

Abstract
Multifunctional vinyl sulfonates and vinyl sulfonamides were conveniently synthesized and assessed in thiol-Michael crosslinking polymerizations. The monomer reactivities, mechanical behavior and hydrolytic properties were analyzed and compared with those of analogous thiol-acrylate polymerizations. Materials with a broad range of mechanical properties and diverse hydrolytic stabilities were obtained.
The growing demand for the convenient synthesis of diverse types of photopolymerizable monomers with inherent novelty of the molecule architectures has provided numerous opportunities to explore the thiol-Michael addition as a click reaction, specifically for photopolymerizations and other network-forming reactions. Progress achieved by utilizing anion-mediated thiol-vinyl additions was summarized in recent work and reviews devoted to thiol-Michael reactions and applications.1–4 The highly exploitable features of this reaction such as orthogonality with radical reactions, impressive reactivity in various media, and general simplicity of synthesis and implementation have made it a popular tool. Complementing the thiol reactants, Michael acceptors have included electron-deficient vinyls such as acrylates, methacrylates, acrylamides, vinyl sulfones, and malemides.5–8 Amongst all of these, the vinyl sulfone group as a Michael acceptor has attracted a great deal of attention due to its stability in aqueous media, biocompatibility and excellent reactivity towards thiols under relatively mild conditions.9 However, except for divinyl sulfone, there are few if any other non-crystalline, multivinyl sulfone monomers commercially available or readily synthesized for use in crosslinking polymerizations. Recently, vinyl sulfonates10,11 and vinyl sulfonamide12,13 have also been employed as Michael acceptors, most frequently as monofunctional reactants, in the decoration of aminated polymers14, in the synthesis of dendrimers15, and in the construction of biofunctional molecules16. The incorporation of vinyl sulfones results in the formation of a stable thioether sulfone17 bond as compared to its acrylate18 counterpart which results in a labile thioether ester bond. Additionally, the formed sulfonamide bonds are observed to be extremely stable, which makes sulfonamide conjugates useful for applications where the molecular stability is important.19
However, there are no reports on neat thiol-Michael network materials that incorporate vinyl sulfonates or vinyl sulfonamides as monomers, least to mention the properties attainable from such thiol-vinyl combinations. Meanwhile, due to the poor electronegativity of methacrylates, multifunctional acrylates are the most common Michael acceptors implemented in thiol-Michael crosslinking systems to date. However, they are limited in the range of material properties achieved. Further, the presence of esters in the resulting polymer, particularly the hydrolytically unstable thioether ester resulting from the thiol-acrylate reaction, is also found to be a limiting factor with respect to the mechanical performance of these materials. Therefore, a need still exists to explore the efficiency and versatility of Michael acceptors which can be introduced by structurally modifying enes to enable suitable Michael-acceptors with tunable reactivities.
In this communication, we introduce the vinyl sulfonate and vinyl sulfonamide groups in designing multifunctional Michael acceptors and exploit their features by systematic study of vinyl reactivity, followed by implementation in thiol-Michael crosslinked polymeric materials. A broad diversity in the mechanical and hydrolytic properties of the resultant network polymers has thus been achieved. Firstly, we synthesized vinyl sulfonate and vinyl sulfonamide based monomers from inexpensive multifunctional alcohols and amines, respectively, following a synthetic methodology based on the substitution of the corresponding alcohols (Table 1) and amines (Table 2) with 2-chloroethanesulfonyl chloride in the presence of Et3N.10 To examine the reaction kinetics performance of the new vinyl compounds, the relative reactivities of the vinyl sulfonate and vinyl sulfonamide functional groups were compared with other model compounds. 1-Hexyl acrylate (HA), 1-hexyl vinyl sulfonate (HVS), 1-hexyl acrylamide (HAA), and 1-hexyl vinyl sulfonamide (HVSA), and ethyl vinyl sulfone (EVS) were purchased (or synthesized) and reacted with butyl 3-mercaptopropionate (BMP) in model compound studies, wherein the vinyl (thiol) conversion during the irradiation was monitored using real-time Fourier transform infrared (FTIR) spectroscopy. The photo thiol-Michael model reactions were performed in 2M solution of ethylene glycol diethyl ether as the solvent of choice with NPPOC-TMG photobase (20 mg/mL) at a thiol-to-vinyl stoichiometric ratio of 1:1. The vinyl conversion kinetic profiles are shown in Fig. 1.
Table 1.
Synthesis of vinyl sulfonate 4a-g from multifunctional alcohols 1a-g with 2-chloroethanesulfonyl chloride.a,b
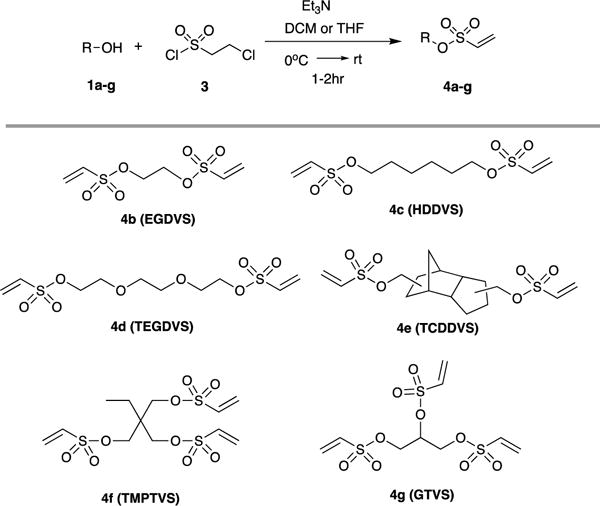
|
All reactions were carried out with 1(1mmol), 3(1.1mmol) and Et3N(3mmol).
Dichloromethane was used as the solvent for alcohols 1a-f and tetrahydrofuran was used as the solvent for alcohol 1g.
Table 2.
Synthesis of vinyl sulfonamide 7a-e from multifunctional amines 5a-e with 2-chloroethanesulfonyl chloride.a,b
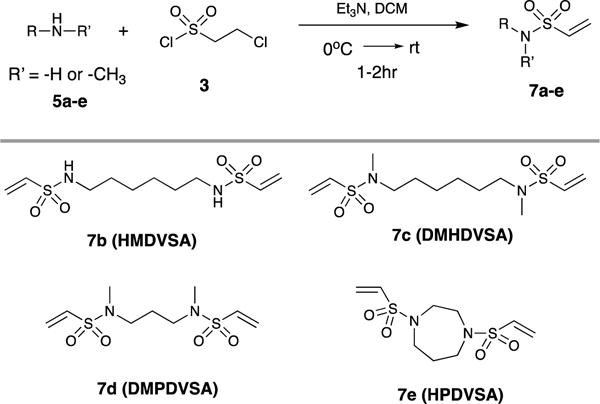
|
All reactions were carried out with 5(1mmol), 3(1.1mmol) and Et3N(2.1mmol).
Dichloromethane was used as the solvent in all cases.
Fig. 1.
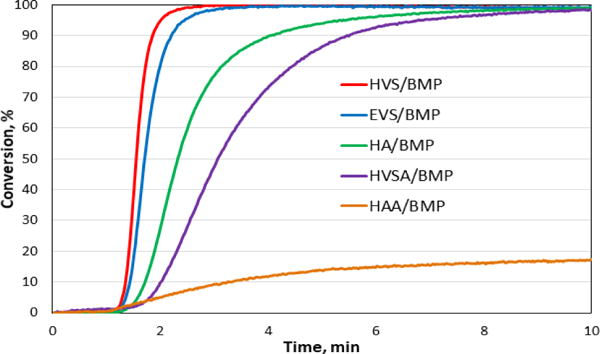
Vinyl conversions for the model thiol–Michael addition reactions. Reactions were initiated with UV light in the range of 320-390 nm, 10 mW/cm2 turned on after 1min, and continued for 5 min.
The initial kinetic studies demonstrated that the vinyl sulfonate is even more reactive than the vinyl sulfone group where both of these groups were found to be much more reactive than an acrylate (Fig. 1). The relative rates of the Michael addition to α,β– unsaturated sulfonate, sulfone, acrylate ester, and amides could be related by considering the electrophilicity of the enoyl β-carbon, which accounts for the energy of the transition state which then leads to the formation of an enolate intermediate upon addition of the nucleophile to the Michael acceptor. Recently, Reddick et al. demonstrated the relative reactivity of Michael reactions of 2′-(phenethyl)thiol with vinyl sulfones, vinyl sulfonate esters, and vinyl sulfonamides relevant to vinyl sulfonyl cysteine protease inhibitors. Indeed, the mechanistic behavior could also be interpreted by electronic effects arising from the linked oxygen or nitrogen atoms of the sulfonate ester and sulfonamide groups. The presence of these group introduces an inductive effect, wherein the poor sp3/d orbital can overlap between the oxygen or nitrogen and the sulfur, precluding an electron-donating resonance effect. Therefore, the sulfonate ester group is expected to introduce a strong inductive σ–electron withdrawing effect which would then be expected to stabilize α–carbanion formation.20 Such an interpretation accurately explains the reasoning for the higher reactivity of vinyl sulfonate as a Michael acceptor as compared to the conventional acrylates and vinyl sulfones. It has been well reported in the literature that primary vinyl sulfonamides are particularly unreactive in aza-Michael reactions. This behavior has been ascribed to the deprotonation of the acidic sulfonamide protons to a certain extent which leads to a reduction in the electrophilicity.
The lower reactivity of the vinyl sulfonamide as compared to vinyl sulfonate could be attributed to the lower electronegativity of the nitrogen as compared to oxygen.21 At the same time, the vinyl sulfonamide reacts more rapidly than acrylamide, for similar reasons as indicated for the reactivity of the vinyl sulfonates relative to acrylates.
Initiation of polymerizations in bulk, solventless conditions was also conducted for various model monomeric systems. Specifically, mixtures of thiols with either vinyl sulfonates or vinyl sulfones were cured by using thermal initiation (80°C, TEMPO, 1-2 wt. %)22 whereas mixtures incorporating other vinyls were cured photochemically with NPPOC-TMG (2 wt. %) as a photobase. Thermal polymerizations were implemented because the inherent basicity of the photobase did not allow time-stable compositions to be formed from the two most reactive vinyls. Before any mechanical testing, all compositions were thermally postcured (100°C for 1 hr) to assure complete functional group conversions. Exemplary real-time kinetic profiles for photocuring of DMHDVSA/PETMP and DMHDVSA/SiTSH are in the †ESI in Fig. S1. These, as well as other photo-thiol-Michael systems exhibited nearly identical characteristic kinetic behavior for both reactive groups, suggesting a stoichiometric reaction. Because the synthesized vinyl sulfonates were found to be quite reactive (and unstable) with thiols in the presence of NPPOC-TMG, i.e., with quantities less than 0.5 wt.% catalyst gelation was observed only 30 sec after photobase addition, their reactivity was also probed in radical thiol-ene reactions (†ESI Fig. S2b). Unexpectedly, radical thiol-vinyl sulfonate reactions were also found to proceed exceptionally well. Although, these systems are less reactive than their radical thiol-acrylate counterparts, they still reach high conversions with less than 10 % off-stoichiometric ene consumption as measured at the end of irradiation. For example, HDDA/PETMP when cured radically results in 100 % conversion of acrylate and 66 % thiol conversion whereas HDDVS/PETMP cured analogously reaches 95 % vinyl conversion and 87 % thiol conversion (†ESI Fig.S2a and b). Contrary to observations of the thiol-vinyl sulfonates, the thiol-vinyl sulfones and thiol-vinyl sulfonamides are sluggish reactions when initiated radically. More off-stoichiometric ene consumption and low functional group conversions were measured in these systems (†ESI Fig. 2c and 2d). Therefore, even though the thiol-vinyl sulfonate reaction is too reactive to initiate with the photobase used here, it is readily photoinitiated (radically), resulting in materials with equivalent or improved mechanical properties as compared to those resulting from a stoichiometrically reacting thiol-Michael system (compare DMA †ESI results).
Network polymers were prepared from neat stoichiometric mixtures (per functional group concentration) of a commercial thiol PETMP and an ester free thiol SiTSH and reacted with the synthesized multifunctional vinyl sulfonate and vinyl sulfonamide derivatives. These results were then compared with analogous acrylate derivatives. Using divinyl Michael acceptors, representative DMA plots were obtained for a range of new materials showing the differences in Tg values between structurally analogues thiol-vinyl systems, i.e. thiol-acrylate, thiol-vinyl sulfonate, and thiol-vinyl sulfonamide (secondary and tertiary amides). As can be seen from Table 3 and Fig. 2, thiol-vinyl sulfonate (HDDVS/PETMP) and thiol-vinyl sulfonamide based on the primary amine (HMDVSA/PETMP) both exhibited similar and significantly higher Tg’s than the HDDA/PETMP. As reported previously, the incorporation of sulfone groups8 with their dipole-dipole interactions improve the mechanical performance of these materials. Additionally, with the incorporation of a rigid bicyclic core as in TCDDVS/PETMP or by increasing the overall monomer functionality as in TMPTVS/PETMP or GTVS/PETMP, the resultant thiol-vinyl sulfonate network polymers were expected to exhibit further thermomechanical enhancement as compared with analogously crosslinked thiol-acrylates. Indeed, as in the sulfone-based networks, the sulfonate and sulfonamide networks showed similar property enhancements when compared to analogous acrylates. Furthermore, in the case of the thiol-sulfonamide network polymer incorporating the secondary amide (HMDVSA/PETMP), a higher Tg was measured than in the network incorporating a tertiary amide (DMHDVSA/PETMP).
Table 3.
Summary of DMA results for neat thiol-Michael networks. All samples were prepared from mixtures containing equivalent amount of thiol and vinyl functional groups. Values in parentheses represent standard deviations of three replicates.
| Acrylates with PETMP (Tg/°C) | Vinyl sulfonates with PETMP (Tg/°C) | Vinyl sulphonamide (Tg/°C) |
|---|---|---|
| EGDA (−5.1(0.8)) | EGDVS (35(2)) | HMDVSA/PETMP (46(3)) |
| HDDA (−9(0)) | HDDVS (43(3)) | HMDVSA/SiTSH (58(6)) |
| TEGDA(−15.7(0.8)) | TEGDVS (11(0.8)) | DMHDVSA/PETMP (34(1)) |
| TCDDA (22(1)) | TCDDVS (45(0.7)) | DMHDVSA/SiTSH (29(3)) |
| TMPTA (22.4(0)) | TMPTVS (50(2)) | DMPDVSA/SiTSH (57(0.6)) |
| GTA (23(2)) | GTVS (58(3)) | HPDVSA/SiTSH (95(0.3)) |
Fig. 2.
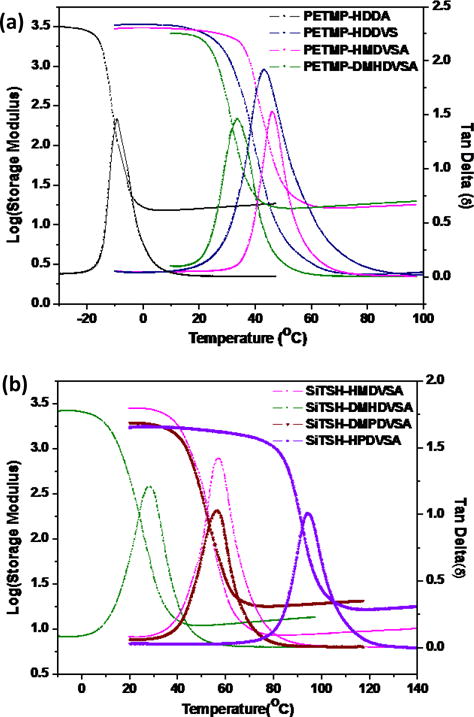
Storage modulus and tan delta data with neat PETMP as the thiol monomer (a) acrylate (PETMP-HDDA), vinyl sulfonate (PETMP/HDDVS), vinyl sulfonamide (2° amine) (PETMP-HMDVSA) and vinyl sulphonamide (3° amine)(PETMP-DMHDVSA). (b) SiTSH as ester-free thiol monomer in thiol-vinylsulfonamide network polymers. The monomer ratios are: thiol:vinyl = 1:1 based on the functional group content.
This difference could be attributed to the presence of an amide proton in the secondary sulfonamide group that participates in hydrogen bonding along with the oxygen atom of the sulfonamide participating in C-H…O interactions.23 As reported in the literature, the elimination, or reduction in the amount, of esters in the network results in higher Tg materials. It is therefore expected that incorporation of ester-free thiol monomers in mixtures with vinyl sulfonamides would also yield hydrolytically stable, tough and glassy polymers.22 Accordingly, we introduced a shorter chain length tertiary vinyl sulfonamide (DMPDVSA/SiTSH) and structurally rigid cyclic vinyl sulfonamide (HPDVSA/SiTSH) into the mixtures with the ester free thiol SiTSH. The resulting networks reach an impressive glassy Tg of over 90°C. In contrast, due to very poor reactivity of acrylamides towards the photo thiol-Michael addition, design of a network polymer based on such systems is highly ineffective.
Furthermore, a series of thiol-Michael polymers based on structural vinyl analogues was investigated for their hydrolytic stability and interactions (Table 4). Thiol-Michael materials of similar crosslinking densities exhibit drastically different susceptibility to basic and acidic environments. Among the materials tested, several polymers hydrolyzed in the order of minutes whereas others were found to be practically non-degradable. It is not surprising that the sulfonate esters degrade in both acidic and basic conditions; however, even as compared to acrylate esters, their hydrolysis is evidently accelerated. On the other hand, tertiary vinyl sulfonamides do not degrade under these conditions upon exposure to either acids or bases. Acidic secondary amides are sensitive to bases and hydrolyze rapidly in dilute basic solutions but are stable in acidic conditions. This broad range of hydrolytic stability and characteristics will likely allow for a design of materials with tunable hydrolysis/degradation profiles for a variety of applications, from soft to hard hydrogel scaffolds, micro-, and nanoparticles and macroscopic bulk network polymers.
Table 4.
Hydrolytic properties of a series of Thiol-Michael polymers.
| Thiol-Michael Network | Sample mass change after 7 days in 5wt% HCl | Sample mass change after 7 days in 5wt% NaOH |
|---|---|---|
| HDDA/PETMP | −0.7 (0.3) | −15.1 (1) |
| HDDVS/PETMP | 24 (8) | Hydrolyzed in 1 day |
| HMDVSA/PETMP | 1.8 (1.1) | Hydrolyzed in 10 min |
| DMHDVSA/PETMP | 1.0 (0.4) | 0 (0) |
In summary, we demonstrated the utilization of two rarely considered families of multifunctional vinyl monomers (sulfonates and sulfonamides) for the crosslinking photo-, and thermal thiol-Michael addition polymerization. This extended library of vinyls now offers multiple way of initiation as well as a broad range of thermomechanical properties that are conveniently generated from anionic (and radical) thiol-vinyl step-growth reactions. The expanded range of available vinyl monomers is expected to further popularize the thiol-Michael addition-based polymerization and functionalization in various material science disciplines. One extraordinary feature of these materials is an impressive tunability of the hydrolytic properties of thiol-Michael networks. Ranging from photo-, to thermally degradable and non-degradable macromolecular architectures, useful functional materials can be facilely fabricated fulfilling the existing gaps in applied polymer and material science.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from National Institutes of Health for this research (NIH: 1U01DE023777-01)
Footnotes
Electronic Supplementary Information (ESI) available: See DOI:10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Allen CFH, Humphlett WJ, Fournier JO. Can J Chem. 1964;42:2616–2620. [Google Scholar]
- 2.Nair DP, Podgorski M, Chatani S, Gong T, Xi WX, Fenoli CR, Bowman CN. Chem Mater. 2014;26:724–744. [Google Scholar]
- 3.Xi WX, Scott TF, Kloxin CJ, Bowman CN. Adv Funct Mater. 2014;24:2572–2590. [Google Scholar]
- 4.Mather BD, Viswanathan K, Miller KM, Long TE. Prog Polym Sci. 2006;31:487–531. [Google Scholar]
- 5.Li GZ, Randev RK, Soeriyadi AH, Rees G, Boyer C, Tong Z, Davis TP, Becer CR, Haddleton DM. Polym Chem. 2010;1:1196–1204. [Google Scholar]
- 6.Chan JW, Hoyle CE, Lowe AB, Bowman M. Macromolecules. 2010;43:6381–6388. [Google Scholar]
- 7.Chatani S, Nair DP, Bowman CN. Polym Chem. 2013;4:1048–1055. [Google Scholar]
- 8.Podgorski M, Chatani S, Bowman CN. Macromol Rapid Commun. 2014;35:1497–1502. doi: 10.1002/marc.201400260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stenzel MH. ACS Macro Lett. 2013;2:14–18. doi: 10.1021/mz3005814. [DOI] [PubMed] [Google Scholar]
- 10.Cruz CM, Ortega-Munoz M, Lopez-Jaramillo FJ, Hernandez-Mateo F, Blanco V, Santoyo-Gonzalez F. Adv Synth Catal. 2016;358:3394–3413. [Google Scholar]
- 11.Caddick S, Bush HD. Org Lett. 2003;5:2489–2492. doi: 10.1021/ol0347388. [DOI] [PubMed] [Google Scholar]
- 12.Dadova J, Vrabel M, Adamik M, Brazdova M, Pohl R, Fojta M, Hocek M. Chem Eur J. 2015;21:16091–16102. doi: 10.1002/chem.201502209. [DOI] [PubMed] [Google Scholar]
- 13.Tong K, Tu JC, Qi XY, Wang M, Wang YJ, Fu HZ, Pittman CU, Zhou AH. Tetrahedron. 2013;69:2369–2375. [Google Scholar]
- 14.Haamann D, Keul H, Klee D, Moller M. Macromolecules. 2010;43:6295–6301. [Google Scholar]
- 15.Chatani S, Podgorski M, Wang C, Bowman CN. Macromolecules. 2014;47:4894–4900. [Google Scholar]
- 16.Brzozowski Z, Saczewski F, Neamati N. Bioorg Med Chem Lett. 2006;16:5298–5302. doi: 10.1016/j.bmcl.2006.07.089. [DOI] [PubMed] [Google Scholar]
- 17.Morales-Sanfrutos J, Lopez-Jaramillo J, Ortega-Munoz M, Megia-Fernandez A, Perez-Balderas F, Hernandez-Mateo F, Santoyo-Gonzalez F. Org Biomol Chem. 2010;8:667–675. doi: 10.1039/b920576d. [DOI] [PubMed] [Google Scholar]
- 18.Schoenmakers RG, van de Wetering P, Elbert DL, Hubbell JA. J Controlled Release. 2004;95:291–300. doi: 10.1016/j.jconrel.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Koniev O, Wagner A. Chem Soc Rev. 2015;44:5495–5551. doi: 10.1039/c5cs00048c. [DOI] [PubMed] [Google Scholar]
- 20.Reddick JJ, Cheng JM, Roush WR. Org Lett. 2003;5:1967–1970. doi: 10.1021/ol034555l. [DOI] [PubMed] [Google Scholar]
- 21.Ceppi E, Eckhardt W, Grob CA. Tetrahedron Lett. 1973:3627–3630. [Google Scholar]
- 22.Podogorski M, Becka E, Chatani S, Claudino M, Bowman CN. Polym Chem. 2015;6:2234–2240. doi: 10.1039/C4PY01552E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singhamahapatra A, Sahoo L, Varghese B, Loganathan D. RSC Adv. 2014;4:18038–18043. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.