

Abstract
Congenital sodium diarrhea is a rare and life-threatening disorder characterized by a severe, secretory diarrhea containing high concentrations of sodium, leading to hyponatremia and metabolic acidosis. It may occur in isolation or in association with systemic features such as facial dysmorphism, choanal atresia, imperforate anus, and corneal erosions. Mutations in the serine protease inhibitor, Kunitz-type 2 (SPINT2) gene have been associated with congenital sodium diarrhea and additional syndromic features. We present a child with congenital sodium diarrhea, cleft lip and palate, corneal erosions, optic nerve coloboma, and intermittent exotropia who was found to have biallelic mutations in SPINT2. One mutation, c.488A>G, predicting p.(Tyr163Cys), has been previously associated with a syndromic form of congenital sodium diarrhea. The other mutation, c.166_167dupTA, predicting p.(Asn57Thrfs*24) has not previously been reported and is likely a novel pathogenic variant for this disorder. We found only one other report of an optic nerve coloboma associated with SPINT2 mutations and this occurred in a patient with congenital tufting enteropathy. Our patient confirms an association of ocular coloboma with presumed loss of SPINT2 function.
Keywords: SPINT2, congenital sodium diarrhea, congenital tufting enteropathy, ocular findings with SPINT2, eye findings with SPINT2, keratitis, corneal erosions, coloboma
INTRODUCTION
Congenital sodium diarrhea (CSD) is a rare, life-threatening diarrhea that affects children in infancy. It is characterized by a severe secretory diarrhea containing high concentrations of sodium, hyponatremia, and metabolic acidosis in the absence of an infectious, autoimmune or endocrine cause [Heinz-Erian et al., 2009]. It can occur in isolation (“classic CSD”), or as part of a syndromic presentation with choanal atresia, anal atresia, wide-spaced eyes and other dysmorphic facial features, cleft palate, polydactyly and thumb anomalies, genitourinary defects including ureteral or renal duplications and rectovaginal fistula, and corneal erosions [Muller et al., 2000; Heinz-Erian et al., 2009]. Patients are frequently dependent on parenteral nutrition and the prognosis is often poor.
The SPINT2 gene encodes a transmembrane protein with two extracellular Kunitz domains that was originally described as a hepatocyte growth factor activator inhibitor. The protein functions as an inhibitor of various gastrointestinal serine proteases [Faller et al., 2014]. Biallelic, loss of function mutations in SPINT2 cause a syndromic form of CSD with choanal atresia, wide-spaced eyes, corneal erosions/superficial punctate keratitis, and bone malformations with hexadactyly and thumb anomalies [Heinz-Erian et al., 2009; Faller et al., 2014]. In mice, Spint2 plays a key role in development, and knockout mice suffer from early lethality at or after embryonic day (E) 10.5 [Szabo et al., 2016], with defects in their ectoderm, neural tube, and placenta [Szabo et al., 2009]. In humans, the gene is hypothesized to play a role in organogenesis and intestinal ionic homeostasis [Szabo et al., 2009].
Few reports have focused specifically on the ocular features of patients with SPINT2 sequence variants [Heinz-Erian et al., 2009; Salomon et al., 2014]. We present a child with biallelic SPINT2 mutations who had CSD, bilateral cleft lip and palate, corneal erosions, optic nerve coloboma, and intermittent exotropia.
CASE REPORT
A two-month-old male with failure to thrive, hyponatremia, hypovolemia, and cleft lip and palate was referred to the ophthalmology service for evaluation. He was born to healthy, unrelated parents and had three unaffected siblings. A cleft lip was noted on prenatal ultrasound and a repeat ultrasound confirmed bilateral cleft lip and palate. Fetal echocardiogram was normal. The remainder of the pregnancy was uncomplicated and he was born at 39 weeks via spontaneous vaginal delivery with a birthweight of 2,953 grams (10–25th centile). He spent two days in the hospital before being discharged home. At seven weeks of age, he was hospitalized for dehydration and electrolyte abnormalities after seven days of vomiting and diarrhea. An orogastric tube was placed to supplement his oral feeds. However, two weeks later, he was readmitted for emesis and discomfort with feeds. Given his recurrent episodes of failure to thrive and his cleft lip and palate, the decision was made to supplement his parenteral nutrition with enteral nutrition through a gastrostomy tube.
On examination at two months of life, weight was 3.88 kilograms (<2nd centile), length was 57 centimeters (25th centile) and occipitofrontal circumference was 39 centimeters (50th centile). Craniofacial features included a prominent and square forehead, a prominent occiput, bilateral preauricular pits, and bilateral cleft lip and palate with distortion of the nares (Figure 1). He was light averse in both eyes, but had normal intraocular pressures by palpation. The anterior segment appeared normal, but he had a large chorioretinal coloboma involving the optic nerve and macula in his left eye (Figure 2). On review in ophthalmology clinic six months later, his mother reported he was sensitive to light. At this time, he was noted to fix and follow with each eye. He had normal pupillary responses and intraocular pressures of 9 mmHg in each eye by iCare. He demonstrated an intermittent left exotropia with grossly full extraocular movements. The anterior segment was notable for irregularity of the corneal surface with significant punctate staining of both corneas when examined with fluorescein eye drops and cobalt blue light. Although there were diffuse punctate epithelial erosions in both eyes, there were no frank epithelial defects. The right fundus was normal and the left fundus demonstrated the same extensive optic disc coloboma, unchanged from prior examination. A cycloplegic refraction was also performed and revealed +2.00 sphere/+1.00 cylinder × 090 for the right eye and +0.50 diopter sphere for the left.
Fig. 1.
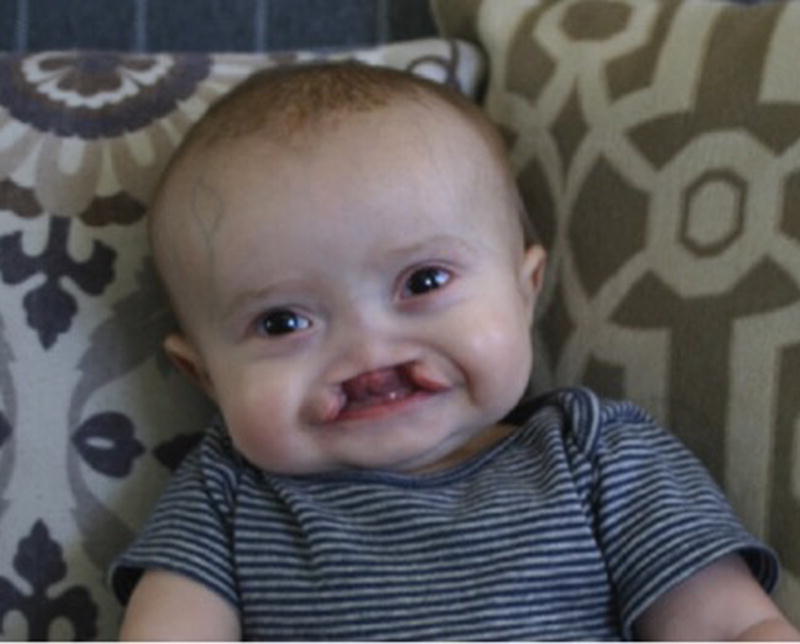
External photograph of patient demonstrating a prominent square forehead and bilateral cleft lip and palate with distortion of the nares.
Fig. 2.
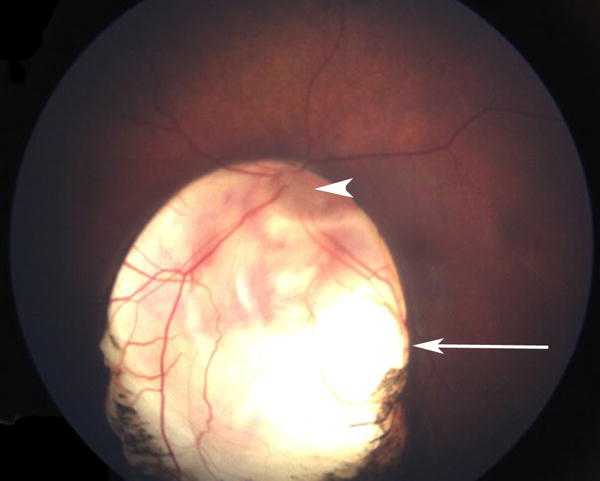
Fundus photograph of the left eye demonstrating a large chorioretinal coloboma involving the optic nerve (arrowhead) and macula (arrow).
Genetic analysis was conducted using clinical whole-genome sequencing that included clinical interpretation for all single nucleotide variants, small deletions, small insertions and copy number variants greater than 10kb [Illumina, CA; Raczy et al., 2013; Saunders et al., 2012; Roller et al., 2016; Richards et al., 2015]. DNA was extracted from peripheral blood using the Qiagen DSP DNA kit with a QIAsymphony robot (Qiagen, Germany). The sequencing library was prepared using the Illumina TruSeq PCR-free kit and sequenced on a HiSeqX instrument with paired-end 150bp reads. Samples were sequenced to an average of ≥30 fold coverage. Reads were aligned to human reference genome assembly GRCh37 using the ISAAC aligner [Raczy et al., 2013]. Single nucleotide and copy number variant calling were performed with Strelka and Canvas, respectively [Saunders et al., 2012; Kim et al., 2017; Roller et al., 2016]. Over 99% of each genome was covered at 10-fold coverage or more and 96.5% of the genome was callable. Variants were filtered using a set of in-house developed software and bioinformatics tools that utilize inheritance patterns, allele frequency, and molecular consequence. Variants were manually curated and classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines [Richards et al., 2015].
Two heterozygous sequence variants in the SPINT2 gene were identified - c.488A>G, predicting p.(Tyr163Cys), and c.166_167dupTA, predicting p.(Asn57Thrfs*24) (NM_021102.3). The p.(Tyr163Cys) variant was maternally inherited and has previously been described as pathogenic [Heinz-Erian et al., 2009; Sivagnanam et al., 2010; Slae et al., 2013; Salomon et al., 2014; D’Apolito et al., 2016]. The p.(Asn57Thrfs*24) variant was paternally inherited and predicts a frameshift alteration that results in a premature stop codon. This sequence variant has not previously been reported and was classified as pathogenic based on the American College of Medical Genetics and Genomics variant interpretation guidelines (predicted null, found in trans with a known pathogenic variant) [Richards et al., 2015] and the consistent features of this patient with the phenotype reported in other patients with biallelic SPINT2 sequence variants. There were no other deleterious sequence variants identified in any gene known to cause any other recessive Mendelian disorder or coloboma.
Pathologic analysis of the duodenum demonstrated unremarkable small intestinal mucosa with an intact brush border. Electron microscopy verified an intact unremarkable surface epithelium, without evidence of tufting or microvillus inclusions, although it can be difficult to detect the characteristic “tufts” associated with tufting enteropathy on routine biopsies of the small bowel. The cell junctions between the surface epithelial cells were intact and of normal configuration and the epithelial cell organelles were of normal morphology. These findings paired with the fact that there were no variants in the EpCam gene on genetic testing makes the diagnosis of syndromic CSD most likely.
DISCUSSION
We report a male with CSD, bilateral cleft lip and palate, and striking eye findings, including an extensive coloboma involving the left optic disc and macula. There are few papers that discuss the eye findings in patients with SPINT2 mutations. From the initial description of SPINT2 mutations in ten families with syndromic CSD, ocular features included recurrent, painful, punctate epithelial corneal erosions, peripheral neovascularization of the cornea, hyperopia, and astigmatism [Heinz-Erian et al., 2009]. Coloboma of the optic nerve has not previously been described in a patient with CSD, but it has been described once before in a patient with congenital tufting enteropathy and a SPINT2 mutation. Interestingly, the patient was heterozygous for p.(Tyr163Cys), but a second SPINT2 variant was not identified [Salomon et al., 2014]. Thus the ocular manifestations of corneal erosions and coloboma seen in our patient are consistent with the eye findings previously reported in patients with SPINT2 mutations (Table I). Note that the patients represented in Table I are a compilation of patients with CSD and congenital tufting enteropathy, as both are associated with deleterious SPINT2 variants.
Table I.
Eye findings in patients with biallelic, deleterious SPINT2 sequence variants
| Nucleotide Alteration (NM_ 021102.3) | Protein alteration | Protein domain | Ocular Findings | Reference |
|---|---|---|---|---|
| c.2T>C | removal start codon | Kunitz inhibitor type I domain | Superficial punctate keratitis | Salomon et al., 2014 |
| c.172dupG | p.(Val58Glyfs*3) | Kunitz inhibitor type I domain | Superficial punctate keratitis, hypertelorism | Salomon et al., 2014 |
| c.247G>T | p.(Glu83*) | Kunitz inhibitor type I domain | Superficial punctate keratitis | Salomon et al., 2014 |
| c.337+2T>C | Corneal erosions | Heinz-Erian et al., 2009 | ||
| c.442C>T | p.(Arg148Cys) | Kunitz inhibitor type I domain | Superficial punctate keratitis | Salomon et al., 2014 |
| c.488A>G | p.(Tyr163Cys) | Corneal erosions, lacrimal duct imperforation, optic nerve coloboma | Heinz-Erian et al., 2009; Sivagnanam et al., 2010; Salomon et al., 2014 | |
| c.502G>A | p.(Gly168Ser) | Kunitz inhibitor type I domain | Superficial punctate keratitis | Salomon et al., 2014 |
| c.553+2T>A | Corneal erosions | Heinz-Erian et al., 2009 | ||
| c.593-1G>A | Corneal erosions, hypertelorism | Heinz-Erian et al., 2009 |
Spint2 is expressed in epithelial cells and is thought to regulate protease, serine 8 (PRSS8, or prostasin) that, together with matripase, is essential for terminal epidermal differentiation and tight junction formation [Szabo et al., 2016]. It is possible to speculate that aberrant prostasin function could result in severe defects of epidermal barrier function that could compromise the function of corneal epithelial cells and cause surface erosions. However, the mechanism for the colobomas is less clear. SPINT2 is expressed in the human adult brain [Kaaguchi et al., 1997], but we could not find studies that show that it is expressed in the eye. Our patient also manifested bilateral cleft lip and palate that is also rare in CSD, although cleft palate has previously been described [Muller et al., 2000]. For reference, the prevalence of cleft lip with or without cleft palate independent of any other rare disease is 9.9 per 10,000. The prevalence of isolated cleft lip is 3.28 per 10,000, and that of cleft lip and palate is 6.64 per 10,000 [IPDTOC Working Group, 2011].
One of our patient’s sequence variants, c.488A>G, predicting p.(Tyr163Cys), has been previously reported as a pathologic variant associated with the syndromic form of CSD. This missense mutation has been reported in 9 patients with CSD, including 6 homozygotes and 3 compound heterozygotes [Heinz-Erian et al., 2009]. Additionally, this variant has been reported in four other studies describing 12 patients with congenital tufting enteropathy, including 5 homozygotes, 5 compound heterozygotes, and 2 heterozygotes in whom a second variant was not identified [Sivagnanam et al., 2010; Slae et al., 2013; Salomon et al., 2014; d’Apolito et al., 2016]. The p.(Tyr163Cys) variant affects the second Kunitz domain of SPINT2.
It is still unclear how mutations in certain domains of the SPINT2 protein affect function. Qin et al. [1998] characterized each of the two Kunitz domains by introducing a point mutation into the reactive site of each domain and then assaying the mutant protein for its HGF activator inhibitory activity. Their results showed that a mutated carboxy (COOH) terminus did not affect the activity of HAI-2 (SPINT2), whereas a mutated amino (NH2) terminus markedly reduced protein activity, suggesting that the NH2 terminal Kunitz domain is largely responsible for the inhibitory activity of SPINT2 against the hepatocyte growth factor (HGF) activator [Qin et al., 1998].
In conclusion, we present a patient with syndromic CSD who had cleft lip and palate, corneal erosions, optic nerve coloboma, and intermittent exotropia. Our patient had both the recurrent SPINT2 sequence variant, c.488A>G, predicting p.(Tyr163Cys), and a novel variant, c.166_167dupTA, predicting p.(Asn57Thrfs*24). We could find only one other report of an optic nerve coloboma associated with a SPINT2 mutation, but this was in a patient with congenital tufting enteropathy. This confirms and further characterizes the association of coloboma with presumed loss of function for this gene.
Acknowledgments
We would like to thank the patient’s family for their contribution to this report. The following authors have potential conflicts of interest: Dr. Mendelsohn is on the advisory board of Clear Genetics and Dr. Taft, Dr. Chawla, and Ms. Perry are employees and shareholders of Illumina Inc. Clinical whole genome sequencing for the patient in this work was provided by the Illumina iHope program.
References
- Bird LM, Sivagnanam M, Taylor S, Newbury RO. A new syndrome of tufting enteropathy and choanal atresia, with ophthalmologic, hematologic and hair abnormalities. Clin Dysmorphol. 2007;16(4):211–221. doi: 10.1097/MCD.0b013e328274264b. [DOI] [PubMed] [Google Scholar]
- d’Apolito M, Pisanelli D, Faletra F, Giardino I, Gigante M, Pettoello-Mantovani M, Goulet O, Gasparini P, Campanozzi A. Genetic analysis of Italian patients with congenital tufting enteropathy. World journal of pediatrics : WJP. 2016;12(2):219–224. doi: 10.1007/s12519-015-0070-y. [DOI] [PubMed] [Google Scholar]
- Faller N, Gautschi I, Schild L. Functional analysis of a missense mutation in the serine protease inhibitor SPINT2 associated with congenital sodium diarrhea. PloS one. 2014;9(4):e94267. doi: 10.1371/journal.pone.0094267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz-Erian P, Muller T, Krabichler B, Schranz M, Becker C, Ruschendorf F, Nurnberg P, Rossier B, Vujic M, Booth IW, Holmberg C, Wijmenga C, Grigelioniene G, Kneepkens CMF, Rosipal S, Mistrik M, Kappler M, Michaud L, Doczy LC, Siu VM, Krantz M, Zoller H, Utermann G, Janecke AR. Mutations in SPINT2 cause a syndromic form of congenital sodium diarrhea. Am J Hum Genet. 2009;84(2):188–196. doi: 10.1016/j.ajhg.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi T, Qin L, Shimomura T, Kondo J, Matsumoto K, Denda K, Kitamura N. Purification and cloning of hepatocyte growth factor activator inhibitor type 2, a Kunitz-type serine protease inhibitor. J Biol Chem. 1997;272(44):27558–27564. doi: 10.1074/jbc.272.44.27558. 1997. [DOI] [PubMed] [Google Scholar]
- Muller T, Wijmenga C, Phillips AD, Janecke A, Houwen RH, Fischer H, Ellemunter H, Fruhwirth M, Offner F, Hofer S. Congenital sodium diarrhea is an autosomal recessive disorder of sodium/proton exchange but unrelated to known candidate genes. Gastroenterology. 2000;119:1506–1513. doi: 10.1053/gast.2000.20514. [DOI] [PubMed] [Google Scholar]
- Qin L, Denda K, Shimomura T, Kawaguchi T, Kitamura N. Functional characterization of Kunitz domains in hepatocyte growth factor activator inhibitor type 2. FEBS Lett. 1998;436(1):111–114. doi: 10.1016/s0014-5793(98)01105-3. [DOI] [PubMed] [Google Scholar]
- Roche O, Putterman M, Salomon J, Lacaille F, Brousse N, Goulet O, Dufier JL. Superficial punctate keratitis and conjunctival erosions associated with congenital tufting enteropathy. Am J Ophthalmol. 2010;150(1):116–121.e111. doi: 10.1016/j.ajo.2010.01.034. [DOI] [PubMed] [Google Scholar]
- Salomon J, Goulet O, Canioni D, Brousse N, Lemale J, Tounian P, Coulomb A, Marinier E, Hugot JP, Ruemmele F, Dufier JL, Roche O, Bodemer C, Colomb V, Talbotec C, Lacaille F, Campeotto F, Cerf-Bensussan N, Janecke A, Mueller T, Koletzko S, Bonnefont JP, Lyonnet S, Munnich A, Poirier F, Smahi A. Genetic characterization of congenital tufting enteropathy: epcam associated phenotype and involvement of SPINT2 in the syndromic form. Hum Genet. 2014;133(3):299–310. doi: 10.1007/s00439-013-1380-6. [DOI] [PubMed] [Google Scholar]
- Sivagnanam M, Janecke AR, Muller T, Heinz-Erian P, Taylor S, Bird LM. Case of syndromic tufting enteropathy harbors SPINT2 mutation seen in congenital sodium diarrhea. Clin Dysmorphol. 2010;19(1):48. doi: 10.1097/MCD.0b013e328331de38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slae MA, Saginur M, Persad R, Yap J, Lacson A, Salomon J, Canioni D, Huynh H. Syndromic congenital diarrhea because of the SPINT2 mutation showing enterocyte tufting and unique electron microscopy findings. Clin Dysmorphol. 2013;22(3):118–120. doi: 10.1097/MCD.0b013e328361d42f. [DOI] [PubMed] [Google Scholar]
- Szabo R, Hobson JP, Christoph K, Kosa P, List K, Bugge TH. Regulation of cell surface protease matriptase by HAI2 is essential for placental development, neural tube closure and embryonic survival in mice. Development. 2009;136(15):2653–2663. doi: 10.1242/dev.038430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo R, Lantsman T, Peters DE, Bugge TH. Delineation of proteolytic and non-proteolytic functions of the membrane-anchored serine protease prostasin. Development. 2016;143(15):2818–2828. doi: 10.1242/dev.137968. [DOI] [PMC free article] [PubMed] [Google Scholar]