12, mitochondrial function13, the vascular system14,15, and placental function15. The breakthroughs in these systems, however, are being made as epigenetic underpinnings are identified. Epigenetics is defined as heritable changes in gene expression that do not involve changes to the underlying DNA sequence — a change in phenotype without a change in genotype. One class of epigenetic mechanisms is chromatin modifications such as DNA methylation and histone methylation and acetylation. In most incidences, DNA or histone methylation prevents transcription factor binding and thus inhibits gene transcription. DNA methylation has been linked to some heritable changes seen with DOHAD, but the role of histone methylation in DOHAD is still debatable 16. Other epigenetic mechanisms include noncoding RNAs and microRNAs17, but whether these play a role in DOHAD has not been determined. The focus of current DOHAD research is on defining the epigenetic mechanisms involved in transmitting developmentally programmed traits to subsequent generations.
Dietary alterations, epigenetics, and intergenerational transmission in rodents
Several well-studied examples in rodents have linked offspring phenotypes to epigenetic modifications thought to be induced by adverse maternal diet and other exposures. Obesity and metabolic disorders have been induced in rodent and human offspring by maternal global undernutrition 18-21, maternal over nutrition, maternal protein restriction22, maternal uterine artery ligation23,24, maternal synthetic glucocorticoid treatment 25, maternal anemia 26, or prenatal cytokine exposure27. In this section, we will discuss the effect of these stressors on offspring outcomes and their adult phenotypic consequences.
First, one study revealed that poor maternal diet during pregnancy can affect appetite in offspring 10. In rats and mice, the hypothalamic proopiomelanocortin (POMC) gene promoter region is a critical target of epigenetic changes involved in appetite. The POMC promoter was found to be less methylated in rat offspring from dams fed a low-protein diet28, whereas it showed hypermethylation in neonatal overfed animals. These epigenetic modifications impair hormonal effects on POMC expression and are thought to be responsible for hypothalamic leptin/insulin resistance 29. In white adipose tissue, adult mice from dams fed a low-protein diet showed hypomethylation of the leptin promoter. Juvenile rat offspring from the same mothers exhibited an increase in miRNA-483-3p which leads to the translational repression of growth/differentiation factor-3 (GDF-3). GDF-3 normally facilitates lipid storage in adipose tissue, and thus elevated miRNA-483-3p as a result of early life nutrition changes leads to lipotoxicity and insulin resistance and thus increasing susceptibility to metabolic disease.30.
Another classic example of maternal diet influencing epigenetic changes in the offspring is the agouti mice 31,32. In mice carrying the viable yellow allele, Avy, mice can either have a brown coat color and be healthy, or they can have a yellow coat color and be obese, hyperinsulinemic, hyperglycemic, and at increased risk of cancer. 33 This phenotypic difference occurs as a result of DNA methylation; mice with normal levels of methylation of Avy are brown and healthy, whereas those with low levels are yellow and unhealthy. If a pregnant Avy yellow mouse is fed a diet supplemented with extra folate, vitamin B-12, choline, and betaine for two weeks before mating and throughout gestation and lactation, her pups will have methylated Avy alleles and a brown coat color and will be healthy 32 31.
In a third example, rats fed a protein-restricted diet delivered pups that, as adults, had reduced hepatic expression of several transcription factors that regulate developmental and metabolic processes, such as the glucocorticoid receptor (GR) and peroxisomal proliferator-activated receptor (PPAR). This loss of gene expression was caused by a significant loss of DNA methyltransferase 1 activity34 and DNA methylation and could be prevented by supplementing the mother's diet with folate35. Together, these findings suggest that feeding a protein-restricted diet during pregnancy alters regulation of energy balance within the liver, increasing capacity for gluconeogenesis and peroxisomal fatty acid p-oxidation. These effects may explain the non-alcoholic fatty liver disease that another group reported in the offspring of protein-restricted dams36.
In addition to nutrition, other models of early life hardships have been shown to affect epigenetic regulation of gene expression. In one such example, researchers artificially created intrauterine growth restriction (IUGR) in rats by ligating the uterine artery on day 19 of pregnancy (rats deliver on day 22). The resulting pups were born small but eventually caught up with controls and were obese and developed diabetes symptoms as adults. The diabetic phenotype resulting from IUGR occurred because of reduced expression of the gene Pancreatic and duodenal homeobox 1 (Pdx1), a transcription factor required for development and function of insulin-producing pancreatic 5 cells. 24,37 This intergenerational silencing of Pdx1 was the result of a complex cluster of epigenetic modifications characterized by loss of upstream stimulatory factor-1 binding at the proximal promoter of Pdx1, recruitment of the histone deacetylase 1 and the corepressor Sin3A, and deacetylation of histones H3 and H437.
In another example of the effects of early-life exposure, mice that received low levels of maternal licking and grooming as pups developed into adults that showed increased levels of stress and were less likely to lick and groom their own offspring. An important gene in this effect was GR, which, among its many roles, regulates the stress response. Expression of GR was decreased in the hippocampus of mice that received less maternal care as a result of altered DNA methylation at specific CpGs in the GR promoter in the hippocampus38-40.
From all this work in rodents, we have learned that early life exposures, ranging from extreme diets and environmental stressors to inadequate maternal behavior during the weaning period, have long-term epigenetic consequences on the offspring. The next question is whether the same thing occurs in humans.
Dietary alterations, epigenetics, and intergenerational transmission in humans
In humans, few studies have connected maternal exposures during the periconception and in utero periods to metabolic disease or disease risk in the offspring. Similarly, we have only limited evidence linking maternal exposures to epigenetic changes in offspring. However, studies of the effects of the Dutch famine of 1944 provide evidence for inheritance of methylation patterns41. The Dutch famine is often considered equivalent to a rodent experimental study allowing investigation of the effects of in utero undernutrition in humans 42. Women (F1) whose mothers (F0) were exposed to famine during the periconceptual period later had offspring (F2) with birth weights lower than offspring of women not exposed to famine in utero. This effect of F0 famine on F2 birth weight persisted after control for potential confounding and intervening variables 43. Interestingly, the F1s who had been exposed to famine during periconception had less DNA methylation of the insulin-like growth factor II gene 60 years later than did their unexposed, same-sex siblings44.
Several examples exist supporting the hypothesis that developmental changes in DNA methylation are correlated with childhood obesity. DNA from umbilical cord tissue was isolated from two cohorts of patients who were either obese or non-obese at nine years of age. Methylation status of the promoters of five candidate genes that had been shown to correlate with obesity in animal models was analyzed. 45-47 In both cohorts, methylation of retinoid X receptor in the umbilical cord cells was associated with adiposity at age 9, and in one cohort, methylation of endothelial nitric oxide synthase showed this correlation. These observations suggest that epigenetics is involved in fetal programming of later obesity 45.
Transgenerational transmission of cardiometabolic traits through the germline
Most of the previously discussed generational animal and human studies did not examine the F3 generation. This is important because without F3 inheritance, a phenotype in the F1 and F2 can only be considered multigenerational, not transgenerational48. For example, when a pregnant mother is subjected to a low-protein diet, three generations are effectively exposed simultaneously to this dietary insult; the pregnant mother (F0), her fetal offspring (F1), and the primordial germ cells—the precursors of sperm and eggs that will become the F2—within the F1 fetus (Figure 1). Only if the F3 is affected can we conclude that the phenotype was transmitted epigenetically through the germline.
Fig. 1.
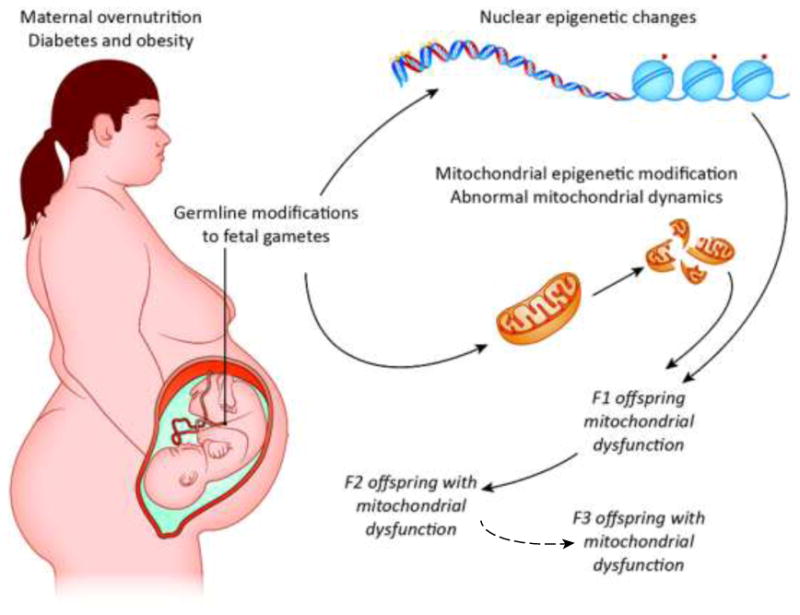
A schematic of possible maternal overnutrition influences on mitochondrial dysfunction in all tissues of the offspring. In a maternal obesogenic environment, offspring cells could inherit in a germline fashion malfunctioning mitochondria and/or deregulated epigenetic signatures. These alterations contribute to a phenotype of impaired mitochonodrial function for several generations. To be considered transgenerational transmission due to maternal obesity and overnutrition in this example, the inherited traits must be apparent in the F3 generation since the F1 embryo and F2 primordial germ cells are directly exposed to a given environmental factor in utero. In such cases, a change must occur in the germline to allow the effect to persist through three generations.
One well-established form of epigenetic transgenerational transmission is genomic imprinting, which involves DNA methylation or histone methylation without changing the genetic sequence. These epigenetic marks are established in sperm or oocytes of the parents and are maintained through mitotic cell divisions in the somatic cells of an organism49. The epigenetic marks are parent-of-origin-specific, meaning that if the imprinted allele is inherited from the father, it is thereby silenced, and only the allele from the mother is expressed. As of 2014, approximately 150 imprinted genes were known in the mouse and about half that in humans50.
Imprinting is a dynamic process. It must be possible to erase and re-establish imprints through each generation so that genes that are imprinted in an adult may still be expressed in that adult's offspring. In oocytes and sperm, the imprint is erased and then reestablished according to the sex of the individual. For instance during spermatogenesis, the paternal imprint is established, and in developing oocytes oogenesis, a maternal imprint is established51. This process of erasure and reprogramming is required so that the germ cell imprinting is relevant to the sex of the individual.
Whereas genomic imprinting has long been known to epigenetically regulate developmental gene expression52, the importance and mechanism of non-imprinted transgenerational inheritance of epigenetic marks that are not erased and reset is only now coming to light. Although many methylation marks are erased in mouse primordial germ cells via conversion of 5mC to 5-hydroxymethylcytosine (5hmC) and subsequent dilution during cell division 53, rare regulatory elements escape systematic DNA demethylation, thus providing a potential mechanism for true non-imprinted transgenerational epigenetic inheritance. In another recent landmark study, researchers showed that a small number (1%) of histone marks—located at key sequences with gene regulatory functions—remain in mouse sperm after differentiation54.
Several rodent studies have suggested that maternal high-fat or low-protein diets during gestation lead to transgenerational inheritance of metabolic syndrome. In one of the first studies, the investigators reported that feeding F0 rat dams a low-protein diet throughout pregnancy and lactation resulted in reduced insulin secretion in F1 animals and insulin resistance in their F2 offspring through the maternal line55. F3 generation males were also more insulin resistant than controls, although they were more insulin sensitive than F2 animals. In a second rat study, researchers found that a maternal (F0) high-fat diet affected offspring body size and glucose-insulin metabolism over three consecutive generations through both the maternal and paternal lines. Both F1 and F2 generation animals were more insulin resistant and had longer body length than control animals, even though they consumed control diets after weaning. In the F3 generation, animals were no longer insulin resistant, but females inherited increased body length through the paternal line 56.
A great deal of evidence supports the notion that early life exposure to maternal obesity alters the nuclear epigenome of the exposed offspring and may promote metabolic disease57. However, effects on oocyte mitochondria should be considered as well, especially because mitochondrial DNA (mtDNA) is inherited exclusively from the mother (Figure 1). Such an idea is supported by a recent study in which female mice (F0) were fed a high-fat/high-sugar diet beginning four to six weeks before conception and continuing throughout gestation and lactation. Female F1-F3 offspring were fed a regular diet beginning at weaning, but all exhibited impaired peripheral insulin signaling that was associated with mitochondrial dysfunction and altered expression of mitochondrial dynamics and electron transport chain proteins in skeletal muscle, demonstrating that metabolic dysfunction and aberrant mitochondria can be passed through the maternal germline to second and third-generation offspring.58 The transfer of aberrant oocyte mitochondria to subsequent generations may contribute to the increased risk for developing insulin resistance.
Two types of epigenetic modifications have been detected in mtDNA. First, studies in mouse oocytes demonstrated that methylation occurring at both CpG and non-CpG sites in mtDNA positively correlates with gene expression 59. Second, the recently discovered epigenetic mark 5hmC54 is widely distributed in all types of tissues with varying degrees of abundance, and the highest density of 5hmC was found in the mtDNA60. Although the above mentioned study did not explore the epigenetic mechanisms leading to inheritance of abnormal mitochondrial phenotypes, they did report altered mitochondrial metabolism, morphology, and expression of the mitochondrial dynamics protein OPA1 in F1, F2, and F3 germ cells, suggesting that both modification of nuclear-encoded mitochondrial genes and altered mtDNA play a role in this transgenerational effect.
Potential intervention strategies to prevent or reverse developmental programming
Transgenerational inheritance of epigenetic modifications that lead to long-term health effects is thought to have been selected for in evolution because it allows phenotypes to adapt to meet the demands of the later-life environment16,61. For example, teleologically, the transgenerational effects of the Dutch famine could have ensured that subsequent generations were more able to store energy if the famine situation had continued. But because it did not, the next generations instead were at risk of becoming obese. Now that we are beginning to define the epigenetic mechanisms of transgenerational inheritance, we may be able to develop strategies to prevent deleterious outcomes. However, doing so will require first answering many key questions 16: How plastic is the system, and what are the critical windows of development at which strategies should be targeted? How many generations are required to reverse an epigenetic mark? Can surrogate markers be used to predict disease?
Several studies in experimental animal models have indicated that some components of the metabolic syndrome that are induced as a consequence of developmental programming are potentially reversible by nutritional or targeted therapeutic interventions during windows of developmental plasticity. First, in a study in which pregnant female mice were undernourished, offspring fed a control diet postnatally demonstrated hypertension, obesity, hyperphagia, hyperinsulinemia, and hyperleptinemia. In the presence of a postnatal high-fat diet, the effects were markedly amplified20. However, when adult offspring were treated with growth hormone, their systolic blood pressure and fat mass were normalized, but hyperinsulinemia was enhanced as a result of the diabetogenic actions of growth hormone 62. Second, it was demonstrated that female offspring from mothers subjected to undernutrition during gestation experienced aberrations in adiposity, appetite, fasting plasma insulin, and leptin concentrations21. This group also determined that IGF-I infusion in these adult females reversed these effects. These studies suggest a role of the somatotropic axis in metabolic derangements, although the long-term efficacy of such treatment regimens is not known.
Mouse and human studies have shown that early nutrient restriction leads to glucose intolerance and a predisposition for T2DM in the offspring. Additionally, it has been shown that treatment of neonatal rats with the glucagon-like peptide (GLP)-1 analog Exendin-4 (EX-4) reversed the adverse consequences of nutrient deprivation-induced growth restriction and prevented the development of diabetes in adulthood 37,63,64. Treatment with EX-4 during the neonatal period also prevented the loss in insulin-producing β-cell mass that is observed in IUGR rats over time. Most interestingly, this treatment normalized the expression of Pdx1. More recently, it has been reported that EX-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat 65. Although adiposity was not examined in this study, GLPs are known to modify food intake, increase satiety, delay gastric emptying, and suppress glucagon release. Further studies are needed to explore these signaling pathways.
Cross-fostering of pups which alters post natal nutrition has been shown in several studies to improve the outcomes of offspring exposed to various conditions of in utero stress. It has been 66 demonstrated that pregnant rats treated with the synthetic glucocorticoid Dexamethasone from embryonic day 13 to term give birth to offspring with reduced birthweight. In this instance, they were cross-fostered with untreated rats and fed a standard diet, however, they still experienced delayed puberty, hyperleptinemia, and increased fat mass by six months of age. However, if instead the offspring were fostered to mothers fed a diet high in omega-3 fatty acids and consumed this diet post-weaning, they did not develop hyperleptinemia and increased fat mass. In an IUGR model induced by Bilateral uterine vessel ligation, IUGR pups when cross-fostered to dams receiving sham surgery do not experience the abnormalities in skeletal muscle development and mitochondria biogenesis that are seen in the IUGR pups that are kept with the original dams during lactation67. Cross-fostering with unaffected dams also significantly improves.
If rats are fed a low-protein diet during pregnancy, their offspring are born small for gestational age and develop pancreatic β-cell dysregulation and endocrine cell apoptosis, leading to IUGR, hyperglycemia, and cardiometabolic disease in the offspring. If the rat dams were supplemented with taurine, an amino acid important in the metabolism of fats, during either pregnancy or lactation, these defects were prevented68 The mechanism by which maternal taurine regulates fetal pancreatic function69 is not completely understood. Maternal supplementation with taurine leads to increased expression of VEGF and fetal liver kinase 1 and may increase fetal islet vascularization, thus preventing the programmed β-cell dysregulation 68. Recent findings suggest that a maternal low-protein diet causes long-lasting mitochondrial changes in β-cells, with abnormal appearing mitochondria, abnormal insulin secretion, and decreased expression of Cytochrome C oxidase, a key mitochondrial electron transporter complex protein. All these abnormalities were reversed when the mothers were fed taurine along with the low-protein diet during pregnancy. This led the authors to speculate that the dysfunctional mitochondria were due to taurine deficiency.70
Overnutrition studies in rodents have revealed that maternal exercise before and during pregnancy reduces the metabolic risk in offspring. Maternal exercise significantly reduces insulin and glucose concentrations in males pups from obese dams. In addition, maternal pre pregnancy exercise continued throughout gestation reversed the changes in mRNA expression seen in muscle myogenic differentiation 1 (MYOD1), tumor necrosis factor-alpha (TNF-α), and peroxisome proliferator activated receptor gamma coactivator 1 alpha (PGC1α)71. Other studies have similarly shown that maternal exercise before and during gestation ameliorated the detrimental effect of a maternal high-fat diet on the metabolic profile of offspring72,73. Interestingly, Maternal exercise appeared to decrease the metabolic risk induced by maternal obesity, improving insulin/glucose metabolism, with greater effects in male than female offspring71,73.
Concluding Remarks
Early life exposures, ranging from extreme diets and environmental stressors to inadequate maternal behavior, have long-term epigenetic consequences on the offspring and may in part be responsible for the increasing incidence of cardiometabolic disease and obesity in the world today. A large body of work done in rodents and humans supports this conclusion, and the current task will be to define the epigenetic mechanisms involved in transmitting developmentally programmed traits to subsequent generations (See Outstanding Questions Box). Several studies in experimental animal models have indicated that some components of the metabolic syndrome that are induced as a consequence of developmental programming are potentially reversible by nutritional or targeted therapeutic interventions during windows of developmental plasticity. The pursuit of studies examining prevention and reversibility of these events is critical if the wave of obesity-related health disorders is to be flattened.
Trends Box.
Studies performed in both rodents and humans suggest that adverse maternal conditions during the pre- and perinatal period can cause long term transgenerational epigenetic modifications in the offspring which mediate susceptibility to cardiometabolic disease.
An adverse maternal environment exposes three generations and is transmitted epigenetically through the germline only if the F3 offspring are affected.
Rare regulatory sequences that escape systematic DNA demethylation and others which are able to maintain histone marks in the primordial germ cells represent a potential mechanism for non-imprinted transgenerational epigenetic inheritance.
Inheritance of damaged oocyte mitochondria is an important potential transgenerational mechanism to consider because they are passed exclusively from the mother and contain mtDNA subject to epigenetic regulation.
Outstanding Questions.
As highlighted in our review, an adverse maternal environment can result in the transgenerational inheritance of cardiometabolic disease. Multiple pieces of evidence from rodent and human studies suggest transgenerational inheritance has epigenetic underpinnings. However, the exact molecular mechanisms of inheritance remain to be elucidated. In order to decrease the prevalence of obesity and cardiovascular disease caused by transgenerational exposures, the following questions should be explored.
What role does epigenetic regulation of mtDNA play in transgenerational inheritance?
What are the critical windows of exposure that can modify epigenetic marks within the primordial germ cells and alter F3 offspring phenotype?
How many generations are required to reverse an epigenetic mark?
Are there specific epigenetic markers within the germ cells that predict susceptibility to cardiometabolic disease in future generations?
How plastic is the system? Are there interventions that can reverse epigenetic modifications? Can we prevent deleterious outcomes caused by adverse maternal environments?
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WHO. Obesity and overweight. Organization WH, ed. 2016 [Google Scholar]
- 2.Loos RJ. Genetic determinants of common obesity and their value in prediction. Best Pract Res Clin Endocrinol Metab. 2012;26:211–26. doi: 10.1016/j.beem.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35:595–601. doi: 10.1007/BF00400248. [DOI] [PubMed] [Google Scholar]
- 4.Barker DJ. Sir Richard Doll Lecture. Developmental origins of chronic disease. Public Health. 2012;126:185–9. doi: 10.1016/j.puhe.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 5.Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–7. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DI, Roseboom TJ. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG. 2008;115:1243–9. doi: 10.1111/j.1471-0528.2008.01822.x. [DOI] [PubMed] [Google Scholar]
- 7.Veenendaal MV, Painter RC, de Rooij SR, et al. Transgenerational effects of prenatal exposure to the 1944-45 Dutch famine. BJOG. 2013;120:548–53. doi: 10.1111/1471-0528.12136. [DOI] [PubMed] [Google Scholar]
- 8.Rogers I, Group E-BS. The influence of birthweight and intrauterine environment on adiposity and fat distribution in later life. Int J Obes Relat Metab Disord. 2003;27:755–77. doi: 10.1038/sj.ijo.0802316. [DOI] [PubMed] [Google Scholar]
- 9.Gluckman PD, Hanson MA. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab. 2004;15:183–7. doi: 10.1016/j.tem.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Breton C. The hypothalamus-adipose axis is a key target of developmental programming by maternal nutritional manipulation. J Endocrinol. 2013;216:R19–31. doi: 10.1530/JOE-12-0157. [DOI] [PubMed] [Google Scholar]
- 11.Cheong JN, Cuffe JS, Jefferies AJ, Anevska K, Moritz KM, Wlodek ME. Sex-Specific Metabolic Outcomes in Offspring of Female Rats Born Small or Exposed to Stress During Pregnancy. Endocrinology. 2016;157:4104–20. doi: 10.1210/en.2016-1335. [DOI] [PubMed] [Google Scholar]
- 12.Feng B, Zhang T, Xu H. Human adipose dynamics and metabolic health. Ann N Y Acad Sci. 2013;1281:160–77. doi: 10.1111/nyas.12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.M JML. A mitochondrial component of of developmental programming. Cambridge UK: Cambridge University Press; 2006. [Google Scholar]
- 14.Thompson JA, Regnault TR. In utero origins of adult insulin resistance and vascular dysfunction. Semin Reprod Med. 2011;29:211–24. doi: 10.1055/s-0031-1275522. [DOI] [PubMed] [Google Scholar]
- 15.M L, R V. Placental mechanisms and developmental origins of health and disease. Cambridge UK: Cambridge University Press; 2006. [Google Scholar]
- 16.Tang WY, Ho SM. Epigenetic reprogramming and imprinting in origins of disease. Rev Endocr Metab Disord. 2007;8:173–82. doi: 10.1007/s11154-007-9042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodall SM, Breier BH, Johnston BM, Gluckman PD. A model of intrauterine growth retardation caused by chronic maternal undernutrition in the rat: effects on the somatotrophic axis and postnatal growth. J Endocrinol. 1996;150:231–42. doi: 10.1677/joe.0.1500231. [DOI] [PubMed] [Google Scholar]
- 19.McArdle HJ, Andersen HS, Jones H, Gambling L. Fetal programming: causes and consequences as revealed by studies of dietary manipulation in rats -- a review. Placenta. 2006;27(Suppl A):S56–60. doi: 10.1016/j.placenta.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 20.Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD. Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am J Physiol Endocrinol Metab. 2000;279:E83–7. doi: 10.1152/ajpendo.2000.279.1.E83. [DOI] [PubMed] [Google Scholar]
- 21.Vickers MH, Ikenasio BA, Breier BH. IGF-I treatment reduces hyperphagia, obesity, and hypertension in metabolic disorders induced by fetal programming. Endocrinology. 2001;142:3964–73. doi: 10.1210/endo.142.9.8390. [DOI] [PubMed] [Google Scholar]
- 22.Langley-Evans SC, Welham SJ, Jackson AA. Fetal exposure to a maternal low protein diet impairs nephrogenesis and promotes hypertension in the rat. Life Sci. 1999;64:965–74. doi: 10.1016/s0024-3205(99)00022-3. [DOI] [PubMed] [Google Scholar]
- 23.Rajakumar PA, He J, Simmons RA, Devaskar SU. Effect of uteroplacental insufficiency upon brain neuropeptide Y and corticotropin-releasing factor gene expression and concentrations. Pediatr Res. 1998;44:168–74. doi: 10.1203/00006450-199808000-00005. [DOI] [PubMed] [Google Scholar]
- 24.Simmons RA, Templeton LJ, Gertz SJ. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes. 2001;50:2279–86. doi: 10.2337/diabetes.50.10.2279. [DOI] [PubMed] [Google Scholar]
- 25.Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest. 1998;101:2174–81. doi: 10.1172/JCI1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis RM, Petry CJ, Ozanne SE, Hales CN. Effects of maternal iron restriction in the rat on blood pressure, glucose tolerance, and serum lipids in the 3-month-old offspring. Metabolism. 2001;50:562–7. doi: 10.1053/meta.2001.22516. [DOI] [PubMed] [Google Scholar]
- 27.Dahlgren J, Nilsson C, Jennische E, et al. Prenatal cytokine exposure results in obesity and gender-specific programming. Am J Physiol Endocrinol Metab. 2001;281:E326–34. doi: 10.1152/ajpendo.2001.281.2.E326. [DOI] [PubMed] [Google Scholar]
- 28.Coupe B, Amarger V, Grit I, Benani A, Parnet P. Nutritional programming affects hypothalamic organization and early response to leptin. Endocrinology. 2010;151:702–13. doi: 10.1210/en.2009-0893. [DOI] [PubMed] [Google Scholar]
- 29.Plagemann A, Harder T, Brunn M, et al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J Physiol. 2009;587:4963–76. doi: 10.1113/jphysiol.2009.176156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferland-McCollough D, Fernandez-Twinn DS, Cannell IG, et al. Programming of adipose tissue miR-483-3p and GDF-3 expression by maternal diet in type 2 diabetes. Cell Death Differ. 2012;19:1003–12. doi: 10.1038/cdd.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolff GL, Kodell RL, Moore SR, Cooney CA. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998;12:949–57. [PubMed] [Google Scholar]
- 32.Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miltenberger RJ, Mynatt RL, Bruce BD, Wilkison WO, Woychik RP, Michaud EJ. An agouti mutation lacking the basic domain induces yellow pigmentation but not obesity in transgenic mice. Proc Natl Acad Sci U S A. 1999;96:8579–84. doi: 10.1073/pnas.96.15.8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr. 2005;135:1382–6. doi: 10.1093/jn/135.6.1382. [DOI] [PubMed] [Google Scholar]
- 35.Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr. 2007;97:1064–73. doi: 10.1017/S000711450769196X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li M, Reynolds CM, Segovia SA, Gray C, Vickers MH. Developmental Programming of Nonalcoholic Fatty Liver Disease: The Effect of Early Life Nutrition on Susceptibility and Disease Severity in Later Life. Biomed Res Int. 2015;2015:437107. doi: 10.1155/2015/437107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest. 2008;118:2316–24. doi: 10.1172/JCI33655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meaney MJ, Brake W, Gratton A. Environmental regulation of the development of mesolimbic dopamine systems: a neurobiological mechanism for vulnerability to drug abuse? Psychoneuroendocrinology. 2002;27:127–38. doi: 10.1016/s0306-4530(01)00040-3. [DOI] [PubMed] [Google Scholar]
- 39.Meaney MJ, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci. 2005;7:103–23. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Champagne FA, Meaney MJ. Transgenerational effects of social environment on variations in maternal care and behavioral response to novelty. Behav Neurosci. 2007;121:1353–63. doi: 10.1037/0735-7044.121.6.1353. [DOI] [PubMed] [Google Scholar]
- 41.Silva AJ, White R. Inheritance of allelic blueprints for methylation patterns. Cell. 1988;54:145–52. doi: 10.1016/0092-8674(88)90546-6. [DOI] [PubMed] [Google Scholar]
- 42.Roseboom TJ, Painter RC, van Abeelen AF, Veenendaal MV, de Rooij SR. Hungry in the womb: what are the consequences? Lessons from the Dutch famine Maturitas. 2011;70:141–5. doi: 10.1016/j.maturitas.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 43.Lumey LH. Decreased birthweights in infants after maternal in utero exposure to the Dutch famine of 1944-1945. Paediatr Perinat Epidemiol. 1992;6:240–53. doi: 10.1111/j.1365-3016.1992.tb00764.x. [DOI] [PubMed] [Google Scholar]
- 44.Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–9. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Godfrey KM, Sheppard A, Gluckman PD, et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes. 2011;60:1528–34. doi: 10.2337/db10-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burdge GC, Lillycrop KA. Nutrition, epigenetics, and developmental plasticity: implications for understanding human disease. Annu Rev Nutr. 2010;30:315–39. doi: 10.1146/annurev.nutr.012809.104751. [DOI] [PubMed] [Google Scholar]
- 47.Lillycrop KA, Rodford J, Garratt ES, et al. Maternal protein restriction with or without folic acid supplementation during pregnancy alters the hepatic transcriptome in adult male rats. Br J Nutr. 2010;103:1711–9. doi: 10.1017/S0007114509993795. [DOI] [PubMed] [Google Scholar]
- 48.Skinner MK. What is an epigenetic transgenerational phenotype? F3 or F2 Reprod Toxicol. 2008;25:2–6. doi: 10.1016/j.reprotox.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wood AJ, Oakey RJ. Genomic imprinting in mammals: emerging themes and established theories. PLoS Genet. 2006;2:e147. doi: 10.1371/journal.pgen.0020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peters J. The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet. 2014;15:517–30. doi: 10.1038/nrg3766. [DOI] [PubMed] [Google Scholar]
- 51.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–93. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 52.Reik W, Collick A, Norris ML, Barton SC, Surani MA. Genomic imprinting determines methylation of parental alleles in transgenic mice. Nature. 1987;328:248–51. doi: 10.1038/328248a0. [DOI] [PubMed] [Google Scholar]
- 53.Hackett JA, Sengupta R, Zylicz JJ, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339:448–52. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Erkek S, Hisano M, Liang CY, et al. Molecular determinants of nucleosome retention at CpG-rich sequences in mouse spermatozoa. Nat Struct Mol Biol. 2013;20:868–75. doi: 10.1038/nsmb.2599. [DOI] [PubMed] [Google Scholar]
- 55.Benyshek DC, Johnston CS, Martin JF. Glucose metabolism is altered in the adequately-nourished grand-offspring (F3 generation) of rats malnourished during gestation and perinatal life. Diabetologia. 2006;49:1117–9. doi: 10.1007/s00125-006-0196-5. [DOI] [PubMed] [Google Scholar]
- 56.Dunn GA, Bale TL. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology. 2011;152:2228–36. doi: 10.1210/en.2010-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng J, Xiao X, Zhang Q, Yu M. DNA methylation: the pivotal interaction between early-life nutrition and glucose metabolism in later life. Br J Nutr. 2014;112:1850–7. doi: 10.1017/S0007114514002827. [DOI] [PubMed] [Google Scholar]
- 58.Saben JL, Asghar Z, Rhee JS, Drury A, Scheaffer S, Moley KH. Excess Maternal Fructose Consumption Increases Fetal Loss and Impairs Endometrial Decidualization in Mice. Endocrinology. 2016;157:956–68. doi: 10.1210/en.2015-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kobayashi H, Sakurai T, Imai M, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun Z, Terragni J, Borgaro JG, et al. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 2013;3:567–76. doi: 10.1016/j.celrep.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ho SM, Tang WY. Techniques used in studies of epigenome dysregulation due to aberrant DNA methylation: an emphasis on fetal-based adult diseases. Reprod Toxicol. 2007;23:267–82. doi: 10.1016/j.reprotox.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vickers MH, Ikenasio BA, Breier BH. Adult growth hormone treatment reduces hypertension and obesity induced by an adverse prenatal environment. J Endocrinol. 2002;175:615–23. doi: 10.1677/joe.0.1750615. [DOI] [PubMed] [Google Scholar]
- 63.Stoffers DA, Desai BM, DeLeon DD, Simmons RA. Neonatal exendin-4 prevents the development of diabetes in the intrauterine growth retarded rat. Diabetes. 2003;52:734–40. doi: 10.2337/diabetes.52.3.734. [DOI] [PubMed] [Google Scholar]
- 64.Raab EL, Vuguin PM, Stoffers DA, Simmons RA. Neonatal exendin-4 treatment reduces oxidative stress and prevents hepatic insulin resistance in intrauterine growth-retarded rats. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1785–94. doi: 10.1152/ajpregu.00519.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pinney SE, Jaeckle Santos LJ, Han Y, Stoffers DA, Simmons RA. Exendin-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat. Diabetologia. 2011;54:2606–14. doi: 10.1007/s00125-011-2250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wyrwoll CS, Mark PJ, Mori TA, Puddey IB, Waddell BJ. Prevention of programmed hyperleptinemia and hypertension by postnatal dietary omega-3 fatty acids. Endocrinology. 2006;147:599–606. doi: 10.1210/en.2005-0748. [DOI] [PubMed] [Google Scholar]
- 67.Laker RC, Wadley GD, McConell GK, Wlodek ME. Stage of perinatal development regulates skeletal muscle mitochondrial biogenesis and myogenic regulatory factor genes with little impact of growth restriction or cross-fostering. J Dev Orig Health Dis. 2012;3:39–51. doi: 10.1017/S204017441100064X. [DOI] [PubMed] [Google Scholar]
- 68.Boujendar S, Arany E, Hill D, Remacle C, Reusens B. Taurine supplementation of a low protein diet fed to rat dams normalizes the vascularization of the fetal endocrine pancreas. J Nutr. 2003;133:2820–5. doi: 10.1093/jn/133.9.2820. [DOI] [PubMed] [Google Scholar]
- 69.Boujendar S, Reusens B, Merezak S, et al. Taurine supplementation to a low protein diet during foetal and early postnatal life restores a normal proliferation and apoptosis of rat pancreatic islets. Diabetologia. 2002;45:856–66. doi: 10.1007/s00125-002-0833-6. [DOI] [PubMed] [Google Scholar]
- 70.Lee YY, Lee HJ, Lee SS, et al. Taurine supplementation restored the changes in pancreatic islet mitochondria in the fetal protein-malnourished rat. Br J Nutr. 2011;106:1198–206. doi: 10.1017/S0007114511001632. [DOI] [PubMed] [Google Scholar]
- 71.Raipuria M, Bahari H, Morris MJ. Effects of maternal diet and exercise during pregnancy on glucose metabolism in skeletal muscle and fat of weanling rats. PLoS One. 2015;10:e0120980. doi: 10.1371/journal.pone.0120980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vega CC, Reyes-Castro LA, Bautista CJ, Larrea F, Nathanielsz PW, Zambrano E. Exercise in obese female rats has beneficial effects on maternal and male and female offspring metabolism. Int J Obes (Lond) 2015;39:712–9. doi: 10.1038/ijo.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stanford KI, Lee MY, Getchell KM, So K, Hirshman MF, Goodyear LJ. Exercise before and during pregnancy prevents the deleterious effects of maternal high-fat feeding on metabolic health of male offspring. Diabetes. 2015;64:427–33. doi: 10.2337/db13-1848. [DOI] [PMC free article] [PubMed] [Google Scholar]